

Abstract
Bacteria comprise the most diverse domain of life on Earth, where they occupy nearly every possible ecological niche and play key roles in biological and chemical processes. Studying the composition and ecology of bacterial ecosystems and understanding their function are of prime importance. High-throughput sequencing technologies enable nearly comprehensive descriptions of bacterial diversity through 16S ribosomal RNA gene amplicons. Analyses of these communities generally rely upon taxonomic assignments through reference data bases or clustering approaches using de facto sequence similarity thresholds to identify operational taxonomic units. However, these methods often fail to resolve ecologically meaningful differences between closely related organisms in complex microbial data sets.
In this paper, we describe oligotyping, a novel supervised computational method that allows researchers to investigate the diversity of closely related but distinct bacterial organisms in final operational taxonomic units identified in environmental data sets through 16S ribosomal RNA gene data by the canonical approaches.
Our analysis of two data sets from two different environments demonstrates the capacity of oligotyping at discriminating distinct microbial populations of ecological importance.
Oligotyping can resolve the distribution of closely related organisms across environments and unveil previously overlooked ecological patterns for microbial communities. The URL http://oligotyping.org offers an open-source software pipeline for oligotyping.
Keywords: 16S, bacterial taxonomy, microbial diversity, OTU clustering, Shannon entropy
Introduction
Bacteria represent the most diverse domain of life on Earth (Pace 1997), with members occupying nearly every natural niche (Rothschild & Mancinelli 2001). They catalyse chemical reactions within biogeochemical cycles that sustain habitability for more complex organisms (Falkowski, Fenchel & Delong 2008). With their diverse metabolic capabilities, bacteria underpin large food webs by utilizing a wide range of energy sources to accessible biomass for other organisms’ consumption (Newman & Banfield 2002). Studying the composition and ecology of microbial ecosystems is of prime importance, not only for understanding their functional roles, but also for developing predictive tools that will allow efficient resource management.
The 16S ribosomal RNA (rRNA) gene commonly serves as a molecular marker for investigating microbial community composition and structure. High-throughput sequencing of 16S rRNA gene hypervariable regions allows microbial ecologists to explore microbial community dynamics over temporal and spatial scales (Huber et al. 2007). Large 16S gene data bases and alignments provide a reference framework for mapping fragmentary sequences, each of which represents the occurrence of a microbial taxon in a sampled community. Such comprehensive studies permit the discovery of fundamental ecological patterns and link microbiomes to ecosystem functioning or to the health and disease states of hosts that harbour them.
The analysis of microbial communities via 16S rRNA gene data generally relies upon classification-based approaches that make taxonomic assignments by comparing each DNA sequence to reference data bases (Wang et al. 2007; Huse et al. 2008; Liu et al. 2008), or clustering-based methods that identify taxon-independent operational taxonomic units (OTUs) using a sequence similarity threshold (Schloss & Handelsman 2005; Schloss et al. 2009; Huse et al. 2010). Both approaches seek to partition large data sets into manageable operational units. The identities and abundances of these units are then commonly used in alpha- and beta-diversity analyses to investigate links between community structures and environmental factors.
Both taxonomic assignment and clustering approaches have critical limitations. Analyses that classify sequence reads by similarity to taxonomic data base entries may provide poorly resolved diversity descriptions, especially for samples collected from high-diversity environments. Reference classifications based on isolated micro-organisms, such as Bergey's Manual [5014 species (Garrity 2004)] or the List of Prokaryotic Names with Standing in Nomenclature [LPSN, 12 822 entries (Euzeby 1997)], represent a small fraction of the estimated microbial diversity in environmental samples (Pace 1997; Sogin et al. 2006; Huse et al. 2010). Despite ongoing efforts to annotate uncultured clades (Quast et al. 2013), large areas of the 16S reference tree offer a poor taxonomic resolution due to lack of such isolated representatives. In contrast, clustering approaches that utilize sequence similarities to define membership in a phylogenetic assemblage have dramatically expanded the number of inferred OTUs. However, researchers are forced to employ relatively low similarity thresholds (such as de facto similarity threshold of 96% or 97%) to minimize inflation of the number of OTUs because of random sequencing errors (Huse et al. 2010; Kunin et al. 2010). Such a requisite makes it impossible to identify organisms in communities that differ from each other by a very small number of nucleotides.
The 16S rRNA gene has limited specificity (e.g. two distant organisms may have identical 16S rRNA genes), yet it is very sensitive, since a single nucleotide difference at the 16S rRNA gene level can predict remarkable genomic variation (Ward et al. 1998; Thompson et al. 2005). Unravelling complex relationships between bacteria and their environments often requires information about microbial diversity at finer scales when closely related but subtly distinct gene sequences represent separate entities in a microbial community. However, comparisons to sequences in annotated data bases and clustering methods will rarely if ever fully resolve very closely related sequences into distinct taxonomic units.
Here we describe the use of oligotyping, a novel supervised computational method that can elucidate concealed diversity within the final operational units of classification or clustering approaches. Oligotyping relies on the information that stems from the entropy analysis of variable sites in sequences that initially map to the same taxon in molecular data bases or that aggregate together in cluster analyses. Unlike classification or clustering methods that compare all positions in sequence reads to assess similarity, oligotyping utilizes only the most discriminating information by focusing on the variable sites revealed by the entropy analysis to identify highly refined taxonomic units (hereafter called oligotypes). We also present a user-friendly open-source software pipeline for oligotyping, which guides the oligotyping analysis and provides output files in standard formats that can be further analysed by third-party software packages.
Through oligotyping, we previously identified meaningful subpopulations of a single species in a human microbiome data set where the variation between different members displayed as little as 0·2% variation in short hypervariable regions of 16S rRNA genes (Eren et al. 2011). In this study, we expand the scope of oligotyping and demonstrate that it can successfully resolve key microbial diversity among numerically and ecologically important microbial taxa. We validate the method by reanalysing Bacteroides diversity in a previously published Human Microbiome Project (HMP) data set and Pelagibacter diversity from an unpublished coastal marine environment data set. We also present a stepwise procedure to facilitate oligotyping analyses by microbial ecologists.
Materials and methods
Oligotyping
After identifying sequences of interest (e.g. sequences assigned to the same taxonomical group or clustered together in one OTU), and optionally performing sequence alignment, oligotyping analysis entails (1) systematically identifying nucleotide positions that represent information-rich variation among closely related sequences, and (2) generating oligotypes. Appendix S1 provides a detailed example.
Performing sequence alignment
The identification of similarities and differences between DNA sequences requires the comparison of nucleotide residues at positions that share a common evolutionary history. For oligotyping, the artificial insertion or deletion of bases (indels) in sequence reads versus naturally occurring length variation imposes different constraints on data analyses. The former requires the use of alignment tools for the insertion of gaps that will dissipate artificial length variations and align sites that share a common evolutionary history. In contrast, oligotyping of sequences that contain few artificially introduced indels only need to start at the same evolutionarily conserved position and extend for the same number of nucleotides. The frequency of indels varies widely for different sequencing platforms (Loman et al. 2012). For instance, the occurrence of homopolymeric region-associated indels, which are common in both untreated and denoised (Quince et al. 2011) sequence reads from Roche GS-FLX or Ion Torrent PGM platforms, requires a DNA sequence alignment step and procedure to trim all reads to the same length prior to oligotyping, because non-biological positional shifts in sequencing reads will hinder the identification of variable sites that can discriminate between closely related taxa and will inflate the number of identified oligotypes in later steps. Luckily, an efficient template-based aligner [such as PyNAST (Caporaso et al. 2010a)] against a curated template [such as Greengenes (McDonald et al. 2012)] enables the alignment of hundreds of thousands of reads within hours on an average laptop computer with sufficient accuracy for oligotyping. In contrast, oligotyping analysis does not require an alignment step for Illumina-generated data since the number of sequencing cycles determines read length, and indels are rare (Loman et al. 2012).
Selecting nucleotide positions that present variation
The concatenation of nucleotides from information-rich, variable positions in sequencing reads defines an oligotype. Oligotypes converge towards the minimal number of nucleotide positions that will explain the maximum amount of biological diversity. Strategies for identifying appropriate variable regions in a collection of reads range from simple measurements of sequence conservation to more sophisticated statistical techniques that employ complex models (Margulies et al. 2003; Cooper et al. 2005; Asthana et al. 2007). The oligotyping software pipeline utilizes Shannon entropy (Shannon 1948) as the default method to identify positional variation to facilitate the identification of nucleotide positions of interest. Shannon entropy lies at the core of widely used diversity indices (Jost 2006) and has a scalable capacity to detect uncertainty in a random variable that has information content. Shannon entropy quantifies the extent to which a discrete distribution (that assigns a probability to some discrete events) deviates from a distribution with a mass concentrated at one event (i.e. with only one event having probability 1, and all other events having probability 0). In particular, Shannon entropy is zero on a distribution whose mass is concentrated on one event and attains its maximum value, log(n), where n is the number of events, on the probability distribution with probability of each event equal to 1/n. Thus, while Shannon entropy of the distribution of different nucleotides in ‘AAAAAAAA’ equals to 0, it would be log(4) for ‘AACCTTGG’. Once the entropy of each column in an alignment is known, the oligotyping process can use nucleotide positions that present the highest entropy values (Fig.1 and Appendix S1). The key advantage of oligotyping is the identification and utilization of only the most discriminating information among reads, instead of depending on nucleotide conservation over their full length to estimate similarity. With this strategy, oligotyping discards redundant information that does not contribute to further identification of different groups and provides improved explanations for the inferred community structure represented by closely related but distinct groups of reads (see Appendix S2 for comparison of oligotyping and OTU clustering results of an E. coli data set with minimal parameters).
Figure 1.
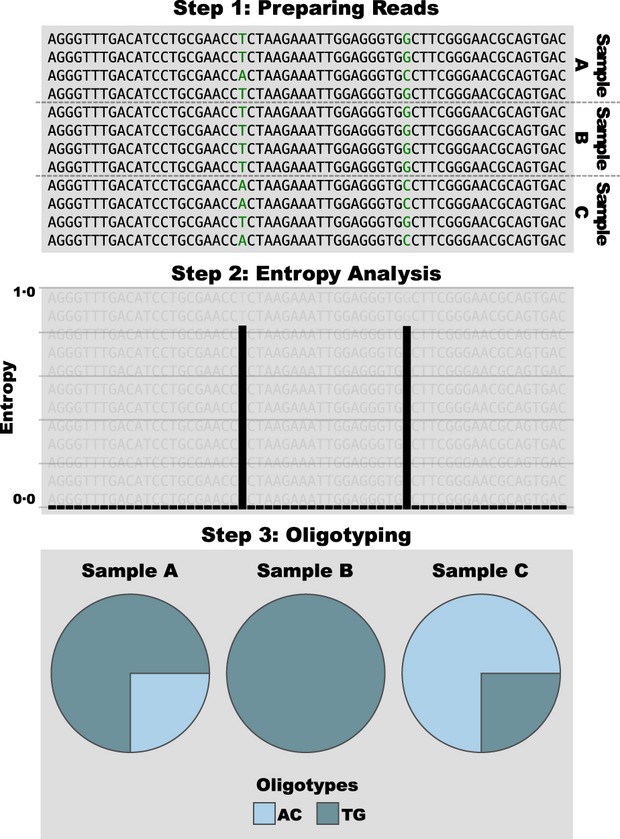
Major steps of oligotyping analysis. In step 1, reads that were identified as one taxon or a single OTU from all samples in a data set are gathered. In the hypothetical example given in the figure, reads with very subtle nucleotide variation (positions of variation are highlighted with green) are shared between three samples, A, B and C. In step 2, the collection of reads is analysed with Shannon entropy, during which the variable positions are recovered. In step 3, each read is affiliated with the base they possess at the high entropy position among the reads, and thus, oligotypes are generated (AC and TG in this mock example), and finally, oligotype profiles, depicted as pie charts, are generated to explain differences among samples.
Generating oligotypes
Entropy profiles identify information-rich nucleotide positions that the user selects and the pipeline concatenates to define oligotypes. Initial entropy analysis may not be sufficient to identify all nucleotide positions that would resolve all oligotypes. However, after the initial run, a supervised strategy can identify variable sites that will allow decomposition into additional oligotypes. Iterative analyses can further resolve diversity patterns through the inclusion of additional nucleotide positions. Upon completion, the process generates for each sample in the data set oligotype profiles and distribution patterns (AC and TG in Fig.1) for beta-diversity analyses. The oligotyping pipeline generates a comprehensive static HTML output, through which the user can evaluate oligotyping results and supervise the oligotyping process until all oligotypes have converged. An oligotype has converged if additional decomposition does not generate new oligotypes that exhibit differential abundances in different samples (or environments). See Appendix S1 for detailed workflow of oligotyping, stop criteria, best practices and example oligotyping outputs that facilitate user supervision.
Reducing the impact of errors
Oligotyping assumes that quality-filtering techniques have corrected or eliminated most reads that contain sequencing errors. However, even the most effective quality filtering (Qu, Hashimoto & Morishita 2009; Schroder et al. 2009; Bravo & Irizarry 2010; Leek et al. 2010; Meacham et al. 2011; Minoche, Dohm & Himmelbauer 2011; Quince et al. 2011; Benjamini & Speed 2012; Victoria et al. 2012) will not produce error-free data sets. Oligotyping, by using only a fraction of each read to define closely related but distinct organisms, drastically diminishes the actual number of nucleotides used for read comparison. However, during the generation of oligotypes, any sequencing error that may have occurred at one of the selected sites will indeed spawn a new oligotype. The pipeline implements various parameters that help to identify and discard such noisy oligotypes and reduce the impact of sequencing errors on results. These include (s) the minimum number of samples in which an oligotype is expected to be present, (a) the minimum per cent abundance of an oligotype in at least one sample, (A) the minimum actual abundance of an oligotype across all samples and (M) the minimum count of the most abundant unique sequence in an oligotype. The pipeline can also incorporate machine-reported quality scores to set (q) the minimum quality threshold for bases to be used for oligotyping. As with the selection of variable positions for oligotyping, the noise removal step requires user input. Default values are set at s = 1, a = 0, A = 0 and M = 4. These values perform well for data sets that contain 1000–10 000 reads and 1–10 samples. However, data set size and the number of samples should be considered when setting the value of each parameter. Our empirical tests with the oligotyping pipeline showed that the criteria s and M eliminate noise most efficiently. For instance, if there are biological or technical replicates in the experiment, setting s to match the number of replicates will eliminate oligotypes that appear in fewer than s samples. For very large data sets, setting M to equal the average number of reads per sample divided by 1000 will eliminate oligotypes with very low substantive abundance. Although they are similar, M is more efficient than A at reducing noise. Parameter A is comparable to the ‘minimum OTU size’ parameter used by OTU clustering pipelines. However, the actual number of reads that form an OTU rarely indicates the robustness of an OTU alone. For instance, two OTUs, one with 10 unique reads with the abundance of 1 and another with 1 unique read with the abundance of 10, would have the same abundance, but different authenticity. Both would have a parameter value of 10, but the first has a substantive abundance, M, of 1 and the latter a substantive abundance, M, of 10. Hence, we suggest M serve as a noise reduction step instead of the more conventional parameter A. The oligotyping pipeline tracks the read fate throughout the process to inform the user of the number of reads lost by quality-filtering criterion and sample, which makes it possible to detect potential biases in eliminated reads among samples.
The open-source software pipeline, tutorials and example analyses are available from http://oligotyping.org.
Bacteroides in human gut microbiomes
Sample collection, sequencing, quality filtering and data availability
Sample collection, sequencing and quality filtering are described in detail in a previously published study (Yatsunenko et al. 2012).
Oligotyping analysis
We used 1 093 740 274 quality-controlled single 101 nucleotide long Illumina HiSeq reads from 531 human gut microbiome samples for oligotyping (data available through the NCBI Sequence Read Archive, accession number ERX115504). We assigned taxonomy for 785 534 577 reads with minimal sequence length of 101 nucleotides with GAST (Huse et al. 2008). Of the 91 990 654 reads that were classified as Bacteroides, we randomly selected up to 100 000 reads from each sample. The total data set for oligotyping included 30 637 709 Bacteroides reads from 529 samples (two of the samples lacked Bacteroides sequences). Since homopolymer region-associated insertion/deletion errors are not common in Illumina data, we did not perform an alignment. After the initial entropy analysis, we performed oligotyping using 31 variable sites (Fig. S1). To reduce the noise in the results, we required that each oligotype must (1) appear in at least three samples, (2) occur in more than 0·5% of the reads of at least one sample and (3) represent a minimum of 500 reads in all samples combined. We arrived at these values by starting with default suggestions and then testing a range of values. After removal of oligotypes that did not meet these criteria, the analysis retained 28 966 870 reads (94·5%), an average of 54 757 reads per sample. However, samples from Malawi and the Amazon had an average of only 8445 and 18 931 Bacteroides reads, respectively, while US samples had an average of 82 891. Oligotyping analysis identified 385 oligotypes, 197 of which perfectly matched sequences in NCBI's nr data base over the entire length of their representative sequence.
Generating the cladogram
Presence or absence of oligotypes in data sets from Malawi, Amazon and the US was determined based on a minimum abundance of 0·01% in a data set, and results were superimposed on the cladogram of oligotypes generated using MrBayes (version 3.1.2, http://mrbayes.sourceforge.net/) (Ronquist & Huelsenbeck 2003) and depicted using the Interactive Tree of Life (Letunic & Bork 2007).
Pelagibacter succession patterns in Little Sippewissett Marsh
Sample collection, sequencing and quality filtering
Surface water samples were collected in sterile 1-L PET bottles during low tide at seven stations (Fig. S2) in Little Sippewissett Marsh (Massachusetts, USA). The samples were collected weekly from 31 May to 4 September 2007 and then monthly until September 2008. Water samples were kept on ice and brought back to the laboratory for filtration through polyethersulphone membrane capsule filters (0·22 µm pore size Sterivex, Millipore, Billerica, MA) followed by DNA extraction and purification using a modified salt precipitation method (PUREGENE, Gentra Systems, Minneapolis, MN, USA) as described in (Sinigalliano et al. 2007). Bacterial 16S rRNA amplicons spanning the V4 through V6 regions were amplified using fusion primers, sequenced from the V6 end on a Roche GS-FLX 454 instrument using Titanium protocols, and quality-filtered and trimmed as described in (Marteinsson et al. 2013).
Oligotyping analysis and noise reduction
For oligotyping analysis, we used 239 887 quality-controlled Pelagibacter V6-V4 reads from 189 samples classified by GAST (Huse et al. 2008). The PyNAST algorithm (Caporaso et al. 2010a) aligned the 454 reads against the Greengenes (McDonald et al. 2012) multiple sequence alignment template (97% OTUs, 6 October 2010 release). We identified 11 high entropy locations for oligotyping. Due to read length (>450 nt), error towards the end of reads was extremely high. To reduce the noise in the results, we required that each oligotype must (1) appear in at least three samples and (2) have a minimum of 50 copies of the most abundant unique sequence. After the removal of oligotypes that did not meet these criteria, the analysis retained 223 631 reads (93·22%), an average of 1895 reads per sample. This analysis identified 22 oligotypes, 16 of which had at least one perfect match for their representative sequences in rRNA entries in NCBI's non-redundant (nr) data base.
Clustering analyses and biomarker discovery
Clustering of Bacteroides and Pelagibacter data sets was carried out using a 97% similarity threshold for OTU formation. Clustering was done with QIIME (v1.5) (Caporaso et al. 2010b) using the default UCLUST method (Edgar 2010). We used LEfSe to identify biomarkers in both clustering and oligotyping results (Segata et al. 2011).
Results
We used oligotyping to explain bacterial diversity in two genera (Bacteroides and Pelagibacter) in data sets for two distinct environments (human gut and saltmarsh) using different sequencing technologies (Illumina and Roche/454). The previously published human gut data set (Yatsunenko et al. 2012) represented cross-sectional sampling of human populations across three continents. In contrast, the previously unpublished saltmarsh data set included temporally distributed samples. We also benchmarked oligotyping with a data set that contained reads from one E. coli strain (Appendix S2).
Bacteroides in human gut microbiomes
Oligotyping analysis of reads classified as Bacteroides in human gut microbiomes using published V4 region 16S rRNA sequences (Yatsunenko et al. 2012) from 531 individuals from three different continents revealed 385 different oligotypes. Despite Bacteroides being strongly overrepresented in individuals from the United States compared to the individuals from Malawi or Venezuela, some Bacteroides oligotypes were only present in the Malawi and Venezuela samples, revealing fine-scale biogeographical patterns between closely related Bacteroides (Fig.2). We explored whether oligotyping could enhance the structural description of the data set with respect to Bacteroides reads. To investigate the recovery of region-specific OTUs and oligotypes, we focused on 316 samples collected from individuals in the US who represent one of five different demographics (Boulder residents, Missouri-born but now living elsewhere in the United States, Philadelphia residents, St. Louis residents and residents of the greater St. Louis area). We also clustered reads that mapped to Bacteroides using the same quality control filtering parameters as employed for oligotyping and a 97% cut-off value to identify 246 OTUs. We then used LEfSe (Segata et al. 2011), a biomarker discovery package, to investigate the presence and effect size of region-specific OTUs and oligotypes. LEfSe identifies an OTU or an oligotype as a biomarker only if they are consistently abundant in a group of samples collected from a specific region, and it estimates the effect size of such biomarkers. Effect size is the quantification of the magnitude of a biomarker with respect to its differential mean abundance between groups of samples (Segata et al. 2011). Briefly, identification of a biomarker depends on its statistically significant presence in one group, and the high effect size would indicate the larger difference between the mean abundance of the biomarker in distinct groups. We used the default values suggested for both statistical significance and minimum effect size threshold for biomarker identification. When applied to Bacteroides data, LEfSe detected higher number of oligotype biomarkers for each region in the United States compared to the OTU biomarkers (Fig. S3). The only category in which the mean effect size of OTU biomarkers identified by LEfSe was larger than that of oligotypes was Boulder, with no statistical significance (Wilcoxon rank-sum test, P = 0·75). In the remaining four categories, the mean effect size of discriminant oligotypes was larger than the mean effect size of OTUs and significantly larger in three of them (Missouri-born, Philadelphia and St. Louis with P < 0·05, metropolitan area of St. Louis with P = 0·69). This result suggests that the oligotypes identified in Bacteroides reads offer a comparable or higher level of resolution than OTUs at 97% and oligotypes have increased power for recovering information about distribution patterns.
Figure 2.

Bacteroides oligotype distribution inferred from the study published by Yatsunenko et al. (2012). Bars indicate the presence of an oligotype in a given community; a full-length bar represents oligotypes that occur in 100% of the analysed samples. The lower panel magnifies numbered regions in the cladogram. Numbers 1, 2 and 3 are Bacteroides oligotypes that are more than 97% similar in full length, yet exhibit noteworthy differences in their geographical distribution. Light yellow background colour on the cladogram marks the oligotypes with perfect matches in NCBI's non-redundant nucleotide sequence data base. Number 4 demonstrates several oligotypes that consistently occur in samples from the Malawian and Amerindian communities but not in samples from the United States. None of the oligotypes in Number 4 have perfect matches in NCBI's nr data base. Number 5, on the other hand, shows several oligotypes with similar occurrence patterns in Malawian and Amerindian communities with the ones shown in Number 4, but with a remarkably larger presence in the samples collected from the United States. In contrast to Number 4, 3 out of 4 oligotypes listed in Number 5 have perfect matches in NCBI's nr data base.
Pelagibacter succession patterns in Little Sippewissett Marsh
Oligotyping analysis of Pelagibacter (a genus in the SAR11 clade) in Little Sippewissett Marsh (LSM) revealed 22 oligotypes and displayed remarkable seasonal variation (Fig.3). The two most abundant Pelagibacter oligotypes (Fig.3a) differed from each other by only two nucleotides, which is equivalent to 99·57% sequence identity across the entire amplicon read (459 nt). BLAST searches for representative sequences of two oligotypes revealed that they are identical to the 16S rRNA gene of two genome-sequenced Pelagibacter strains at the V6-V4 region: Candidatus Pelagibacter ubique HTCC1062 and Pelagibacter strain HTCC7211. These strains are members of the SAR11 clade subgroup S1a (Morris et al. 2002), and they are further grouped into internal transcribed spacer (ITS)-based phylotype P1a.1 and P1a.3, respectively (Stingl, Tripp & Giovannoni 2007). Recent studies showed that phylotype P1a.1 predominates in polar regions, while the phylotype P1a.3 represents the dominant Pelagibacter in tropical regions (Brown et al. 2012). In the LSM data set, we observed the dominance of the oligotype that matched the polar phylotype P1a.1 from December to June, while the dominant oligotype from July to November matched the tropical phylotype P1a.3. The emergence of the dominant tropical-like oligotype lags the increased temperature shift (Fig.3a) similar to that reported for shifts in archaeal and protistan networks in other marine environments (Steele et al. 2011; Gilbert et al. 2012).
Figure 3.

Pelagibacter oligotype and OTU distribution in samples from Little Sippewissett Marsh. In panel (a), seasonal variation of two Pelagibacter oligotypes is shown based on their relative abundance. The representative sequence of Oligotype 1 is identical to HTCC1062 (predominant in polar regions) through the V4-V6 region, and the representative sequence of Oligotype 2 is identical to HTCC7211 (more abundant in tropical regions) at the V4-V6 region. These oligotypes are 99·57% identical to each other over their 459 nt amplicon lengths. The water temperature observed during the sampling is superimposed on the figure. In panel (b), the distribution of Pelagibacter oligotypes and 3% OTUs across all sampling stations is compared side by side. Data from each station consisted of temporal samples spanning a 17-month time period between May 2007 and September 2008. Each colour represents a different oligotype and OTU. Colour range order is defined by the relative abundance; therefore, identical colours do not suggest any correlation across panels.
Discussion
We analysed two separate data sets to demonstrate the capacity of oligotyping to discriminate distinct microbial populations of ecological importance. Oligotyping analysis of Illumina and 454 amplicon sequences for Bacteroides from the human gastrointestinal tract, and Pelagibacter from Little Sippewissett Marsh, respectively, facilitated the recovery of ecological information that taxonomical classification and OTU clustering at 97% identity level cannot detect.
Bacteroides account for a major fraction of the human microbiome (Ley, Peterson & Gordon 2006) and represent one of the most diverse genera in the gastrointestinal tract (Arumugam et al. 2011). Our oligotyping analysis revealed that the relative abundance or simple presence–absence patterns of amplicon sequences that differ from each other by only two nucleotides in the V4 region show remarkable geographical specificity (Fig.2). Why an organism that is present in the vast majority of samples from rural communities is virtually absent from the US population poses an important question that cannot be answered through the analysis of reads from the 16S rRNA gene alone. However, such questions may emerge from observations of microbial diversity at very fine scales and could easily be overlooked using standard binning methods. We also used LEfSe (Segata et al. 2011) to investigate whether oligotyping recovered information that separates different environments more efficiently compared to OTU clustering. Among the OTUs and oligotypes identified in Bacteroides reads, LEfSe detected more biomarkers for region-specific oligotypes than OTUs for the samples collected from the United States. More discriminants may be due to the fact that the analysis of Bacteroides reads resulted in more oligotypes (385) than OTUs (246). However, the mean effect size of oligotypes was larger in four out of five categories as well [significantly higher in three out of five categories (P < 0·05)] (Fig. S3), suggesting that oligotyping results were comparable or better than clustering analysis at explaining the structure of the Bacteroides data set.
SAR11 dominates aerobic bacterial phylotypes in the oceans (Morris et al. 2002). This group includes Pelagibacter ubique, which through its abundance and photoheterotrophic metabolism plays a critical role in the carbon cycle. Only few isolates are available due to the challenging cultivation procedures (Connon & Giovannoni 2002; Rappe et al. 2002; Stingl, Tripp & Giovannoni 2007; Carini et al. 2013); hence, the depth of taxonomic classification for proper identification of different SAR11 organisms in environmental samples is limited. Oligotyping of the large number of Pelagibacter reads from samples collected over an eighteen-month time frame from LSM demonstrated remarkable seasonal variation in the abundance of closely related Pelagibacter organisms (Fig.3). The most abundant two oligotypes that together comprised more than half of the Pelagibacter population in the samples analysed were more than 99·5% similar to each other over the sequenced region, yet their relative abundance exhibits statistically significant negative association throughout the changing seasons (Kendall's rank correlation tau: −1; P < 0·001) (Fig.3a). Since the levels of sequence similarity between most Pelagibacter organisms were beyond the de facto 97% threshold, we did not detect the seasonal phenomenon analysing the same data set with OTU clustering (Fig.3b). Oligotyping of high-throughput sequencing data identified very closely related organisms occupying ecological niches separated by season and warranting further study.
The final operational units of taxonomic classification or clustering methods provide the initial input for oligotyping analyses. Therefore, this technique works with existing common methodologies and offers an analytical technology that allows researchers to investigate diversity within the specific taxa or OTUs rather than a method to be applied to the entire data set. As was the case with Pelagibacter in LSM, the steady presence of an operational unit may simply reflect unexplained diversity concealed in an OTU that can only be further explored through with oligotyping. This makes oligotyping most rewarding when it is applied to reads with the same taxonomic assignments or OTUs that occur in all samples in a data set despite the changing environmental parameters.
The user guidance that oligotyping requires does not end with selecting the operational unit upon which to focus. The user must also consider which nucleotide positions will explain the diversity most effectively in any group of reads. This step starts with identifying variable positions following the entropy analysis and usually requires oligotyping to be repeated with an increasing number of nucleotide positions until each oligotype converges (with little or no entropy left in the group), or until the user accepts the level of resolution. Having no fixed similarity threshold in any step of the analysis has the advantage of making oligotyping more suitable for explaining varying degrees of diversity. However, it has the disadvantage of requiring the investigator to supervise the optimal solution for a given group of reads. Therefore, our oligotyping pipeline offers a user-friendly interface to facilitate the necessary steps of supervision (see Materials and methods).
In summary, oligotyping is a supervised computational method to investigate and reveal microbial diversity concealed within final operational units of canonical approaches. It relies on the position-specific information in high-throughput reads obtained from 16S rRNA gene amplicons to exploit subtle nucleotide variations for identification of closely related but distinct taxa. By focusing only on the variable sites among reads that contain the most discriminating information, oligotyping can reveal previously unobserved ecological patterns in a data set by identifying highly refined operational units to elaborate differences among high-throughput sequencing reads. The open-source software pipeline for oligotyping, user tutorials and example analyses are available from http://oligotyping.org.
Acknowledgments
This work was supported by the National Institutes of Health [1UH2DK083993 to M.L.S.] and the Alfred P. Sloan Foundation.
Data Accessibility
Sequences for The Little Sippewissett Marsh have been deposited in the NCBI Sequence Read Archive as ‘SRA062454’. Sequences from (Yatsunenko et al. 2012) for human gut have been deposited in MG-RAST (http://metagenomics.anl.gov/) as ‘qiime:850’.
Author contributions
A.M.E. designed and implemented the oligotyping method; A.M.E., L.M., W.J.S and S.L.G. analysed the data; A.M.E., L.M, S.L.G. and W.J.S. wrote the manuscript; L.G.M. collected Little Sippewissett Marsh samples; S.L.G, H.G.M and L.G.M. performed the sequencing and gave technical support. M.L.S. and H.G.M. supervised all analyses and edited the manuscript.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Entropy analysis results on ∽30 million 101 nucleotide long Bacteroides reads. Bars show the Shannon entropy value for each position.
Aerial map of seven sampling stations at Little Sippewissett Marsh, Massachusetts, USA.
LEfSe analysis results for five categories used to define the origin of samples collected from individuals live in the United States.
Oligotyping flowchart and an example analysis to highlight best practices.
Benchmarking oligotyping with a data set that contained reads from one E. coli strain.
References
- Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asthana S, Roytberg M, Stamatoyannopoulos J. Sunyaev S. Analysis of sequence conservation at nucleotide resolution. PLoS Computational Biology. 2007;3:e254. doi: 10.1371/journal.pcbi.0030254. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y. Speed TP. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Research. 2012;40:e72. doi: 10.1093/nar/gks001. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo HC. Irizarry RA. Model-based quality assessment and base-calling for second-generation sequencing data. Biometrics. 2010;66:665–674. doi: 10.1111/j.1541-0420.2009.01353.x. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MV, Lauro FM, DeMaere MZ, Muir L, Wilkins D, Thomas T, et al. Global biogeography of SAR11 marine bacteria. Molecular Systems Biology. 2012;8:595. doi: 10.1038/msb.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL. Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010a;26:266–267. doi: 10.1093/bioinformatics/btp636. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods. 2010b;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carini P, Steindler L, Beszteri S. Giovannoni SJ. Nutrient requirements for growth of the extreme oligotroph ‘Candidatus Pelagibacter Ubique’ HTCC1062 on a defined medium. The ISME Journal. 2013;7:592–602. doi: 10.1038/ismej.2012.122. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connon SA. Giovannoni SJ. High-throughput methods for culturing microorganisms in very-low-nutrient media yield diverse new marine isolates. Applied and Environment Microbiology. 2002;68:3878–3885. doi: 10.1128/AEM.68.8.3878-3885.2002. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S. Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Research. 2005;15:901–913. doi: 10.1101/gr.3577405. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Eren AM, Zozaya M, Taylor CM, Dowd SE, Martin DH. Ferris MJ. Exploring the diversity of Gardnerella vaginalis in the genitourinary tract microbiota of monogamous couples through subtle nucleotide variation. PLoS ONE. 2011;6:e26732. doi: 10.1371/journal.pone.0026732. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euzeby JP. List of Bacterial Names with Standing in Nomenclature: a folder available on the Internet. International Journal of Systematic Bacteriology. 1997;47:590–592. doi: 10.1099/00207713-47-2-590. [DOI] [PubMed] [Google Scholar]
- Falkowski PG, Fenchel T. Delong EF. The microbial engines that drive Earth's biogeochemical cycles. Science. 2008;320:1034–1039. doi: 10.1126/science.1153213. &. [DOI] [PubMed] [Google Scholar]
- Garrity GM. Taxonomic Outline of the Prokaryotes. In: Lilburn TG, editor; Garrity GM, Bell JA, editors. Bergey's Manual of Systematic Bacteriology. New York: Springer; 2004. p. 399. & ), 2nd Edn ( [Google Scholar]
- Gilbert JA, Steele JA, Caporaso JG, Steinbruck L, Reeder J, Temperton B, et al. Defining seasonal marine microbial community dynamics. ISME Journal. 2012;6:298–308. doi: 10.1038/ismej.2011.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber JA, Mark Welch DB, Morrison HG, Huse SM, Neal PR, Butterfield DA. Sogin ML. Microbial population structures in the deep marine biosphere. Science. 2007;318:97–100. doi: 10.1126/science.1146689. &. [DOI] [PubMed] [Google Scholar]
- Huse SM, Dethlefsen L, Huber JA, Mark Welch D, Relman DA. Sogin ML. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genetics. 2008;4:e1000255. doi: 10.1371/journal.pgen.1000255. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse SM, Welch DM, Morrison HG. Sogin ML. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environmental Microbiology. 2010;12:1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost L. Entropy and diversity. Oikos. 2006;113:363–375. [Google Scholar]
- Kunin V, Engelbrektson A, Ochman H. Hugenholtz P. Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environmental Microbiology. 2010;12:118–123. doi: 10.1111/j.1462-2920.2009.02051.x. &. [DOI] [PubMed] [Google Scholar]
- Leek JT, Scharpf RB, Bravo HC, Simcha D, Langmead B, Johnson WE, Geman D, Baggerly K. Irizarry RA. Tackling the widespread and critical impact of batch effects in high-throughput data. Nature Reviews Genetics. 2010;11:733–739. doi: 10.1038/nrg2825. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I. Bork P. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 2007;23:127–128. doi: 10.1093/bioinformatics/btl529. &. [DOI] [PubMed] [Google Scholar]
- Ley RE, Peterson DA. Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124:837–848. doi: 10.1016/j.cell.2006.02.017. &. [DOI] [PubMed] [Google Scholar]
- Liu Z, DeSantis TZ, Andersen GL. Knight R. Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers. Nucleic Acids Research. 2008;36:e120. doi: 10.1093/nar/gkn491. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, Wain J. Pallen MJ. Performance comparison of benchtop high-throughput sequencing platforms. Nature Biotechnology. 2012;30:434–439. doi: 10.1038/nbt.2198. &. [DOI] [PubMed] [Google Scholar]
- Margulies EH, Blanchette M, Haussler D. Green ED. Identification and characterization of multi-species conserved sequences. Genome Research. 2003;13:2507–2518. doi: 10.1101/gr.1602203. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marteinsson VT, Runarsson A, Stefansson A, Thorsteinsson T, Johannesson T, Magnusson SH, et al. Microbial communities in the subglacial waters of the Vatnajokull ice cap, Iceland. ISME Journal. 2013;7:427–437. doi: 10.1038/ismej.2012.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R. Hugenholtz P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME Journal. 2012;6:610–618. doi: 10.1038/ismej.2011.139. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham F, Boffelli D, Dhahbi J, Martin DI, Singer M. Pachter L. Identification and correction of systematic error in high-throughput sequence data. BMC Bioinformatics. 2011;12:451. doi: 10.1186/1471-2105-12-451. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minoche AE, Dohm JC. Himmelbauer H. Evaluation of genomic high-throughput sequencing data generated on Illumina HiSeq and genome analyzer systems. Genome Biology. 2011;12:R112. doi: 10.1186/gb-2011-12-11-r112. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RM, Rappe MS, Connon SA, Vergin KL, Siebold WA, Carlson CA. Giovannoni SJ. SAR11 clade dominates ocean surface bacterioplankton communities. Nature. 2002;420:806–810. doi: 10.1038/nature01240. &. [DOI] [PubMed] [Google Scholar]
- Newman DK. Banfield JF. Geomicrobiology: how molecular-scale interactions underpin biogeochemical systems. Science. 2002;296:1071–1077. doi: 10.1126/science.1010716. &. [DOI] [PubMed] [Google Scholar]
- Pace NR. A molecular view of microbial diversity and the biosphere. Science. 1997;276:734–740. doi: 10.1126/science.276.5313.734. [DOI] [PubMed] [Google Scholar]
- Qu W, Hashimoto S. Morishita S. Efficient frequency-based de novo short-read clustering for error trimming in next-generation sequencing. Genome Research. 2009;19:1309–1315. doi: 10.1101/gr.089151.108. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J. Glockner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Research. 2013;41:D590–D596. doi: 10.1093/nar/gks1219. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quince C, Lanzen A, Davenport RJ. Turnbaugh PJ. Removing noise from pyrosequenced amplicons. BMC Bioinformatics. 2011;12:38. doi: 10.1186/1471-2105-12-38. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappe MS, Connon SA, Vergin KL. Giovannoni SJ. Cultivation of the ubiquitous SAR11 marine bacterioplankton clade. Nature. 2002;418:630–633. doi: 10.1038/nature00917. &. [DOI] [PubMed] [Google Scholar]
- Ronquist F. Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. &. [DOI] [PubMed] [Google Scholar]
- Rothschild LJ. Mancinelli RL. Life in extreme environments. Nature. 2001;409:1092–1101. doi: 10.1038/35059215. &. [DOI] [PubMed] [Google Scholar]
- Schloss PD. Handelsman J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Applied and Environment Microbiology. 2005;71:1501–1506. doi: 10.1128/AEM.71.3.1501-1506.2005. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environment Microbiology. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder J, Schroder H, Puglisi SJ, Sinha R. Schmidt B. SHREC: a short-read error correction method. Bioinformatics. 2009;25:2157–2163. doi: 10.1093/bioinformatics/btp379. &. [DOI] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS. Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biology. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon CE. A mathematical theory of communication. The Bell System Technical Journal. 1948;27:379–423. [Google Scholar]
- Sinigalliano CD, Gidley ML, Shibata T, Whitman D, Dixon TH, Laws E, et al. Impacts of Hurricanes Katrina and Rita on the microbial landscape of the New Orleans area. Proc Natl Acad Sci U S A. 2007;104:9029–9034. doi: 10.1073/pnas.0610552104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, Arrieta JM. Herndl GJ. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc Natl Acad Sci U S A. 2006;103:12115–12120. doi: 10.1073/pnas.0605127103. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele JA, Countway PD, Xia L, Vigil PD, Beman JM, Kim DY, et al. Marine bacterial, archaeal and protistan association networks reveal ecological linkages. ISME Journal. 2011;5:1414–1425. doi: 10.1038/ismej.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingl U, Tripp HJ. Giovannoni SJ. Improvements of high-throughput culturing yielded novel SAR11 strains and other abundant marine bacteria from the Oregon coast and the Bermuda Atlantic Time Series study site. ISME Journal. 2007;1:361–371. doi: 10.1038/ismej.2007.49. &. [DOI] [PubMed] [Google Scholar]
- Thompson JR, Pacocha S, Pharino C, Klepac-Ceraj V, Hunt DE, Benoit J, Sarma-Rupavtarm R, Distel DL. Polz MF. Genotypic diversity within a natural coastal bacterioplankton population. Science. 2005;307:1311–1313. doi: 10.1126/science.1106028. &. [DOI] [PubMed] [Google Scholar]
- Victoria XW, Blades N, Ding J, Sultana R. Parmigiani G. Estimation of sequencing error rates in short reads. BMC Bioinformatics. 2012;13:185. doi: 10.1186/1471-2105-13-185. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM. Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environment Microbiology. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward DM, Ferris MJ, Nold SC. Bateson MM. A natural view of microbial biodiversity within hot spring cyanobacterial mat communities. Microbiology and Molecular Biology Reviews. 1998;62:1353–1370. doi: 10.1128/mmbr.62.4.1353-1370.1998. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Entropy analysis results on ∽30 million 101 nucleotide long Bacteroides reads. Bars show the Shannon entropy value for each position.
Aerial map of seven sampling stations at Little Sippewissett Marsh, Massachusetts, USA.
LEfSe analysis results for five categories used to define the origin of samples collected from individuals live in the United States.
Oligotyping flowchart and an example analysis to highlight best practices.
Benchmarking oligotyping with a data set that contained reads from one E. coli strain.
Data Availability Statement
Sequences for The Little Sippewissett Marsh have been deposited in the NCBI Sequence Read Archive as ‘SRA062454’. Sequences from (Yatsunenko et al. 2012) for human gut have been deposited in MG-RAST (http://metagenomics.anl.gov/) as ‘qiime:850’.