

Abstract
Wee1, a protein kinase, regulates the G2 checkpoint in response to DNA damage. Preclinical studies have elucidated the role of wee1 in DNA damage repair and the stabilization of replication forks, supporting the validity of wee1 inhibition as a viable therapeutic target in cancer. MK-1775, a selective and potent small-molecule inhibitor of wee1, is under clinical development as a potentiator of DNA damage caused by cytotoxic chemotherapies. We present a review of the role of wee1 in the cell cycle and DNA replication and summarize the clinical development to date of this novel class of anticancer agents.
Keywords: cyclin-dependent kinase, G2 checkpoint, cell cycle, DNA damage, MK 1775
Introduction
Wee1 belongs to a family of protein kinases involved in the terminal phosphorylation and inactivation of cyclin-dependent kinase 1-bound cyclin B, resulting in G2 cell cycle arrest in response to DNA damage. Wee1 was first identified in fission yeast,1 where wee1 deficiency resulted in premature mitotic entry and replication of smaller-sized yeast. It is the major kinase responsible for the inhibitory phosphorylation of the tyrosine15 residue on Cdk1/Cdc2, near its ATP-binding pocket.2 Wee1 also plays a critical role in the proper timing of cell division, modulating the activity of cyclin-dependent kinases 1 and 2 through inhibitory phosphorylation of conserved tyrosine15 residues on both kinases, thereby controlling entry into mitosis and DNA replication during S phase. As a consequence of its key role in the regulation of Cdk1/Cdc2 activity, early preclinical and clinical studies have focused on the modulation of wee1 activity and abrogation of the G2 checkpoint in the presence of DNA damaging agents, utilizing the concept of mitotic lethality as a mechanism of antitumor activity. Cells depend upon cell cycle checkpoints (G1, S, and G2) to allow time for repair of DNA damage prior to cell division (Fig. 1). Inhibition of cell cycle checkpoints in the setting of DNA damage effectively results in the perpetuation of the damage, culminating in cell death as a result of irreparable genetic lesions. More recent in vitro studies implicate wee1 in the stabilization of replication forks and homologous recombination (HR) repair, highlighting a greater role for wee1 in DNA repair and further supporting the validity of wee1 inhibition as a viable therapeutic target in cancer therapy.
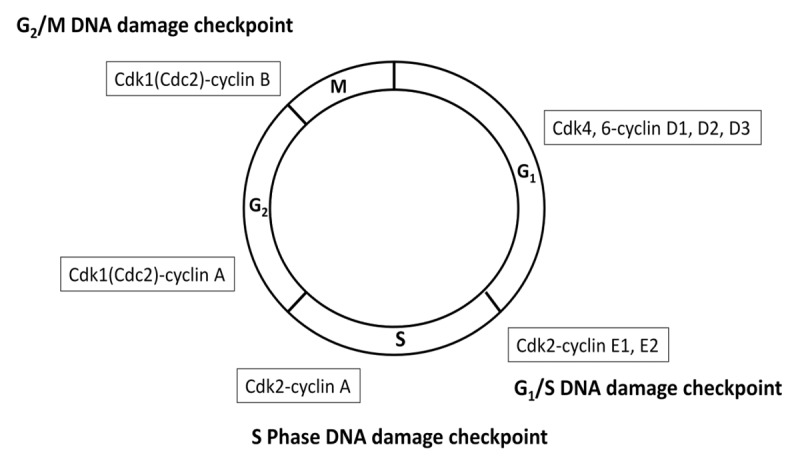
Figure 1. Cyclin-dependent kinase regulation of the cell cycle. Cell cycle checkpoints serve to halt progression of the cell cycle in response to DNA damage, allowing time for DNA repair and the maintenance of genomic integrity. Cyclin-dependent kinases regulate the progression through cell cycle checkpoints. Abbreviations: Cdk, cyclin-dependent kinase.
Cell Cycle and DNA Repair
Cell cycle checkpoints serve to halt progression of the cell cycle in response to DNA damage, allowing time for repair and for maintenance of genomic integrity. p53, a key regulator of the G1 checkpoint via p21Cip1, is frequently mutated in malignancies. Tumors that are defective in p53 function are often defective in arresting cells in G1 in response to DNA damage and rely on an intact G2 checkpoint. DNA damage detected at the G2 checkpoint is relayed via 2 well-characterized parallel and interconnected pathways that culminate in the inhibitory phosphorylation of cyclin-dependent kinase 1 (Cdk1/Cdc2) and subsequent delay of entry into mitosis.3 Depending on the type of genotoxic stress, either ataxia-telangectasia mutated (ATM) protein kinase or ataxia-telangiectasia-related (ATR) protein kinase, is preferentially activated (Fig. 2).
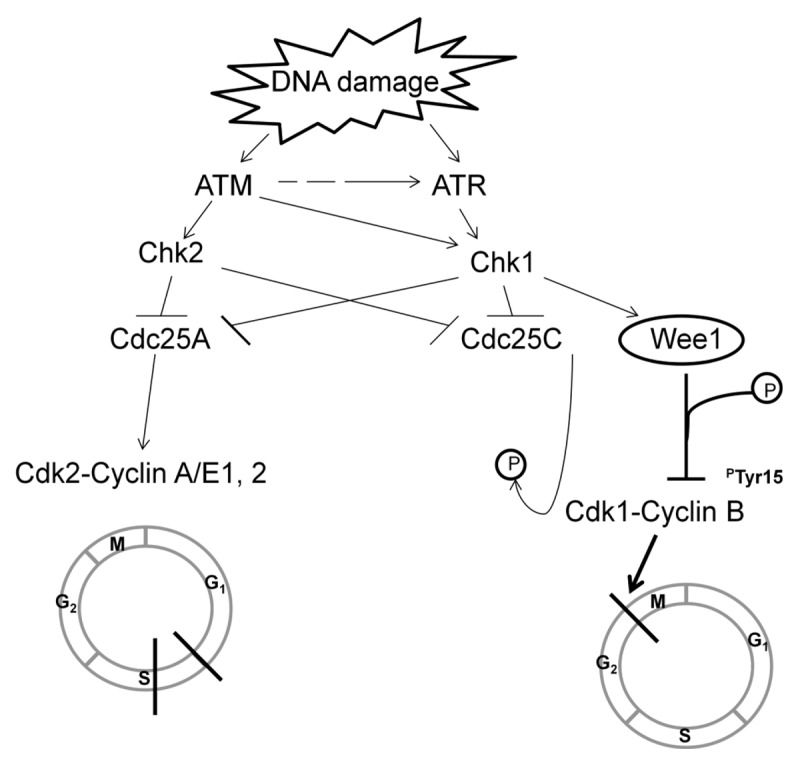
Figure 2. DNA damage detected at the G2 checkpoint. Depending on the type of genotoxic stress, either ataxia-telangectasia mutated (ATM) protein kinase or ataxia-telangiectasia-related (ATR) protein kinase are preferentially activated. ATR is the main kinase responsible for the phosphorylation and activation of checkpoint kinase 1 (Chk1). Chk1, in turn, concomitantly phosphorylates wee1 and Cdc25C, thereby activating wee1 kinase activity and inactivating Cdc25C phosphatase activity. Wee1 phosphorylates and inactivates Cdk1/Cdc2-bound cyclin B on its tyrosine15 residue, resulting in cell cycle arrest at G2, allowing time for DNA repair. Abbreviations: ATM, ataxia-telangectasia mutated protein kinase; ATR, ataxia-telangiectasia-related protein kinase; Chk, checkpoint kinase; Cdk, cyclin-dependent kinase; Cdc, cell division cycle phosphatase.
ATM is recruited and activated in response to ionizing radiation, radiomimetic agents, and agents which cause double-strand DNA breaks. ATM phosphorylates and activates checkpoint kinase 2 (Chk2), which, in turn, phosphorylates cell division cycle 25C phosphatase (Cdc25C) at Ser216, creating a binding site for the 14-3-3σ protein.4 This leads to nuclear export and cytoplasmic sequestration of Cdc25C, effectively inactivating its phosphatase activity. Suppression of Cdc25C phosphatase activity, in turn, results in unopposed inhibitory phosphorylation of the Cdk1/Cdc2-bound cyclin B complex, maintaining Cdk1 in an inert form and preventing entry into mitosis.
ATR is activated by a broader range of genotoxic stimuli that result in single-strand DNA breaks.5,6 In addition, ATM-dependent activation of ATR also occurs during the processing of double-stranded DNA breaks, where 5′ to 3′ resection of double-strand break ends results in the generation of single-strand DNA ends, which are stabilized and coated by replication protein A (RPA), serving as a scaffold and activation point for ATR.6 ATR is the main kinase responsible for the phosphorylation and activation of checkpoint kinase 1 (Chk1). In contrast to Chk2, Chk1 can be activated by both ATM and ATR, and activation can occur during normal cell cycle progression or in response to replicative stress, such as during stalled replication forks. Chk1, in turn, concomitantly phosphorylates wee1 and Cdc25C, thereby activating wee1 kinase activity and inactivating Cdc25C phosphatase activity. Wee1, in turn, phosphorylates and inactivates Cdk1/Cdc2-bound cyclin B on its tyrosine15 residue, resulting in cell cycle arrest at G2, allowing time for DNA repair.
Recent evidence demonstrates that wee1 is also involved in the coordination of DNA replication and the maintenance of stalled replication forks through regulation of cyclin-dependent kinase 2 (Cdk2).7 Wee1 phosphorylates and inactivates Cdk2-bound cyclin E, thereby controlling regulation of DNA replication during S phase. Enhanced activation of Cdk2 in Wee1-depleted cells is proposed to result in a loss of control of coordination of origins of replication, resulting in aberrant DNA structures that require cleavage by Mus81. The Mus81/Eme1 enzyme complex is required for reversion of stalled replication forks, induction of double-stranded breaks to allow for replication recovery, and initiation of homologous recombination repair. Martin et al. demonstrated a direct role for wee1 in the regulation of the Mus81-Eme1 endonuclease activity.8 Deregulation of Mus81 activity would result in unchecked cleavage of substrates, further destabilizing the chromatin structure.
Wee 1 as a Target for Cancer Therapy
Overexpression of wee1 has been observed in several malignancies, including hepatocellular carcinoma,9 luminal and HER-2 positive breast cancers,10 glioblastoma,11 and malignant melanoma, where high expression has been shown to correlate with poor disease-free survival.12 In cancer cells with aberrant G1 checkpoint, inhibition of wee1 with resultant abrogation of the G2 checkpoint allows for continued cell cycle progression while still harboring unrepaired damaged DNA, culminating in mitotic lethality. Inhibition of wee1 either by the pyrido-pyrimidine derivative (PD0166285) or via siRNA gene knockdown has been shown to sensitize ovarian, colon, cervical, osteosarcoma, glioblastoma, and lung cancer cells to DNA damage by irradiation and topoisomerase inhibition.13-15 Cytotoxicity was more pronounced in the p53-deficient compared with p53-intact cell lines in these studies, consistent with the expectation that tumor cells harboring p53 mutations with aberrant G1 checkpoint would be more sensitive to the abrogation of the G2 checkpoint. Xenografts of glioblastoma cells transduced with short-hairpin RNA against WEE1 implanted into the brain of nude mice, followed by a single dose of 6 Gy of irradiation at 2 weeks, resulted in tumor regression at 6 wk with a survival advantage compared with controls.13
MK-1775, a recently developed pyrazolo-pyrimidine derivative, is a potent and selective small-molecule inhibitor of wee1 with an IC50 of 5 nmol/L. Co-administration of MK-1775 in preclinical models demonstrated enhanced antitumor efficacy of various classes of DNA damaging agents with different modes of action, including anti-metabolites (gemcitabine, 5-FU, capecitabine, pemetrexed), topoisomerase inhibition (camptothecin, doxorubicin), and DNA cross-linking agents (carboplatin, cisplatin, and mitomycin C) in p53-deficient colon, cervical, lung, and pancreatic cell lines.16-18 In these initial studies, investigators demonstrated dose-dependent direct substrate inhibition of Cdk1 by MK-1775, based on colorimetric ELISA measurement of levels of phosphorylated tyrosine15 on Cdc2 (pCDC2-Tyr15). Premature mitotic entry was demonstrated by a concomitant increase in the percentage of cells expressing serine10-phosphorylated histone H3 (pHH3) at the same doses.
Subsequent xenograft studies using oral administration of MK-1775 in nude rats bearing colorectal, pancreatic, cervical, ovarian, and breast cancers demonstrated objective tumor regression in combination with various DNA damaging agents, including gemcitabine, carboplatin, cisplatin, and capecitabine.17,18 These studies demonstrate that chemical inhibition of wee1 with MK-1775 allows for potentiation of cytotoxicity of various DNA damaging agents with different modes of action, and that this effect is more pronounced in p53-deficient cancers, supporting the notion that tumors with defective G1 checkpoint are heavily dependent upon the G2 checkpoint for escape from mitotic lethality. The antitumor effect also correlated with increased levels of pHH3 and caspase-3/7 activation, confirming that potentiation of cytotoxicity was due to both early entry into mitosis and cell death via apoptosis.
Wee 1 and Radiation Therapy
Based on the promising preclinical data demonstrating potentiation of cytotoxicity in combination with various DNA damaging agents, MK-1775 was tested as a radiosensitizer in human tumor cells derived from lung, breast, and prostate cancers.19 Double-strand breaks (DSBs) induced by radiation require cell cycle delay at the G2 checkpoint, as the primary mechanism for repair of DSBs through HR requires use of the sister chromatid as a template, which is only available after DNA replication during S phase. Upon the induction of DSBs, phosphorylation of histone H2AX also occurs at serine 139 sites flanking the areas of DSBs, forming γH2AX foci. Measurement of γH2AX levels can therefore serve as a dosimeter of the degree of DSBs. γH2AX foci formation has also been proposed to serve as a focal point for the recruitment of mediators and effectors of the DNA damage repair response.20 Chemical inhibition of wee1 has been shown to result in an increase of γH2AX levels in breast carcinoma cells 4 h after administration of the inhibitor, with concomitant accumulation of cells in S-phase.21 Similar results have also been demonstrated with siRNA depletion of Wee1 in U2OS, human osteosarcoma cells.22 In clonogenic survival assays, MK-1775 was shown to preferentially radiosensitize p53-defective tumor cells compared with wild-type controls, with concomitant decrease in pCDC2Tyr15 levels within 1 h following administration of MK-1775. In these studies, investigators additionally tested the effect of pre-treatment of the same p53-defective tumor cells with MK-1775 prior to irradiation. Both irradiated and non-irradiated cells were accelerated into mitosis prematurely, reflected in the percentage of cells expressing pHH3, though higher levels of γH2AX, reflecting unrepaired DSBs, were seen within the fraction of p53-defective mitotic cells which had undergone irradiation. The fact that un-irradiated cells also demonstrated high levels of γH2AX suggests inhibition of wee1 alone results in propagation of inherent DNA damage. Bridges et al. also tested the in vivo efficacy of combination of MK-1775 and radiation using the human non-small cell lung cancer cell line, Calu-6, in a xenograft model in nude mice. Using fractionated radiotherapy, they demonstrated MK-1775 enhanced xenograft tumor response when given on the same days as irradiation. In addition to demonstrating clinical utility in combination with radiotherapy, this study was the first to demonstrate MK-1775 activity in un-irradiated cells, highlighting the potential for MK-1775 as a single-agent therapy.
Single Agent Activity of Wee1 Inhibition
Recent preclinical data using sarcoma cell lines and patient-derived tumor explants provide additional supportive evidence for single-agent cytotoxicity of MK-1775.23 Interestingly, these cytotoxic effects were independent of p53 status, as cell death was demonstrated both in p53 wild-type sarcoma cell lines as well as p53 mutant cell lines. At doses ranging from 100 to 550 nM, single-agent MK-1775 treatment resulted in decrease in pCDC2Tyr15 levels, elevation of pHH3 levels, and increase in γH2AX, comparable to that of previous data with combination therapy with DNA damaging agents and MK-1775 doses of 200 nM. Additionally, investigators demonstrated a significant increase in cleaved PARP and caspase-3 correlating with the decrease in pCDC2Tyr15 levels, indicating that the mechanism of cell death was by induction of apoptosis. Patient-derived tumors from undifferentiated high-grade sarcoma, malignant peripheral nerve sheath tumor, and pleomorphic spindle cell tumor, were also treated ex vivo with MK-1775 at doses of 500 nM, with evidence of decrease in pCDC2Tyr15 levels and histologic evidence of cell death in all samples. More recent studies by Guertin et al. alternatively propose that the predominant effect of wee1 inhibition by MK-1775 may be due to direct DNA damage rather than premature entry into mitosis, demonstrating cytotoxicity in various cell lines regardless of the mitotic indices.24 In these studies, they also show tumor growth inhibition of non-small cell lung cancer and colorectal cancer xenografts treated with MK-1775 alone. Collectively, the results of these studies provide strong support for further development of MK-1775 as a single agent anticancer therapy.
Clinical Studies
Preliminary data from phase I studies testing escalating single and multiple doses of MK-1775 in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors have shown promising activity, with stable disease in 14 of 28 evaluable patients with a manageable toxicity profile.25,26 Two other phase I trials have recently completed enrollment: one evaluating MK-1775 in combination with either 5-FU in patients with advanced solid tumors (NCT01047007)27 or 5-FU with cisplatin in patients with advanced head and neck, esophageal, or gastric cancer, and a second phase I/II study evaluating the combination of MK-1775 with cisplatin and topotecan in patients with advanced cervical cancer (NCT01076400).28 Two phase II studies are currently underway: one sponsored by the Netherlands Cancer Institute currently evaluating the combination of MK-1775 and carboplatin in p53-mutated epithelial ovarian cancer patients whose disease has progressed through prior paclitaxel/carboplatin therapy (NCT01164995),29 and a randomized study evaluating the combination of MK-1775 with paclitaxel + carboplatin vs. placebo with paclitaxel + carboplatin in p53-mutated ovarian, fallopian tube, and primary peritoneal tumors (NCT01357161).30 A phase I study is currently evaluating the safety and maximum tolerated dose of MK-1775 as a single-agent in advanced refractory solid tumors (NCT01748825).31
Future Directions for Combination with Other Molecular Targeted Agents
The initial clinical development of the Wee1 inhibitor, MK-1775, has been to chemopotentiate the DNA damaging effects of chemotherapy, utilizing the concept of mitotic lethality as a mechanism for antitumor activity based on early in vitro studies demonstrating its role in regulation of Cdk1 and the G2 checkpoint. More recent evidence implicates a role for wee1 in stabilization of replication forks and DNA repair, highlighting the potential for monotherapy and in combination with other targeted agents. Cdk1, the main target of wee1, additionally plays a key role in HR and double-strand DNA repair. Early studies in yeast showed Cdk1 is required for the efficient 5′ to 3′ resection of double-stranded break ends to produce single-stranded DNA and the subsequent recruitment of the RPA complex and the Rad51 recombination protein to sites of DNA damage.32 Subsequent studies of whole-cell extracts of HeLa cells demonstrated a role for Cdk1 in the phosphorylation of the Rad51 binding domain of BRCA2 at serine 3291 (S3291).33 BRCA2 modulation of Rad51 activity is essential for strand exchange and HR repair. In vitro studies have shown that chemical inhibition of Cdk1 sensitizes BRCA-proficient cancers to PARP inhibition.34 In contrast, chemical inhibition of wee1 with resultant aberrant forced activation of Cdk1 also appears to impair HR, as demonstrated by an increase in phosphorylation of BRCA2 at S3291.35 Low levels of Cdk activity are required for proper induction of HR repair, and inhibition of Cdk activity impairs the DNA damage response. For reasons which remain poorly understood, induction of Cdk activity to aberrantly high levels, achieved through wee1 inhibition, also appears to result in a comparable impairment of HR DNA damage response. Given the role of wee1 in HR, the induction of a HR deficient state through wee1 inhibition presents a unique opportunity to evaluate the dual inhibition of wee 1 and poly (ADP-ribose) polymerase (PARP) in clinical trials. PARPs are a family of nuclear enzyme proteins that play a key role in the DNA damage response, including base excisional repair, homologous recombination, and non-homologous end-joining.36-39 PARP inhibition results in trapping of PARP-1 on single-strand DNA repair intermediates that require BRCA-dependent HR for resolution.40 Inhibition of multiple DNA repair pathways prevents the ability of cells to compensate for DNA damage and provides a rationale for targeting both wee1 and PARP in cancer therapy.
Hyperactivation of Cdk activity through combined inhibition of both Chk1 and wee1 has also been shown to result in synergistic potentiation of inhibition of cellular proliferation in multiple cell lines.41 Given the roles of Chk1 and wee1 in the regulation of cell cycle checkpoints, investigators have sought to combine inhibition of both kinases and demonstrated a greater than additive effect in cytotoxicity and induction of apoptosis in multiple cell lines and colorectal and ovarian carcinoma xenografts. In addition to regulation of cell cycle checkpoints through inhibitory phosphorylation of Cdc25, Chk1 is also involved in DNA replication and initiation of HR repair through direct interaction with Rad51.42 In these studies, investigators demonstrated non-overlapping distinct activity of each agent, as Chk1 inhibition alone affects DNA synthesis with accumulation of cells in early S-phase, as demonstrated by flow cytometry of labeled nucleotide, whereas wee1 inhibition alone affects DNA synthesis in late S-phase.41 The combined inhibition with both inhibitors resulted in greater than additive increase in DNA damage, reflected as an increase in γH2AX levels with resultant induction of apoptosis, as evidenced by an increase in cleaved caspase levels. This synergy was independent of p53 status, consistent with known activity of Chk1 in regulation of the G1/S checkpoint through inhibitory phosphorylation of Cdc25A.
Wee1 is known to be a client protein of the molecular chaperone Hsp90, which is involved in stabilizing and maintaining the functional conformation of newly synthesized and misfolded proteins, including oncoproteins.43 Studies in yeast have demonstrated wee1-dependent tyrosine phosphorylation of the N-terminal domain of Hsp90, allowing for regulation of chaperone function and reinforcement of its own interaction with the molecular chaperone.44 Subsequent studies have demonstrated that chemical inhibition of wee1 sensitized prostate and cervical cancer cells to the Hsp90 inhibitor, 17-allylamino-17-demethoxygeldanamycin (17-AAG), which binds to the ATP-binding pocket of Hsp90 at the N-terminal domain, interfering with the function of the molecular chaperone.45 Inhibition of wee1 has also been shown to sensitize tumor xenografts to Hsp90 inhibition, invoking activation of the intrinsic apoptotic pathway as the main mechanism of action.46 Combined inhibition of wee1 and Hsp90 provides not only an opportunity to augment the efficacy of Hsp90 inhibitors, but may also provide an opportunity to achieve more complete inhibition of wee1 activity, further enhancing the antitumor activity.
Wee1 inhibition has also been shown to enhance caspase-dependent apoptosis in combination with TNF-related apoptosis-inducing ligand (TRAIL) in triple-negative breast cancer cell lines.47 Pre-treatment with the pan-caspase inhibitor Z-VAD-FMK abrogated the induction of cell death by the combination of TRAIL and wee1 inhibition. Chemical inhibition of wee1 alone was also shown to upregulate transcription of TRAIL receptors in a p53-independent manner. Interestingly, the rise in γH2AX levels induced upon inhibition of wee1 in these studies was not abrogated by the pan-caspase inhibitor, suggesting that the DNA damage is independent of caspase activation. Collectively, these data highlight the potential for combining wee1 inhibitors with other molecular targeted agents and presents a viable strategy for enhancing the therapeutic ratio of these various molecular targeted agents.
Conclusion
The inherent genetic instability and the enhanced proliferation index make tumor cells sensitive to DNA damaging agents. With continued therapy, cancer cells acquire the ability to repair the DNA damage and survive in spite of cytotoxic chemotherapy. The rationale behind development of checkpoint kinase inhibitors lies in the fact that abrogation of cell cycle checkpoints allows for progression of the cell cycle in the face of damaged DNA, perpetuation, and accumulation of fatal defective lesions, and premature entry into cell division, ultimately resulting in cell death. Development of a selective wee1 inhibitor, MK-1775, presents a viable and novel therapeutic approach to enhancing the antitumor efficacy of traditional chemotherapeutic agents. Recent evidence implicates a greater role for wee1 in DNA repair, highlighting the potential for combination with other targeted agents. Future clinical development of wee1 inhibitors should focus on further defining their role as potentiators of DNA damaging therapies, evaluating combinations with other targeted agents and as a single agent in tumors with DNA repair defects.
Glossary
Abbreviations:
- Chk1
checkpoint kinase 1
- Chk2
checkpoint kinase 2
- CDK
cyclin-dependent kinase
- pHH3
serine10 phosphorylated histone H3
- HR
homologous recombination
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26062
References
- 1.Russell P, Nurse P. Negative regulation of mitosis by wee1+, a gene encoding a protein kinase homolog. Cell. 1987;49:559–67. doi: 10.1016/0092-8674(87)90458-2. [DOI] [PubMed] [Google Scholar]
- 2.Parker LL, Piwnica-Worms H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science. 1992;257:1955–7. doi: 10.1126/science.1384126. [DOI] [PubMed] [Google Scholar]
- 3.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–9. doi: 10.1016/S1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 4.Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci U S A. 2000;97:10389–94. doi: 10.1073/pnas.190030497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson N, Cai D, Kennedy RD, Pathania S, Arora M, Li YC, D’Andrea AD, Parvin JD, Shapiro GI. Cdk1 participates in BRCA1-dependent S phase checkpoint control in response to DNA damage. Mol Cell. 2009;35:327–39. doi: 10.1016/j.molcel.2009.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 7.Domínguez-Kelly R, Martín Y, Koundrioukoff S, Tanenbaum ME, Smits VA, Medema RH, Debatisse M, Freire R. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J Cell Biol. 2011;194:567–79. doi: 10.1083/jcb.201101047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martín Y, Domínguez-Kelly R, Freire R. Novel insights into maintaining genomic integrity: Wee1 regulating Mus81/Eme1. Cell Div. 2011;6:21. doi: 10.1186/1747-1028-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masaki T, Shiratori Y, Rengifo W, Igarashi K, Yamagata M, Kurokohchi K, Uchida N, Miyauchi Y, Yoshiji H, Watanabe S, et al. Cyclins and cyclin-dependent kinases: comparative study of hepatocellular carcinoma versus cirrhosis. Hepatology. 2003;37:534–43. doi: 10.1053/jhep.2003.50112. [DOI] [PubMed] [Google Scholar]
- 10.Iorns E, Lord CJ, Grigoriadis A, McDonald S, Fenwick K, Mackay A, Mein CA, Natrajan R, Savage K, Tamber N, et al. Integrated functional, gene expression and genomic analysis for the identification of cancer targets. PLoS One. 2009;4:e5120. doi: 10.1371/journal.pone.0005120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mir SE, De Witt Hamer PC, Krawczyk PM, Balaj L, Claes A, Niers JM, Van Tilborg AA, Zwinderman AH, Geerts D, Kaspers GJ, et al. In silico analysis of kinase expression identifies WEE1 as a gatekeeper against mitotic catastrophe in glioblastoma. Cancer Cell. 2010;18:244–57. doi: 10.1016/j.ccr.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Magnussen GI, Holm R, Emilsen E, Rosnes AKR, Slipicevic A, Flørenes VA. High expression of Wee1 is associated with poor disease-free survival in malignant melanoma: potential for targeted therapy. PLoS One. 2012;7:e38254. doi: 10.1371/journal.pone.0038254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Li J, Booher RN, Kraker A, Lawrence T, Leopold WR, Sun Y. Radiosensitization of p53 mutant cells by PD0166285, a novel G(2) checkpoint abrogator. Cancer Res. 2001;61:8211–7. [PubMed] [Google Scholar]
- 14.Wang Y, Decker SJ, Sebolt-Leopold J. Knockdown of Chk1, Wee1 and Myt1 by RNA interference abrogates G2 checkpoint and induces apoptosis. Cancer Biol Ther. 2004;3:305–13. doi: 10.4161/cbt.3.3.697. [DOI] [PubMed] [Google Scholar]
- 15.PosthumaDeBoer J, Würdinger T, Graat HC, van Beusechem VW, Helder MN, van Royen BJ, Kaspers GJ. WEE1 inhibition sensitizes osteosarcoma to radiotherapy. BMC Cancer. 2011;11:156. doi: 10.1186/1471-2407-11-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, Kimura T, Kaneko N, Ohtani J, Yamanaka K, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther. 2009;8:2992–3000. doi: 10.1158/1535-7163.MCT-09-0463. [DOI] [PubMed] [Google Scholar]
- 17.Hirai H, Arai T, Okada M, Nishibata T, Kobayashi M, Sakai N, Imagaki K, Ohtani J, Sakai T, Yoshizumi T, et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol Ther. 2010;9:514–22. doi: 10.4161/cbt.9.7.11115. [DOI] [PubMed] [Google Scholar]
- 18.Rajeshkumar NV, De Oliveira E, Ottenhof N, Watters J, Brooks D, Demuth T, et al. MK-1775, a potent Wee1 inihibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin Cancer Res. 2011;17:1–8. doi: 10.1158/1078-0432.CCR-10-2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bridges KA, Hirai H, Buser CA, Brooks C, Liu H, Buchholz TA, Molkentine JM, Mason KA, Meyn RE. MK-1775, a novel Wee1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Clin Cancer Res. 2011;17:5638–48. doi: 10.1158/1078-0432.CCR-11-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murrow LM, Garimella SV, Jones TL, Caplen NJ, Lipkowitz S. Identification of WEE1 as a potential molecular target in cancer cells by RNAi screening of the human tyrosine kinome. Breast Cancer Res Treat. 2010;122:347–57. doi: 10.1007/s10549-009-0571-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beck H, Nähse V, Larsen MS, Groth P, Clancy T, Lees M, Jørgensen M, Helleday T, Syljuåsen RG, Sørensen CS. Regulators of cyclin-dependent kinases are crucial for maintaining genome integrity in S phase. J Cell Biol. 2010;188:629–38. doi: 10.1083/jcb.200905059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kreahling JM, Gemmer JY, Reed D, Letson D, Bui M, Altiok SMK. MK1775, a selective Wee1 inhibitor, shows single-agent antitumor activity against sarcoma cells. Mol Cancer Ther. 2012;11:174–82. doi: 10.1158/1535-7163.MCT-11-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guertin AD, Li J, Liu Y, Hurd MS, Schuller AG, Long B, Hirsch HA, Feldman I, Benita Y, Toniatti C, et al. Preclinical Evaluation of the WEE1 Inhibitor MK-1775 as Single-Agent Anticancer Therapy. Mol Cancer Ther. 2013;12:1442–52. doi: 10.1158/1535-7163.MCT-13-0025. [DOI] [PubMed] [Google Scholar]
- 25.Schellens JH, Leijen S, Shapiro GI, Pavlick AC, Tibes R, O’Day SJ, et al. ASCO Annual Meeting Proceedings A phase I and pharmacologic study of MK-1775, a Wee1 tyrosine kinase inhibitor, in both monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J Clin Oncol. 2009 doi: 10.1200/JCO.2016.67.5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.A Phase I Dose Escalation Study Evaluating MK1775 in Both Monotherapy and in Combination With Either Gemcitabine, Cisplatin, or Carboplatin in Adult Subjects With Advanced Solid Tumors (MK-1775-001 AM7). Clinical Trials. gov Identifier: NCT00648648 [Google Scholar]
- 27.A Phase I Dose Escalation Study of MK1775 in Monotherapy, in Combination With 5-Fluorouracil, and in Combination With 5-Fluorouracil and Cisplatin in Patients With Advanced Solid Tumor (1775-005). Clinical Trials. gov Identifier: NCT01047007 [Google Scholar]
- 28.A Two Part, Phase I-IIa Study Evaluating MK1775 in Combination With Topotecan/Cisplatin in Adult Patients With Cervical Cancer (1775-008). Clinical Trials. gov Identifier: NCT01076400 [Google Scholar]
- 29.Phase II Pharmacological Study With Wee-1 Inhibitor MK-1775 Combined With Carboplatin in Patients With p53 Mutated Epithelial Ovarian Cancer and Early Relapse (< 3 Months) or Progression During Standard First Line Treatment (M10MKO). Clinical Trials. gov Identifier: NCT01164995 [Google Scholar]
- 30.A Randomized, Phase II Study Evaluating MK-1775 in Combination With Paclitaxel and Carboplatin Versus Paclitaxel and Carboplatin Alone in Adult Patients With Platinum Sensitive p53 Mutant Ovarian Cancer. Clinical Trials. gov Identifier: NCT01357161 [Google Scholar]
- 31.A Phase I Study of Single-agent MK-1775, a Wee1 Inhibitor, in Patients With Advanced Refractory Solid Tumors. Clinical Trials. gov Identifier: NCT01748825 [Google Scholar]
- 32.Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth NM, et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 2004;431:1011–7. doi: 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Esashi F, Christ N, Gannon J, Liu Y, Hunt T, Jasin M, West SC. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature. 2005;434:598–604. doi: 10.1038/nature03404. [DOI] [PubMed] [Google Scholar]
- 34.Johnson N, Li YC, Walton ZE, Cheng KA, Li D, Rodig SJ, Moreau LA, Unitt C, Bronson RT, Thomas HD, et al. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat Med. 2011;17:875–82. doi: 10.1038/nm.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krajewska M, Heijink AM, Bisselink YJ, Seinstra RI, Silljé HH, de Vries EG, et al. Forced activation of Cdk1 via wee1 inhibition impairs homologous recombination. Oncogene. 2012 doi: 10.1038/onc.2012.296. [DOI] [PubMed] [Google Scholar]
- 36.Houtgraaf JH, Versmissen J, van der Giessen WJ. A concise review of DNA damage checkpoints and repair in mammalian cells. Cardiovasc Revasc Med. 2006;7:165–72. doi: 10.1016/j.carrev.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 37.El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003;31:5526–33. doi: 10.1093/nar/gkg761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haince JF, McDonald D, Rodrigue A, Déry U, Masson JY, Hendzel MJ, Poirier GG. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J Biol Chem. 2008;283:1197–208. doi: 10.1074/jbc.M706734200. [DOI] [PubMed] [Google Scholar]
- 39.Wang M, Wu W, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34:6170–82. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol. 2011;5:387–93. doi: 10.1016/j.molonc.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davies KD, Cable PL, Garrus JE, Sullivan FX, von Carlowitz I, Huerou YL, Wallace E, Woessner RD, Gross S. Chk1 inhibition and Wee1 inhibition combine synergistically to impede cellular proliferation. Cancer Biol Ther. 2011;12:788–96. doi: 10.4161/cbt.12.9.17673. [DOI] [PubMed] [Google Scholar]
- 42.Sørensen CS, Hansen LT, Dziegielewski J, Syljuåsen RG, Lundin C, Bartek J, Helleday T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol. 2005;7:195–201. doi: 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
- 43.Aligue R, Akhavan-Niak H, Russell P. A role for Hsp90 in cell cycle control: Wee1 tyrosine kinase activity requires interaction with Hsp90. EMBO J. 1994;13:6099–106. doi: 10.1002/j.1460-2075.1994.tb06956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mollapour M, Tsutsumi S, Neckers L. Hsp90 phosphorylation, Wee1 and the cell cycle. Cell Cycle. 2010;9:2310–6. doi: 10.4161/cc.9.12.12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mollapour M, Tsutsumi S, Donnelly AC, Beebe K, Tokita MJ, Lee MJ, Lee S, Morra G, Bourboulia D, Scroggins BT, et al. Swe1Wee1-dependent tyrosine phosphorylation of Hsp90 regulates distinct facets of chaperone function. Mol Cell. 2010;37:333–43. doi: 10.1016/j.molcel.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iwai A, Bourboulia D, Mollapour M, Jensen-Taubman S, Lee S, Donnelly AC, Yoshida S, Miyajima N, Tsutsumi S, Smith AK, et al. Combined inhibition of Wee1 and Hsp90 activates intrinsic apoptosis in cancer cells. Cell Cycle. 2012;11:3649–55. doi: 10.4161/cc.21926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garimella SV, Rocca A, Lipkowitz S. WEE1 inhibition sensitizes basal breast cancer cells to TRAIL-induced apoptosis. Mol Cancer Res. 2012;10:75–85. doi: 10.1158/1541-7786.MCR-11-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]