

Abstract
The highly evolutionarily conserved 70 kDa heat shock protein (HSP70) family was first understood for its role in protein folding and response to stress. Subsequently additional functions have been identified for it in regulation of organelle interaction, of the inflammatory response, and of cell death and survival. Overexpression of HSP70 family members is associated with increased resistance to and improved recovery from cerebral ischemia. MicroRNAs (miRNAs) are important post-transcriptional regulators that interact with multiple target messenger RNAs (mRNA) coordinately regulating target genes, including chaperones. The members of the HSP70 family are now appreciated to work together as networks to facilitate organelle communication and regulate inflammatory signaling and cell survival after cerebral ischemia. This review will focus on the new concept of the role of the chaperone network in the organelle network, and its novel regulation by miRNA.
Keywords: chaperone, mitochondria, endoplasmic reticulum, microRNA, stroke
Introduction
Although many clinical trials have been completed in stroke, none have demonstrated clinical protective efficacy. Suggested reasons for the many failures include the complex interplay among signaling pathways and the potentially short therapeutic window for acute neuroprotection. Increasing evidence supports the involvement of microRNAs (miRNA) in the response to cerebral ischemia, as we have reviewed recently [1]. The faster post-transcriptional effect of miRNAs, and their ability to simultaneously regulate many target genes, suggests that miRNAs may have greater therapeutic potential as candidates for the treatment of stroke than therapies targeting a single gene by direct transcriptional control [1]. Further increasing their potential for translation is the fact that miRNAs are already in clinical trials, suggesting that formulation and administration will be straightforward in a new disease setting or for a new miRNA target.
In animal models, focal ischemia/stroke and global ischemia have many similar underlying injury mechanisms, including excitotoxicity, mitochondrial dysfunction, calcium dysregulation, oxidative stress [2, 3] and inflammation, though with regard to inflammation fewer studies have been performed in the setting of global ischemia [4, 5]; see more detailed discussion in section 3 below. Molecular chaperones or stress proteins can protect mitochondrial function, reduce oxidative stress, regulate inflammation, and protect from cerebral ischemia [3, 6, 7]. The importance of the chaperone network in the context of endoplasmic reticulum (ER)-mitochondrial crosstalk during cerebral ischemia was reviewed recently [8]. This review summarizes current knowledge about the roles of chaperones as regulators of the organelle network and of the inflammatory network, focusing on the regulation of the chaperone network by miRNAs.
1. The HSP70 family in cerebral ischemia- a functional network
Molecular chaperones are a functionally related group of proteins that assist protein folding in bacterial, plant, and animal cells. These highly evolutionarily conserved proteins are also called heat shock proteins (HSPs) due to the original identification of several family members as induced by heat stress, and are classified by size. The 70 kDa family is the most extensively studied ATP-dependent chaperone family, and includes a cytosolic form HSP73 (also Hsc70), an inducible cytosolic form HSP72, a mitochondrial form HSP75/mortalin, and an endoplasmic reticulum (ER) form, HSP78/BIP. These proteins facilitate correct folding of nascent and unfolded proteins and, importantly, regulate the assembly of protein complexes involved in specific organelle functions such as protein import and folding in the ER and mitochondria. Work from our lab and several others has demonstrated neuroprotection from ischemic brain injury with overexpression of chaperones and co-chaperones, both in animal stroke models and in in vitro cultures as described in the next sections.
A. HSP72
The Hspa1a and Hspa1b genes encode stress-inducible 72-kDa heat shock proteins (HSP72), which are highly conserved from bacteria to mammals: the amino acid sequences of the E. coli and human homologs are −50% identical [9]. HSP72 is a largely cytosol localized member of the HSP70 family but can also be released and function as an extracellular signal (for a review, see Giffard, Han [6]). HSP72 consists of a 44-kDa ATPase domain at the amino terminal end, an 18-kDa peptide or substrate binding domain toward the carboxy terminal end, and a 10-kDa domain terminating in the highly conserved EEVD sequence at the carboxy terminus [10].
Overexpression of HSP72 has been shown to protect both in animal and cell models of cerebral ischemia [6, 11–18]. Interestingly, studies have demonstrated that the carboxyl-terminal half of HSP72 is sufficient for protection from cerebral ischemia in vivo and in vitro [19, 20]. Our recent findings indicate that astrocyte targeted overexpression of HSP72 reduces neuronal vulnerability to forebrain ischemia [3], improves long term recovery after focal cerebral ischemia [21], and has an effect on the evolution of astrocyte activation following stroke [22].
As described previously, multiple mechanisms are involved in the protective role of HSP72 in cerebral ischemia [6, 23, 24]. As a chaperone, HSP72 can bind and sequester nascent peptide sequences or partially denatured proteins to prevent harmful aggregation and can facilitate refolding of denatured proteins to active conformations. HSP72 is known to protect from both necrotic and apoptotic cell death, and affects several different steps in the apoptosis cascade including reducing mitochondria-dependent apoptotic signaling (see Fig. 1 in Giffard, Han [6]). Viral vector-mediated HSP72 overexpression was associated with increased levels of BCL2 protein, a key anti-apoptotic protein, and protection from global ischemia [25]. HSP72 also plays an important role in modulating inflammation caused by cerebral ischemia [6, 18, 26]. Inflammatory responses include the activation of resident microglia and astrocytes, as well as recruitment of peripheral inflammatory cells. HSP70 family members play a crucial role in modulating these responses; one key aspect of HSP72’s modulatory effects on inflammation is its regulation of the NF-kB pathway (Fig. 2 in Giffard, Han [6]; Sheppard, Sun [27]). We will discuss this further in the section about chaperone control of inflammatory signaling.
B. GRP75/mortalin
GRP75 (also HSP75/mtHSP70/mortalin) is a constitutively expressed glucose regulated protein largely localized to mitochondria. GRP75 is also called heat shock 70kDa protein 9 and is encoded by the Hspa9 gene. GRP75 is an essential protein as knockout is embryonic lethal. It is a mitochondrial chaperone, and a vital component of the mitochondrial protein import machinery [28]. It is increased in response to some stresses, including ischemia and glucose deprivation. GRP75 mRNA increases in the ischemic region within 24 h of transient (30 min) focal brain ischemia [29]. Several studies have shown that overexpression of GRP75 reduces damage in both in vitro and in vivo models of ischemic stroke [16, 30]. The mechanisms of GRP75 protection against ischemia include: attenuated oxidative stress, preservation of mitochondrial function, inhibition of apoptosis, and enhanced neurogenesis. For the protective effect of GRP75 on ischemic brain injury and the mechanisms involved, the reader is referred to a recent review [31].
C. GRP78/BIP
GRP78 (also HSP78/BIP) is a constitutively expressed glucose regulated protein and also referred to as immunoglobulin heavy chain binding protein (BIP). GRP78 is largely localized to the endoplasmic reticulum (ER), is strongly induced by ER stress and is a master regulator of the unfolded protein response. GRP78 is encoded by the Hspa5 gene. Due to recent findings that significant amounts of GRP78 are present on the surface of cancer cells, it has emerged as an important regulator of tumor cell viability signaling, and cell surface GRP78 is now being used for therapeutic targeting [32]. GRP78 plays a critical role in physiologic and pathologic stress [33], including developmental and neurological disorders [34]. As a multifunctional receptor on the cell surface [35], GRP78 may be associated with the AKT and ERK signaling pathways [32]. Several studies suggest that GRP78 plays a role in the regulation of cell death, including both apoptotic Purkinje cell death in the cerebellum [36] and autophagy [37], both relevant for brain cell loss following ischemia. Two reports show that prior induction of increased levels of GRP78 with a pharmacological inducer reduces neuronal loss in both forebrain [38] and focal cerebral ischemia [39]. We recently showed that GRP78 overexpression protects primary cultured astrocytes against ischemic injury in vitro [7]. We further found that increasing GRP78 protein by downregulating miR-181 protects cerebral ischemia in vitro and in vivo [40].
D. HSP70 family functions in a chaperone network
Recently a more complex, integrating role of these heat shock proteins has been recognized, that of stabilizing intracellular morphological and functional networks through protein-protein interactions with numerous client proteins [41–43]. This chaperoning network concept is increasingly accepted as a basic regulatory mechanism involved in diverse cellular functions [43, 44]. These networks allow the cell to change phenotype by releasing client proteins from chaperones allowing them to be activated, or in some cases released and degraded. These functional adjustments are rapid, do not require protein synthesis, and facilitate calibrated and integrated adaptation to changing conditions. Changes in binding partners leading to changes in outcome are well captured in computational modeling. Examples of these networks include the organelle network and inflammation network as detailed below.
2. Chaperones participate in the organelle network
Just as our view of the interactions of proteins within the cell is evolving, our appreciation of the interdependence of organelle function within the cell is changing. Mounting evidence has identified communication between organelles which allows them to work effectively together. This produces a coordinated response to changing environmental and intracellular conditions. Chaperones facilitate organelle interactions; their induction with stress increases their capacity. By translocating between organelles, chaperones either couple the stress response with increased communication, or if the chaperones are titrated away by unfolded proteins, this may reduce organelle communication. A major example of organelle coordination of particular relevance to ischemia is the cooperation between the ER and mitochondria in the regulation of intracellular calcium (Fig. 1).
Fig. 1.
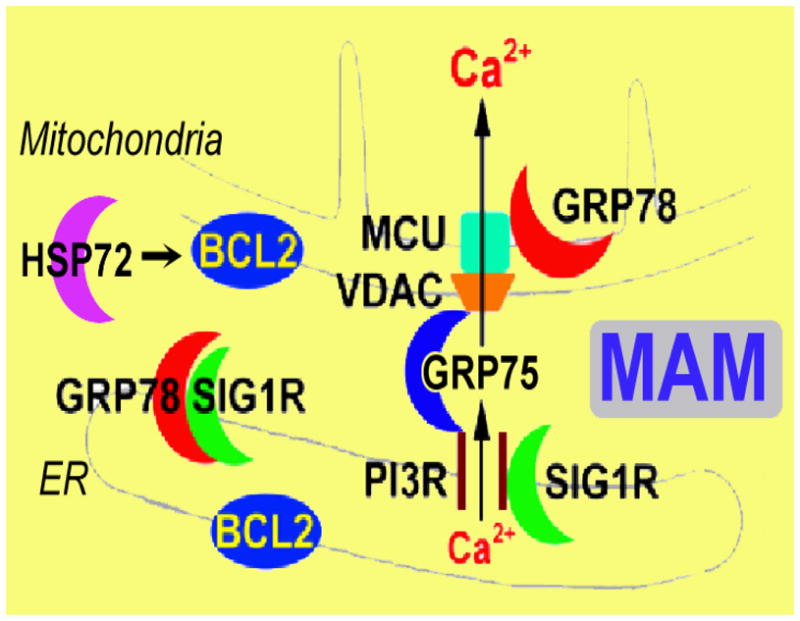
Chaperone machinery regulates ER-mitochondria Ca2+ crosstalk at the MAM. Both GRP75 and GRP78 are directly associated with MAM in the control of Ca2+ signaling between ER and mitochondria and HSP72 might regulate MAM indirectly through BCL2 which is directly associated with MAM. Cyt c: cytochrome c; IP3R: inositol trisphosphate receptor; MCU: mitochondrial Ca2+ uniporter; SIG1R: sigma-1 receptor; VDAC: voltage-dependent anion channel.
The ER is a multifunctional organelle central to Ca2+ homeostasis, protein synthesis, protein trafficking and secretion, and the regulation of apoptosis [45, 46]. Mitochondria are the site of oxidative phosphorylation dependent ATP generation, and they integrate and transduce apoptotic signals and also help regulate intracellular Ca2+. Recent work demonstrates the close association of mitochondria with specialized regions of the ER, the mitochondria-associated ER membrane, MAM. This coupling is regulated by cytosolic Ca2+ levels [47]. Signaling from the ER to mitochondria can be critical in the induction of mitochondrial dependent cell death pathways [48–51].
A. Mitochondria and MAM in cerebral ischemia
Mitochondria play a central role in normal neuronal cell function by controlling cellular energy metabolism and producing reactive oxygen species (ROS), but also as a central regulator of cell death both via release of apoptotic factors into the cytosol and acting as a target for apoptosis regulatory proteins [52–55]. Furthermore, mitochondrial function has a direct effect on inflammation and neurogenesis [56, 57]. One accepted cell death mechanism triggered by cerebral ischemia is mitochondrial permeability transition (MPT) pore opening, and excessive mitochondrial matrix Ca2+ accumulation and its release to the cytosol is a critical step in this cell death pathway (for a recent review, see Ouyang and Giffard [8]).
Mitochondria can accumulate large amounts of calcium through a Ca2+-selective channel known as the mitochondrial Ca2+ uniporter (MCU) [58, 59]. MCU has a relatively low Ca2+ affinity [60] but in response to cytosolic Ca2+ transients not exceeding concentrations of 1–3 μM, mitochondrial Ca2+ concentrations rise almost simultaneously to values above 10 μM [61].
The existence of close contact points between the ER and mitochondria, MAM, is thought to provide a selective direct pathway for calcium to transit from the ER to mitochondria. MAM has been demonstrated for rat brain [62]. The inositol trisphosphate receptor (IP3R) and the ryanodine receptor, Ca2+ release channels in the ER, and the voltage dependent anion channel (VDAC) in the mitochondrial outer membrane are important nodes of this interaction network [63–66], critical participants in MAM that define the main calcium transfer route between ER and mitochondria. Upon cell stimulation, the release of high concentrations of Ca2+ at the MAM leads to the formation of microdomains of high Ca2+ concentration that are crucial for efficient Ca2+ uptake by mitochondria [67, 68].
B. Chaperone network control of MAM
Several important chaperones including several HSP70 family members are enriched in MAM (Fig. 1) and may play a key role in regulating Ca2+ signaling between ER and mitochondria. HSP75 directly interacts with both VDAC and IP3R, playing a central role in scaffolding this ER-mitochondrial complex [69]. It was demonstrated that isoform 1 of VDAC is physically linked to the ER Ca2+-release channel IP3R through GRP75, highlighting chaperone-mediated conformational coupling between the IP3R and the mitochondrial Ca2+ uptake machinery. It was found that the mitochondrial chaperone GRP75 regulates IP3R-mediated mitochondrial Ca2+ signaling [69]. Overexpressing GRP75 protected ATP levels and mitochondrial function, and reduced ROS accumulation during glucose deprivation in neuronal cells [70]. We have found that overexpression of GRP75 improved mitochondrial function after in vivo and in vitro cerebral ischemia [16, 30]. The improved mitochondrial function includes protection of complex IV activity, marked reduction of ROS, reduction of lipid peroxidation, increased preservation of ATP levels in vivo [16], and preserved mitochondrial membrane potential, decreased ROS production, and preserved ATP levels in vitro [30].
Normally, ER chaperone GRP78/BIP forms a complex at the MAM with SIG1R, a Ca2+-sensitive and ligand-operated receptor chaperone at the MAM [71]. Upon ER Ca2+ depletion or after ligand stimulation, GRP78 can dissociate from SIG1R. GRP78 has been found to be one of the VDAC interactors (Table 1 in Szabadkai, Bianchi [69]) together with GRP75. We recently found that a fusion protein consisting of green fluorescent protein (eGFP) fused with GRP78 retargets to mitochondria within a short period of ischemia-like stress [8] and overexpressing GRP78 preserves respiratory activity and mitochondrial membrane potential, reduces ROS production, reduces mitochondria Ca2+ overload, and increases Ca2+ uptake capacity in isolated mitochondria after stress [7]. A prior report in 9L tumor cells demonstrated relocalization of GRP78 to mitochondria after induction of ER stress by thapsigargin [72].
HSP72 participates in protein import/sorting at MAM [1, 73] and regulates BCL2, an anti-apoptotic protein associated with Ca2+ homeostasis in MAM [74, 75]. HSP72 overexpression increases the expression of the anti-apoptotic protein BCL2 in vitro and in vivo [25]. While overexpression of HSP72 and its mutants is associated with maintenance of mitochondrial physiology during ischemia-like stress [19], HSP72 may also directly interfere with cell death pathways and inflammatory signaling [6, 24, 76]. Fig. 1 summarizes the chaperone control of MAM Ca2+ signaling.
3. Chaperones participate in the inflammatory network
A. Immune response following stroke
Recent clinical and experimental studies have highlighted a complex role for the immune system in the pathophysiological changes that occur after stroke [77–80]. The concentration of various cytokines is increased in the cerebrospinal fluid [81] and blood [82, 83] of acute stroke patients, and these changes are associated with clinical events including infection [84], level of functional outcome, and mortality [48, 81, 85, 86]. Both the sympathetic and parasympathetic arms of the autonomic nervous system play key roles in immune regulation, and in the setting of stroke, communicating to the peripheral immune system that a stroke has occurred, leading to direct modulation of peripheral immune cell function [78, 87, 88].
Cerebral ischemia in animal models induces acute and prolonged inflammatory processes. Even though multiple cell types including astrocytes, microglia and neurons may be involved in the inflammatory response in the brain after stroke we will focus on microglia and astrocytes in this review. Inflammation can be detected within a few hours after the onset of cerebral ischemia and the initial immune response is mostly innate, including activation of microglia and astrocytes [89–93]. Microglia display a ramified appearance while in the resting state, but when activated, undergo a series of morphologic changes often leading to an amoeboid morphology. Microglial activation is the initial step in the CNS inflammatory response; depending on the stimulus, this step may be followed by infiltration of macrophages, monocytes, neutrophils, T-cells and other inflammatory cells, and by reactive astrocytosis [76, 94]. Astrocytes also respond to and produce inflammatory signals [37] and through their interaction with microglia, neurons, and endothelial cells help determine the outcome from injury. Astrocyte activation is diminished in brains of HSP72 overexpressing mice subjected to focal ischemia [22].
Acute brain insult triggers an innate immune response via several mechanisms. Cellular injury leads to release of danger associated molecular patterns (DAMPs) which are recognized by and trigger the innate immune response. Activation of toll-like receptors (TLR) by DAMPs leads to activation of pro-inflammatory signaling by activation of inflammasomes, with activation of both transcription factor nuclear factor-kappa B (NF-kB), and activation of caspase 1 leading to release of processed IL-1β [95]. TLR2 or TLR4, but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia [96, 97]. Recent studies indicate that mitochondrial ROS also act as signaling molecules to trigger proinflammatory cytokine production [98–101] through NF-kB transcriptional activation. ROS cause phosphorylation of IKK [102, 103] and phosphorylated IKK causes the phosphorylation of IkB, leading to the ubiquitination and degradation of IkB, and the release of NF-kB.
The NF-kB transcriptional activation pathway is considered to be a “master regulator” of inflammation and critical to the regulation of apoptosis [104, 105] (Fig. 2). NF-kB is a family of dimeric transcription factors that regulate the transcription of hundreds of genes in a coordinated manner in response to an inducing signal. In resting cells NF-kB is found primarily in the cytosol bound to its inhibitor IkB proteins. Upon stimulation by cytokines or other inducers, IkB proteins are targeted for proteasomal degradation by the IkB kinase (IKK). Once IkB degrades, NF-kB translocates to the nucleus and binds DNA at kB sites in the regulatory region of pro-inflammatory genes and promotes their transcription [106, 107]. Its target genes also include its own inhibitors and other regulatory proteins that form a complex network that tightly regulates the dynamic response and gene transcription. An ordinary differential equation computational model of NF-κB activation specific for microglia has been developed recently to better understand the regulation of NF-κB activation at a systems level in this individual cell type [27].
Fig. 2.
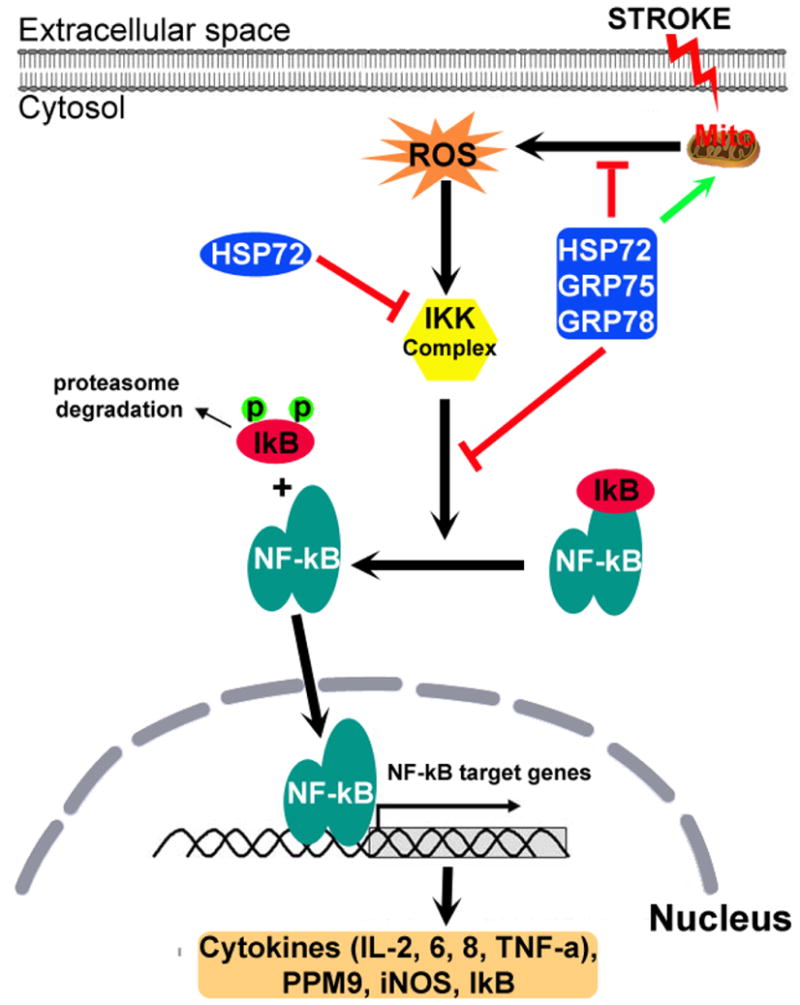
Chaperone machinery influences the NF-kB pro-inflammatory signaling pathway. NF-kB, a dimer consisting of p50/p65 subunits is normally resident in the cytosol and is maintained in an inactive form by its inhibitor IkB. Stroke stimulates mitochondria to release reactive oxygen species (ROS) that activate the IkB kinase (IKK) complex. The activated IKK complex phosphorylates IkB and initiates its ubiquitination and degradation, exposing the nuclear localization signal on NF-kB. NF-kB then translocates to the nucleus and binds to the promoter region of genes expressing pro-inflammatory cytokines and IkB. HSP72 interacts with the IKK complex and several HSP70 family members (HSP70, GRP75 and GRP78), protects mitochondrial function, inhibits ROS production and NF-kB activation.
B. Chaperones regulate inflammation signaling
The HSP70 family modulates inflammation via several mechanisms as reviewed previously [6, 26, 76, 108]. In this section we focus on its relation with NF-kB pro-inflammatory signaling. HSP72 interacts with TLR pathway activation [109] and the NF-kB signaling pathway. Intracellular overexpression of HSP72 or its intracellular induction by heat stress has been shown to decrease NF-kB activation in astrocytes [110]. Activation of NF-kB was inhibited significantly in HSP72 overexpressing microglia and transgenic mice [18]. HSP72 binds to the NF-kB:IkB complex preventing IkB phosphorylation by IKK and NF-kB dissociation [18]. In another context HSP72 was found to bind IKK to impair NFkB signaling [111].
Potential anti-inflammatory effects of GRP75 remain largely unexplored. We recently found that overexpression of GRP75 is able to modulate the LPS-induced pro-inflammatory response of microglial cells [112]. We observed that LPS treatment promoted significant increases in mitochondrial ROS levels as well as the proinflammatory cytokines TNF-a and IL-6, which were significantly reduced by GRP75 overexpression. These observations are consistent with a known role for oxidative metabolism in anti-inflammatory activation compared to the role of glycolytic metabolism in pro-inflammatory activation [113]. Thus GRP75 may be acting either indirectly via maintenance of oxidative metabolism, or perhaps also by an unknown direct mechanism to inhibit NF-kB activation and reduce proinflammatory cytokine production.
GRP78, also known as immunoglobulin heavy chain binding protein (BIP), is closely related to autoimmune and inflammatory diseases [114]. GRP78 can act as an anti-inflammatory factor. Several studies have suggested that GRP78 stimulates the production of the anti-inflammatory cytokines IL-4 and IL-10 through specific T lymphocytes [115–120]. Furthermore, the pre-administration of GRP78 protein to mice prevents the in vivo induction of adjuvant arthritis or collagen-induced arthritis, both of which are well-known models of autoimmune diseases [117, 119]. In some cell types, GRP78 might be important in the suppression of NF-kB [121]. It was found that, in murine podocytes, acute ablation of GRP78 by SubAB caused transient activation of NF-kB. Furthermore, transient transfection with GRP78 significantly inhibited activation of NF-kB by TNF-a [122].
Since both GRP75 and 78 have effects on oxidative stress and mitochondrial function as discussed above, they might also have anti-inflammatory effects in stroke by reducing mitochondrial ROS production and supporting anti-inflammatory activation of immune cells. Fig. 2 summarizes the chaperone control of the NF-kB inflammation signaling pathway.
4. miRNAs regulate the chaperone network
The study of miRNAs is rapidly growing and recent studies have revealed that miRNAs have a significant role in ischemic disease. miRNAs are especially important candidates for stroke therapeutics because of their ability to simultaneously regulate many target genes, as targeting single genes for therapeutic intervention has not yet succeeded in the clinic (for a recent review, see [1]). Further, miRNA based therapeutics are already in clinical trials, suggesting that translation to new diseases or miRNAs will be relatively straightforward.
A. Translational inhibition of the HSP70 family in cerebral ischemia
Cerebral ischemia/reperfusion (I/R) injury induces multiple genes [123], which activate molecular cascades leading to both necrotic cell death in the anoxic core, and delayed apoptotic cell death in the surrounding penumbra [124, 125]. While the fate of brain cells in the anoxic core is likely fixed early following the initial insult, cells in the peri-ischemic penumbra represent targets for rescue from delayed cell death. There is thought to be a temporal window in which reversal or prevention of induction of the apoptotic cascade can occur, and such prevention of cell death would be predicted to improve functional outcome. Transient arrest of protein translation is appreciated to be a stereotypical response to ischemia and a variety of other cell stresses, including the heat shock response and the unfolded protein response which may also occur in the setting of ischemia. Transient translational arrest limits the increase in unfolded/misfolded proteins and allows for rapid induction of a stress response with selective translation of stress proteins before synthesis of constitutive proteins resumes. Several biochemical pathways and sequestration of ribosomes are well studied components of ischemic translational arrest. Prior studies have focused on biochemical mechanisms of transient translational arrest, particularly phosphorylation of eukaryotic Initiation Factor 2 α(eIF2α), and sequestration of ribosomes in persistent translational arrest [126, 127].
More than twenty years ago regional and cellular distributions of HSP72 were investigated after cerebral ischemia [128]. After global ischemia, HSP72 was induced primarily in CA3 pyramidal neurons and dentate granule cell neurons that survived the ischemic episode, whereas HSP72 was not induced in CA1 pyramidal neurons destined to die [129]. After focal cerebral ischemia HSP72 was induced in the penumbra surrounding the infarction core [130, 131]. Thus HSP72 protein is primarily induced in cells that survive the ischemic injury and in this setting they function to protect the cells from subsequent lethal injury. These early studies also reported the interesting finding that induction of HSP72 mRNA was not always associated with induction of HSP72 protein after cerebral ischemia. Following global ischemia hsp72 mRNA was induced in CA1 hippocampal pyramidal neurons that failed to express the HSP72 protein [132]. Similarly Hsp72 mRNA is induced within the MCAO infarction but HSP72 protein is not [131].
Post-mortem study of brains of stroke victims found a relatively early increase in GRP78 in the penumbra [133]. Recently we reported a failure to elevate levels of GRP78 protein in the core, while GRP78 is induced in the penumbra after transient focal ischemia [40]. Following 1 h middle cerebral artery occlusion, Grp78 mRNA was induced in both ischemic core and penumbra, while GRP78 protein declined in the core. At present, even though there are some answers to the question of why translational block of Hsp70 genes occurs in the ischemic area or in cells destined to die, few reports are available about miRNA regulation of HSP70 family chaperones, yet another mechanism of translational inhibition.
B. miRNA and chaperones
Several studies have demonstrated alterations in the cerebral “miRNA-ome” following ischemia reperfusion [134–136] suggesting that miRNA mediated translational arrest may be an important factor in modulating the gene expression cascade that occurs in response to ischemia and reperfusion. Studies of the regulation of molecular chaperones by miRNA are just beginning. miR-320 has been shown to be involved in the regulation of heart ischemia/reperfusion injury by targeting HSP20 [137]. The target was validated experimentally using a luciferase/green fluorescent protein reporter activity assay and examining the expression of HSP20 with miR-320 overexpression and knockdown in cardiomyocytes.
Injection of miRNA extracted from the hearts of mice following ischemic preconditioning protected naive hearts against ischemia/reperfusion injury, possibly through upregulating HSP72 and the HSP72 transcription factor HSF-1 [138]. miRNA-1, miRNA-21, miRNA-24, and some additional miRNAs may be linked to increased expression of the cytoprotective proteins in this study, though no targets were validated. Recent publications have shown that the level of muscle-specific miR-1 changes in the ischemic myocardium [139–141] and two of miR-1’s targets are HSP60 and HSP72 [142]. This is the only validated HSP72 miRNA at present.
No reports are available regarding GRP75 targeting miRNA. Using computational miRNA target prediction algorithms from TargetScan (http://targetscan.org), we identified that 3′UTRs of Hspa9 (GRP75) have conserved sites for the miR-200 family, one of the miRNA families broadly conserved among vertebrates (Fig. 3A). Another target prediction website, Microcosm Targets (http://www.ebi.ac.uk/enright-srv/microcosm), lists miR-200c as the number one miRNA targeting Hspa9 (Fig. 3B). Interestingly, like miRNA-181 [1], miR-200 can also potentially target the 3′UTR of Bcl2 (Fig. 3C). Still more interesting is that the miRNA family has already been identified in profiling studies as selectively upregulated in ischemic cortex 3 h after a 15 min preconditioning MCAO insult in mice [143], after MCAO in rat [135], and after endothelial cell oxidative stress and hind limb ischemia [144]. Opposite effects of miR-200 on cell survival have however been reported. miR-200c overexpression induced apoptosis of endothelial cells [144], but increased survival of Neuro-2a cells [143]. In the case of endothelial cells, effects were attributed to the target being ZEB1, while in the second case effects were suggested to be due to effects on prolyl hydroxylase 2. Since miRNAs can regulate different genes in different cell types and exert different effects under different conditions, this oxidative stress inducible miRNA is an interesting one to pursue in the setting of cerebral ischemia.
Fig. 3.

Sequences showing the unique sites of miRNA::mRNA complementarity between miR- 200 or miR-181 and chaperones
A–C. Potential miR-200 target sites in GRP75 and BCL2 3′UTRs.
D–F. Potential miR-181 target sites in HSP72, GRP75, and GRP78 3′UTRs.
Recently we demonstrated that a brain-enriched miRNA, miR-181a, regulates GRP78 expression and outcome from cerebral ischemia [40]. A reciprocal expression of miR-181a and GRP78 protein was found in both core and penumbra. In vitro and in vivo experiments show that miR-181a mimic decreases and its inhibitor/antagomir increases GRP78 protein expression [40]. Interestingly miR-181a also targets the anti-apoptotic protein BCL2 [75], which also exists in MAM and affects ER and mitochondrial calcium homeostasis (Fig. 1). Using computational miRNA target prediction algorithms TargetScan (http://targetscan.org, Release 5.1) and Microcosm Targets (http://www.ebi.ac.uk/enright-srv/microcosm), we found that miR-181 can target GRP78 but could potentially target the 3′UTRs of two other HSP70 family members, Hspa1a (HSP72) and Hspa9 (GRP75) (Fig. 3D–F). Therefore one miRNA like miR-181 could potentially target multiple chaperones and apoptotic proteins as BCL2 and efficiently regulate cell death pathways after cerebral ischemia.
In summary, a single miRNA (for example miR-200 or miR-181) is able to simultaneously regulate many target genes in both the organelle and inflammation regulating chaperone networks, which are key players in the mechanisms of cerebral ischemia (Fig. 4). Therefore, miRNAs are exciting new candidates for stroke therapeutics.
Fig. 4.
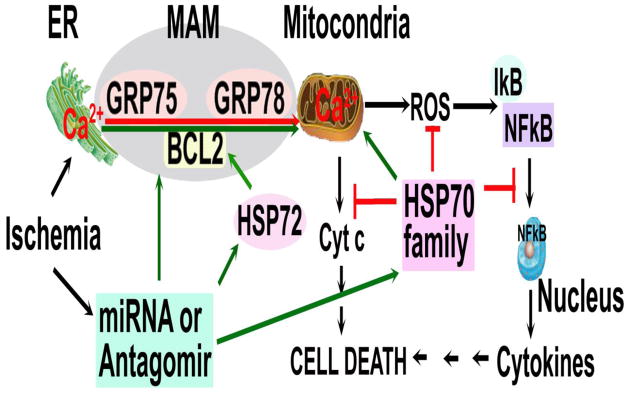
miRNA control of chaperone networks. Ischemia leads to mitochondrial Ca2+ overload through MAM and then dysfunction of mitochondria releases ROS, a key element linking the organelle network to the inflammatory network of chaperone. One miRNA (miR-200 or miR-181) or its antagomir/inhibitor can target HSP70/GRP75/GRP78 and BCL2 at the same time and thus efficiently regulate cerebral ischemic cell death pathways.
Acknowledgments
This work was supported in part by NIH grants NS053898, GM49831, and NS080177 to RGG. The authors would like to thank William Magruder for help preparing the paper.
Footnotes
Compliance with Ethics Requirements
Yi-Bing Ouyang declares that she has no conflict of interest.
Rona Giffard declares that she has no conflict of interest.
All institutional and national guidelines for the care and use of laboratory animals were followed.
The authors have no conflicting financial interests.
References
- 1.Ouyang YB, et al. microRNAs: Innovative Targets for Cerebral Ischemia and Stroke. Current drug targets. 2013;14(1):90–101. doi: 10.2174/138945013804806424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ouyang YB, et al. Selective Dysfunction of Hippocampal CA1 Astrocytes Contributes to Delayed Neuronal Damage after Transient Forebrain Ischemia. The Journal of Neuroscience. 2007;27(16):4253–4260. doi: 10.1523/JNEUROSCI.0211-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu L, et al. Astrocyte targeted overexpression of Hsp72 or SOD2 reduces neuronal vulnerability to forebrain ischemia. Glia. 2010;58(9):1042–1049. doi: 10.1002/glia.20985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okuyama S, et al. Anti-inflammatory and neuroprotective effects of auraptene, a citrus coumarin, following cerebral global ischemia in mice. European Journal of Pharmacology. 2013;699(1–3):118–123. doi: 10.1016/j.ejphar.2012.11.043. [DOI] [PubMed] [Google Scholar]
- 5.Xiong X, et al. Increased Brain Injury and Worsened Neurological Outcome in Interleukin-4 Knockout Mice After Transient Focal Cerebral Ischemia. Stroke. 2011;42(7):2026–2032. doi: 10.1161/STROKEAHA.110.593772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giffard RG, et al. Regulation of Apoptotic and Inflammatory Cell Signaling in Cerebral Ischemia: The Complex Roles of Heat Shock Protein 70. Anesthesiology. 2008;109(2):339–348. doi: 10.1097/ALN.0b013e31817f4ce0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ouyang YB, et al. Overexpressing GRP78 influences Ca2+ handling and function of mitochondria in astrocytes after ischemia-like stress. Mitochondrion. 2011;11(2):279–286. doi: 10.1016/j.mito.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ouyang Y-B, Giffard R. ER-Mitochondria Crosstalk during Cerebral Ischemia: Molecular Chaperones and ER-Mitochondrial Calcium Transfer. International Journal of Cell Biology. 2012;2012:493934–493934. doi: 10.1155/2012/493934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mayer MP, Bukau B. Hsp70 chaperone systems: diversity of cellular functions and mechanism of action. Biological Chemistry. 1998;379(3):261–268. [PubMed] [Google Scholar]
- 10.Kiang JG, Tsokos GC. Heat Shock Protein 70 kDa: Molecular Biology, Biochemistry, and Physiology. Pharmacology & Therapeutics. 1998;80(2):183–201. doi: 10.1016/s0163-7258(98)00028-x. [DOI] [PubMed] [Google Scholar]
- 11.Hoehn B, et al. Overexpression of HSP72 After Induction of Experimental Stroke Protects Neurons From Ischemic Damage. J Cereb Blood Flow Metab. 2001;21(11):1303–1309. doi: 10.1097/00004647-200111000-00006. [DOI] [PubMed] [Google Scholar]
- 12.Lee WC, et al. Heat shock protein 72 overexpression protects against hyperthermia, circulatory shock, and cerebral ischemia during heatstroke. Journal of Applied Physiology. 2006;100(6):2073–2082. doi: 10.1152/japplphysiol.01433.2005. [DOI] [PubMed] [Google Scholar]
- 13.Plumier JC, et al. Transgenic mice expressing the human inducible Hsp70 have hippocampal neurons resistant to ischemic injury. Cell stress & chaperones. 1997;2(3):162–167. doi: 10.1379/1466-1268(1997)002<0162:tmethi>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rajdev S, et al. Mice overexpressing rat heat shock protein 70 are protected against cerebral infarction. Annals of Neurology. 2000;47(6):782–791. [PubMed] [Google Scholar]
- 15.van der Weerd L, et al. Neuroprotective effects of HSP70 overexpression after cerebral ischaemia—An MRI study. Experimental Neurology. 2005;195(1):257–266. doi: 10.1016/j.expneurol.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 16.Xu L, et al. Overexpression of mitochondrial Hsp70/Hsp75 in rat brain protects mitochondria, reduces oxidative stress, and protects from focal ischemia. J Cereb Blood Flow Metab. 2009;29(2):365–374. doi: 10.1038/jcbfm.2008.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yenari MA, et al. Gene therapy with HSP72 is neuroprotective in rat models of stroke and epilepsy. Annals of Neurology. 1998;44(4):584–591. doi: 10.1002/ana.410440403. [DOI] [PubMed] [Google Scholar]
- 18.Zheng Z, et al. Anti-inflammatory effects of the 70kDa heat shock protein in experimental stroke. J Cereb Blood Flow Metab. 2008;28(1):53–63. doi: 10.1038/sj.jcbfm.9600502. [DOI] [PubMed] [Google Scholar]
- 19.Ouyang YB, et al. Overexpression of inducible heat shock protein 70 and its mutants in astrocytes is associated with maintenance of mitochondrial physiology during glucose deprivation stress. Cell stress & Chaperones. 2006;11(2):180–186. doi: 10.1379/CSC-182R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun Y, et al. The carboxyl-terminal domain of inducible Hsp70 protects from ischemic injury in vivo and in vitro. J Cereb Blood Flow Metab. 2005;26(7):937–950. doi: 10.1038/sj.jcbfm.9600246. [DOI] [PubMed] [Google Scholar]
- 21.Xu L, et al. Heat shock protein 72 (Hsp72) improves long term recovery after focal cerebral ischemia in mice. Neuroscience Letters. 2011;488(3):279–282. doi: 10.1016/j.neulet.2010.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barreto GE, et al. Effects of heat shock protein 72 (Hsp72) on evolution of astrocyte activation following stroke in the mouse. Experimental Neurology. 2012;238(2):284–296. doi: 10.1016/j.expneurol.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giffard RG, et al. Chaperones, protein aggregation, and brain protection from hypoxic/ischemic injury. Journal of Experimental Biology. 2004;207(18):3213–3220. doi: 10.1242/jeb.01034. [DOI] [PubMed] [Google Scholar]
- 24.Giffard RG, Yenari MA. Many Mechanisms for Hsp70 Protection From Cerebral Ischemia. Journal of Neurosurgical Anesthesiology. 2004;16(1):53–61. doi: 10.1097/00008506-200401000-00010. [DOI] [PubMed] [Google Scholar]
- 25.Kelly S, et al. Gene transfer of HSP72 protects cornu ammonis 1 region of the hippocampus neurons from global ischemia: Influence of Bcl-2. Annals of Neurology. 2002;52(2):160–167. doi: 10.1002/ana.10264. [DOI] [PubMed] [Google Scholar]
- 26.Yenari MA, et al. Antiapoptotic and Anti-inflammatory Mechanisms of Heat-Shock Protein Protection. Annals of the New York Academy of Sciences. 2005;1053(1):74–83. doi: 10.1196/annals.1344.007. [DOI] [PubMed] [Google Scholar]
- 27.Sheppard P, et al. Quantitative characterization and analysis of the dynamic NF-kB response in microglia. BMC bioinformatics. 2011;12:276–276. doi: 10.1186/1471-2105-12-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wadhwa R, Taira K, Kaul SC. An Hsp70 family chaperone, mortalin/mthsp70/PBP74/Grp75: what, when, and where? Cell stress & chaperones. 2002;7(3):309–316. doi: 10.1379/1466-1268(2002)007<0309:ahfcmm>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Massa SM, et al. Cloning of rat grp75, an hsp70-family member, and its expression in normal and ischemic brain. Journal of Neuroscience Research. 1995;40(6):807–819. doi: 10.1002/jnr.490400612. [DOI] [PubMed] [Google Scholar]
- 30.Voloboueva LA, et al. Overexpression of mitochondrial Hsp70/Hsp75 protects astrocytes against ischemic injury in vitro. J Cereb Blood Flow Metab. 2008;28(5):1009–1016. doi: 10.1038/sj.jcbfm.9600600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.White R, Ouyang Y-B, Giffard R. Hsp75/mortalin and Protection from Ischemic Brain Injury. In: Kaul SC, Wadhwa R, editors. Mortalin Biology: Life, Stress and Death. Springer; Netherlands: 2012. pp. 179–190. [Google Scholar]
- 32.Zhang LH, et al. Association of elevated GRP78 expression with increased astrocytoma malignancy via Akt and ERK pathways. Brain Research. 2011;1371(0):23–31. doi: 10.1016/j.brainres.2010.11.063. [DOI] [PubMed] [Google Scholar]
- 33.Pfaffenbach KT, Lee AS. The critical role of GRP78 in physiologic and pathologic stress. Current Opinion in Cell Biology. 2011;23(2):150–156. doi: 10.1016/j.ceb.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang M, et al. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxidants & redox signalling. 2009;11(9):2307–2316. doi: 10.1089/ars.2009.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gonzalez Gronow M, et al. GRP78: a multifunctional receptor on the cell surface. Antioxidants & Redox signalling. 2009;11(9):2299–2306. doi: 10.1089/ARS.2009.2568. [DOI] [PubMed] [Google Scholar]
- 36.Wang M, et al. Essential role of the unfolded protein response regulator GRP78/BiP in protection from neuronal apoptosis. Cell Death and Differentiation. 2010;17(3):488–498. doi: 10.1038/cdd.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li J, et al. The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death and Differentiation. 2008;15(9):1460–1471. doi: 10.1038/cdd.2008.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oida Y, et al. Induction of BiP, an ER-resident protein, prevents the neuronal death induced by transient forebrain ischemia in gerbil. Brain Research. 2008;1208(0):217–224. doi: 10.1016/j.brainres.2008.02.068. [DOI] [PubMed] [Google Scholar]
- 39.Kudo T, et al. A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ. 2008;15(2):364–375. doi: 10.1038/sj.cdd.4402276. [DOI] [PubMed] [Google Scholar]
- 40.Ouyang YB, et al. miR-181 regulates GRP78 and influences outcome from cerebral ischemia in vitro and in vivo. Neurobiology of Disease. 2012;45(1):555–563. doi: 10.1016/j.nbd.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barabasi AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004;5(2):101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 42.Csermely P. Strong links are important, but weak links stabilize them. Trends in Biochemical Sciences. 2004;29(7):331–334. doi: 10.1016/j.tibs.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 43.Sõti C, et al. Molecular chaperones as regulatory elements of cellular networks. Current Opinion in Cell Biology. 2005;17(2):210–215. doi: 10.1016/j.ceb.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 44.Csermely P, Vigh L, editors. Molecular Aspects of the Stress Response: Chaperones, Membranes and Networks. Springer; Berlin: 2007. [Google Scholar]
- 45.Brostrom MA, Brostrom CO. Calcium dynamics and endoplasmic reticular function in the regulation of protein synthesis: implications for cell growth and adaptability. Cell Calcium. 2003;34(4–5):345–363. doi: 10.1016/s0143-4160(03)00127-1. [DOI] [PubMed] [Google Scholar]
- 46.Sitia R, Braakman I. Quality control in the endoplasmic reticulum protein factory. Nature. 2003;426(6968):891–894. doi: 10.1038/nature02262. [DOI] [PubMed] [Google Scholar]
- 47.Wang HJ, et al. Calcium Regulates the Association between Mitochondria and a Smooth Subdomain of the Endoplasmic Reticulum. The Journal of Cell Biology. 2000;150(6):1489–1498. doi: 10.1083/jcb.150.6.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deniaud A, et al. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene. 2007;27(3):285–299. doi: 10.1038/sj.onc.1210638. [DOI] [PubMed] [Google Scholar]
- 49.Hetz C. ER stress signaling and the BCL-2 family of proteins: from adaptation to irreversible cellular damage. Antioxidants & redox signalling. 2007;9(12):2345–2355. doi: 10.1089/ars.2007.1793. [DOI] [PubMed] [Google Scholar]
- 50.Hom JR, et al. Thapsigargin induces biphasic fragmentation of mitochondria through calcium-mediated mitochondrial fission and apoptosis. Journal of Cellular Physiology. 2007;212(2):498–508. doi: 10.1002/jcp.21051. [DOI] [PubMed] [Google Scholar]
- 51.Scorrano L, et al. BAX and BAK Regulation of Endoplasmic Reticulum Ca2+: A Control Point for Apoptosis. Science. 2003;300(5616):135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 52.Beal MF. Energetics in the pathogenesis of neurodegenerative diseases. Trends in Neurosciences. 2000;23(7):298–304. doi: 10.1016/s0166-2236(00)01584-8. [DOI] [PubMed] [Google Scholar]
- 53.Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6(5):513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 54.Murphy KM, Streips UN, Lock RB. Bax membrane insertion during Fas(CD95)-induced apoptosis precedes cytochrome c release and is inhibited by Bcl-2. Oncogene. 1999;18(44):5991–5999. doi: 10.1038/sj.onc.1203001. [DOI] [PubMed] [Google Scholar]
- 55.Ravagnan L, Roumier T, Kroemer G. Mitochondria, the killer organelles and their weapons. Journal of Cellular Physiology. 2002;192(2):131–137. doi: 10.1002/jcp.10111. [DOI] [PubMed] [Google Scholar]
- 56.Voloboueva LA, Giffard RG. Inflammation, mitochondria, and the inhibition of adult neurogenesis. Journal of Neuroscience Research. 2011;89(12):1989–1996. doi: 10.1002/jnr.22768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Voloboueva LA, et al. Mitochondrial Protection Attenuates Inflammation-Induced Impairment of Neurogenesis In Vitro and In Vivo. The Journal of Neuroscience. 2010;30(37):12242–12251. doi: 10.1523/JNEUROSCI.1752-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427(6972):360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- 59.Nicholls DG, Budd SL. Mitochondria and Neuronal Survival. Physiological Reviews. 2000;80(1):315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- 60.Bernardi P. Mitochondrial Transport of Cations: Channels, Exchangers, and Permeability Transition. Physiological Reviews. 1999;79(4):1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- 61.Rizzuto R, Pozzan T. Microdomains of Intracellular Ca2+: Molecular Determinants and Functional Consequences. Physiological Reviews. 2006;86(1):369–408. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- 62.Camici O, Corazzi L. Phosphatidylserine translocation into brain mitochondria: involvement of a fusogenic protein associated with mitochondrial membranes. Molecular and cellular biochemistry. 1997;175(1–2):71–80. doi: 10.1023/a:1006889328983. [DOI] [PubMed] [Google Scholar]
- 63.Colombini M. VDAC: The channel at the interface between mitochondria and the cytosol. Molecular and cellular biochemistry. 2004;256–257(1–2):107–115. doi: 10.1023/b:mcbi.0000009862.17396.8d. [DOI] [PubMed] [Google Scholar]
- 64.Patterson R, Boehning D, Snyder S. Inositol 1,4,5-trisphosphate receptors as signal integrators. Annual review of biochemistry. 2004;73:437–465. doi: 10.1146/annurev.biochem.73.071403.161303. [DOI] [PubMed] [Google Scholar]
- 65.Rostovtseva T, Tan W, Colombini M. On the Role of VDAC in Apoptosis: Fact and Fiction. Journal of Bioenergetics and Biomembranes. 2005;37(3):129–142. doi: 10.1007/s10863-005-6566-8. [DOI] [PubMed] [Google Scholar]
- 66.Vyssokikh M, Brdiczka D. VDAC and peripheral channelling complexes in health and disease. Molecular and cellular biochemistry. 2004;256–257(1–2):117–126. doi: 10.1023/b:mcbi.0000009863.69249.d9. [DOI] [PubMed] [Google Scholar]
- 67.Rizzuto R, et al. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science. 1993;262(5134):744–747. doi: 10.1126/science.8235595. [DOI] [PubMed] [Google Scholar]
- 68.Rizzuto R, et al. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science. 1998;280(5370):1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- 69.Szabadkai G, et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. The Journal of Cell Biology. 2006;175(6):901–911. doi: 10.1083/jcb.200608073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu Y, et al. Effect of GRP75/mthsp70/PBP74/mortalin overexpression on intracellular ATP level, mitochondrial membrane potential and ROS accumulation following glucose deprivation in PC12 cells. Molecular and cellular biochemistry. 2005;268(1–2):45–51. doi: 10.1007/s11010-005-2996-1. [DOI] [PubMed] [Google Scholar]
- 71.Hayashi T, Su TP. Sigma-1 Receptor Chaperones at the ER- Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell. 2007;131(3):596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- 72.Sun FC, et al. Localization of GRP78 to mitochondria under the unfolded protein response. Biochem J. 2006;396(1):31–39. doi: 10.1042/BJ20051916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Szabadkai G, Rizzuto R. Chaperones as parts of organelle networks. In: Csermely P, Vigh L, editors. Molecular Aspects of the Stress Response: Chaperones, Membranes and Networks. Springer; Berlin: 2007. [DOI] [PubMed] [Google Scholar]
- 74.Foyouzi-Youssefi R, et al. Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proceedings of the National Academy of Sciences. 2000;97(11):5723–5728. doi: 10.1073/pnas.97.11.5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ouyang YB, et al. miR-181 targets multiple Bcl-2 family members and influences apoptosis and mitochondrial function in astrocytes. Mitochondrion. 2012;12(2):213–219. doi: 10.1016/j.mito.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim N, Kim J, Yenari M. Anti-inflammatory properties and pharmacological induction of Hsp70 after brain injury. Inflammopharmacology. 2012;20(3):177–185. doi: 10.1007/s10787-011-0115-3. [DOI] [PubMed] [Google Scholar]
- 77.Chamorro A, et al. The immunology of acute stroke. Nat Rev Neurol. 2012;8(7):401–410. doi: 10.1038/nrneurol.2012.98. [DOI] [PubMed] [Google Scholar]
- 78.Ouyang YB. Inflammation and stroke. Neurosci Lett. 2013;548:1–3. doi: 10.1016/j.neulet.2013.05.031. [DOI] [PubMed] [Google Scholar]
- 79.Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17(7):796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kamel HIC. Brain-immune interactions and ischemic stroke: Clinical implications. Archives of Neurology. 2012;69(5):576–581. doi: 10.1001/archneurol.2011.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tarkowski E, et al. Early Intrathecal Production of Interleukin-6 Predicts the Size of Brain Lesion in Stroke. Stroke. 1995;26(8):1393–1398. doi: 10.1161/01.str.26.8.1393. [DOI] [PubMed] [Google Scholar]
- 82.Beamer NB, et al. Interleukin-6 and interleukin-1 receptor antagonist in acute stroke. Annals of Neurology. 1995;37(6):800–805. doi: 10.1002/ana.410370614. [DOI] [PubMed] [Google Scholar]
- 83.Fassbender K, et al. Proinflammatory cytokines in serum of patients with acute cerebral ischemia: kinetics of secretion and relation to the extent of brain damage and outcome of disease. Journal of the Neurological Sciences. 1994;122(2):135–139. doi: 10.1016/0022-510x(94)90289-5. [DOI] [PubMed] [Google Scholar]
- 84.Chamorro Á, Urra X, Planas AM. Infection After Acute Ischemic Stroke: A Manifestation of Brain-Induced Immunodepression. Stroke. 2007;38(3):1097–1103. doi: 10.1161/01.STR.0000258346.68966.9d. [DOI] [PubMed] [Google Scholar]
- 85.Vila N, et al. Proinflammatory cytokines and early neurological worsening in ischemic stroke. Stroke. 2000;31(10):2325–2329. doi: 10.1161/01.str.31.10.2325. [DOI] [PubMed] [Google Scholar]
- 86.Vila N, et al. Levels of Anti-Inflammatory Cytokines and Neurological Worsening in Acute Ischemic Stroke. Stroke. 2003;34(3):671–675. doi: 10.1161/01.STR.0000057976.53301.69. [DOI] [PubMed] [Google Scholar]
- 87.Andersson U, Tracey KJ. Neural reflexes in inflammation and immunity. The Journal of Experimental Medicine. 2012;209(6):1057–1068. doi: 10.1084/jem.20120571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lafargue M, et al. Stroke-induced activation of the α7 nicotinic receptor increases Pseudomonas aeruginosa lung injury. The FASEB Journal. 2012;26(7):2919–2929. doi: 10.1096/fj.11-197384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.del Zoppo GJ, et al. Microglial Activation and Matrix Protease Generation During Focal Cerebral Ischemia. Stroke. 2007;38(2):646–651. doi: 10.1161/01.STR.0000254477.34231.cb. [DOI] [PubMed] [Google Scholar]
- 90.Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50(4):427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- 91.Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol. 2009;9(6):429–439. doi: 10.1038/nri2565. [DOI] [PubMed] [Google Scholar]
- 92.Sofroniew M, Vinters H. Astrocytes: biology and pathology. Acta Neuropathologica. 2010;119(1):7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. Journal of Neuroimmunology. 2007;184(1–2):53–68. doi: 10.1016/j.jneuroim.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zheng Z, Yenari MA. Post-ischemic inflammation: molecular mechanisms and therapeutic implications. Neurological Research. 2004;26(8):884–892. doi: 10.1179/016164104X2357. [DOI] [PubMed] [Google Scholar]
- 95.Trendelenburg G. Acute neurodegeneration and the inflammasome: central processor for danger signals and the inflammatory response? J Cereb Blood Flow Metab. 2008;28(5):867–881. doi: 10.1038/sj.jcbfm.9600609. [DOI] [PubMed] [Google Scholar]
- 96.Hyakkoku K, et al. Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience. 2010;171(1):258–267. doi: 10.1016/j.neuroscience.2010.08.054. [DOI] [PubMed] [Google Scholar]
- 97.Ziegler G, et al. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochemical and Biophysical Research Communications. 2007;359(3):574–579. doi: 10.1016/j.bbrc.2007.05.157. [DOI] [PubMed] [Google Scholar]
- 98.Bulua AC, et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS) The Journal of Experimental Medicine. 2011;208(3):519–533. doi: 10.1084/jem.20102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Naik E, V, Dixit M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. The Journal of Experimental Medicine. 2011;208(3):417–420. doi: 10.1084/jem.20110367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nakahira K, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhou R, et al. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 102.Ortego M, et al. HMG-CoA Reductase Inhibitors Reduce I[kappa]B Kinase Activity Induced by Oxidative Stress in Monocytes and Vascular Smooth Muscle Cells. Journal of Cardiovascular Pharmacology. 2005;45(5):468–475. doi: 10.1097/01.fjc.0000159042.50488.e5. [DOI] [PubMed] [Google Scholar]
- 103.Song YS, Lee YS, Chan PH. Oxidative stress transiently decreases the IKK complex (IKK[alpha], [beta], and [gamma]), an upstream component of NF-[kappa]B signaling, after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2005;25(10):1301–1311. doi: 10.1038/sj.jcbfm.9600123. [DOI] [PubMed] [Google Scholar]
- 104.Hoffmann A, Baltimore D. Circuitry of nuclear factor κB signaling. Immunological Reviews. 2006;210(1):171–186. doi: 10.1111/j.0105-2896.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- 105.Harari OA, Liao JK. NF-κB and innate immunity in ischemic stroke. Annals of the New York Academy of Sciences. 2010;1207(1):32–40. doi: 10.1111/j.1749-6632.2010.05735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ghosh S, Karin M. Missing Pieces in the NF-κB Puzzle. Cell. 2002;109(2, Supplement 1):S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 107.Hayden MS, Ghosh S. Shared Principles in NF-κB Signaling. Cell. 2008;132(3):344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 108.Jones Q, et al. Heat shock proteins protect against ischemia and inflammation through multiple mechanisms. Inflamm Allergy Drug Targets. 2011;10(4):247–259. doi: 10.2174/187152811796117726. [DOI] [PubMed] [Google Scholar]
- 109.Asea A. Heat Shock Proteins and Toll-Like Receptors. In: Bauer S, Hartmann G, editors. Toll-Like Receptors (TLRs) and Innate Immunity. Springer; Berlin Heidelberg: 2008. pp. 111–127. [Google Scholar]
- 110.Feinstein DL, et al. Heat Shock Protein 70 Suppresses Astroglial-inducible Nitric-oxide Synthase Expression by Decreasing NFκB Activation. Journal of Biological Chemistry. 1996;271(30):17724–17732. doi: 10.1074/jbc.271.30.17724. [DOI] [PubMed] [Google Scholar]
- 111.Ran R, et al. Hsp70 promotes TNF-mediated apoptosis by binding IKKγ and impairing NF-κB survival signaling. Genes & Development. 2004;18(12):1466–1481. doi: 10.1101/gad.1188204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Voloboueva LA, et al. Inflammatory response of microglial BV-2 cells includes a glycolytic shift and is modulated by mitochondrial glucose-regulated protein 75/mortalin. FEBS Letters. 2013;587(6):756–762. doi: 10.1016/j.febslet.2013.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nareika A, et al. Sodium lactate increases LPS-stimulated MMP and cytokine expression in U937 histiocytes by enhancing AP-1 and NF-κB transcriptional activities. American Journal of Physiology - Endocrinology And Metabolism. 2005;289(4):E534–E542. doi: 10.1152/ajpendo.00462.2004. [DOI] [PubMed] [Google Scholar]
- 114.Morito D, Nagata K. ER stress proteins in autoimmune and inflammatory diseases. Frontiers in Immunology. 2012:3. doi: 10.3389/fimmu.2012.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bläß S, et al. The stress protein BiP is overexpressed and is a major B and T cell target in rheumatoid arthritis. Arthritis & Rheumatism. 2001;44(4):761–771. doi: 10.1002/1529-0131(200104)44:4<761::AID-ANR132>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 116.Bodman-Smith MD, et al. BiP, a putative autoantigen in rheumatoid arthritis, stimulates IL-10-producing CD8-positive T cells from normal individuals. Rheumatology. 2003;42(5):637–644. doi: 10.1093/rheumatology/keg204. [DOI] [PubMed] [Google Scholar]
- 117.Brownlie RJ, et al. Treatment of murine collagen-induced arthritis by the stress protein BiP via interleukin-4–producing regulatory T cells: A novel function for an ancient protein. Arthritis & Rheumatism. 2006;54(3):854–863. doi: 10.1002/art.21654. [DOI] [PubMed] [Google Scholar]
- 118.Corrigall VM, et al. Inhibition of antigen-presenting cell function and stimulation of human peripheral blood mononuclear cells to express an antiinflammatory cytokine profile by the stress protein BiP: Relevance to the treatment of inflammatory arthritis. Arthritis & Rheumatism. 2004;50(4):1164–1171. doi: 10.1002/art.20134. [DOI] [PubMed] [Google Scholar]
- 119.Corrigall VM, et al. The Human Endoplasmic Reticulum Molecular Chaperone BiP Is an Autoantigen for Rheumatoid Arthritis and Prevents the Induction of Experimental Arthritis. The Journal of Immunology. 2001;166(3):1492–1498. doi: 10.4049/jimmunol.166.3.1492. [DOI] [PubMed] [Google Scholar]
- 120.Panayi GS, V, Corrigall M. BiP regulates autoimmune inflammation and tissue damage. Autoimmunity Reviews. 2006;5(2):140–142. doi: 10.1016/j.autrev.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 121.Kitamura M. Biphasic, bidirectional regulation of NF-kappaB by endoplasmic reticulum stress. Antioxidants & redox signalling. 2009;11(9):2353–2364. doi: 10.1089/ars.2008.2391. [DOI] [PubMed] [Google Scholar]
- 122.Okamura M, et al. Suppression of cytokine responses by indomethacin in podocytes: a mechanism through induction of unfolded protein response. American Journal of Physiology - Renal Physiology. 2008;295(5):F1495–F1503. doi: 10.1152/ajprenal.00602.2007. [DOI] [PubMed] [Google Scholar]
- 123.Kernagis DN, Laskowitz DT. Evolving role of biomarkers in acute cerebrovascular disease. Annals of Neurology. 2012;71(3):289–303. doi: 10.1002/ana.22553. [DOI] [PubMed] [Google Scholar]
- 124.Chopp M, Li Y. Apoptosis in focal cerebral ischemia. Acta Neurochirurgica Supplementum. 1996;66:21–26. doi: 10.1007/978-3-7091-9465-2_4. [DOI] [PubMed] [Google Scholar]
- 125.Mattson MP, Culmsee C, Yu ZF. Apoptotic and antiapoptotic mechanisms in stroke. Cell and Tissue Research. 2000;301(1):173–187. doi: 10.1007/s004419900154. [DOI] [PubMed] [Google Scholar]
- 126.DeGracia D, et al. Translation arrest and ribonomics in post-ischemic brain: layers and layers of players. Journal of neurochemistry. 2008;106(6):2288–2301. doi: 10.1111/j.1471-4159.2008.05561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.DeGracia DJ, Hu BR. Irreversible translation arrest in the reperfused brain. J Cereb Blood Flow Metab. 2007;27(5):875–893. doi: 10.1038/sj.jcbfm.9600388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sharp FR, et al. HSP70 heat shock gene regulation during ischemia. Stroke. 1993;24(12 Suppl):I72–I75. [PubMed] [Google Scholar]
- 129.Vass K, Welch WJ, Nowak TS. Localization of 70-kDa stress protein induction in gerbil brain after ischemia. Acta Neuropathologica. 1988;77(2):128–135. doi: 10.1007/BF00687422. [DOI] [PubMed] [Google Scholar]
- 130.Kinouchi H, et al. Induction of 70-kDa Heat Shock Protein and hsp70 mRNA Following Transient Focal Cerebral Ischemia in the Rat. J Cereb Blood Flow Metab. 1993;13(1):105–115. doi: 10.1038/jcbfm.1993.13. [DOI] [PubMed] [Google Scholar]
- 131.Kinouchi H, et al. Induction of heat shock hsp70 mRNA and HSP70 kDa protein in neurons in the ‘penumbra’ following focal cerebral ischemia in the rat. Brain Research. 1993;619(1–2):334–338. doi: 10.1016/0006-8993(93)91630-b. [DOI] [PubMed] [Google Scholar]
- 132.Nowak TS. Localization of 70 kDa Stress Protein mRNA Induction in Gerbil Brain After Ischemia. J Cereb Blood Flow Metab. 1991;11(3):432–439. doi: 10.1038/jcbfm.1991.84. [DOI] [PubMed] [Google Scholar]
- 133.Duan S-r, et al. Ischemia induces endoplasmic reticulum stress and cell apoptosis in human brain. Neuroscience Letters. 2010;475(3):132–135. doi: 10.1016/j.neulet.2010.03.058. [DOI] [PubMed] [Google Scholar]
- 134.Jeyaseelan K, Lim KY, Armugam A. MicroRNA Expression in the Blood and Brain of Rats Subjected to Transient Focal Ischemia by Middle Cerebral Artery Occlusion. Stroke. 2008;39(3):959–966. doi: 10.1161/STROKEAHA.107.500736. [DOI] [PubMed] [Google Scholar]
- 135.Liu DZ, et al. Brain and blood microRNA expression profiling of ischemic stroke, intracerebral hemorrhage, and kainate seizures. J Cereb Blood Flow Metab. 2010;30(1):92–101. doi: 10.1038/jcbfm.2009.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tan K, et al. Expression profile of MicroRNAs in young stroke patients. PLoS ONE. 2009;4(11):e7689–e7689. doi: 10.1371/journal.pone.0007689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ren XP, et al. MicroRNA-320 Is Involved in the Regulation of Cardiac Ischemia/Reperfusion Injury by Targeting Heat-Shock Protein 20. Circulation. 2009;119(17):2357–2366. doi: 10.1161/CIRCULATIONAHA.108.814145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yin C, Salloum FN, Kukreja RC. A Novel Role of MicroRNA in Late Preconditioning: Upregulation of Endothelial Nitric Oxide Synthase and Heat Shock Protein 70. Circulation Research. 2009;104(5):572–575. doi: 10.1161/CIRCRESAHA.108.193250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shan ZX, et al. Upregulated expression of miR-1/miR-206 in a rat model of myocardial infarction. Biochemical and Biophysical Research Communications. 2009;381(4):597–601. doi: 10.1016/j.bbrc.2009.02.097. [DOI] [PubMed] [Google Scholar]
- 140.Tang Y, et al. MicroRNA-1 Regulates Cardiomyocyte Apoptosis by Targeting Bcl-2. International Heart Journal. 2009;50(3):377–387. doi: 10.1536/ihj.50.377. [DOI] [PubMed] [Google Scholar]
- 141.Yang B, et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med. 2007;13(4):486–491. doi: 10.1038/nm1569. [DOI] [PubMed] [Google Scholar]
- 142.Xu C, et al. The muscle-specific microRNAs miR-1 and miR-133 produce opposing effects on apoptosis by targeting HSP60, HSP70 and caspase-9 in cardiomyocytes. Journal of Cell Science. 2007;120(17):3045–3052. doi: 10.1242/jcs.010728. [DOI] [PubMed] [Google Scholar]
- 143.Lee ST, et al. MicroRNAs Induced During Ischemic Preconditioning. Stroke. 2010;41(8):1646–1651. doi: 10.1161/STROKEAHA.110.579649. [DOI] [PubMed] [Google Scholar]
- 144.Magenta A, et al. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death and Differentiation. 2011;18(10):1628–1639. doi: 10.1038/cdd.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]