

Abstract
A third-generation polyamidoamine dendrimer (PAMAM G3) was used as a macromolecular carrier for pyridoxal and biotin. The binary covalent bioconjugate of G3, with nine molecules of biotin per one molecule of G3 (G39B), and the ternary covalent bioconjugate of G3, with nine biotin and ten pyridoxal molecules (G39B10P), were synthesized. The biotin and pyridoxal residues of the bioconjugate were available for carboxylase and transaminase enzymes, as demonstrated in the conversion of pyruvate to oxaloacetate and alanine to pyruvate, respectively, by in vitro monitoring of the reactions, using 1H nuclear magnetic resonance spectroscopy. The toxicity of the ternary bioconjugate (BC-PAMAM) was studied in vitro on BJ human normal skin fibroblasts and human squamous cell carcinoma (SCC-15) cell cultures in comparison with PAMAM G3, using three cytotoxicity assays (XTT, neutral red, and crystal violet) and an estimation of apoptosis by confocal microscopy detection. The tests have shown that BC-PAMAM has significantly lower cytotoxicity compared with PAMAM. Nonconjugated PAMAM was not cytotoxic at concentrations up to 5 μM (NR) and 10 μM (XTT), and BC-PAMAM was not cytotoxic up to 50 μM (both assays) for both cell lines. It has been also found that normal fibroblasts were more sensitive than SCC to both PAMAM and BC-PAMAM. The effect of PAMAM and BC-PAMAM on the initiation of apoptosis (PAMAM in fibroblasts at 5 μM and BC-PAMAM at 10 μM in both cell lines) corresponded with cytotoxicity assays for both cell lines. We concluded that normal fibroblasts are more sensitive to the cytotoxic effects of the PAMAM G3 dendrimer and that modification of its surface cationic groups by substitution with biologically active molecules significantly decreases that effect, confirming that PAMAM G3 is a useful candidate as a carrier for active biocompound delivery.
Keywords: ternary bioconjugate, pyridoxal, biotin, enzymatic transamination, enzymatic carboxylation, apoptosis, normal fibroblasts, squamous carcinoma cells
Introduction
The current search for nanomolecular carriers for medical application is a main goal of nanomedicine. Polyamidoamine dendrimers (PAMAM dendrimers) belong to the vehicles most explored as a result of their specific properties: strictly defined size and molecular weight, spherical shape, low intrinsic viscosities, high solubility in various solvents, and high density of surface-localized functional groups. Full-generation PAMAM dendrimers are furnished with a number of surface amine groups, depending on generation, which are accessible for the covalent attachment of nanoscopic and subnanoscopic reagents; for example, prodrug molecules. Such binary bioconjugates can be further functionalized to obtain ternary, quaternary, and higher multicomponent, biologically active molecules. Recently, so-called “tecto-dendrimers” have been obtained by the introduction of covalent linkage between building blocks of dendrimers to obtain higher-level architectural structures.1 Tecto-dendrimer PAMAM G5 core-PAMAM G2.5 shells constructed by Schilrreff et al revealed a specific and selective toxicity for human epidermal melanoma (SK-Mel-28) cells.2
The PAMAM dendrimers are promising nanoscale carriers for small drug–guest molecules, as they are encapsulated within the dendritic structure and are particularly nonpolar, increasing their solubility and bioavailability, as well as their toxicity. Both drug encapsulation and bioconjugation strategies have been extensively tested with various drugs.3,4 Much attention was drawn to the study of their fate in mammalian tissues, and the current state of the art was extensively reviewed.5–9 Dendrimer biological properties are dependent on the chemistry of the core but are most strongly influenced by the nature of its surface. The biocompatibility and toxicity of dendrimers are found to be generation-dependent, with higher generations being the most toxic. Comparative toxicity studies on Caco-2 cells have shown a significantly lower cytotoxicity of the anionic than cationic PAMAM dendrimers, as cationic PAMAM dendrimers are interacting with the cell membrane, causing increases in permeability.
Full-generation PAMAM dendrimers furnished with surface amine groups are relatively strong bases and are polycationic at physiological pH. Therefore, they are able to interact with negatively charged biological membranes, as well as nucleic acids, disturbing their biological function. It is known that toxicity of the PAMAM dendrimers decreases on blocking amine groups. Reduction of the toxicity of cationic dendrimers was achieved by partial surface derivatization with chemically inert functionalities, including polyethylene glycol, fatty acids, or amino acids.7,10,11 In addition, dendrimer–drug complexes may be used to reduce the cytotoxicity of dendrimers, with improvements in the stability, permeability, and cellular uptake of the drug.
Recent studies have shown that possible mechanisms of PAMAM cationic and anionic dendrimer permeability involved enhancement of paracellular transport by modulation of the tight junctions of proteins, together with an associated increase in membrane porosity and transcellular translocation by endocytosis, via caveolae-dependent, clathrin-dependent, and macropinocytosis pathways, depending on size, charge, surface modifications, and concentration.12–14
Particular attention has been given to the study of transepithelial transport of PAMAM dendrimers to evaluate their role as drug carriers penetrating cellular barriers (eg, intestinal or the blood–brain barrier). In particular, biotin has been chosen as a targeting molecule for small molecule transport through the blood–brain barrier in central nervous system disorder diagnosis and therapy.15,16
The targeting of cancer cells with biotin–dendrimer conjugates for cancer therapy and cancer diagnosis is a promising field of investigation because of the much higher cellular uptake of the cisplatin-loaded biotinylated PAMAM dendrimers and the bifunctional conjugate (dendrimer–biotin– fluorescein isothiocyanate) into ovarian cancer cell lines17,18 and HeLa cells than seen for the conjugate without biotin.19 It has been reported that biotin uptake is provided by the sodium-dependent multivitamin transporter present in many cells, including human keratinocytes, blood mononuclear cells, and epithelial cells of intestine, liver, and kidney.18
The application of dendrimers as topical enhancers for transdermal drug delivery also has been considered, as modified PAMAM dendrimers can significantly improve the transdermal bioavailability of compounds such as nonsteroidal anti-inflammatory drugs and antiarthritic and anticancer drugs, which often cause gastrointestinal adverse effects.20–22
Some hyperproliferative skin disorders, such as psoriasis and atopic eczema, are treated by photochemotherapy with oral psoralens such as a combination of 8-methoxypsoralene (8-MOP) and ultraviolet A radiation (PUVA therapy). Oral or topical administration of 8-MOP causes many adverse effects. It has been proven, however, that full-generation G3 and G4 PAMAM dendrimers may serve as useful carriers for 8-MOP, increasing its permeability, as evidenced in vitro and in vivo.23–25 The enhancement of transdermal delivery of tamsulosin,26 as well as the promotion of the transdermal diffusion of riboflavin by G2-G5 PAMAM,27 were also shown.
The aim of this study was the investigation of the biological properties of multicomponent bioconjugate PAMAM G3, which provides 32 amine groups accessible on the surface to which to covalently attach drug and enzyme cofactor molecules. A ternary bioconjugate was constructed in which 60% of the amine group of G3 was substituted with biotin and pyridoxal, with the remaining amine groups still rendering the bioconjugate cationic macromolecule. Comparative studies of biocompatibility and cytotoxicity of the G3 PAMAM dendrimer and its ternary bioconjugate were performed, and in vitro cytotoxicity also was evaluated, using normal and transformed human cell cultures. Cellular morphology and apoptosis were visualized by confocal microscopy.
Materials and methods
All solvents were distilled before use. Biochemical reagents, including acetyl coenzyme A trilithium salt, (+)–biotin N-hydroxysuccinimide ester, adenosine-5′-triphosphate magnesium salt (ATP), α-Ketoglutaric acid, L-alanine (L-Ala), and pyruvic acid (Pyr), as well as enzymes, including glutamic-pyruvic transaminase from porcine heart (EC 2.6.1.2, activity of 75 U/mg protein) and pyruvate carboxylase (CL) from bovine liver (EC 6.4.1.1, activity of 23 U/mg protein) in buffered glycerol solution, were purchased from Sigma-Aldrich (St Louis, MO, USA). Other compounds were synthesized using high vacuum-nitrogen lines to operate with synthesized materials at ambient temperatures.
Stock cultures of BJ human normal fibroblasts (ATCC-CRL-2522) and human squamous cell carcinoma (SCC-15) cells (ATCC-CRL-1623) were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). Eagle’s minimum essential medium, Dulbecco’s Modified Eagle’s Medium and Ham’s (DMEM-F12), fetal calf serum, penicillin and streptomycin, trypsinethylenediaminetetraacetic acid solution, and phosphate-buffered saline (PBS) with and without magnesium and calcium ions also were products of ATCC. Hydrocortisone, XTT sodium salt (2,3-bis[2-methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxanilide inner salt), Phenazine methosulfate (PMS), N-methyl dibenzopyrazine methyl sulfate, crystal violet (CV) (4-[(4-dimethylaminophenyl)-phenyl-methyl]-N,N-dimethyl-aniline), neutral red (NR; 3-amino-m-dimethylamino-2-methyl-phenazine hydrochloride), trypan blue, and other chemicals and buffers were of cellular grade and provided by Sigma-Aldrich (St Louis, MO, USA) or Roth (Carlo Roth GmbH+Co, KG, Karlsruhe, Germany). Fluorescent marker DAPI (4′,6-diamidino-2-phenylindole, dihydrochloride) was purchased from Life Technologies (Carlsbad, CA, USA). Mouse antibody (immunoglobulin G [IgG] κ) human-specific against cleaved poly(ADP-ribose) polymerase (PARP; Asp 214) conjugated with AlexaFluor 647 were from BD Pharmingen (BD Biosciences, San Jose, CA, USA). All other cell culture materials were purchased from Corning Incorporated (Corning, NY, USA) or Sigma-Aldrich (St Louis, MO, USA).
Synthesis of the PAMAM G3–biotin and PAMAM G3–pyridoxal–biotin ternary bioconjugates
PAMAM G3 was synthesized according to the Tomalia et al protocol, adding methyl acrylate into the ethylenediamine core in an initial step, followed by further alternate condensation with ethylenediamine and the addition of methyl acrylate steps to finally obtain G3 at a 30 g product scale.28
The crude product was purified by extensive dialysis of water solution against water (a cellulose 4–6-kD cutoff ZelluTrans/Roth membrane was used), as before,29 to remove low-molecular-weight substrates as well as traces of lower-generation dendrimers.
The G39B (where “B” stand for biotin) bioconjugate was synthesized as follows: 223 mg N-hydroxysuccinimide (NHS)–biotin (0.653 mmoL) dissolved in 5 cm3 dimethyl sulfoxide was added dropwise with vigorous stirring into a solution of 450 mg G3 (0.065 mmoL) in 20 cm3 methanol. The mixture was stirred for 4 hours at room temperature, and then the solvents were removed in vacuo, the solid residue was dissolved in water (15 cm3), and this solution was triple-dialyzed against water, as before. Water was removed under vacuum, and the solid residue was identified as a G3 conjugate with an average of nine biotin substituents per single G3 molecule (G39B average molecular weight =8,946 g/moL) by 1H nuclear magnetic resonance (NMR) spectroscopy (yield, 540 mg, 92.9%).
NHS-biotin: 1H NMR (300 MHz, dimethyl sulfoxide-d6): δ (ppm) 6.42 (10b, s, 1H); 6.36 (9b, s, 1H); 4.30 (8b, m, 1H); 4.15 (9b, m, 1H); 3.12 (6b, m, 1H); 2.84–2.56 (7b, AB spectrum, m, 2H); 2.68 (2b, t, 2H); 1.7–1.4 (3b, 4b, 5b, m, 6H).
G39B 1H NMR (300 MHz, dimethyl sulfoxide-d6): δ (ppm) 6.48 (10b, s, 1H); 6.39 (9b, s, 1H); 4.30 (8b, m, 1H); 4.14 (8b, m, 1H); 2.07 (2b, t, 2H); 1.68–1.21 (3b, 4b, 5b, m, 6H). Resonances of 6b and 7b are overlapped with high intensity resonances from CH2 of PAMAM G3 dendrimer (2.77–2.20).
G39B 1H NMR (300 MHz, deuterium oxide): δ (ppm) 4.45 (8b, m, 1H); 4.27 (8b, m, 1H); 2.09 (2b, t, 2H); 1.62–1.20 (3b, 4b, 5b, m, 6H).
Ternary bioconjugate G39B10P (where P stands for pyridoxal) was obtained by the condensation of pyridoxal hydrochloride with amine groups of G39B bioconjugate, as follows: 70.8 mg pyridoxal hydrochloride (0.347 mmoL) in 10 cm3 methanol was added dropwise into a solution of 310 mg G39B (0.0346 mmoL) in 25 cm3 methanol, with vigorous stirring. The mixture was heated to 50°C (0.5 hour) and then dialyzed against methanol two times within 24 hours. The solvents were removed under extensive evaporation under vacuum, and 336 mg solid G39B10P was obtained (yield, 336 mg, 93.0%; average molecular weight =10,437 g/moL), as identified by 1H NMR spectroscopy. 1H NMR (300 MHz, methanol-d4): δ (ppm) 8.92 (7p, s, 10H); 7.86 (6p, s, 10H); 4.78 (C=NH-CH2−CH2 PAMAM, s, 20 H); 4.51 (9b, m, 9H); 4.33 (8b, m, 9H); 3.85 (C=NH-CH2−CH2 PAMAM, s, 20 H); 2.23 (2b, t, 18H). Resonances of 6b and 7b are overlapped with high intensity resonances from CH2 of PAMAM G3 dendrimer (2.45–2.25). 1H NMR (300 MHz, deuterium oxide): δ (ppm) 8.57 (7p, s, 10H); 7.36 (6p, s, 10H); 4.42 (8b, m, 9H); 4.21 (8b, m, 9H); 2.02 (2b, t, 18H); 1.58–1.08 (3b, 4b, 5b, m, 48H).
Enzymatic tests
Carboxylation of pyruvate into oxaloacetate
The carboxylation of pyruvate with bicarbonate catalyzed by pyruvate CL from bovine liver in glycerol and aqueous buffer at pH 7.4 (EC 6.4.1.1) was performed in D2O on the 1H NMR scale (Figure 1B). A solution containing 40 mM Pyr, 40 mM HCO3−, 25 mM ATP, and 0.25 mM acetyl coenzyme A (AcCoA) was prepared and divided into two portions. A biotin (2.5 mM) was added to the first portion of the solution, and G39B was added to the second part (0.25 mM). Next, the pH of both solutions was adjusted to 7.4, a CL solution (10 μL, 0.2 units) was injected in to both samples, and conversion of Pyr was monitored by disappearance of pyruvate methyl group resonance at 2.27 ppm. The enzymatic system was active by 1 day; an alternate addition of ATP and Pyr into the NMR tube triggered conversion 3 times within 16 hours in both systems (ie, the control experiment with free biotin and biotin conjugate G39B). The concentration of reagents was about ten times higher than those used by Utter and Keech.30
Figure 1.

Enzyme-catalyzed pathways of amine groups transfer from L-alanine (Ala) to α-ketoglutarate (α-KG) into L-glutamate (Glu) and pyruvate (Pyr) (part A), and carboxylation of pyruvate into oxaloacetate (OxAl) (part B).
Notes: Part A adapted with permission from Filipowicz A, Wołowiec S. Bioconjugates of PAMAM dendrimers with trans-retinal, pyridoxal, and pyridoxal phosphate. Int J Nanomedicine. 2012;7:4819–4828. © Dovepress 2012.31
Abbreviations: CL, carboxylase; TA, transaminase.
Carboxylation of in situ–generated pyruvate into oxaloacetate
The ternary bioconjugate G39B10P was used in a double enzyme experiment in which pyruvate was formed from L-Ala in transaminase (TA) and α-ketoglutarate (α-KG) as a substrate for amine group transfer31 and further carboxylation of Pyr into oxaloacetate catalyzed by CL as before (Figure 1A and B). The following substrates for the double enzymatic system were placed in the NMR tube: L-Ala, α-KG, bicarbonate (HCO3−) (all 40 mM), 25 mM ATP, and0.25 mM AcCoA. The pH of the solution was adjusted to 7.4. The CL solution (0.2 units) was added into this mixture, which was allowed to equilibrate for about 15 minutes before transaminase (TA, 0.2 units) was injected. The progress of reaction was monitored by 1H NMR spectroscopy.
Cell culture
BJ human normal skin fibroblasts (CRL-2522), doubling time 1.9, were cultured in Eagle’s minimum essential medium containing nonessential amino acids, supplemented with 10% heat-inactivated fetal calf serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. SCC-15 human squamous cell carcinoma (CRL-1623), doubling time 7.7, was propagated in a 1:1 mixture of DMEM-F12 medium containing 15 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid and supplemented with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 400 ng/mL hydrocortisone. Cells were kept at 37°C in an atmosphere of 5% CO2 and 95% humidity with growth medium changed every 2 days and passaged at 70–85 confluence, using 0.25% trypsin-0.03% ethylenediaminetetraacetic acid in PBS (calcium- and magnesium-free). Cell morphology was checked under a Nikon TS100 Eclipse Inverted Microscope (Tokyo, Japan) with phase contrast. The number and viability of cells were estimated by the trypan blue exclusion test, using hemocytometer or Automatic Cell Counter TC10™ (Bio-Rad Laboratories, Hercules, CA, USA).
In vitro cytotoxicity assays
The XTT reduction assay and NR and CV uptake quantitative spectrophotometric assays were performed for assessment of the investigated dendrimers (PAMAM) and PAMAM bioconjugate (BC-PAMAM) cytotoxicity to the two cell lines. Applied methods determine different parameters associated with cell damage, proliferation, and death. Preliminary tests were performed, including estimation of cell population doubling time and cell density range, giving a curve with a linear response for each assay and both cell lines to optimize the greatest sensitivity to changes induced by the experimental parameters. For each assay, the four independent cell cultures were used for various exposure treatments. Mean values from triplicates were calculated. Results are presented as percentage of control (nontreated) cells set at 100%.
XTT reduction assay
XTT sodium salt assay is one of the mostly used cytotoxicity assays and shows linearity over a broad range of cell densities. In this assay, the slightly yellow tetrazolium salt XTT is reduced by mitochondrial and microsomal dehydrogenases and oxidases (mitochondrial succinoxidase and cytochrome P450 systems, as well as flavoprotein oxidases) of viable cells to a water-soluble orange formazan derivative, which is measured in cellular media. The reaction is potentiated in the presence of an electron-coupling agent such as PMS.32
For assay, cells were cultured in a flat-bottom 96-well microtiter plate in triplicate (200 μL cell suspension per well). The optimum cell concentration, as determined by the growth profile of each cell line, was 2 × 104 cells/well and 4 × 104 cells/well for BJ and SCC-15 cells, respectively. Cells were allowed to attach for 12 hours before treatment with dendrimers. The stock solution of 1 mM dendrimers in H2O was filtered (syringe filters, 0.22 μm), and a working solution in corresponding culture media was prepared. Cell monolayers were treated with PAMAM and BC-PAMAM solutions within a range of increasing concentrations from 0 to 100 μM for 24 hours in 5% CO2 in a humidified incubator. After that time, media were exchanged (100 μL/well), 50 μL/well of 5 mM XTT and 25 μM PMS mixture was added, and plates were returned to the incubator. After either 1 hour (BJ cells) or 2.5 hours (SCC-15), absorbance was measured at the 450-nm wavelength against 620 nm against a blank sample (200 μL of complete growth medium containing XTT and PMS), using a microtiter plate reader (FilterMax F5 Multi-Mode Microplate Reader; Molecular Devices LLC, Sunnyvale, CA, USA).
NR uptake assay
The NR assay is used as indicator of cytotoxicity and determines the accumulation of the NR dye in the lysosomes of viable, uninjured cells. The procedure is considered to be more sensitive than other cytotoxicity tests, including tetrazolium salts or enzyme leakage assays for different cell lines, and provides a strong signal of both cell integrity and growth inhibition.33,34
Cells (both lines) were seeded at a density of 4 × 104 per well and treated with dendrimers, as described earlier. After exposure to dendrimers, medium was removed, 200 μL NR solution (100 μg/mL in culture medium) was added per well, and cells were incubated for 3 hours in a CO2 incubator. After washing once with PBS, 100 μL/well of fixative (50% ethanol, 49% H2O, and 1% glacial acetic acid) was added and plates were shaken at 400 rpm for 10 minutes until complete dissolution of dye was achieved. Absorbance was measured at 540 nm against 620 nm in a microtiter plate reader against blank (fixative mixture).
CV assay
In a CV assay, the dye stains DNA in the nuclei of intact cells. On solubilization, the amount of dye taken up by the cell growing as a monolayer can be quantitated spectrophotometrically, and the intensity of the color produced is proportional to an adherent cell number.35–37 Assay cells were seeded at 4 × 104 (BJ) and 2 × 104 (SCC-15) per well, and incubation and dendrimer treatment were performed as described earlier. After incubation, cells were washed with PBS (three times using automatic cell washer type ELx50; BioTek Instruments, Winooski, VT, USA), followed by the addition of 0.1% CV in PBS (50 μL/well), and incubated for 30 minutes at room temperature. After washing six times with PBS, cell solubilization was performed by adding 10% acetic acid (50 μL/well), and incubation continued with shaking (10 minutes, 400 rpm, room temperature). Absorbance was measured at 595 nm against 450 nm against a blank sample (10% acetic acid).
Confocal microscopy analysis of apoptosis
Morphological detection of apoptotic cells was performed using Carl Zeiss Axio Observer Z1 LSM 700 confocal microscopy with ZEN2010 software (Carl Zeiss MicroImaging GmbH, Jena, Germany). Apoptosis was estimated as the presence of cleaved fragments of PARP 1 (Asp 214) present in the nuclei by specific antibody conjugated to AlexaFluor 647. PARP fragmentation is an early hallmark of apoptosis. To visualize nuclear morphology, DNA was stained with DAPI.
The BJ and SCC-15 cells were seeded in 96-well clear-bottom microplates (Corning Inc, Corning, NY, USA) at 104 cells/well, cultured for 24 hours, and treated with increasing concentrations (0, 0.1, 1, 5, and 10 μM) of PAMAM and BC-PAMAM for 24 hours before staining. After each step of the procedure, cells were washed three times with PBS. All incubations were performed at room temperature. After incubation, cells were fixed with 3.7% formaldehyde (200 μL/well, 15 minutes), and permeabilized with 0.1% Triton X-100/PBS (200 μL/well for 30 minutes). After washing, antibody (1:10 in PBS) against PARP (Asp 214) was added (50 μL/well), and incubation was performed in darkness for 1 hour at room temperature. After washing, 300 nM DAPI/PBS was added (200 μL/well), and incubation continued for 15 minutes. Images were collected from each canal (DAPI: 405 nm/461 nm; AlexaFluor: 639 nm/665 nm). Signal was collected from 12 randomly chosen images of three independent experiments. Results are presented as mean of the average of the PARP/DAPI signal intensity ratio.
Statistical analysis
Statistical analysis was carried out using nonparametric statistics: Kruskal–Wallis test for differences between dendrimer-treated and nontreated control, and paired U Mann–Whitney tests for differences between PAMAM- and BC-PAMAM-treated cells (P,0.05 was considered significant).38
NMR spectra
The 1H and 13C NMR, as well as two-dimensional 1H-1H correlation spectroscopy, heteronuclear single-quantum correlation spectroscopy, and heteronuclear multiple-bond correlation spectroscopy spectra, were recorded with a 300-MHz instrument (Bruker Corporation, Billerica, MA, USA). For quantitative measurements of catalytic substrate conversions based on integration of 1H NMR resonances, the 30° pulse was used with 3 seconds of relaxation delay (T1 longitudinal relaxation time measured by inversion recovery technique was #0.6 seconds for the protons of Ala, α-KG, Pyr, and oxaloacetate). The signal-to-noise ratio was 60 or higher after 64 scans. The integration of relevant resonances was preceded by baseline correction procedure. The intensity of resonances was linear within 1–40 mM concentration within an estimated 10% error at the lowest concentration detected (1 mM).
Results and discussion
Syntheses and purification of bioconjugates
The G3 PAMAM dendrimer was chosen as a potential carrier for biotin and pyridoxal. The level of substitution of what were originally 32 amine groups was intentionally high both in G39B and G39B10P. Dimethyl sulfoxide-soluble NHS-biotin was used as substrate to bind to G3 through the amide bond. The average number of biotin molecules attached to G3 was estimated on the basis of the integration of 8b and 9b resonances of G39B related to the intensity of PAMAM G3 resonance at 2.20 ppm. The assignment of resonances in the spectrum of G39B was performed by two-dimensional 1H-1H correlation spectroscopy, heteronuclear single-quantum correlation spectroscopy, and heteronuclear multiple-bond correlation spectroscopy experiments to characterize the species used later for enzymatic experiments, followed in situ by 1H NMR spectroscopy. Replacement of the N-hydroxysuccinimide group in NHS-biotin by the amine group in G39B resulted in a considerable shift of 2b triplet from 2.68 ppm in NHS-biotin to 2.07 ppm in G39B (Figure 2), although the pattern of the remaining proton resonances remained almost unchanged. The 1H NMR spectrum G39B in D2O remained unchanged for at least 1 day at room temperature, indicating that the bioconjugate is stable in such conditions.
Figure 2.
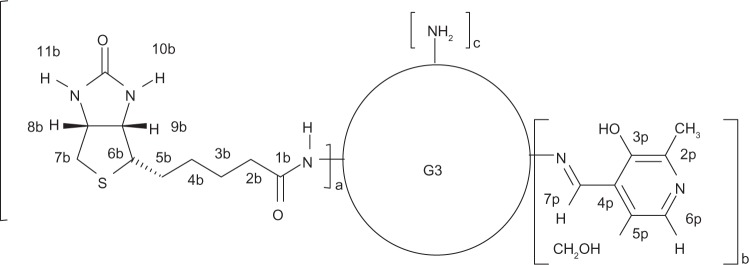
Schematic representation of bioconjugate molecules: G39B (a=9, b=0, c=23); G39B10P (a=9, b=10, c=13) with atom numbering.
The ternary G39B10P bioconjugate (Figure 2) was obtained by condensation of pyridoxal with G39B, as described earlier.31 The diagnostic for aldimine bond formation was a large shift of aldimine protons from 10.5 ppm (for pyridoxal) to 8.92 ppm (7p in G39B10P) in the 1H NMR spectrum in methanol-d4. Integration of resonances of 7p and 6p in aromatic regions related to PAMAM resonance indicated that an average of 10 molecules of pyridoxal were attached to G39B.
Both bioconjugates were extensively purified at every step of synthesis by the evaporation of methanol and dialysis to remove side products of condensation and traces of lower-generation dendrimers.
Enzymatic catalysis in situ
Pyruvate CL from bovine lever was used to monitor the carboxylation of Pyr in deuterium oxide by 1H NMR spectroscopy on a submicromolar level in a NMR tube. Injection of 0.2 units CL into the reaction mixture containing 40 mM Pyr, about 40 mM HCO3−, 25 mM ATP, AcCoA, and Mg2+ (the reagent concentrations were chosen according to pioneer experiment by Utter and Keech30) triggered the carboxylation of Pyr into oxaloacetate. The conversion was determined by disappearance of CH3(Pyr) resonance at 2.27 ppm in the 1H NMR spectrum (Figure 3, left a–g spectra). The substrate Pyr itself underwent slow deuterating on the methyl group as a result of the keto–enol equilibrium with solvent assistance; therefore, the starting spectrum of Pyr (Figure 3, trace a) was accompanied by the presence of multiplets from CH2D(Pyr) and CHD2(Pyr) slightly shifted highfield in relation to starting CH3(Pyr) resonance at 2.27 ppm (Figure 3, trace a′). Traces b–g in Figure 3 show the disappearance of CH3(Pyr) resonance accompanied by the appearance of oxaloacetate methylene proton resonance at 2.14 ppm, although the intensity of the latter increased deficiently as a result of the deuterating of both the Pyr substrate and oxaloacetate by keto–enol fast equilibrium with solvent deuterium. The 50% conversion of Pyr was observed within 1.5 hour, with the enzymatic system still active. The addition of a new portion of Pyr caused an increase of intensity in CH3(Pyr) resonance in the 1H NMR spectrum; further addition of ATP resulted in its disappearance. A similar picture was obtained when G39B conjugate was used instead of biotin in the reaction system (Figure 3, right side spectra, traces i–m). The broad, high-intensity resonances within the 2.4–2.3-ppm region originate from PAMAM G3 methylene protons, as well as a series of lower-intensity signals within the 2.20–2.10-ppm region. The half-time of Pyr conversion was about 1 hour, and the system remained active for 1 day, as checked via the addition of new portions of Pyr and ATP. It can be concluded that biotin covalently attached to a G3 carrier serves as a cofactor for pyruvate CL in vitro.
Figure 3.

The relevant regions of 1H nuclear magnetic resonance spectra of Pyr (trace a) and reaction mixtures composed of (left stack) pyruvate (Pyr, 40 mM) bicarbonate (HCO3−, 40 mM), adenosine-5′-triphosphate (ATP; 25 mM), Mg2+ (both 40 mM), acetylcoenzyme A (AcCoA, 0.25 mM), biotin (2.5 mM), and pyruvate carboxylase (CL, 0.2 units); traces b–g (b, starting spectrum; c–g, after 5 minutes [c], 15 minutes [d], 40 minutes [e], 90 minutes [f], and 120 minutes [g] of reaction); (right stack) the concentration of Pyr, HCO −, ATP, Mg2+3, and AcCoA as specified (series b-g), the 0.25 mM G39B, and 0.2 units of CL; traces i–m (i, starting spectrum; j–m, after 5 minutes [j], 10 minutes [k], 40 minutes [i], and 90 min [m] of reaction).
Our earlier, similar studies demonstrated comparable activity of G34P as donor of cofactor for transaminase.31 The amine group transfer from L-Ala to α-KG catalyzed by glutamic–pyruvic transaminase from porcine heart was followed by 1H NMR spectroscopy. Pyridoxal and pyridoxal phosphate both were active cofactors for the enzyme when released from G34P or G34PLP (where “PLP” stands for pyridoxal phosphate). The products of that conversion were glutamic acid and pyruvate. Therefore, we have synthesized the ternary bioconjugate containing both potential cofactors, namely, pyridoxal and biotin, as G39B10P and used it as double donor of cofactors for two enzymes: TA and CL. The reagents composition was quite complicated; the reaction mixture included both Ala and α-KG as substrates for TA; and the remaining auxiliary reagents were ATP, HCO3−, AcCoA, and Mg2+. Fortunately, both enzymes operate at pH 7.4, and thus the pH of the reaction mixture was adjusted to that pH before the injection of the TA and CL enzymes.
The double-enzymatic system was activated in two different sequences. First, the TA was injected into the reaction mixture and formation of pyruvate and L-Glu was determined by the appearance of characteristic 1H NMR spectra (Figure 4, trace b) (multiplets from two magnetically nonequivalent 4-CH2 were centered at 1.96 ppm and 3-CH2 at 2.24 ppm, and a singlet from CH3[Pyr] was centered at 2.27 ppm). Then CL was injected, and the disappearance of CH3(Pyr) resonance at 2.27 ppm was noticed. When the sequence of enzyme addition was inverted (eg, when CL was added before the injection of TA), the growth of CH3(Pyr) resonance was unnoticed, unlike the growth of L-Glu resonances. The spectra of reaction mixtures from the first experiment are presented in Figure 4. The double enzymatic system remained active for 12 hours, as determined by the addition of L-Ala and α-KG, or by addition of Pyr and ATP, as in the experiment with CL (vide supra).
Figure 4.

The relevant regions of the 1H nuclear magnetic resonance spectra of solutions containing L-alanine (Ala), α-ketoglutarate (α-KG), bicarbonate (HCO3−) (all 40 mM), 25 mM adenosine-5′-triphosphate (ATP), 0.25 mM acetylcoenzyme A (AcCoA), and 0.25 mM G39B10P (full spectrum shown at trace a′); 0.2 units of transaminase and carboxylase were added and the spectra were recorded after (b) 5 minutes, (c) 15 minutes, (d) 30 minutes, (e) 60 minutes, and (g) 120 minutes of reaction. The resonance of CH3 protons of pyruvate was identified by in situ addition of a new portion of pyruvic acid into the reacting system.
Thus, the ternary bioconjugate G39B10P provides active cofactors for transaminase and CL enzymes. To determine the potential usefulness of such a bioconjugate, we have further studied its toxicity in vitro in fibroblast and epithelial cells. The G3 originally had 32 basic amine groups, which are cationic at physiological pH and potentially bind to negatively charged biological membranes. Therefore, we have constructed ternary bioconjugate with 60% of amine groups blocked with pyridoxal and biotin and studied the involvement of both compounds (G3 and G39B10P) on cell cultures.
Comparison of cytotoxicity of PAMAM (G3) and BC-PAMAM (G39B10P)
Cytotoxicity data for PAMAM and BC-PAMAM obtained with BJ and SCC-15 cells are presented in Figure 5A–C. For all assays, a dose-dependent response was observed, which revealed a structurally dependent cytotoxic response for both cell lines.
Figure 5.

BJ normal human fibroblast and squamous cell carcinoma (SCC-15) cells cytotoxicity after 24 hours of treatment with polyamidoamine dendrimer of third generation (PAMAM) and bioconjugate-PAMAM measured by (A) XTT assay (2 × 104 and 4 × 104 cells/well, BJ and SCC-15), (B) NR assay (4 × 104 cells/well, both cell lines), (C) Crystal violet assay (4 × 104 and 2 × 104 cells/well, BJ and SCC-15). Results are mean of triplicate assays of four independent experiments. *P,0.05; Kruskal–Wallis test (against nontreated control); ▼P,0.05; Mann–Whitney U test (PAMAM against bioconjugate-PAMAM).
The data demonstrate that normal fibroblasts were more sensitive than squamous cell carcinoma to both PAMAM and BC-PAMAM. Staining with CV shows that PAMAM are nontoxic at concentrations of 10 μM, and BC-PAMAM at 50 μM, for both cell lines (Figure 5C). NR and XTT assays revealed a higher cytotoxicity of both dendrimers. Measuring XTT reduction shows that PAMAM is nontoxic at concentrations up to 5 μM, and BC-PAMAM up to 10 μM, for both cell lines (Figure 5A). Similarly, the NR uptake test reveals that PAMAM is not significantly toxic at a concentration of 2.5 μM for fibroblasts, and BC-PAMAM at10 μM for SCC-15 cells. Although the XTT assay indicates the mitochondrial and endoplasmic reticulum membrane enzyme activity corresponding to metabolic potential, the NR assay indicates the lysosomal and cell membrane integrity.
All three tests (CV, XTT, and NR) confirmed the significantly lower cytotoxicity of biotin–pyridoxal–conjugated BC-PAMAM compared with PAMAM. For normal fibroblasts, PAMAM is two-, four-, and fivefold more toxic than BC-PAMAM, as evaluated by XTT, NR, and CV, respectively. The squamous cell carcinoma line is more resistant to the toxic effects of PAMAM, but the lower toxicity of BC-PAMAM is also evident in the significantly higher viability of cells treated with the same concentrations of both dendrimers, as shown in Figure 5A–C.
Our results correspond well with other authors’ data obtained for various generations of nonsubstituted PAMAM dendrimers with different cell lines after 24 hours of exposition. Winnicka et al reported half maximal inhibitory concentration (IC50) values for PAMAM toxicity to cancer cells (MCF-7 and MDA-MB-231 cells) in the range of 100–140 μM.39 Mukherjee et al obtained half maximal effective concentration (EC50) for various PAMAM generations (G4, G5, G6) in HaCaT immortalized human keratinocytes and the SW480 cancer cell line of from 1 to 23 μM, using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and NR assays.8 Shao et al investigated the cytotoxicity of PAMAM with A549 and MCF-7 human carcinoma cell lines and found that no toxicity is observed up to 74 μM.40 PAMAM dendrimers also were investigated with the L929 mouse fibroblast line by Fischer et al, and a very limited cytotoxicity effect was observed by MTT assay after 24 hours.41 Screening studies by Malik et al show that cationic dendrimers were cytotoxic for B16F10 mice melanoma, CCRF human leukemic cells, and HepG2 human hepatocellular carcinoma displaying IC50 values of 50–300 μg/mL after 72 h incubation, depending on dendrimer type and generation and cell line.42
No increased cytotoxicity of biotin–dendrimer conjugates compared with parent PAMAM dendrimers was found by Yellepeddi et al, using a human embryonic kidney (HEK 293T) cell line.17 Further studies concerning the biotinylated PAMAM dendrimers loaded with cisplatin have shown that the IC50s of dendrimer–cisplatin complexes were similar to those of nonbiotinylated dendrimers in the range of 20–70 μM for anionic dendrimers and 25–50 μM for cationic dendrimers, as estimated with various ovarian cancer cell lines (OVCAR-3, SKOV-3, and A2780).18 In the studies by Yang et al of the bifunctional conjugate (dendrimer–biotin– fluorescein isothiocyanate), no significant cytotoxicity with MTT assay was observed at 8 μM concentration, despite much higher cellular uptake into HeLa cells than with the conjugate without biotin.43
Apoptotic effect of PAMAM and BC-PAMAM dendrimers
Apoptosis was detected in cells treated with PAMAM and BC-PAMAM in a dose-dependent manner, as measured by confocal microscope analysis using anti-PARP (Asp 214) antibody coupled with AlexaFluor 647 (Figure 6A and B). An increase in PARP fragmentation at the early stage of apoptosis, as a result of the fibroblasts’ exposure to PAMAM and CB-PAMAM, was evident at a concentration of 5 μM (Figure 6A), and at 10 μM for SCC-15 cells (Figure 6B). Presented representative pictures from confocal microscopy analysis of apoptosis induced by 10 μM PAMAM G3 in fibroblasts (Figure 6A) reveal less-adherent cells compared with the control and sample with 5 μM PAMAM G3. The majority of the remaining cells are positively stained with used apoptotic marker (PARP). Because washed-out cells, which lose their adhesion, were certainly dead, counting them will increase the number of dead cells but not change the final conclusion. The apoptotic signal was always significantly lower for biotin–pyridoxal-conjugated BC-PAMAM compared with nonconjugated PAMAM (Figure 7).
Figure 6.
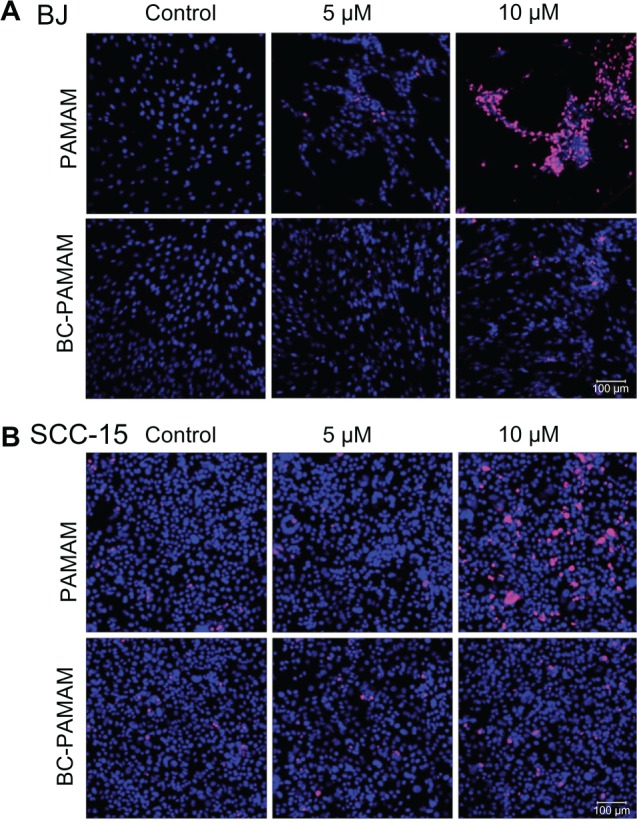
Confocal microscopy images of cleaved poly (ADP-ribose) polymerase (PARP; Asp 214) protein (pink) in nuclei (blue) of (A) BJ and (B) squamous cell carcinoma (SCC)-15 cells treated with polyamidoamine dendrimer and bioconjugate–polyamidoamine dendrimer dendrimers. Cells 104/well were incubated with dendrimers for 24 hours and double-stained with AlexaFluor 647-conjugated anti-PARP antibody (639/665 nm) and 4′,6-diamidino-2-phenylindole, dihydrochloride (405/461 nm). Signal was collected from 12 randomly chosen images of three independent experiments.
Figure 7.

Comparison of proapoptotic effect of polyamidoamine dendrimer (PAMAM) and bioconjugate-PAMAM for BJ and squamous cell carcinoma (SCC)-15 cells. Cells 104/well were treated with dendrimers for 24 hours and double-stained with AlexaFluor 647-conjugated anti-PARP antibody and DAPI. *P,0.05. Kruskal–Wallis test (against nontreated control); ▼P,0.05; Mann–Whitney U test (PAMAM against bioconjugate-PAMAM).
The cytotoxic effects of PAMAM and BC-PAMAM on apoptosis correspond very well with the results of XTT and NR cytotoxicity assays for both cell lines and support the observation that normal fibroblasts are more sensitive to the cytotoxic effects of investigated dendrimers and that modification of the surface cationic groups by the substitution of biologically active molecules significantly decreases that effect.
Our results differ from those published by Mukherjee et al, showing significantly higher sensitivity to PAMAM G4-, G5-, and G6-induced cytotoxicity, as measured by alamar-Blue, NR, and MTT assays in SW480 colon adenocarcinoma cells, compared with a HaCaT human immortal noncancerous keratinocyte cell line. Mukherjee et al suggest this difference can be explained by different intrinsic antioxidant levels of both cell lines, as estimated by intracellular reactive oxygen species assays.44 Higher cytotoxicity also was observed for biotinylated compared with nonbiotinylated G4 PAMAM dendrimers by a lactate dehydrogenase release assay performed with the bEnd.3 immortalized murine brain capillary endothelial cell line, but not with normal keratinocytes, by Bullen et al.45 The authors investigated the permeability of the blood–brain barrier for the delivery of therapeutic drugs to the brain by means of biotinylated PAMAM dendrimers, as biotin has been shown to cross the barrier through carrier-mediated endocytosis. The blood–brain barrier creates a permeability barrier caused by tight junctions of specialized brain capillary endothelial cells building brain capillary walls. These findings were confirmed by biophysical measurements of the disruption of the monolayer lipid model membrane by Langmuir Blodgett techniques and atomic force microscopy. The explanation of higher cytotoxicity of biotinylated G4 PAMAM dendrimers may be a result of more effective uptake and accumulation mechanisms in this particular cell culture system.
A significant difference in sensitivity to dendrimer-induced damage between the two investigated cell lines might be a result of various specific properties of cancer cells; mainly, the impairment of cell cycle control and apoptotic signals, which allows even damaged cancer cells to proliferate,46 or the self-correcting ability of the DNA repair systems,47 as well as the increased activity of telomerases.48 It is also known that lysosomes play an important role in the progression of apoptosis through the release of proteases.49 PAMAM- and BC-PAMAM-dependent apoptosis might be a result of damage to lysosomes, as shown by NR assay (Figure 5B). Mukherjee et al have found increased lysosomal activity in HaCaT cells treated with PAMAM for 24 hours, but not in SW480 cells.44 Recent investigations by Appelqvist et al also have shown that lysosome-dependent cell death is directly regulated by lysosomal cholesterol content, with a protective effect of cholesterol.50 Because increased intracellular cholesterol levels and elevated cholesterol synthesis were found in cancer cells, the higher resistance of cancer cells (compared with normal fibroblasts) to the cytotoxic effects of PAMAM and BC-PAMAM observed in current studies could be a result of this mechanism.51,52
Many studies have shown that the biocompatibility and biodistribution of dendrimers as a result of interaction with cell membranes and cytotoxicity depend on the dendrimers’ composition and architecture and, most strongly, on the nature of their surface. Cationic dendrimers both interact with phospholipids in cell membranes, causing their disturbance and damage, which results in leakage of intracellular components, and penetrate into cells through an energy-dependent combination of paracellular and endocytic mechanisms.53,54 The intracellular fate of dendrimers plays a major part in determining their biological properties, including cytotoxicity and antigenicity. It is known that cationic dendrimers interact with mitochondrial membrane, leading to the generation of free radicals, induction of oxidative stress, and apoptotic signals.55 Damage to genomic DNA was also described.8,56,57 Lysosomal membrane disturbance and accumulation of nondegradable and slowly degrading dendrimers in lysosomes create other mechanisms of cytotoxicity.8,58 Further studies are needed to recognize and verify the molecular mechanisms of these findings.
It has been well documented that surface modifications of PAMAM dendrimers reduce their toxicity.7,59 Recent studies by Sadekar and Ghandehari have shown a linear correlation for cell viability with the surface density of amine groups, irrespective of dendrimer generation.60 The substitution of PAMAM with two biologically active biomolecules (pyridoxal and biotin) in the current study decreases the number of amine groups per dendrimer, which does correlate well with the cytotoxic response of both normal and cancer cells.
Conclusion
In a search for safe and efficacious dendrimer-based delivery systems and biologically active molecules, the bioconjugate of PAMAM G3 with pyridoxal and biotin (BC-PAMAM) was obtained. The vitamins were proven by 1H NMR spectral monitoring to release from the bioconjugate and participate in enzymatic conversions catalyzed by CL and transaminase in vitro. The cytotoxicity of PAMAM and BC-PAMAM was compared, using three cytotoxicity assays (XTT, NR, and CV) and an estimation of apoptosis by confocal microscopy detection. Human BJ normal fibroblasts and SCC-15 squamous endothelial cell carcinoma lines were used as experimental models. All three cytotoxicity tests (XTT, NR, and CV) confirmed the significantly lower cytotoxicity of BC-PAMAM compared with PAMAM. Nonconjugated PAMAM is not significantly cytotoxic at concentrations up to 2.5 μM (NR), 5 μM (XTT), and 10 μM, whereas the BC-PAMAM was not toxic up to 10 μM (XTT and NR) and 50 μM (CV) for both cell lines. The data also demonstrate that normal fibroblasts are more sensitive than squamous cell carcinoma to both PAMAM and BC-PAMAM because of their significantly lower viability during treatment with the same concentrations of both dendrimers. The effect of PAMAM and BC-PAMAM on initiation of apoptosis (PAMAM and BC-PAMAM in fibroblasts at 5 μM and 10 μM in SCC-15 cells) corresponds very well with the results of cytotoxicity assays for both cell lines, as the apoptotic signal was always significantly higher in fibroblasts at the same dendrimer concentrations. Obtained data support the conclusion that the modification of surface cationic groups of PAMAM dendrimers by substituting biologically active molecules (biotin and pyridoxal) significantly decreases cytotoxicity, giving bioconjugated dendrimers the opportunity to be used as carriers for various biocompounds in delivery to normal and cancer cells. However, considering the possibility of bioaccumulation of nanoparticles in various organs, both understanding their cytotoxicity (depending on cell or tissue type) and the search for less cytotoxic bioconjugated dendrimer generations are essential before using them for in vivo applications.61,62
Acknowledgments
The work was supported by grant N N302 432839 from the Ministry of Higher Education and Research, Poland.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Uppuluri S, Swanson DR, Piehler LT, Li J, Hagnauer GL, Tomalia DA. Core-shell tecto(dendrimers): I. Synthesis and characterization of saturated shell models. Adv Mater. 2000;12(11):796–800. [Google Scholar]
- 2.Schilrreff P, Mundiña-Weilenmann C, Romero EL, Morilla MJ. Selective cytotoxicity of PAMAM G5 core – PAMAM G2.5 shell tecto-dendrimers on melanoma cells. Int J Nanomedicine. 2012;7:4121–4133. doi: 10.2147/IJN.S32785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.D’Emanuele A, Attwood D. Dendrimer-drug interactions. Adv Drug Deliv Rev. 2005;57(15):2147–2162. doi: 10.1016/j.addr.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 4.Najlah M, D’Emanuele A. Crossing cellular barriers using dendrimer nanotechnologies. Curr Opin Pharmacol. 2006;6(5):522–527. doi: 10.1016/j.coph.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 5.Jain K, Kesharwani P, Gupta U, Jain NK. Dendrimer toxicity: Let’s meet the challenge. Int J Pharm. 2010;394(1–2):122–142. doi: 10.1016/j.ijpharm.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 6.Svenson S, Tomalia DA. Dendrimers in biomedical applications – reflections on the field. Adv Drug Deliv Rev. 2005;57(15):2106–2129. doi: 10.1016/j.addr.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 7.Duncan R, Izzo L. Dendrimer biocompatibility and toxicity. Adv Drug Deliv Rev. 2005;57(15):2215–2237. doi: 10.1016/j.addr.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 8.Mukherjee SP, Davoren M, Byrne HJ. In vitro mammalian cytotoxicological study of PAMAM dendrimers – towards quantitative structure activity relationships. Toxicol In Vitro. 2010;24(1):169–177. doi: 10.1016/j.tiv.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 9.Labieniec M, Watalia C. PAMAM dendrimers – diverse biomedical applications. Facts and unresolved questions. Centr Eur J Biol. 2009;4:434–451. [Google Scholar]
- 10.Pisal DS, Yellepeddi VK, Kumar A, et al. Permeability of surface-modified polyamidoamine (PAMAM) dendrimers across Caco-2 cell monolayers. Int J Pharm. 2008;350(1–2):113–121. doi: 10.1016/j.ijpharm.2007.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kolhatkar RB, Kitchens KM, Swaan PW, Ghandehari H. Surface acetylation of polyamidoamine (PAMAM) dendrimers decreases cytotoxicity while maintaining membrane permeability. Bioconjug Chem. 2007;18(6):2054–2060. doi: 10.1021/bc0603889. [DOI] [PubMed] [Google Scholar]
- 12.Teow HM, Zhou Z, Najlah M, Yusof SR, Abbott NJ, D’Emanuele A. Delivery of paclitaxel across cellular barriers using a dendrimer-based nanocarrier. Int J Pharm. 2013;441(1–2):701–711. doi: 10.1016/j.ijpharm.2012.10.024. [DOI] [PubMed] [Google Scholar]
- 13.Sadekar S, Ghandehari H. Transepithelial transport and toxicity of PAMAM dendrimers: implications for oral drug delivery. Adv Drug Deliv Rev. 2012;64(6):571–588. doi: 10.1016/j.addr.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saovapakhiran A, D’Emanuele A, Attwood D, Penny J. Surface modification of PAMAM dendrimers modulates the mechanism of cellular internalization. Bioconjug Chem. 2009;20(4):693–701. doi: 10.1021/bc8002343. [DOI] [PubMed] [Google Scholar]
- 15.Shi F, Bailey C, Malick AW, Audus KL. Biotin uptake and transport across bovine brain microvessel endothelial cell monolayers. Pharm Res. 1993;10(2):282–288. doi: 10.1023/a:1018903330985. [DOI] [PubMed] [Google Scholar]
- 16.Beg S, Samad A, Alam MI, Nazish I. Dendrimers as novel systems for delivery of neuropharmaceuticals to the brain. CNS Neurol Disord Drug Targets. 2011;10(5):576–588. doi: 10.2174/187152711796235023. [DOI] [PubMed] [Google Scholar]
- 17.Yellepeddi VK, Kumar A, Palakurthi S. Biotinylated poly(amido)amine (PAMAM) dendrimers as carriers for drug delivery to ovarian cancer cells in vitro. Anticancer Res. 2009;29(8):2933–2943. [PubMed] [Google Scholar]
- 18.Yellepeddi VK, Kumar A, Maher DM, Chauhan SC, Vangara KK, Palakurthi S. Biotinylated PAMAM dendrimers for intracellular delivery of cisplatin to ovarian cancer: role of SMVT. Anticancer Res. 2011;31(3):897–906. [PubMed] [Google Scholar]
- 19.Yang W, Cheng Y, Xu T, Wang X, Wen LP. Targeting cancer cells with biotin-dendrimer conjugates. Eur J Med Chem. 2009;44(2):862–868. doi: 10.1016/j.ejmech.2008.04.021. [DOI] [PubMed] [Google Scholar]
- 20.Cheng Y, Man N, Xu T, et al. Transdermal delivery of nonsteroidal anti-inflammatory drugs mediated by polyamidoamine (PAMAM) dendrimers. J Pharm Sci. 2007;96(3):595–602. doi: 10.1002/jps.20745. [DOI] [PubMed] [Google Scholar]
- 21.Singh P, Gupta U, Asthana A, Jain NK. Folate and folate-PEG-PAMAM dendrimers: synthesis, characterization, and targeted anticancer drug delivery potential in tumor bearing mice. Bioconjug Chem. 2008;19(11):2239–2252. doi: 10.1021/bc800125u. [DOI] [PubMed] [Google Scholar]
- 22.Venuganti VV, Perumal OP. Poly(amidoamine) dendrimers as skin penetration enhancers: Influence of charge, generation, and concentration. J Pharm Sci. 2009;98(7):2345–2356. doi: 10.1002/jps.21603. [DOI] [PubMed] [Google Scholar]
- 23.Borowska K, Laskowska B, Magoń A, Mysliwiec B, Pyda M, Wołowiec S. PAMAM dendrimers as solubilizers and hosts for 8-methoxypsoralene enabling transdermal diffusion of the guest. Int J Pharm. 2010;398(1–2):185–189. doi: 10.1016/j.ijpharm.2010.07.019. [DOI] [PubMed] [Google Scholar]
- 24.Borowska K, Wołowiec S, Rubaj A, Głowniak K, Sieniawska E, Radej S. Effect of polyamidoamine dendrimer G3 and G4 on skin permeation of 8-methoxypsoralene – in vivo study. Int J Pharm. 2012;426(1–2):280–283. doi: 10.1016/j.ijpharm.2012.01.041. [DOI] [PubMed] [Google Scholar]
- 25.Borowska K, Wołowiec S, Głowniak K, Sieniawska E, Radej S. Transdermal delivery of 8-methoxypsoralene mediated by polyamidoamine dendrimer G2.5 and G3.5 – in vitro and in vivo study. Int J Pharm. 2012;436(1–2):764–770. doi: 10.1016/j.ijpharm.2012.07.067. [DOI] [PubMed] [Google Scholar]
- 26.Wang Z, Itoh Y, Hosaka Y, et al. Mechanism of enhancement effect of dendrimer on transdermal drug permeation through polyhydroxyalkanoate matrix. J Biosci Bioeng. 2003;96(6):537–540. doi: 10.1016/S1389-1723(04)70146-2. [DOI] [PubMed] [Google Scholar]
- 27.Filipowicz A, Wołowiec S. Solubility and in vitro transdermal diffusion of riboflavin assisted by PAMAM dendrimers. Int J Pharm. 2011;408(1–2):152–156. doi: 10.1016/j.ijpharm.2011.01.033. [DOI] [PubMed] [Google Scholar]
- 28.Tomalia DA, Naylor AM, Goddard WA. Starburst dendrimers: Molecular-level control of size, shape, surface chemistry, topology, and flexibility from atoms to macroscopic matter. Angew Chem Int Ed Engl. 1990;29(2):138–175. [Google Scholar]
- 29.Subik P, Welc B, Wisz B, Wołowiec S. Stable hemiaminals attached to PAMAM dendrimers. Tetrahedron Lett. 2009;50:6512–6514. [Google Scholar]
- 30.Utter MF, Keech DB. Pyruvate carboxylase. I. Nature of the reaction. J Biol Chem. 1963;238:2603–2608. [PubMed] [Google Scholar]
- 31.Filipowicz A, Wołowiec S. Bioconjugates of PAMAM dendrimers with trans-retinal, pyridoxal, and pyridoxal phosphate. Int J Nanomedicine. 2012;7:4819–4828. doi: 10.2147/IJN.S34175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berridge MV, Herst PM, Tan AS. Tetrazolium dyes as tools in cell biology: new insights into their cellular reduction. Biotechnol Annu Rev. 2005;11:127–152. doi: 10.1016/S1387-2656(05)11004-7. [DOI] [PubMed] [Google Scholar]
- 33.Repetto G, del Peso A, Zurita JL. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat Protoc. 2008;3(7):1125–1131. doi: 10.1038/nprot.2008.75. [DOI] [PubMed] [Google Scholar]
- 34.Test Method Protocol for the NHK Neutral Red Uptake Cytotoxicity Assay Phase III – Validation Study: November 4, 2003. [Accessed October 23, 2013]. Available from http://iccvam.niehs.nih.gov/methods/acutetox/invidocs/phIIIprot/nhkphIII.pdf.
- 35.Vega-Avila E, Pugsley MK. An overview of colorimetric assay methods used to assess survival or proliferation of mammalian cells. Proc West Pharmacol Soc. 2011;54:10–14. [PubMed] [Google Scholar]
- 36.Saotome K, Morita H, Umeda M. Cytotoxicity test with simplified crystal violet staining method using microtitre plates and its application to injection drugs. Toxicol In Vitro. 1989;3(4):317–321. doi: 10.1016/0887-2333(89)90039-8. [DOI] [PubMed] [Google Scholar]
- 37.BD Bioscence Cell Adhesion Protocol Manual. [Accessed October 23, 2013]. Available from: http://www.bdbiosciences.com/documents/Cell_Adhesion_protocol_manual.pdf.
- 38.Hollander M, Wolfe DA. Nonparametric Statistical Methods, Wiley Series in Probability and Statistics. 2nd ed. New York: John Wiley and Sons, Inc; 1999. [Google Scholar]
- 39.Winnicka K, Bielawski K, Rusak M, Bielawska A. The effect of generation 2 and 3 poly(amidoamine) dendrimers on viability of human breast cancer cells. J Health Science. 2009;55:169–177. [Google Scholar]
- 40.Shao N, Su Y, Hu J, Zhang J, Zhang H, Cheng Y. Comparison of generation 3 polyamidoamine dendrimer and generation 4 polypropylenimine dendrimer on drug loading, complex structure, release behavior, and cytotoxicity. Int J Nanomedicine. 2016:3361–3372. doi: 10.2147/IJN.S27028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fischer D, Li Y, Ahlemeyer B, Krieglstein J, Kissel T. In vitro cytotoxicity testing of polycations: influence of polymer structure on cell viability and hemolysis. Biomaterials. 2003;24(7):1121–1131. doi: 10.1016/s0142-9612(02)00445-3. [DOI] [PubMed] [Google Scholar]
- 42.Malik N, Wiwattanapatapee R, Klopsch R, et al. Dendrimers: relationship between structure and biocompatibility in vitro, and preliminary studies on the biodistribution of 125I-labelled polyamidoamine dendrimers in vivo. J Control Release. 2000;65(1–2):133–148. doi: 10.1016/s0168-3659(99)00246-1. [DOI] [PubMed] [Google Scholar]
- 43.Yang W, Cheng Y, Xu T, Wang X, Wen LP. Targeting cancer cells with biotin-dendrimer conjugates. Eur J Med Chem. 2009;44(2):862–868. doi: 10.1016/j.ejmech.2008.04.021. [DOI] [PubMed] [Google Scholar]
- 44.Mukherjee SP, Lyng FM, Garcia A, Davoren M, Byrne HJ. Mechanistic studies of in vitro cytotoxicity of poly(amidoamine) dendrimers in mammalian cells. Toxicol Appl Pharmacol. 2010;248(3):259–268. doi: 10.1016/j.taap.2010.08.016. [DOI] [PubMed] [Google Scholar]
- 45.Bullen HA, Hemmer R, Haskamp A, et al. Evaluation of biotinylated pamam dendrimer toxicity in models of the blood brain barrier: a biophysical and cellular approach. J Biomater Nanobiotechnol. 2011;2(5A):485–493. [Google Scholar]
- 46.LaCasse EC, Baird S, Korneluk RG, MacKenzie AE. The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene. 1998;17(25):3247–3259. doi: 10.1038/sj.onc.1202569. [DOI] [PubMed] [Google Scholar]
- 47.Liu J, Renault L, Veaute X, Fabre F, Stahlberg H, Heyer WD. Rad51 paralogues Rad55-Rad57 balance the antirecombinase Srs2 in Rad51 filament formation. Nature. 2011;479(7372):245–248. doi: 10.1038/nature10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blackburn EH. Telomerase and Cancer: Kirk A. Landon – AACR prize for basic cancer research lecture. Mol Cancer Res. 2005;3(9):477–482. doi: 10.1158/1541-7786.MCR-05-0147. [DOI] [PubMed] [Google Scholar]
- 49.Ivanova S, Repnik U, Bojic L, Petelin A, Turk V, Turk B. Lysosomes in apoptosis. Methods Enzymol. 2008;442:183–199. doi: 10.1016/S0076-6879(08)01409-2. [DOI] [PubMed] [Google Scholar]
- 50.Appelqvist H, Sandin L, Björnström K, et al. Sensitivity to lysosome-dependent cell death is directly regulated by lysosomal cholesterol content. PLoS One. 2012;7(11):e50262. doi: 10.1371/journal.pone.0050262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Freeman MR, Solomon KR. Cholesterol and prostate cancer. J Cell Biochem. 2004;91(1):54–69. doi: 10.1002/jcb.10724. [DOI] [PubMed] [Google Scholar]
- 52.Li YC, Park MJ, Ye SK, Kim CW, Kim YN. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am J Pathol. 2006;168(4):1107–1118. doi: 10.2353/ajpath.2006.050959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.El-Sayed M, Rhodes CA, Ginski M, Ghandehari H. Transport mechanism(s) of poly (amidoamine) dendrimers across Caco-2 cell monolayers. Int J Pharm. 2003;265(1–2):151–157. doi: 10.1016/s0378-5173(03)00391-0. [DOI] [PubMed] [Google Scholar]
- 54.Kitchens KM, Kolhatkar RB, Swaan PW, Eddington ND, Ghandehari H. Transport of poly(amidoamine) dendrimers across Caco-2 cell monolayers: Influence of size, charge and fluorescent labeling. Pharm Res. 2006;23(12):2818–2826. doi: 10.1007/s11095-006-9122-2. [DOI] [PubMed] [Google Scholar]
- 55.Lee JH, Cha KE, Kim MS, et al. Nanosized polyamidoamine (PAMAM) dendrimer-induced apoptosis mediated by mitochondrial dysfunction. Toxicol Lett. 2009;190(2):202–207. doi: 10.1016/j.toxlet.2009.07.018. [DOI] [PubMed] [Google Scholar]
- 56.Qamhieh K, Nylander T, Ainalem ML. Analytical model study of dendrimer/DNA complexes. Biomacromolecules. 2009;10(7):1720–1726. doi: 10.1021/bm9000662. [DOI] [PubMed] [Google Scholar]
- 57.Naha PC, Byrne HJ. Generation of intracellular reactive oxygen species and genotoxicity effect to exposure of nanosized polyamidoamine (PAMAM) dendrimers in PLHC-1 cells in vitro. Aquat Toxicol. 2013:132–133. 61–72. doi: 10.1016/j.aquatox.2013.01.020. [DOI] [PubMed] [Google Scholar]
- 58.Thomas TP, Majoros I, Kotlyar A, Mullen D, Holl MM, Baker JR., Jr Cationic poly(amidoamine) dendrimer induces lysosomal apoptotic pathway at therapeutically relevant concentrations. Biomacromolecules. 2009;10(12):3207–3214. doi: 10.1021/bm900683r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zuckerman ST, Kao WJ. Nanomaterials and biocompatibility: BioMEMS and dendrimers. In: de Kwon GS, DeVilliers M, editors. Nanotechnology in Drug Delivery. New York: Springer; 2009. pp. 193–227. [Google Scholar]
- 60.Sadekar S, Ghandehari H. Transepithelial transport and toxicity of PAMAM dendrimers: implications for oral drug delivery. Adv Drug Deliv Rev. 2012;64(6):571–588. doi: 10.1016/j.addr.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen J, Dong X, Zhao J, Tang G. In vivo acute toxicity of titanium dioxide nanoparticles to mice after intraperitioneal injection. J Appl Toxicol. 2009;29(4):330–337. doi: 10.1002/jat.1414. [DOI] [PubMed] [Google Scholar]
- 62.Revell PA. The biological effect of nanoparticles. Nanotechnol Perceptions. 2006;2:283–298. [Google Scholar]