

Abstract
Mitochondria dysfunction plays a significant role in the apoptosis of retinal cells. Diabetes activates retinal matrix metalloproteinases (MMP-9 and MMP-2), damages retinal mitochondria and activates the apoptotic machinery. This study is to investigate the temporal relationship between the activation of retinal MMPs and mitochondria damage in the development of diabetic retinopathy. Time course of activation of cytosolic MMP-9 and MMP-2 was investigated in the retinal endothelial cells incubated in high glucose for 6–96 h, and correlated with their mitochondrial accumulation and mitochondrial damage. This was confirmed in the retina from rats diabetic for 15 days to ~12 months (streptozotocin-induced). The results show that the activation of cytosolic MMP-9 and MMP-2 is an early event, which is followed by their accumulation in the mitochondria. Increased mitochondrial MMPs dysfunction them and begin to damage their DNA, which initiates a vicious cycle of reactive oxygen species. Thus, modulation of these gelatinase MMPs by pharmacological agents during the early stages of diabetes could provide a strategy to inhibit the development of diabetic retinopathy.
Keywords: Diabetes, Matrix metalloproteinase, Mitochondria, Retinopathy
1. Introduction
Matrix metalloproteinases (MMPs), a family of proteolytic enzymes which were originally characterized only by their cytosolic localization and ability to degrade extracellular matrix proteins, have now been identified in many intracellular organelles including nucleus and mitochondria [1–4]. The gelatinase MMPs (MMP-9 and MMP-2) are secreted as proenzymes, and their activation requires proteolytic processing. In diabetes increased levels of these MMPs are reported in many tissues, including cardiomyoctes, kidney, retina, plasma, fibrovascular tissues and lung [1,4–7], and are considered to be involved in the neovascularization. Our recent work has shown that these MMPs, in addition to contributing to the neovascularization associated with diabetic retinopathy, in the background stage of the disease their activation facilitates apoptosis of retinal capillary cells [8–11].
Diabetes damages the retinal mitochondria and elevates superoxide levels [12–14]. Our recent work has shown that in pathogenesis of diabetic retinopathy, activated MMP-9 and MMP-2 damage the mitochondria disturbing the integrity, and activating the apoptotic machinery [4,9,10]. Furthermore, the retinal vasculature of diabetic mice with MMP-9 gene abrogated is protected from accelerated apoptosis and the development of histopathology characteristic of diabetic retinopathy [4]. In the development of diabetic retinopathy, increase in oxidative stress in the retina precedes mitochondria damage and capillary cell apoptosis [12], and a compensatory mechanism involving mitochondria biogenesis and repair system helps protect the mitochondria in the initial stages of the hyperglycemia. However, sustained hyperglycemic insult overwhelms this compensatory mechanism damaging the mitochondria and initiating the apoptotic machinery [12,15]. Whether the activation of retinal MMPs in diabetes precedes mitochondria damage remains to be established.
In this study, we have investigated the temporal relationship between the activation of retinal gelatinase MMPs (MMP-9 and MMP-2) and the damage of mitochondria in diabetes using isolated retinal endothelial cells, exposed to high glucose insult for various time points, and also in the retina of rats diabetic for 15 days to ~12 months.
2. Methods
2.1. Retinal endothelial cells
Endothelial cells isolated from calf retina (BRECs) were cultured on polystyrene dishes coated with 0.1% gelatin. Cells were incubated for 6–96 h in Dulbecco’s Modified Eagle Medium (DMEM) containing 2% heat inactivated fetal bovine serum, 10% Nu-serum, 50 μg/ml heparin and 1 μg/ml endothelial growth factor with or without 20 mM glucose for 6–96 h [9,12,15]. At the end of the incubation, the cells were trypsinized, rinsed with PBS, and used for further analysis. Cells incubated in 20 mM mannitol, instead of 20 mM glucose, served as osmotic control.
2.2. Rats
Wistar rats (male, ~200 g) were made diabetic by streptozotocin injection and maintained diabetic for 15 days to ~12 months [8,12]. At the end of the desired duration of diabetes, rats were sacrificed and the retina was immediately isolated. Age-matched normal rats served as control, and each group had 7 or more rats. Treatment of rats was carried out as per the guidelines of the National Institute of Health Principles of Laboratory Animal Care, the Association for Research in Vision and Ophthalmology resolution on the use of animals in research, and the institutional guidelines.
2.3. RNA isolation and gene expression
RNA was extracted from BRECs or retina by Trizol reagent (Invitrogen, Carlsbad, CA, USA). RNA (1 μg) was utilized to make cDNA using the High Capacity cDNA reverse transcription kit from Applied Biosystem (Foster City, CA, USA). Gene expression of MMP- 9 in BRECs was assessed by TaqMan assay (primer NM_174744.2 – Applied Biosystems) using 18s rRNA as an internal control. Rest of the genes, including mtDNA encoded genes Cytb and ND6, were quantified by SYBR green-based real time PCR (qPCR) with melting curve analysis on ABI 7500 (Applied Biosystems) using gene-specific primers and β – actin as a housekeeping gene (Table 1). The quantification of the transcripts was performed by ddCt method as routinely performed in our laboratory [4,9,16].
Table 1.
Primers for the target genes.
| Target | Sequence | |
|---|---|---|
| BRECs mRNA | ||
| TIMP-1 | Forward | 5′-GTACCTGCGTCCCACCCCACC-3′ |
| Reverse | 5′-GGCAGGCAGGCCAGGTGGCGG-3′ | |
| Cytb | Forward | 5′-CGATACATACACGCAAACGG-3′ |
| Reverse | 5′-AGAATCGGGTAAGGGTTGCT-3′ | |
| ND6 | Forward | 5′-CGTGATAGGTTTTGTGGGGT-3′ |
| Reverse | 5′-GCCAGTAACAAATGCCCCTA-3′ | |
| β – actin | Forward | 5′-CGCCATGGATGATGATATTGC-3′ |
| Reverse | 5′-AAGCCGGCCTTGCACAT-3′ | |
| Rat-mRNA | ||
| MMP-9 | Forward | 5′-AAGGACGGTCGGTATTGGAAGTTCT-3′ |
| Reverse | 5′-CAGCAGCGTCTGGCCTGTGTA-3′ | |
| TIMP-1 | Forward | 5′-TGCCCCAACCCACCCACAGA-3′ |
| Reverse | 5′-GCTGGGGTGTAGGCGAACCG-3′ | |
| MMP-2 | Forward | 5′-CTGGCCGGAGTGACGGCTTC-3′ |
| Reverse | 5′-GTGCCCTGGAAGCGGAACGG-3′ | |
| TIMP-2 | Forward | 5′-AAGCCCTCCCTGAGCCGTGT-3′ |
| Reverse | 5′-GCCAAGCACCCCTCACCACA-3′ | |
| CytB | Forward | 5′-CCCACAGGATTAAACTCCGA-3′ |
| Reverse | 5′-GTTGGGAATGGAGCGTAGAA-3′ | |
| ND6 | Forward | 5′-CCCAGCCACCACTATCATTC-3′ |
| Reverse | 5′-CATCGTACTCCTGCTTGCTG-3′ | |
| β – actin | Forward | 5′-CCTCTATGCCAACACAGTGC-3′ |
| Reverse | 5′-CATCGTACTCCTGCTTGCTG-3′ | |
2.4. Isolation of mitochondria
Mitochondria were isolated using Mitochondria Isolation kit from Invitrogen (Pierce, Rockford, IL, USA) [4,17]. Mitochondrial pellet was rinsed with PBS and re-suspended in buffer containing protease inhibitor. Protein was quantified by the bicinchoninic acid protein assay (Sigma–Aldrich St. Louis, MO, USA).
2.5. Protein expression
Homogenate or mitochondria fraction (30–60 μg protein) was separated on an 8–20% SDS–PAGE and transferred to a nitrocellulose membrane. After blocking the membrane with 5% nonfat milk for 1 h, the membrane was incubated with the antibody against the protein of interest. The loading controls included β-actin for homogenate and Cox IV for mitochondria.
2.6. Activation of MMP-9 and MMP-2
Activation of MMP-9 and MMP-2 was quantified by in situ zymography using 8% non-reducing SDS–PAGE containing 0.1% gelatin, as previously reported by us [4,8,16,18]. The intensity of the pro- and active-bands was quantified using Kodak digitizing software.
Activation of MMP-9 in mitochondria was assessed by fluorescence kit (SensoLyte® Plus 520 MMP-9 Assay Kit, ANAspec Fremont, CA, USA) using 30–40 μg protein. The MMP-9 induced cleavage of a fluorogenic peptide was measured at 490 nm excitation and 520 nm emission wavelengths.
2.7. Mitochondrial reactive oxygen species (ROS)
Mitochondrial ROS were quantified in BRECs using MitoTracker Red (CM-H2XROS; Molecular Probes), a mitochondria-selective dye that emits fluorescence when oxidized. The cells exposed to 5 or 20 mM glucose for 6–96 h were incubated with 400 nm Mito-Tracker Red for 30 min and washed with PBS, followed by quantification of ROS at 579 nm excitation and 599 nm emission wavelengths [4,17].
2.8. Statistical analysis
Statistical analysis was performed using Sigma Stat software, and the data are expressed as means ± standard deviation. The Shapiro–Wilk test was used to test for normal distribution of the data, and the data that did not present normal distribution, Kruskal– Wallis test followed by Dunn’s test was applied. p value <0.05 was considered as statistically significant.
3. Results
3.1. Retinal endothelial cells
Within 6 h of high glucose exposure of BRECs, the activity of MMP-9 was increased by 30%, and the increase was almost 2-fold when the duration was extended to 24 h and beyond compared to the values obtained from cells incubated in normal glucose. Consistent with this, MMP-9 gene and protein expressions were also elevated as early as 6 h of glucose insult, and they remained elevated throughout the experiment (96 h). Increase in MMP-9 was accompanied by decrease in the mRNA level of TIMP1, an endogenous tissue inhibitor of MMP-9; within 6 h of high glucose exposure TIMP1 was decreased by ~40%, and at 24 h by >60% (Fig. 1A). In contrast, the accumulation of MMP-9 and its activity in the mitochondria, as determined, was not affected at 6 h of high glucose insult, but was slightly elevated (not significantly) at 24 h compared to the values obtained from cells incubated in normal glucose. However, at 48 h MMP-9 activity was significantly increased (Fig. 1B). In the same cell preparations, although increase in MMP-2 activity was not observed at 6 h of glucose exposure, at 24 h it was increased significantly, and continued to increase with extension of the duration. Increase in MMP-2 activity was over 60% at 48 h compared to the values obtained from the cells incubated in 5 mM glucose (data not shown). As reported previously [4,18], incubation of BRECs with mannitol did not increase these MMPs, suggesting that the activation of by these enzymes englucose was not due to increase in the osmolarity.
Fig. 1.
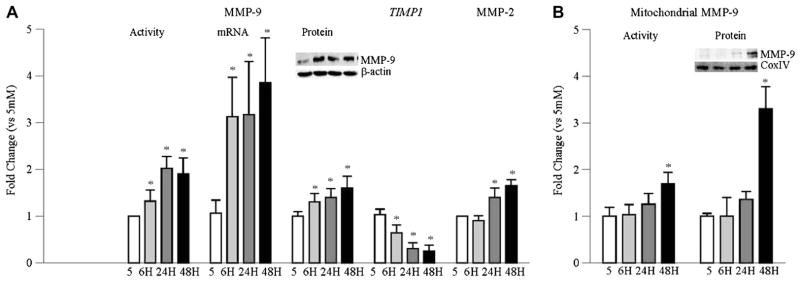
Time course of glucose-induced activation of MMP-9 and MMP-2 in retinal endothelial cells. MMP-9 activity was quantified in BRECs incubated in high glucose for 6– 96 h by in situ zymography, and the ratio of the active and the pro-active MMP-9 bands was plotted in the accompanying histogram. mRNA levels of MMP-9 and TIMP1 were quantified by real time qPCR. Protein expression of MMP-9 was determined by western blot technique. MMP-2 activity was performed by in situ zymography. (B) MMP-9 accumulation in mitochondria was assessed by western blot using Cox IV as loading control; the accompanying blot is representative of 3 different experiments. Its activity was assessed in 30–40 μg mitochondria fraction by fluorescence kit. Each measurement was performed in duplicate using cells from 3–5 different preparations, and the values are represented as mean ± SD 5 = 5 mM glucose; 6 h, 24 h, 48 h = 20 mM glucose treatment for 6 h, 24 h, and 48–96 h respectively.* p < 0.05 compared to 5 mM glucose.
To investigate the effect of glucose on mitochondrial ROS, fluorescence emitted by a mitochondria-specific dye was measured. As shown in Fig. 2, levels of mitochondrial ROS, quantified by fluorescence emitted by a mitochondria-specific dye, at 24 h of high glucose insult were slightly higher compared to the values obtained from cells incubated in normal glucose, but the values didn’t achieve statistical significance. However, after 48 h of high glucose insult, mitochondrial ROS were increased by 80%. Consistent with this, mtDNA transcription was not altered at 24 h, but was decreased by 25–35% at 96 h as shown by decreased expressions of mtDNA encoded proteins, ND6 of complex I and Cytb of complex III (Fig. 2).
Fig. 2.
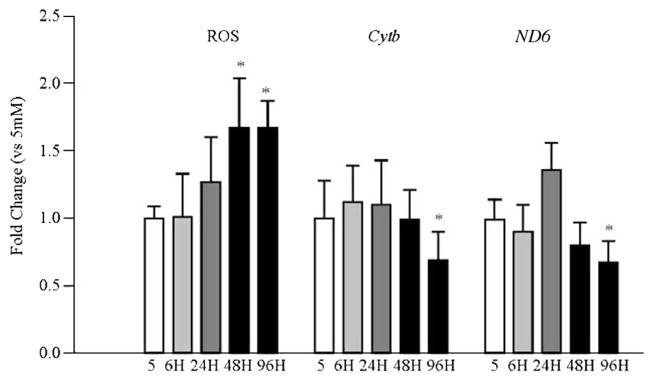
Temporal relationship between high glucose exposure and mitochondrial ROS and mtDNA encoded proteins in retinal endothelial cells. Mitochondrial ROS were quantified in BRECs incubated with MitoTracker Red at the end of the desired duration of high glucose insult. The resultant fluorescence was quantified at 579 nm excitation and 599 nm emission wavelengths. The transcripts of mtDNA-encoded proteins Cytb and ND6 were quantified by real time qPCR using β-actin as an internal control. Values are represented as mean ± SD obtained from 3–4 cells preparations, and each measurement is made at least in duplicate. *p < 0.05 compared with 5 mM glucose.
3.2. Rat retina
Both mRNA and protein expression of MMP-9 in the rat retina was significantly elevated as early as 15 days of diabetes, and remained elevated during the entire duration of the experiment (~12 months of diabetes) compared to the values obtained from age-matched normal rats. This increase in MMP-9 was accompanied by ~50% decrease in TIMP1 mRNA at 2 months of diabetes (Fig. 3A). However, in the retinal mitochondria, MMP-9 levels remained unchanged at 15 days of diabetes, but at 2 months mitochondrial MMP-9 was slightly elevated, and at 6 months the values were increased by 2-fold compared to those obtained from age-matched normal rat retina (Fig. 3B).
Fig. 3.
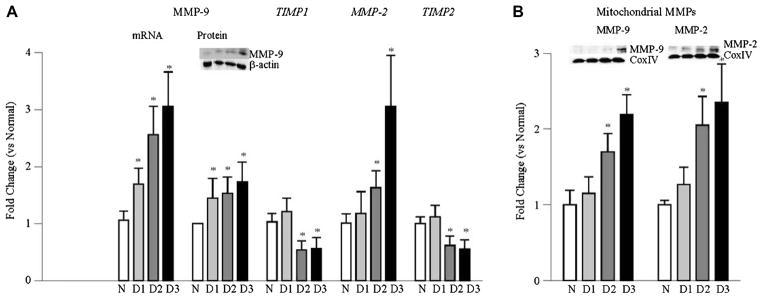
Effect of duration of diabetes in rats on retinal MMP-9 and MMP2: (A) Gene expressions of MMP-9, MMP-2 and their intracellular inhibitors TIMP1 and TIMP2 respectively, were quantified by real time qPCR in the retina from rats diabetic from 15 days to 6–12 months using gene specific rat primers and β-actin as an internal control. Protein expression of MMP-9 was quantified by western blot technique. (B) Accumulation of MMP-9 and MMP-2 in the mitochondria was quantified by western blot technique using Cox IV as loading control. Each measurement was made in 6–12 rats/group, and the values are represented as mean ± SD *p < 0.05 compared to normal. Values obtained from normal rats are considered as 1. N = normal, D1, D2 and D3 = rats diabetic for 15 days, 2 months and 6–12 months respectively.
Consistent with the results from retinal endothelial cells, 70% increase in retinal MMP-2 was observed at 2 months and ~3-fold at 6–12 months of diabetes. Increase in MMP-2 was accompanied by 40% decrease in TIMP2, an internal inhibitor of MMP-2 (Fig. 3A). In concordance with MMP-9, MMP-2 in the mitochondria was elevated significantly at 2 months of diabetes, and continued to be elevated by at 6–12 months of diabetes compared to the values obtained from age-matched normal rats (Fig. 3B). Furthermore, in accordance with our previous results demonstrating normal retinal mtDNA at 2 months of diabetes in rats despite increase in oxidative stress [15,19], transcripts of Cyt b and ND6, the proteins encoded by mtDNA, also remained unchanged, but at 6–12 months of diabetes, they were decreased by 40% compared to the values obtained from age-matched normal rats.
4. Discussion
Activation of MMPs is implicated in the development of a number of chronic diseases such as cancer, atherosclerosis and diabetes [1,4,7,20–22]. Although MMP-2 and MMP-9 have gelatinase activity and angiogenic properties, we have shown that these MMPs have possibly dual role in the development of diabetic retinopathy; in the early stages of the disease (pre-neovascularization), MMP-2 and MMP-9 facilitate the apoptosis of retinal capillary cells by damaging the mitochondria, and during the later stages of the disease, they help in neovascularization [4,8–10]. In the pathogenesis of the disease, although increase in ROS is an early event, the damage of retinal mitochondria is seen at later stages of the disease [12,15]. Here we show that the activation of cytosolic MMP-9 and MMP-2 in the retina is an early event, however, their accumulation in the mitochondria is not seen till the duration of diabetes is extended to two months. Furthermore, despite increase in the mitochondrial MMPs, mitochondrial dysfunction and damage to mtDNA are not observed till the duration of diabetes is further extended to 6 months. Similar phenomenon is also observed in retinal endothelial cells exposed to high glucose. These results suggest that diabetes activates cytosolic MMPs in the retina, and these MMPs damage the mitochondria membrane, and with time, MMPs accumulate in the mitochondria and begin to damage their structure and mtDNA, which initiates a vicious cycle of ROS.
Diabetic retinopathy is a progressive disease, and increase in retinal oxidative stress precedes mitochondrial dysfunction and capillary cell apoptosis. Despite increased oxidative stress at 2 months of diabetes [15,23], retinal mitochondria remain intact, but at 6 months of diabetes they are dysfunctional and capillary cells becomes apoptotic suggesting the role of mitochondrial dysfunction in the accelerated apoptosis of retinal capillary cells, a phenomenon that can predict the development of microvascular histopathology of diabetic retinopathy [12,24,25]. Activation of MMP-9 is modulated by a complex mechanism involving H-Ras mediated signaling steps, H-Ras-ERK-NF-kB pathway [8,18,26,27], and activation of redox-sensitive NF-kB in the retina in diabetes is an early event which eventually contributes to vascular apoptosis [23]. Diabetic mice with MMP-9 gene abrogated have normal retinal mitochondria, and their vasculature is protected from accelerated apoptosis and the pathology associated with diabetic retinopathy [4]. Similarly, activation of MMP-2 also facilitates apoptosis of retinal capillary cells by disrupting mitochondrial membranes further corroborating the role of mitochondrial MMPs in the development of diabetic retinopathy [9,10].
The results presented here show that even a short duration of high glucose insult (15 days in in vivo and 6 h in in vitro model) increases total MMP-9 and activates the enzyme, and the enzyme remains active throughout the experiment (6–12 months in rodents and 96 h in cells). This suggests that activated MMPs tries to enter into the mitochondria membrane, possibly by damaging connexin 43 [10], and once they enter into the mitochondria, they damage their structure and integrity. However, in the initial stages of the damage, increased mtDNA biogenesis and repair mechanisms compensate for the ROS-induced damage and protect the mitochondria. However, sustained hyperglycemic insult overwhelms the protective mechanism and mitochondrial ROS levels begin to increase damaging mtDNA, and ultimately resulting in dysfunctional electron transport system [15].
Thus, in conclusion, this study suggests that in the pathogenesis of diabetic retinopathy activation of gelatinase MMPs (MMP-9 and MMP-2) is an early event. Activated MMPs enter into the mitochondria, structure and integrity of the mitochondria are damaged and cytochrome C leaks out into the cytosol, and this activates the apoptotic machinery resulting in the loss of capillary cells and the development of diabetic retinopathy [12,13]. Due to damaged mitochondria, ROS levels continue to increase, and sustained increase in ROS begins to damage mtDNA. The damaged mtDNA continues to dysfunction the ETC, and the vicious cycle of ROS continues (Fig. 4). Thus, our results further strengthens the importance of early glucose control for diabetic patients, and suggests that modulation of these gelatinase MMPs by pharmacological agents during the early stages of diabetes could provide a strategy to prevent mitochondrial dysfunction and the development of diabetic retinopathy.
Fig. 4.
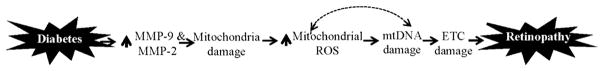
Diabetes activates cytosolic MMP-9 and MMP-2, and activated MMPs damage the mitochondria. ROS production is increased and mtDNA is damaged resulting in cell loss and the development of diabetic retinopathy. To make the bad situation worse, due to damaged mtDNA, transcription of mtDNA-encoded proteins of the electron transport chain (ETC) is impaired further exacerbating ROS production and cell loss.
Acknowledgments
We thank Doug Putt for technical assistance. This study was supported in parts by Grants from the National Institutes of Health, Juvenile Diabetes Research Foundation, the Thomas Foundation, and Research to Prevent Blindness.
References
- 1.Moshal KS, Tipparaju SM, Vacek TP, Kumar M, Singh M, Frank IE, Patibandla PK, Tyagi N, Rai J, Metreveli N, Rodriguez WE, Tseng MT, Tyagi SC. Mitochondrial matrix metalloproteinase activation decreases myocyte contractility in hyperhomocysteinemia. Am J Physiol Heart Circ Physiol. 2008;295:H890–H897. doi: 10.1152/ajpheart.00099.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cauwe B, Opdenakker G. Intracellular substrate cleavage: a novel dimension in the biochemistry, biology and pathology of matrix metalloproteinases. Crit Rev Biochem Mol Biol. 2010;45:351–423. doi: 10.3109/10409238.2010.501783. [DOI] [PubMed] [Google Scholar]
- 3.Klein T, Bischoff R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids. 2011;41:271–290. doi: 10.1007/s00726-010-0689-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kowluru RA, Mohammad G, Dos Santos JM, Zhong Q. Abrogation of MMP9 gene protects against the development of retinopathy in diabetic mice by preventing mitochondrial damage. Diabetes. 2011;60:3023–3033. doi: 10.2337/db11-0816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tyagi SC, Hayden MR. Role of nitric oxide in matrix remodeling in diabetes and heart failure. Heart Fail Rev. 2003;8:23–28. doi: 10.1023/a:1022138803293. [DOI] [PubMed] [Google Scholar]
- 6.Yao XM, Ye S, Zai ZM, Chen Y, Li XC, Yang GW, Wang YX, Chen K. Simvastatin protects diabetic rats against kidney injury through the suppression of renal matrix metalloproteinase-9 expression. J Endocrinol Invest. 2009;33:292–296. doi: 10.1007/BF03346588. [DOI] [PubMed] [Google Scholar]
- 7.Weng Y, Cai M, Zhu J, Geng J, Zhu K, Jin X, Ding W. Matrix metalloproteinase activity in early-stage lung cancer. Onkologie. 2013;36:256–259. doi: 10.1159/000350304. [DOI] [PubMed] [Google Scholar]
- 8.Kowluru RA. Role of matrix metalloproteinase-9 in the development of diabetic retinopathy and its regulation by H-Ras. Invest Ophthalmol Vis Sci. 2010;51:4320–4326. doi: 10.1167/iovs.09-4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mohammad G, Kowluru RA. Matrix metalloproteinase-2 in the development of diabetic retinopathy and mitochondrial dysfunction. Lab Invest. 2010;90:1365–1372. doi: 10.1038/labinvest.2010.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mohammad G, Kowluru RA. Novel role of mitochondrial matrix metalloproteinase-2 in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2011;52:3832–3841. doi: 10.1167/iovs.10-6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kowluru RA, Zhong Q, Santos JM. Matrix metalloproteinases in diabetic retinopathy: potential role of MMP-9. Expert Opin Investig Drugs. 2012;21:797–805. doi: 10.1517/13543784.2012.681043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kowluru RA, Abbas SN. Diabetes-induced mitochondrial dysfunction in the retina. Inves Ophthalmol Vis Sci. 2003;44:5327–5334. doi: 10.1167/iovs.03-0353. [DOI] [PubMed] [Google Scholar]
- 13.Kowluru RA. Diabetic retinopathy: mitochondrial dysfunction and retinal capillary cell death. Antioxid Redox Signal. 2005;7:1581–1587. doi: 10.1089/ars.2005.7.1581. [DOI] [PubMed] [Google Scholar]
- 14.Santos JM, Mohammad G, Zhong Q, Kowluru RA. Diabetic retinopathy, superoxide damage and antioxidant. Curr Pharm Biotechnol. 2011;12:352–361. doi: 10.2174/138920111794480507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santos JM, Tewari S, Kowluru RA. A compensatory mechanism protects retinal mitochondria from initial insult in diabetic retinopathy. Free Rad Biol Med. 2012;53:1729–1737. doi: 10.1016/j.freeradbiomed.2012.08.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhong Q, Kowluru RA. Regulation of matrix metallopeptidase-9 by epigenetic modifications, and the development of diabetic retinopathy. Diabetes. 2013;62:2559–2568. doi: 10.2337/db12-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tewari S, Santos JM, Kowluru RA. Damaged mitochondrial DNA replication system and the development of diabetic retinopathy. Antioxid Redox Signal. 2012;17:492–504. doi: 10.1089/ars.2011.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mohammad G, Kowluru RA. Diabetic retinopathy and signaling mechanism for activation of matrix metalloproteinase-9. J Cell Physiol. 2012;227:1052– 1061. doi: 10.1002/jcp.22822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kowluru RA, Engerman RL, Kern TS. Abnormalities of retinal metabolism in diabetes and galactosemia IV. antioxidant defense system. Free Rad Biol Med. 1997;22:587–592. doi: 10.1016/s0891-5849(96)00347-4. [DOI] [PubMed] [Google Scholar]
- 20.Das A, McGuire PG, Eriqat C, Ober RR, DeJuan E, Williams GA, McLamore A, Biswas J, Johnson DW. Human diabetic neovascular membranes contain high levels of urokinase and metalloproteinase enzymes. Invest Ophthalmol Vis Sci. 1999;40:809–813. [PubMed] [Google Scholar]
- 21.Rosenberg GA. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol. 2009;8:205–2016. doi: 10.1016/S1474-4422(09)70016-X. [DOI] [PubMed] [Google Scholar]
- 22.Huntley GW. Synaptic circuit remodelling by matrix metalloproteinases in health and disease. Nat Rev Neurosci. 2012;13:743–757. doi: 10.1038/nrn3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kowluru RA, Koppolu P, Chakrabarti S, Chen S. Diabetes-induced activation of nuclear transcriptional factor in the retina, and its inhibition by antioxidants. Free Radic Res. 2003;37:1169–1180. doi: 10.1080/10715760310001604189. [DOI] [PubMed] [Google Scholar]
- 24.Mizutani M, Kern TS, Lorenzi M. Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J Clin Invest. 1996;97:2883–2890. doi: 10.1172/JCI118746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kern TS, Tang J, Mizutani M, Kowluru R, Nagraj R, Lorenzi M. Response of capillary cell death to aminoguanidine predicts the development of retinopathy: comparison of diabetes and galactosemia. Invest Ophthalmol Vis Sci. 2000;41:3972–3978. [PubMed] [Google Scholar]
- 26.Lee KW, Kim MS, Kang NJ, Kim DH, Surh YJ, Lee HJ, Moon A. H-Ras selectively up-regulates MMP-9 and COX-2 through activation of ERK1/2 and NF-kappaB: an implication for invasive phenotype in rat liver epithelial cells. Int J Cancer. 2006;119:1767–1775. doi: 10.1002/ijc.22056. [DOI] [PubMed] [Google Scholar]
- 27.Mohammad G, Kowluru RA. The role of Raf-1 kinase in diabetic retinopathy. Expert Opin Ther Targets. 2011;15:537–564. doi: 10.1517/14728222.2011.553604. [DOI] [PMC free article] [PubMed] [Google Scholar]