

Abstract
Akt/PKB is a key signaling molecule in higher eukaryotes and a crucial protein kinase in human health and disease. Phosphorylation, acetylation, and ubiquitylation have been reported as important regulatory post-translational modifications of this kinase. We describe here that Akt is modified by SUMO conjugation, and show that lysine residues 276 and 301 are the major SUMO attachment sites within this protein. We found that phosphorylation and SUMOylation of Akt appear as independent events. However, decreasing Akt SUMOylation levels severely affects the role of this kinase as a regulator of fibronectin and Bcl-x alternative splicing. Moreover, we observed that the Akt mutant (Akt E17K) found in several human tumors displays increased levels of SUMOylation and also an enhanced capacity to regulate fibronectin splicing patterns. This splicing regulatory activity is completely abolished by decreasing Akt E17K SUMO conjugation levels. Additionally, we found that SUMOylation controls Akt regulatory function at G₁/S transition during cell cycle progression. These findings reveal SUMO conjugation as a novel level of regulation for Akt activity, opening new areas of exploration related to the molecular mechanisms involved in the diverse cellular functions of this kinase.
Keywords: signal transduction, post-translational modification, SUMO, Akt/PKB, alternative splicing, cell cycle
Introduction
SUMO (small ubiquitin-related modifier) conjugation or “SUMOylation” is a transient and reversible post-translational modification (PTM) consisting in the covalent attachment of SUMO to a target protein.1 SUMOylation affects activity, structure, sub-cellular localization, and the repertoire of interactions of the target proteins.2 The steps involved in the SUMO conjugation pathway resemble those of the ubiquitin pathway: (1) SUMO needs to be proteolytically cleaved to expose its C-terminal glycine–glycine (GG) motif, a step performed by SUMO-specific proteases of the Sentrin-specific protease (SENP) family; (2) mature SUMO is activated by the SUMO-specific E1-activating enzyme, the heterodimer SAE I/SAE II in mammals, in a Mg2+- and ATP-dependent manner; (3) SUMO is transferred from SAE II to the E2 conjugating enzyme Ubc9, forming a thioester bond; and (4) Ubc9 transfers SUMO to the substrate, catalyzing the formation of an isopeptide bond between the C-terminal glycine of SUMO and the ε-NH2 group of the lysine (K) residue in the target protein.1-4 Although most targets can be SUMOylated in the presence of E1 and E2 in vitro, the process is usually facilitated by SUMO E3 ligases both in vitro and in vivo.5-9 Reversibility of the process is achieved by the action of SENPs that cleave the isopeptide bond and release SUMO for further cycles.10 Although modification by SUMO has been mostly studied in the context of transcription, chromatin remodeling, DNA repair, nucleo-cytoplasmic transport, and mitosis,11 SUMO substrates seem to contribute to every aspect of cell function, including signal transduction.12-14
Akt (also known as protein kinase B or PKB) is a serine/threonine kinase member of the AGC family of protein kinases, conserved from primitive metazoan to humans, and involved in the regulation of a plethora of cellular processes, such as cell growth, proliferation, apoptosis, metabolism, and angiogenesis.15 Remarkably, deregulation of Akt signaling is associated with several human diseases including cardiac hypertrophy, diabetes, neuronal degeneration, vascular disorders, and cancer.16,17 Therefore, Akt-dependent pathways are considered an attractive target for therapeutic intervention, and a deep understanding of the molecular mechanisms underlying the regulation of this kinase activity becomes of paramount importance.18 Three closely related and widely expressed Akt isoforms (Akt1, Akt2, and Akt3) are present in mammals. Akt activation downstream of growth and survival factors as well as other extracellular stimuli comprises recruitment of Akt to the plasma membrane through its binding to phosphoinositide 3 kinase (Pi3K)-generated phosphatidylinositol 3,4,5-trisphosphate (PIP3), and phosphorylation of Akt at a threonine residue within the kinase domain (T308 in Akt1) by PDK1 (phosphoinositide-dependent protein kinase 1), and at a serine residue within the regulatory domain (S473 in Akt1) by mTORC2 (mammalian target of rapamycin [mTOR] complex 2). Once activated by these 2 phosphorylation events, Akt isoforms exert their action by phosphorylating a wide variety of downstream targets at different subcellular compartments.19,20 Phosphorylation of other residues is important also for Akt folding, stability, and activity.21-25
Despite a large body of data dealing with the regulation of Akt by phosphorylation, the information about the influence of other PTMs on Akt activity is less abundant. It has been shown that Akt as well as PDK1 activity is regulated by reversible acetylation.26 Recent reports have also linked ubiquitin conjugation to Akt stability and function. In this context, several E3 ligases responsible for regulating Akt ubiquitylation have been described, some of which trigger ubiquitin-dependent Akt degradation,27,28 while others enhance Akt membrane recruitment and downstream activity.29-31
Among other activities, Akt regulates cell cycle progression impinging at different levels. It has been shown that Akt downregulates transcription of cyclin-dependent kinase (CDK) inhibitors p21Cip1 and p27Kip1 genes through phosphorylation of MDM2 and degradation of p53 in the former case and through phosphorylation and inactivation of FOXO/Forkhead family of transcription factors in the latter. Akt directly phosphorylates GSK3, stimulating cyclin D1 translation. It also phosphorylates p21Cip1 and p27Kip1, causing their nuclear exclusion, thus releasing CDK2 and facilitating G1/S transition. Moreover, activation of Akt overcomes the DNA damage-induced G2/M checkpoint.32-35
Work from our laboratory and others have implicated Akt as a key transducer of extracellular cues into changes in splicing patterns.36-42 We have shown that activation of the Pi3K–Akt pathway stimulates inclusion of EDI (a.k.a. EDA) alternative exon into fibronectin mRNA. This effect is mediated by 2 splicing regulatory factors from the SR protein family, SRSF1 and SRSF7, that we identified as Akt phosphorylation substrates, revealing a role for Akt as a novel SR protein kinase.37 The precise mechanisms involved in this splicing regulatory activity of Akt have remained elusive.
Here we show that Akt is modified by SUMO conjugation both in vitro and in cultured cells. We mapped SUMO conjugation sites within Akt1 and demonstrated that this PTM regulates its activity, in particular its role in alternative splicing and cell cycle regulation.
Results
Akt is modified by SUMO in vitro and in cultured cells
Akt is a known target of different PTMs: phosphorylation, acetylation, and ubiquitylation. In silico analysis of Akt1 protein sequence performed with SUMOsp2.0 (http://sumosp.biocuckoo.org/) and SUMOplot (http://www.abgent.com/sumoplot) software predicted several putative consensus sites for SUMO conjugation, 5 of them residing within the kinase domain (Fig. S1A). To validate Akt1 as a bona-fide SUMOylation target, HEK 293T cells were transfected with expression vectors for 6xHis-SUMO2, the SUMO-E2 conjugating enzyme Ubc9 and HA-tagged Akt1. Whole-cell lysates were analyzed by western blot with anti-HA antibody, revealing several bands that migrated slower than HA-Akt1 and were enhanced upon Ubc9 overexpression, suggesting they correspond to SUMO-modified Akt1 (Fig. 1A). Additionally, SUMO conjugates were enriched from cell lysates by denaturing Ni2+ affinity chromatography and analyzed by western blot. HA-Akt1-SUMO conjugates were clearly detected by this technique only in lysates from 6xHis-SUMO-transfected cells (Fig. 1B). Moreover, the slower migrating bands were sensitive to overexpression of the SUMO-specific proteases SENP1 or 2, corroborating that they, indeed, correspond to SUMO-conjugated forms of Akt1 (Fig. 1C).

Figure 1. Akt1 is a SUMO substrate in cells and in vitro. (A) HEK 293T cells were co-transfected with expression vectors for HA-tagged Akt1 and His-SUMO2, with or without T7-Ubc9 expression vector. Cell lysates were prepared 48 h after transfection and analyzed by SDS/page and western blot with an anti-HA antibody. Slower migrating bands suspected to be SUMO-Akt1 conjugates are indicated with an asterisk. (B) HEK 293T cells were transfected with HA-Akt1 and Ubc9 expression vectors, with or without His-SUMO2. After 48 h, cells were lysed, an aliquot of the lysate was taken as input, and the reminder was subject to denaturing Ni2+ affinity chromatography. Both fractions were analyzed by western blot with an anti-HA antibody. Different lanes from the same gel were put together as indicated by the dividing line. (C) Cells were co-transfected with HA-Akt1, His-SUMO2, Ubc9 and either SENP1 or 2 expression vectors, or the corresponding empty vector (−). SUMO conjugation to Akt1 was analyzed as in (B). (D) Pull-down assay (right panel) was performed combining bacterially expressed and purified GST or GST-Ubc9 with cell lysates from HEK 293T expressing HA-Akt1. An aliquot of the lysate was taken as input (left panel). Samples were analyzed by western blot with an anti-HA antibody. (E) Purified recombinant HA-Akt1 was incubated with Aos1-Uba2 (E1 heterodimer), increasing amounts of Ubc9 (E2 enzyme), and SUMO2. Reactions were stopped by addition of 2X Laemmli sample buffer and analyzed by western blot with an anti-HA antibody.
Akt1 interacts with the SUMOylation machinery, as demonstrated by GST pull-down assays performed by incubating recombinant GST-Ubc9 with lysates from HEK 293T cells expressing HA-Akt1 (Fig. 1D). SUMO conjugation to Akt1 was further corroborated by an in vitro SUMOylation assay (Fig. 1E).
These results indicate that Akt1 is a SUMO conjugation substrate both in vitro and in cultured cells.
Akt1 is modified by SUMO2 at K276 and K301 within its kinase domain
In order to map the target lysine(s) for SUMO conjugation, mutations were introduced at different predicted SUMOylation consensus sites within Akt1 by site-directed mutagenesis designed to replace individual lysine (K) residues by arginine (R). From the different mutants tested, substitution of K276 or K301 led to a decrease in SUMO conjugation levels (Fig. S1B). Moreover, the double mutant (K276/301R) showed a stronger reduction in SUMOylation, suggesting these 2 residues are crucial for Akt1 modification by SUMO (Fig. 2A). Furthermore, the SUMO consensus site corresponding to K276 in Akt1 is conserved in the other mammalian Akt family members (K277 in Akt2 and K272 in Akt3) as well as in Akt orthologous from worms to mammals (Fig. S2). In particular, not only Akt2 is a SUMO conjugation substrate (Fig. S3A), but also the replacement of K277 by R diminished Akt2 SUMOylation levels (Fig. S3B). It is worth noting that the K residue at position 301 in Akt1 is not present in Akt2 or Akt3 isoforms. Additionally, it is absent in phylogenetically distant Akt1 orthologs (Fig. S2).
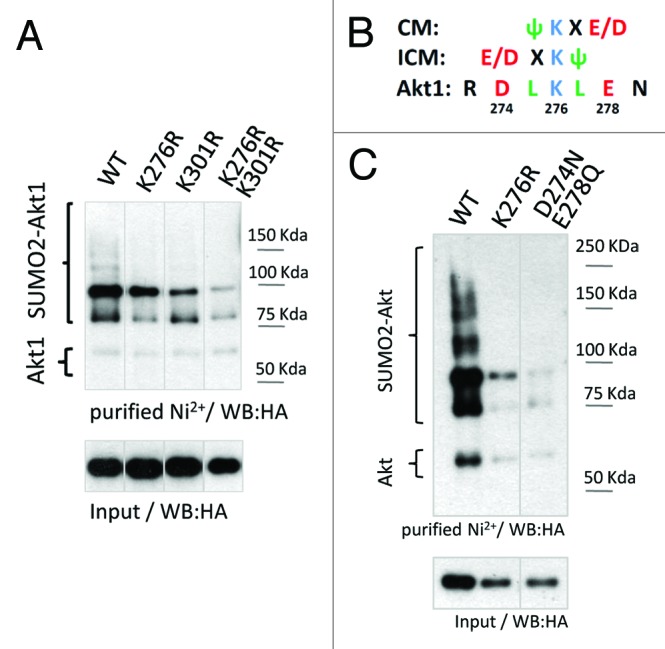
Figure 2. Mutational analysis of Akt1 SUMO conjugation sites. (A) HEK 293T cells were transfected either with wild-type (WT), K276R, K301R, or the double mutant K276R/K301R (“2KR”) HA-tagged Akt1 expression vectors, together with 6xHis-tagged SUMO2 and Ubc9 expression vectors. Cells lysates were subject to denaturing Ni2+ affinity chromatography and analyzed by western blot with an anti-HA antibody as indicated in Figure 1. Different lanes from the same gel were put together as indicated by the dividing lines. (B) Analysis of the SUMO consensus site surrounding K276 in Akt1. CM, canonical consensus motif; ICM, inverted consensus motif; Ψ, bulky, hydrophobic amino acid; X, any amino acid; E, glutamic acid; D, aspartic acid. (C) HEK 293T cells were transfected either with wild-type, K276R, or the double mutant D274N/E278Q HA-tagged Akt1 expression vectors, together with 6xHis-tagged SUMO2 and Ubc9. Cell lysates were subject to denaturing Ni2+ affinity chromatography and analyzed by western blot with an anti-HA antibody as indicated above. Different lanes from the same gel were put together as indicated by the dividing line.
Mutating K276 severely affected Akt1 SUMOylation levels in different cell lines (Fig. S3C and data not shown). However, the effect of mutating K301 on Akt1 SUMOylation seemed to be cell line-dependent. Analyzing the amino acid sequence neighboring K276 within Akt1, the SUMOylation motif appears as a combination of a canonical consensus motif (CM) with and inverted consensus motif (ICM) (Fig. 2B). To further validate K276 as a bona fide SUMO conjugation site within Akt1 and considering this particular feature of the SUMO motif, we mutated both negatively charged residues (D274 and E278) without altering the target K, and observed that this double mutant (D274N/E278Q) showed reduced Akt1 SUMOylation levels, similar to the K276R mutant (Fig. 2C).
Considering that the double mutant K276/301R termed “Akt1 2KR” showed the lowest level of Akt1 SUMOylation in our experimental setting, it was used to further analyze the consequences of SUMO conjugation on different Akt-regulated processes.
Lack of subordination between different Akt PTMs
So far, the best-established parameter of Akt activation within living cells has been its phosphorylation levels at T308 and S473. As for every member of the AGC kinase family, phosphorylation of these 2 residues is necessary for complete activation of Akt1. Given the importance of these modifications on Akt activity, we asked whether any connection between these phosphorylation events and SUMO conjugation to Akt1 exists. To this end, we compared phosphorylation levels of wild-type (WT) HA-Akt1 with those of HA-Akt1 2KR expressed in HEK 293T cells. Diminishing SUMO conjugation to Akt1 does not seem to affect the phosphorylation of this kinase on T308 and S473 (Fig. 3A). Moreover, Akt1 SUMOylation levels were measured in response to inhibition of Akt1 phosphorylation by the Pi3K pharmacological inhibitor LY 294002 (LY). The efficacy of the inhibitor was assessed by western blot analysis of phospho-Akt levels (Fig. 3B, lower panel). Inhibition of Pi3K-dependent Akt phosphorylation did not alter basal or Ubc9-stimulated Akt1 SUMO conjugation levels (Fig. 3B, upper and middle panels). In addition, neither phosphomimetic mutations on T308 and S473 (T308D/S473D) nor the impairment of phosphorylation at T308 (T308A) interfered with Akt1 SUMO conjugation (Fig. 3C).
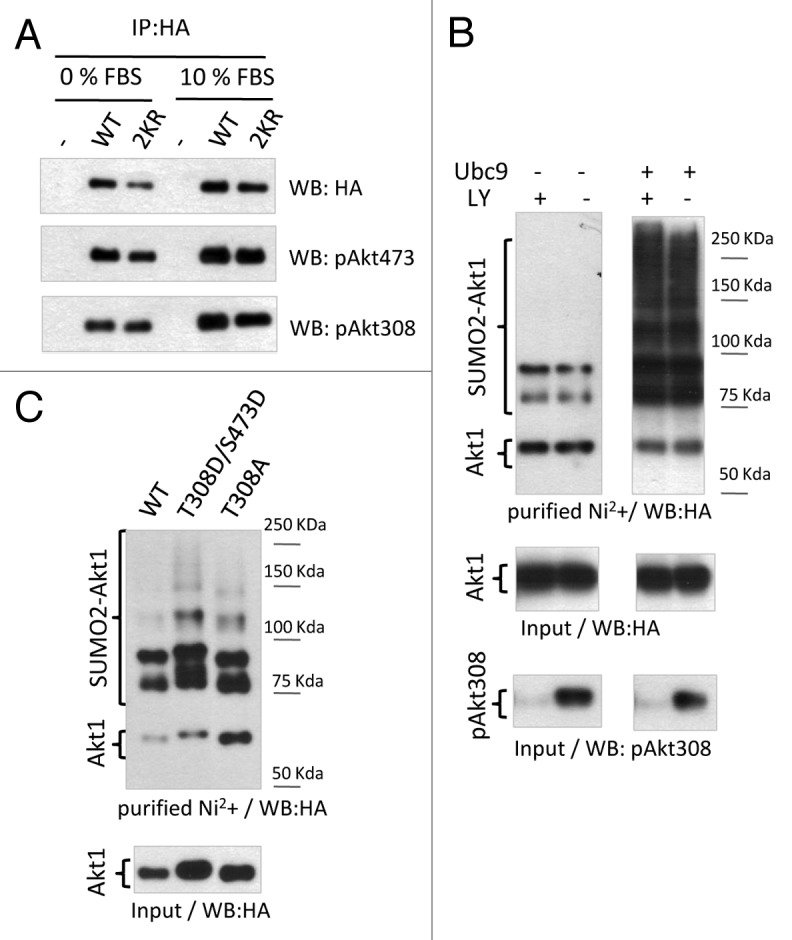
Figure 3. Lack of subordination between phosphorylation and SUMOylation of Akt1. (A) HEK 293T cells were transfected either with wild-type or 2KR HA-tagged Akt1 expression vectors and cultured with 10% fetal bovine serum (FBS) or without serum (0% FBS). Cells lysates were subject to immunoprecipitation with anti-HA antibody and analyzed by western blot with the antibodies indicated on the right side of each panel. (B) HEK 293T cells were transfected with HA-Akt1 and 6xHis-tagged SUMO2, with or without Ubc9 expression vectors, as indicated at the top of the panel. Twenty-four h after transfection cells were treated with the Pi3K inhibitor LY 294002 (LY) for another 24 h before cell lysis. An aliquot of the lysates was taken as input and the reminder was subject to denaturing Ni2+ affinity chromatography. Both fractions were analyzed by western blot with the antibodies indicated at the bottom of each panel. (C) HEK 293T cells were transfected either with wild-type, T308D/S473D, or T308A HA-tagged Akt1, 6xHis-SUMO2, and Ubc9 expression vectors. An aliquot of the lysates was taken as input and the reminder was subject to denaturing Ni2+ affinity chromatography. Both fractions were analyzed by western blot with anti-HA antibody.
These results indicate that modification of Akt by SUMO conjugation is not a prerequisite for Akt1 phosphorylation, nor is Pi3K-dependent Akt phosphorylation required for Akt SUMOylation.
Akt SUMOylation affects alternative splicing regulation
Previous work from our laboratory demonstrated that Akt activation regulates fibronectin splicing patterns in an SR protein-dependent manner.37 Aiming at elucidating possible mechanisms, we wondered whether Akt SUMOylation was involved in the splicing regulatory activity of Akt. To this end, the effect of Akt1 2KR overexpression on fibronectin alternative splicing was monitored by analyzing mRNA isoforms (either containing or lacking the fibronectin alternative exon EDI) derived from a splicing reporter minigene (Fig. 4A).

Figure 4. Akt1 SUMOylation affects alternative splicing patterns. (A) Partial representation of fibronectin (FN EDI) splicing reporter minigene used to analyzed alternative splicing patterns in transfected cells. The scheme shows the 2 mRNA isoforms, either containing (EDI+) or lacking (EDI-) EDI alternative exon, derived from a single pre-mRNA upon transcription of the minigene. (B) HeLa cells were co-transfected with the FN EDI minigene, either with wild-type (WT), 2KR HA-tagged Akt1 expression vectors, or the corresponding empty vector (−), and with an expression vector for a constitutively active form of Ras (RasV12C40) or its corresponding empty vector. Forty-eight h after transfection, RNA was prepared and used to analyze mRNA isoforms derived from the minigene by reverse transcription and radioactive PCR with specific primers. The bar graph corresponds to the ratio between the 2 PCR products shown in the lower panel. The different EDI+/EDI- ratios were standardized against the ratio corresponding to the condition lacking both Akt and Ras isoforms. Different lanes from the same gel are shown. (C) HeLa cells were transfected with siRNA against Akt1 3′UTR (siAkt1) or control siRNA (−). Cells were re-transfected 24 h later with the FN EDI minigene and increasing amounts of HA-Akt1 WT, HA-Akt1 2KR, or the corresponding empty vector (−) and cultured for additional 48 h. EDI splicing patterns were analyzed as indicated in (B). The bar graph corresponds to the ratio between the two PCR products. (D) HeLa cells were transfected either with WT, E17K, or E17K/2KR HA-tagged Akt1 expression vectors together with 6xHis-SUMO2 and Ubc9 expression vectors. An aliquot of the lysates was taken as input and the reminder was subject to denaturing Ni2+ affinity chromatography. Both fractions were analyzed by western blot with anti-HA antibody. Different lanes from the same gel were put together as indicated by the dividing line. (E) HeLa cells were transfected with the FN EDI minigene and either WT, 2KR, E17K, or E17K/2KR HA-Akt1 expression vectors or the corresponding empty vector. Splicing isoforms derived from the minigene were analyzed as in (B). (F) Partial, schematic representation of the Bcl-x splicing reporter minigene and the 2 mRNA isoforms, long (L) and short (S), derived from it. (G) HEK 293T cells were transfected with the Bcl-x splicing reporter minigene together with WT or 2KR HA-Akt1 expression vectors, or the corresponding empty vector. Forty-eight h later, RNA was extracted and mRNA isoforms derived from the transfected minigene were analyzed by reverse transcription and radioactive PCR with specific primers. The bar graph shows the ratio between the PCR products corresponding to the long [Bcl-x(L)] and the short [Bcl-x(S)] mRNA isoforms. In every case, values shown in histograms correspond to mean ± SE of a representative experiment run in duplicate.
Cells were co-transfected with the EDI splicing reporter minigene and either the expression vector for HA-Akt1 WT or HA-Akt1 2KR, with or without an expression vector for a constitutively active Ras mutant that preferentially activates the Pi3K-Akt pathway (RasV12C40).43 As previously shown,37 overexpression of Akt1 stimulates EDI exon inclusion, and this effect is even more pronounced upon activation of the pathway by RasV12C40 (Fig. 4B). Most interestingly, decreasing SUMOylation not only makes Akt unable to stimulate exon inclusion, but also renders it refractory to RasV12C40 overexpression (Fig. 4B; Fig. S4A). To further strengthen this finding, we performed knockdown/rescue experiments. Endogenous Akt1 was knocked-down by an siRNA targeted to the 3′UTR, and the cells were rescued by overexpression of siRNA-resistant wild-type or SUMOylation-deficient Akt1 (Fig. S4B). Figure 4C shows a strong decrease in EDI exon inclusion upon silencing of endogenous Akt1, which is rescued by WT HA-Akt1 but not by HA-Akt1 2KR.
A somatic mutation of Akt1 has been identified in breast, colorectal, and ovarian human cancers. This point mutation at amino acid 17 (E17K) enhances Akt interaction with the plasma membrane, hyperactivating Akt, and therefore stimulating downstream signaling leading to cellular transformation.44 In line with SUMO conjugation being required for Akt activity, hyperactive Akt1 E17K displays a strong increase in SUMO conjugation levels, which is lost upon further introducing the 2 mutations K276R and K301R into this mutant protein (“HA-Akt1 E17K/2KR,” Fig. 4D). We then analyzed the effect of the E17K mutation on Akt splicing regulatory activity. Indeed, Akt1 E17K enhanced inclusion of the EDI exon to a larger extent than wild-type Akt1 (Fig. 4E). Remarkably, this hyperactivity of Akt1 E17K was completely lost upon diminishing SUMO conjugation at K276 and K301 within this mutant protein (Akt1 E17K/2KR, Fig. 4E).
These results point to the dependence on SUMO conjugation for the splicing regulatory function of Akt1 within the Pi3K-Akt-SR protein axis previously described by our laboratory. Furthermore, they suggest a possible regulatory role for SUMO conjugation on Akt tumorigenic activity.
To further explore the influence of Akt SUMO conjugation on splicing regulation, and considering the crucial role that Akt exerts on the balance between cell survival and apoptosis, we analyzed its involvement in the regulation of Bcl-x pre-mRNA splicing. Bcl-x transcripts give rise to 2 different mRNAs, a pro-apoptotic (short) and a pro-survival (long) isoform, by the differential use of two alternative 5′ splice sites. We took advantage of a Bcl-x splicing reporter minigene45 (Fig. 4F) that was co-transfected with expression vectors for RasV12C40 and either HA-Akt1 WT or HA-Akt1 2KR. Consistent with Akt pro-survival role, activation of WT HA-Akt1 altered the balance between Bcl-x mRNA isoforms derived from the minigene, increasing the proportion of the pro-survival mRNA (Bcl-x[L], Fig. 4G). However, Akt1 2KR failed to do so, demonstrating that SUMO conjugation is necessary for Akt-mediated Bcl-x splicing regulation (Fig. 4G). These results implicate Akt in the regulation of Bcl-x pre-mRNA splicing, favoring a pro-survival mRNA isoform, which is consistent with the protective role of this kinase against apoptosis. Furthermore, they expand the relevance of Akt SUMOylation for its involvement in alternative splicing regulation.
Akt SUMOylation regulates cell cycle
Among several actions exerted by Akt to regulate the cell survival/apoptosis balance, it is known that it controls cell cycle progression by facilitating G1/S transition and the initiation of M phase.32 Based on this, we asked whether Akt SUMOylation could have any impact on cell cycle progression by analyzing cell cycle profiles of HEK 293T cells transfected with expression vectors for HA-Akt1 WT or HA-Akt1 2KR. As observed in Figure 5, overexpression of wild-type Akt1 increased the percentage of cells in S and G2 phases while decreasing that of G1 phase compared with control cells. However, overexpression of HA-Akt1 2KR failed to cause these changes.
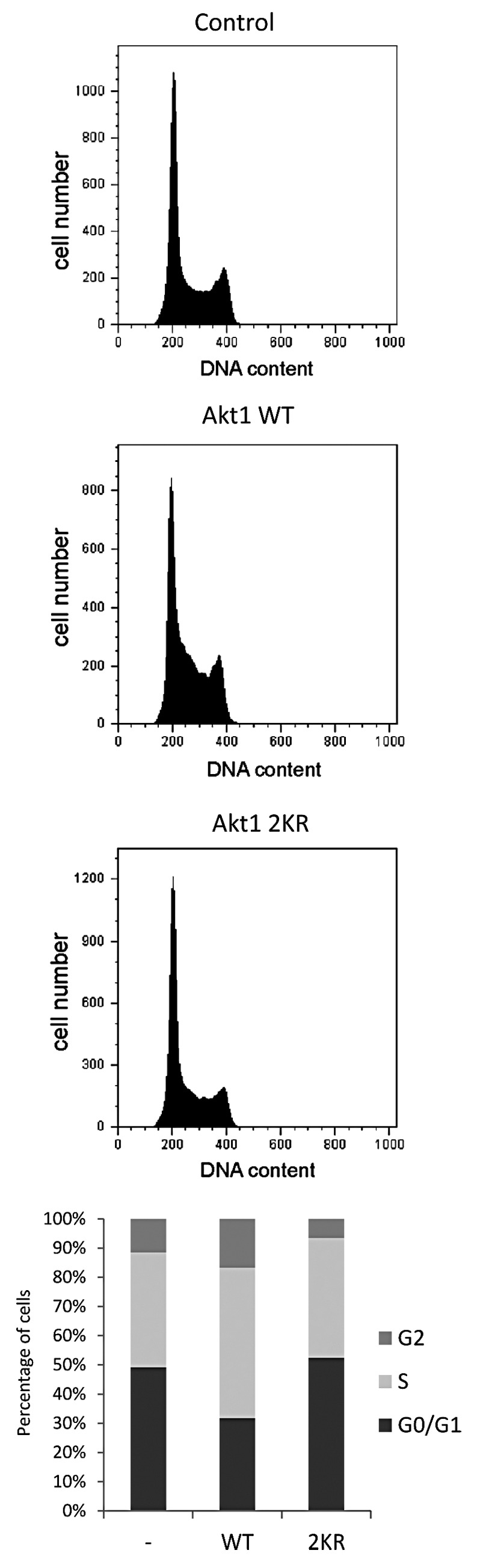
Figure 5. SUMOylation of Akt1 affects cell cycle progression. HEK 293T cells were transfected with empty vector (control), WT or 2KR HA-Akt1 expression vectors and cell cycle profiles (DNA content) were analyzed by flow cytometry 48 h after transfection. The bar graph shows the percentage of cells at the different cell cycle phases for each analyzed condition.
These results indicate that SUMO conjugation to Akt1 is required for the function of this kinase as a regulator of the G1/S transition.
Discussion
Here we reported the post-translational modification of Akt1 by SUMOylation both in vitro and in living cells, and mapped K276 and K301 within its kinase domain as SUMO conjugation sites. Combined replacement of these 2 K residues by R not only diminished Akt SUMOylation levels, but also impaired different Akt activities. In particular, we explored the role of Akt as a regulator of alternative splicing and cell cycle progression, and found that SUMOylation of Akt is required for the activity of this kinase in these different cellular processes. Specifically, the results presented here revealed SUMO conjugation as an Akt PTM controlling the splicing regulatory role of this kinase described by our laboratory several years ago.37
We explored the effect of Akt SUMOylation on some of the cell cycle-related events that have been shown to be dependent on Akt function. Remarkably, SUMO conjugation to Akt is not only relevant for cell cycle progression at the G1/S transition, but also for the production of different mRNA splice variants that are associated with cell proliferation and survival. In particular, EDI-containing fibronectin mRNA isoforms are characteristic of highly proliferative normal tissues and tumors. Moreover, alternative splicing plays a crucial role in the control of apoptosis, as several pre-mRNAs for cell death factors are alternatively spliced, yielding isoforms with opposed functions during programmed cell death. Thus, deciphering the splicing regulators and the signal transduction pathways controlling these alternative splicing events may have important implications for cancer pathophysiology, and, as such, it has been the focus of different laboratories.42,46-51 Here, we determined that Akt is involved in the regulation of Bcl-x pre-mRNA splicing. In agreement with the pro-survival role of Akt, its activation favors the production of the pro-survival mRNA isoform of Bcl-x, and it does so in a SUMOylation-dependent manner. Recent work from Zhou et al. has analyzed endogenous alternative splicing patterns upon cell stimulation with epidermal growth factor and defined the Pi3K-Akt axis as the main signaling pathway involved in alternative splicing regulation downstream of this growth factor.41 Whether different Pi3K-responsive alternative splicing events identified by this high-throughput analysis are also sensitive to Akt SUMOylation remains to be explored.
Four different results presented in Figure 3 indicate that phosphorylation at T308 and S473 and SUMO conjugation to Akt can be considered as independent events: (1) the mutant Akt1 2KR that displays lower SUMOylation levels is equally phosphorylated to wild-type Akt, at least under steady-state conditions; (2) pharmacological blockade of the Pi3K/Akt pathway does not alter SUMO conjugation to Akt; (3) stimulation of the SUMO pathway by Ubc9 overexpression does not alter Akt phosphorylation; and (4) point mutations that abolish or mimic Akt phosphorylation do not interfere with its SUMOylation. However, downstream activities of Akt are drastically impaired in the SUMO-deficient mutant, indicating that this PTM does not regulate the activation of this kinase, but it might regulate its interaction with downstream targets.
Over the last 2 decades, Akt has emerged as a key signaling molecule in higher eukaryotes due to its regulatory function in a wide variety of cellular processes. As such, it is a crucial protein kinase in human physiology and disease. Despite a large body of data dealing with upstream Akt activation and downstream targets, we still have an incomplete understanding of how this vast diversity of Akt cellular actions is differentially regulated. Most of the studies about the regulation of Akt activity have been focused in its phosphorylation, and only recently its acetylation and ubiquitylation have been pointed out as additional regulatory modifications of this protein. During the completion of this manuscript and in agreement with our current data, work from Li et al. reported the modification of Akt by SUMO conjugation at multiple lysine residues, pointing to K276 as the major SUMOylation site within this kinase. Furthermore, they found that this PTM influences Akt activity at cell proliferation and tumor development and, consistent with our findings, showed that diminishing SUMO conjugation to the cancer-associated mutant Akt1 E17K reduces its oncogenic capacity.52 Our results together with these findings reveal a previously unknown regulatory level for the crucial role of this kinase in health and disease and open new areas of exploration for therapeutic intervention. Activation of Akt is opposed by the action of PTEN (the phosphatase and tensin homolog deleted in chromosome 10), which antagonize the action of Pi3K hydrolyzing PIP3. Different laboratories have described recently that SUMO conjugation to PTEN regulates its subcellular distribution and activity.53-55 In this context, it seems that the emerging concept of “group SUMOylation”, which refers to the requirement for simultaneous modification of multiple targets involved in the same biological process,4,56 could apply to the regulation of the Pi3K–Akt signaling axis.
Materials and Methods
Plasmids, siRNAs, and cell transfection
Plasmid DNA and siRNAs were transfected into HEK 293T or HeLa cells with Lipofectamine 2000 as the manufacturer's instructions (Invitrogen). The sequence of the siRNA (Invitrogen) targeting the human Akt1 3′UTR was the following: 5′-GGGUUUACCCAGUGGGACA(dT)(dT)-3′,57 and siRNA targeting luciferase was used as a control, siRNA LUC: 5′-CUUACGCUGAGUACUUCGA(dT)(dT)-3′.58
The plasmids used were HA-Akt1 WT, K20R, K30R, K64R (provided by HK Lin);29 T7-Ubc9 (provided by D wotton); His-SUMO2 (provided by R Hay); Flag-SENP1 (provided by E. Yeh; Addgene plasmid 17357);59 Flag-SENP2 (provided by E Yeh; Addgene plasmid 18047);60 RasV12C40 (provided by JM Rojas);43 Bcl-x minigen (provided by C Chlafant).45
Site-directed mutagenesis
Site-directed mutagenesis was performed by the Dpn I method, based on Stratagene QuickChange specifications. Sequence of the different primers used for this method is available upon request.
Recombinant proteins
GST was expressed in E. coli BL21(DE3) Rosetta, and GST-Ubc9 in M15(pREP4) cells by induction with 0.5 mM IPTG and purified using glutathione Sepharose beads (GE Healthcare). HA-Akt1 was purified from transfected HEK 293T lysates as described.61 Proteins were analyzed by SDS-PAGE and coomassie staining for quantification and purity.
GST pull-down assays
Pull-down assays were performed as described.62 HEK 293T cells were lysed in 1 ml of lysis buffer [20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 1 mM β-glycerophosphate, 1 mM KF, 5% glycerol, and 1× Complete Protease Inhibitor (Roche)] and incubated for 30 min at 4 °C. After centrifugation for 20 min at 4 °C, supernatants were used immediately for pull-down or kept at −80 °C. Lysates were pre-cleared with 2 μg of GST coupled to glutathione Sepharose beads (GE Healthcare), and cleared lysates were incubated with the indicated GST-tagged recombinant protein coupled to glutathione Sepharose beads for 1 h at room temperature. After 3 washes in lysis buffer and one in PBS, proteins were eluted by adding 2× Laemmli sample buffer and analyzed by western blot.
Purification of 6xHis-SUMO-conjugated proteins
HEK 293T or HeLa cells were transfected in 35 mm culture wells with the indicated plasmids. After 48 h, 6xHis-SUMO conjugates were purified under denaturing conditions using Ni-NTA agarose beads according to the manufacturer’s instructions (QIAGEN). Transfected cells were harvested in ice-cold PBS plus 100 mM iodoacetamide. An aliquot was taken as input, and the remaining cells were lysed in 6 M guanidinium-HCl containing 100 mM Na2HPO4/NaH2PO4, 10 mM Tris-HCl pH 8.0, 5 mM imidazole, and 10 mM iodoacetamide. Samples were sonicated to reduce the viscosity, and after centrifugation for 20 min at 12 000 g, proteins in the supernatants were purified using Ni-NTA beads (QIAGEN) according to Tatham et al.63 Samples were subsequently washed with wash buffer I (8 M urea, 10 mM Tris-HCl, 100 mM Na2HPO4/NaH2PO4, 5 mM imidazole, 10 mM iodoacetamide, pH 8), wash buffer II (8 M urea, 10 mM Tris-HCl, 100 mM Na2HPO4/NaH2PO4, 0.2% Triton X-100, 5 mM imidazole, 10 mM iodoacetamide, pH 6.3), and wash buffer III (8 M urea, 10 mM Tris-HCl, 100 mM Na2HPO4/NaH2PO4, 0.1% Triton X-100, 5 mM imidazole, 10 mM iodoacetamide, pH 6.3). Samples were eluted in 2× Laemmli sample buffer containing 300 mM imidazole during 5 min at 95 °C.
In vitro SUMOylation reactions
In vitro SUMOylation reactions were performed as described,62 using HA-Akt as a substrate. The SUMOylation buffer was 20 mM HEPES-KOH pH 7.3, 100 mM KAcO, 0.5 mM EGTA, 1 mM DTT, 0.05% Tween-20, 0.2 mg/ml ovalbumin, and 1× Complete protease inhibitor (Roche). HA-Akt1 (250 ng) was incubated with 150 ng E1, 250 or 500 ng Ubc9, and 1 μg SUMO. Reactions were stopped by addition of 2× Laemmli sample buffer. One-fourth of the reaction was run in SDS-PAGE and analyzed by western blot. Recombinant SUMO, Ubc9 (E2 enzyme) and Aos1-Uba2 (E1 heterodimer) were purchased from Enzo Life Sciences.
Splicing assay
Cells were transfected with 500 ng of the splicing reporter minigene (shown in Fig. 4A and F). Forty-eight hours later, RNA was extracted and subject to RT-PCR as described.64 Radioactive PCR products were run on native 6% polyacrilamide gels, which were subsequently dried and exposed to X-ray films (Agfa). Radioactivity in the corresponding bands was measure by scintillation counter and used to calculate the radio between different mRNA isoforms.
Cell cycle analysis
HEK 293T cells were transfected as indicated above and, 48 h after transfection, were harvested. Cells were fixed with 5 ml of ice cold 70% ethanol and incubated over night at −20 °C. For cell cycle analysis, fixed cells were centrifuged at 1000 rpm for 5 min and washed with PBS. Cells were then incubated with propidium iodide (50 μg/ml) containing RNase A (100 μg/m1) at 37 °C for 60 min. DNA content was determined by flow cytometry (FACS Aria II, BD Biosciences), and the percentage of cells in the different cell cycle phases was quantified by FlowJo software (Tree Star).
Antibodies
The antibodies used were: Akt (#9272), Phospho-Akt Thr308 (#4056), and Phospho-Akt Ser473 (#4060) from Cell Signaling; HA (HA-16B12, Covance).
Supplementary Material
Acknowledgments
We thank Valeria Buggiano, José Clemente and Mariela Veggetti for technical help; Mercedes Tkach for advise with cell cycle experiments; Ana Quaglino, Alberto Kornblihtt and members of his lab for helpful discussion and suggestions. We acknowledge Ron Hay, David Wotton, Eduard Yeh, Hui-Kuan Lin, Charles Chlafant, Claudio Sette, and José Maria Rojas for providing plasmids. This work was supported by grants from: Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT) of Argentina; Universidad de Buenos Aires, Argentina; Fundación Florencio Fiorini, Argentina; L’Oreal-UNESCO-CONICET and European Alternative Splicing Network (EURASNET).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/26183
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26183
References
- 1.Hay RT. SUMO: a history of modification. Mol Cell. 2005;18:1–12. doi: 10.1016/j.molcel.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 2.Wilkinson KA, Henley JM. Mechanisms, regulation and consequences of protein SUMOylation. Biochem J. 2010;428:133–45. doi: 10.1042/BJ20100158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gareau JR, Lima CD. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol. 2010;11:861–71. doi: 10.1038/nrm3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flotho A, Melchior F. Sumoylation: a regulatory protein modification in health and disease. Annu Rev Biochem. 2013;82:357–85. doi: 10.1146/annurev-biochem-061909-093311. [DOI] [PubMed] [Google Scholar]
- 5.Hochstrasser M. SP-RING for SUMO: new functions bloom for a ubiquitin-like protein. Cell. 2001;107:5–8. doi: 10.1016/S0092-8674(01)00519-0. [DOI] [PubMed] [Google Scholar]
- 6.Pichler A, Gast A, Seeler JS, Dejean A, Melchior F. The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell. 2002;108:109–20. doi: 10.1016/S0092-8674(01)00633-X. [DOI] [PubMed] [Google Scholar]
- 7.Kagey MH, Melhuish TA, Wotton D. The polycomb protein Pc2 is a SUMO E3. Cell. 2003;113:127–37. doi: 10.1016/S0092-8674(03)00159-4. [DOI] [PubMed] [Google Scholar]
- 8.Kahyo T, Nishida T, Yasuda H. Involvement of PIAS1 in the sumoylation of tumor suppressor p53. Mol Cell. 2001;8:713–8. doi: 10.1016/S1097-2765(01)00349-5. [DOI] [PubMed] [Google Scholar]
- 9.Weger S, Hammer E, Heilbronn R. Topors acts as a SUMO-1 E3 ligase for p53 in vitro and in vivo. FEBS Lett. 2005;579:5007–12. doi: 10.1016/j.febslet.2005.07.088. [DOI] [PubMed] [Google Scholar]
- 10.Hay RT. SUMO-specific proteases: a twist in the tail. Trends Cell Biol. 2007;17:370–6. doi: 10.1016/j.tcb.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 11.Cubeñas-Potts C, Matunis MJ. SUMO: a multifaceted modifier of chromatin structure and function. Dev Cell. 2013;24:1–12. doi: 10.1016/j.devcel.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kadaré G, Toutant M, Formstecher E, Corvol JC, Carnaud M, Boutterin MC, Girault JA. PIAS1-mediated sumoylation of focal adhesion kinase activates its autophosphorylation. J Biol Chem. 2003;278:47434–40. doi: 10.1074/jbc.M308562200. [DOI] [PubMed] [Google Scholar]
- 13.Kang JS, Saunier EF, Akhurst RJ, Derynck R. The type I TGF-beta receptor is covalently modified and regulated by sumoylation. Nat Cell Biol. 2008;10:654–64. doi: 10.1038/ncb1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kubota Y, O’Grady P, Saito H, Takekawa M. Oncogenic Ras abrogates MEK SUMOylation that suppresses the ERK pathway and cell transformation. Nat Cell Biol. 2011;13:282–91. doi: 10.1038/ncb2169. [DOI] [PubMed] [Google Scholar]
- 15.Matheny RW, Jr., Adamo ML. Current perspectives on Akt Akt-ivation and Akt-ions. Exp Biol Med (Maywood) 2009;234:1264–70. doi: 10.3181/0904-MR-138. [DOI] [PubMed] [Google Scholar]
- 16.Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005;24:7455–64. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- 17.Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res. 2005;94:29–86. doi: 10.1016/S0065-230X(05)94002-5. [DOI] [PubMed] [Google Scholar]
- 18.Hers I, Vincent EE, Tavaré JM. Akt signalling in health and disease. Cell Signal. 2011;23:1515–27. doi: 10.1016/j.cellsig.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 19.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santi SA, Lee H. The Akt isoforms are present at distinct subcellular locations. Am J Physiol Cell Physiol. 2010;298:C580–91. doi: 10.1152/ajpcell.00375.2009. [DOI] [PubMed] [Google Scholar]
- 21.Chen R, Kim O, Yang J, Sato K, Eisenmann KM, McCarthy J, Chen H, Qiu Y. Regulation of Akt/PKB activation by tyrosine phosphorylation. J Biol Chem. 2001;276:31858–62. doi: 10.1074/jbc.C100271200. [DOI] [PubMed] [Google Scholar]
- 22.Conus NM, Hannan KM, Cristiano BE, Hemmings BA, Pearson RB. Direct identification of tyrosine 474 as a regulatory phosphorylation site for the Akt protein kinase. J Biol Chem. 2002;277:38021–8. doi: 10.1074/jbc.M203387200. [DOI] [PubMed] [Google Scholar]
- 23.Facchinetti V, Ouyang W, Wei H, Soto N, Lazorchak A, Gould C, Lowry C, Newton AC, Mao Y, Miao RQ, et al. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 2008;27:1932–43. doi: 10.1038/emboj.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mahajan K, Coppola D, Challa S, Fang B, Chen YA, Zhu W, Lopez AS, Koomen J, Engelman RW, Rivera C, et al. Ack1 mediated AKT/PKB tyrosine 176 phosphorylation regulates its activation. PLoS One. 2010;5:e9646. doi: 10.1371/journal.pone.0009646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hart JR, Vogt PK. Phosphorylation of AKT: a mutational analysis. Oncotarget. 2011;2:467–76. doi: 10.18632/oncotarget.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sundaresan NR, Pillai VB, Wolfgeher D, Samant S, Vasudevan P, Parekh V, Raghuraman H, Cunningham JM, Gupta M, Gupta MP. The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Sci Signal. 2011;4:ra46. doi: 10.1126/scisignal.2001465. [DOI] [PubMed] [Google Scholar]
- 27.Suizu F, Hiramuki Y, Okumura F, Matsuda M, Okumura AJ, Hirata N, Narita M, Kohno T, Yokota J, Bohgaki M, et al. The E3 ligase TTC3 facilitates ubiquitination and degradation of phosphorylated Akt. Dev Cell. 2009;17:800–10. doi: 10.1016/j.devcel.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 28.Bae S, Kim SY, Jung JH, Yoon Y, Cha HJ, Lee H, Kim K, Kim J, An IS, Kim J, et al. Akt is negatively regulated by the MULAN E3 ligase. Cell Res. 2012;22:873–85. doi: 10.1038/cr.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang WL, Wang J, Chan CH, Lee SW, Campos AD, Lamothe B, Hur L, Grabiner BC, Lin X, Darnay BG, et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science. 2009;325:1134–8. doi: 10.1126/science.1175065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang WL, Wu CY, Wu J, Lin HK. Regulation of Akt signaling activation by ubiquitination. Cell Cycle. 2010;9:487–97. doi: 10.4161/cc.9.3.10508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan CH, Li CF, Yang WL, Gao Y, Lee SW, Feng Z, Huang HY, Tsai KK, Flores LG, Shao Y, et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell. 2012;149:1098–111. doi: 10.1016/j.cell.2012.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brazil DP, Yang ZZ, Hemmings BA. Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci. 2004;29:233–42. doi: 10.1016/j.tibs.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 33.Bozulic L, Surucu B, Hynx D, Hemmings BA. PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell. 2008;30:203–13. doi: 10.1016/j.molcel.2008.02.024. [DOI] [PubMed] [Google Scholar]
- 34.Fraser M, Harding SM, Zhao H, Coackley C, Durocher D, Bristow RG. MRE11 promotes AKT phosphorylation in direct response to DNA double-strand breaks. Cell Cycle. 2011;10:2218–32. doi: 10.4161/cc.10.13.16305. [DOI] [PubMed] [Google Scholar]
- 35.Lai VK, Ashraf M, Jiang S, Haider K. MicroRNA-143 is a critical regulator of cell cycle activity in stem cells with co-overexpression of Akt and angiopoietin-1 via transcriptional regulation of Erk5/cyclin D1 signaling. Cell Cycle. 2012;11:767–77. doi: 10.4161/cc.11.4.19211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blaustein M, Pelisch F, Coso OA, Bissell MJ, Kornblihtt AR, Srebrow A. Mammary epithelial-mesenchymal interaction regulates fibronectin alternative splicing via phosphatidylinositol 3-kinase. J Biol Chem. 2004;279:21029–37. doi: 10.1074/jbc.M314260200. [DOI] [PubMed] [Google Scholar]
- 37.Blaustein M, Pelisch F, Tanos T, Muñoz MJ, Wengier D, Quadrana L, Sanford JR, Muschietti JP, Kornblihtt AR, Cáceres JF, et al. Concerted regulation of nuclear and cytoplasmic activities of SR proteins by AKT. Nat Struct Mol Biol. 2005;12:1037–44. doi: 10.1038/nsmb1020. [DOI] [PubMed] [Google Scholar]
- 38.Patel NA, Kaneko S, Apostolatos HS, Bae SS, Watson JE, Davidowitz K, Chappell DS, Birnbaum MJ, Cheng JQ, Cooper DR. Molecular and genetic studies imply Akt-mediated signaling promotes protein kinase CbetaII alternative splicing via phosphorylation of serine/arginine-rich splicing factor SRp40. J Biol Chem. 2005;280:14302–9. doi: 10.1074/jbc.M411485200. [DOI] [PubMed] [Google Scholar]
- 39.Shultz JC, Goehe RW, Wijesinghe DS, Murudkar C, Hawkins AJ, Shay JW, Minna JD, Chalfant CE. Alternative splicing of caspase 9 is modulated by the phosphoinositide 3-kinase/Akt pathway via phosphorylation of SRp30a. Cancer Res. 2010;70:9185–96. doi: 10.1158/0008-5472.CAN-10-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.White ES, Sagana RL, Booth AJ, Yan M, Cornett AM, Bloomheart CA, Tsui JL, Wilke CA, Moore BB, Ritzenthaler JD, et al. Control of fibroblast fibronectin expression and alternative splicing via the PI3K/Akt/mTOR pathway. Exp Cell Res. 2010;316:2644–53. doi: 10.1016/j.yexcr.2010.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou Z, Qiu J, Liu W, Zhou Y, Plocinik RM, Li H, Hu Q, Ghosh G, Adams JA, Rosenfeld MG, et al. The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol Cell. 2012;47:422–33. doi: 10.1016/j.molcel.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vu NT, Park MA, Shultz JC, Goehe RW, Hoeferlin LA, Shultz MD, Smith SA, Lynch KW, Chalfant CE. hnRNP U enhances caspase-9 splicing and is modulated by AKT-dependent phosphorylation of hnRNP L. J Biol Chem. 2013;288:8575–84. doi: 10.1074/jbc.M112.443333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oliva JL, Zarich N, Martínez N, Jorge R, Castrillo A, Azañedo M, García-Vargas S, Gutiérrez-Eisman S, Juarranz A, Boscá L, et al. The P34G mutation reduces the transforming activity of K-Ras and N-Ras in NIH 3T3 cells but not of H-Ras. J Biol Chem. 2004;279:33480–91. doi: 10.1074/jbc.M404058200. [DOI] [PubMed] [Google Scholar]
- 44.Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–44. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 45.Massiello A, Salas A, Pinkerman RL, Roddy P, Roesser JR, Chalfant CE. Identification of two RNA cis-elements that function to regulate the 5′ splice site selection of Bcl-x pre-mRNA in response to ceramide. J Biol Chem. 2004;279:15799–804. doi: 10.1074/jbc.M313950200. [DOI] [PubMed] [Google Scholar]
- 46.Paronetto MP, Achsel T, Massiello A, Chalfant CE, Sette C. The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J Cell Biol. 2007;176:929–39. doi: 10.1083/jcb.200701005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shkreta L, Froehlich U, Paquet ER, Toutant J, Elela SA, Chabot B. Anticancer drugs affect the alternative splicing of Bcl-x and other human apoptotic genes. Mol Cancer Ther. 2008;7:1398–409. doi: 10.1158/1535-7163.MCT-08-0192. [DOI] [PubMed] [Google Scholar]
- 48.Wang Q, Silver PA. Genome-wide RNAi screen discovers functional coupling of alternative splicing and cell cycle control to apoptosis regulation. Cell Cycle. 2010;9:4419–21. doi: 10.4161/cc.9.22.14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chang WH, Liu TC, Yang WK, Lee CC, Lin YH, Chen TY, Chang JG. Amiloride modulates alternative splicing in leukemic cells and resensitizes Bcr-AblT315I mutant cells to imatinib. Cancer Res. 2011;71:383–92. doi: 10.1158/0008-5472.CAN-10-1037. [DOI] [PubMed] [Google Scholar]
- 50.Shkreta L, Michelle L, Toutant J, Tremblay ML, Chabot B. The DNA damage response pathway regulates the alternative splicing of the apoptotic mediator Bcl-x. J Biol Chem. 2011;286:331–40. doi: 10.1074/jbc.M110.162644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Apostolatos A, Song S, Acosta S, Peart M, Watson JE, Bickford P, Cooper DR, Patel NA. Insulin promotes neuronal survival via the alternatively spliced protein kinase CδII isoform. J Biol Chem. 2012;287:9299–310. doi: 10.1074/jbc.M111.313080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li R, Wei J, Jiang C, Liu D, Deng L, Zhang K, Wang P. Akt SUMOylation regulates cell proliferation and tumorigenesis. Cancer Res. 2013 doi: 10.1158/0008-5472.CAN-13-0538. [DOI] [PubMed] [Google Scholar]
- 53.Huang J, Yan J, Zhang J, Zhu S, Wang Y, Shi T, Zhu C, Chen C, Liu X, Cheng J, et al. SUMO1 modification of PTEN regulates tumorigenesis by controlling its association with the plasma membrane. Nat Commun. 2012;3:911. doi: 10.1038/ncomms1919. [DOI] [PubMed] [Google Scholar]
- 54.Bassi C, Ho J, Srikumar T, Dowling RJ, Gorrini C, Miller SJ, Mak TW, Neel BG, Raught B, Stambolic V. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science. 2013;341:395–9. doi: 10.1126/science.1236188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.González-Santamaría J, Campagna M, Ortega-Molina A, Marcos-Villar L, de la Cruz-Herrera CF, González D, Gallego P, Lopitz-Otsoa F, Esteban M, Rodríguez MS, et al. Regulation of the tumor suppressor PTEN by SUMO. Cell Death Dis. 2012;3:e393. doi: 10.1038/cddis.2012.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Psakhye I, Jentsch S. Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell. 2012;151:807–20. doi: 10.1016/j.cell.2012.10.021. [DOI] [PubMed] [Google Scholar]
- 57.Hara S, Nakashiro K, Goda H, Hamakawa H. Role of Akt isoforms in HGF-induced invasive growth of human salivary gland cancer cells. Biochem Biophys Res Commun. 2008;370:123–8. doi: 10.1016/j.bbrc.2008.03.042. [DOI] [PubMed] [Google Scholar]
- 58.Elbashir SM, Martinez J, Patkaniowska A, Lendeckel W, Tuschl T. Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. EMBO J. 2001;20:6877–88. doi: 10.1093/emboj/20.23.6877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheng J, Kang X, Zhang S, Yeh ET. SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell. 2007;131:584–95. doi: 10.1016/j.cell.2007.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kang X, Qi Y, Zuo Y, Wang Q, Zou Y, Schwartz RJ, Cheng J, Yeh ET. SUMO-specific protease 2 is essential for suppression of polycomb group protein-mediated gene silencing during embryonic development. Mol Cell. 2010;38:191–201. doi: 10.1016/j.molcel.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cazalla D, Sanford JR, Cáceres JF. A rapid and efficient protocol to purify biologically active recombinant proteins from mammalian cells. Protein Expr Purif. 2005;42:54–8. doi: 10.1016/j.pep.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 62.Pelisch F, Gerez J, Druker J, Schor IE, Muñoz MJ, Risso G, Petrillo E, Westman BJ, Lamond AI, Arzt E, et al. The serine/arginine-rich protein SF2/ASF regulates protein sumoylation. Proc Natl Acad Sci U S A. 2010;107:16119–24. doi: 10.1073/pnas.1004653107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tatham MH, Rodriguez MS, Xirodimas DP, Hay RT. Detection of protein SUMOylation in vivo. Nat Protoc. 2009;4:1363–71. doi: 10.1038/nprot.2009.128. [DOI] [PubMed] [Google Scholar]
- 64.Cramer P, Cáceres JF, Cazalla D, Kadener S, Muro AF, Baralle FE, Kornblihtt AR. Coupling of transcription with alternative splicing: RNA pol II promoters modulate SF2/ASF and 9G8 effects on an exonic splicing enhancer. Mol Cell. 1999;4:251–8. doi: 10.1016/S1097-2765(00)80372-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.