

Abstract
Fused in sarcoma (FUS) belongs to the group of RNA-binding proteins implicated as underlying factors in amyotrophic lateral sclerosis (ALS) and certain other neurodegenerative diseases. Multiple FUS gene mutations have been linked to hereditary forms, and aggregation of FUS protein is believed to play an important role in pathogenesis of these diseases. In cultured cells, FUS variants with disease-associated amino acid substitutions or short deletions affecting nuclear localization signal (NLS) and causing cytoplasmic mislocalization can be sequestered into stress granules (SGs). We demonstrated that disruption of motifs responsible for RNA recognition and binding not only prevents SG recruitment, but also dramatically increases the protein propensity to aggregate in the cell cytoplasm with formation of juxtanuclear structures displaying typical features of aggresomes. Functional RNA-binding domains from TAR DNA-binding protein of 43 kDa (TDP-43) fused to highly aggregation-prone C-terminally truncated FUS protein restored the ability to enter SGs and prevented aggregation of the chimeric protein. Truncated FUS was also able to trap endogenous FUS molecules in the cytoplasmic aggregates. Our data indicate that RNA binding and recruitment to SGs protect cytoplasmic FUS from aggregation, and loss of this protection may trigger its pathological aggregation in vivo.
Keywords: RNA-binding proteins, cytoplasmic RNP complexes, protein aggregation, proteinopathy, FUSopathy, amyotrophic lateral sclerosis
Introduction
DNA/RNA-binding protein FUS/TLS has been strongly linked to the development of neuronal dysfunction in ALS and related diseases, although the roles of aberrant compartmentalization, alteration of its normal function in RNA metabolism, and pathological aggregation in mediating pathology are still unclear. FUS mutations are found in familial ALS, and mutant FUS loses its normal nuclear localization and forms characteristic cytoplasmic inclusions in motor neurons of these patients.1,2 Moreover, FUS-positive inclusions have been observed in neurons of some patients that did not carry FUS gene mutations but developed ALS,3 frontotemporal lobar degeneration,4 or other, more rare neurodegenerative disorders,5-7 indicating that under certain conditions, genetically unaltered protein may undergo pathogenic conversion and contribute to the pathology that selectively affects the nervous system.
The NLS is located at the C terminus of the FUS molecule, and a significant proportion of missense or nonsense point mutations that cause hereditary ALS are clustered within this domain.8,9 Methylation of arginine residues in the region upstream of the NLS also contributes to nuclear export of FUS.10 In cultured cells, FUS mislocalized to the cytoplasm is recruited to SGs, dense foci composed of stalled pre-initiation complexes assembled together by a specific pool of proteins.11-15 FUS’s ability to enter SGs is closely linked to its RNA binding capacity, and C-terminal RGG boxes and zinc finger of FUS are crucial for this process.16 RRM domain, which plays a pivotal role in binding of RNA by other proteins, including TDP-43,17,18 has been shown to be of limited importance,19,20 or even dispensable,16 for both FUS binding to RNA and SGs recruitment. FUS-containing SGs were recently proposed to serve as “precursors” of insoluble aggregates found in neurons of patients with FUSopathies,13,21,22 although currently there is no direct experimental evidence in support of this idea, and conversion of SGs into insoluble aggregates has never been demonstrated. Moreover, considering that formation of SGs is a protective mechanism; that several aggregation-prone proteins bearing prion-like domains, such as TIA-1, are assembled in SGs11; and that SGs are highly unstable, dynamic structures, rapidly dissipating without leaving residual aggregates of resident proteins after acute stress is removed,23,24 it could be suggested that SGs recruitment might render protection from, rather than stimulate irreversible aggregation of, cytoplasmic FUS in conditions of stress. Further evidence in support of an idea that RNA binding and sequestering into SGs can prevent aggregation of certain types of proteins emerges from recent studies of TDP-43, a protein structurally and functionally related to FUS and involved in pathogenesis of the same set of neurodegenerative diseases.25,26 In affected neurons, TDP-43 undergoes extensive post-translational modifications, including phosphorylation, ubiquitination, and fragmentation.27 Notably, caspase-cleaved 25 kDa C-terminal fragments of TDP-43, lacking NLS and with disrupted RNA-binding motif, were shown to be highly aggregation-prone and accumulated in neurons of TDP-43 proteinopathy patients,25,28-30 suggesting their involvement in the disease pathogenesis. In cells expressing similar C-terminal fragments of TDP-43, RNA depletion triggers its fast aggregation.31 Moreover, cognate RNA or ssDNA are able to ameliorate in vitro aggregation of functional TDP-43 protein but not its variants deficient for RNA binding due to point mutations or N-terminal truncation.32
Here we studied the behavior of cytoplasm-targeted FUS variants with different degrees of disruption of C-terminal domains essential for RNA-binding and SG recruitment, and demonstrated that the inability of cytoplasmic FUS to be recruited to SGs promotes its aggregation.
Results
A human FUS protein variant with disrupted nuclear localization signal and RNA binding motifs readily aggregates in cultured cells
For expression in cultured eukaryotic cells, plasmid constructs encoding human FUS protein variants with an N-terminal GFP tag were created (Fig. 1A). All these proteins were expressed at comparable levels upon transient transfection of neuroblastoma SH-SY5Y cells (Fig. 1B). In agreement with the results obtained in other cultured cells,12-15,33 normal full-length FUS was confined to the nucleus (Fig. 1D), while its variants with a familial ALS-associated R522G substitution and proteins with NLS deletion, FUS 1–513 were predominantly localized to the cytoplasm (Fig. 1C). This is consistent with previous observations that 13 C-terminal amino acids are essential for normal FUS nuclear localization, and that substitution in the position 522 almost completely abrogates nuclear import of the protein.13,34 Later after transfection, in cells that accumulated larger amounts of these FUS variants, they often formed irregularly shaped foci around the cytoplasm (Fig. 1D). A variant with a larger deletion corresponding to the truncated ALS-associated variant, p.G466VfsX14 (FUS 1–466), in which RGG3 domain was almost completely absent, behaved in a similar way (Fig. 1D). However, in cells expressing a further truncated FUS with both C-terminal RGG boxes, zinc finger, and NLS deleted, a principally different pattern was observed. Soon after the onset of expression diffusely distributed protein started to form small, round puncta throughout the cytoplasm (Video S1) that further fused to each other and translocated to the perinuclear area, where they coalesced into a large, irregularly shaped structure (Fig. 1D and F; Video S2), adjacent to but not overlapped with endosomal/lysosomal compartments (Fig. 1F). In some cells FUS 1–359 aggregates were detected as early as 4 h after transfection, when cells expressing other FUS variants showed only diffuse distribution (Fig. 1C). Twenty-four h after transfection, precursors and/or large aggregates were present in a significant fraction of transfected cells (Fig. 1E).

Figure 1. Characterization of mutant FUS proteins transiently expressed in neuroblastoma SH-SY5Y cells. (A) Scheme of FUS domain organization and GFP-fusion constructs. (B) All GFP-labeled FUS variants utilized in the study are expressed at a comparable level in neuroblastoma cells as revealed with anti-GFP antibody. Total protein extracts for western blot analysis were prepared from cells lysed 24 h after transfection with corresponding expression plasmid. C is an unrelated cytosolic GFP-fusion protein used as a control. The size of proteins in kDa is shown. (C) Diffuse distribution of cytoplasmically localized FUS variants FUS R522G, FUS 1–513, and FUS ΔRRMcyt is detected in transfected cells at early stages of the ectopic protein accumulation (4–12 h post-transfection) or in cells with relatively low levels of its expression. In contrast, in most cells expressing FUS 1–359, small aggregates are formed very early after transfection. Scale bar: 20 µm. (D) Intracellular distribution of GFP-FUS fusion proteins and morphology of aggregates formed in the cytoplasm 36 h post-transfection. See also Videos S1 and S2. (E) Twenty-four hours after transfection, the majority of cells expressing FUS 1–359 possess aggregates of some form, and more than half of them display large juxtanuclear structures. (F) Juxtanuclear aggregates formed by FUS 1–359 protein are negative for endosomal/lysosmal compartment markers LAMP-1 and LAMP-2. Scale bars: (D), 15 µm; (C and F), 20 µm.
Large juxtanuclear structures were occasionally observed in cells expressing high levels of the FUS variant with deletion of another domain involved in RNA binding, RRM, which was rendered cytoplasmic by the introduction of R522G substitution (Fig. 1D, ΔRRMcyt), although in the majority of cells ΔRRMcyt formed granule-like aggregates similar to those formed by full-length or slightly truncated variants.
Truncated FUS lacking C-terminal RNA-binding domains forms aggresomes, while RNA binding competent variants form stress granules in cultured cells
Morphology and assembly dynamics of juxtanuclear structures in FUS 1–359-transfected cells resembles those typical for aggresomes, protective cellular structures formed by cultured cells in an attempt to enclose and neutralize potentially deleterious misfolded/aggregated proteins.35 Indeed, immunofluorescent staining for specific markers demonstrated that these structures were adjacent to the centrosome (Fig. 2A), surrounded by a “vimentin cage” (Fig. 2B), and positive for a chaperone protein HSP27 (Fig. S1). Consistent with the importance of microtubule integrity for aggresome formation, treatment of FUS 1–359-expressing cells with a microtubule-disrupting agent nocodazole abolished formation of large aggregates but did not affect small precursors (Fig. 2C).
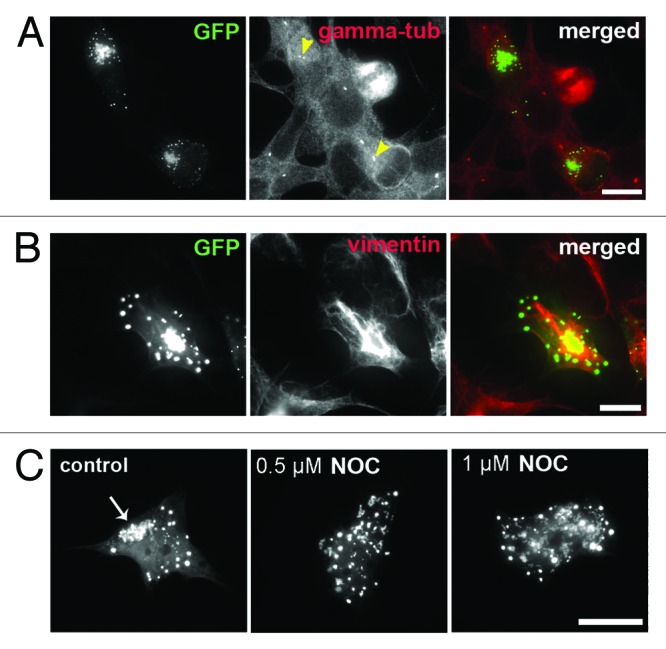
Figure 2. Aggregates formed by truncated FUS 1–359 in SH-SY5Y cells display typical characteristics of aggresomes. (A) Large juxtanuclear structures formed by FUS 1–359 are clustered around the centrosome, visualized with centrosome marker gamma-tubulin (arrowheads). (B) Anti-vimentin staining of cells expressing FUS 1–359 reveals a characteristic “vimentin cage” around large juxtanuclear structures. (C) Formation of juxtanuclear aggregates by FUS 1–359 (arrow) is prevented by nocodazole-induced disruption of microtubules. Scale bar: 15 µm for all panels.
In contrast, cytoplasmic foci formed by FUS R522G or FUS 1–513 proteins did not display any typical aggresome characteristics but perfectly co-localized with SGs markers TIAR (Fig. 3A) and G3BP1 (Fig. 3B). Neither juxtanuclear aggresomes, nor small precursor puncta formed by FUS 1–359 were positive for SGs markers, and these structures did not co-localize with SGs occasionally seen in expressing cells without application of exogenous stress (Fig. 3A and B). Moreover, treatment with emetine, a known inhibitor of translational elongation and suppressor of SG formation, dispersed granules formed by FUS 1–513, but had no effect on FUS 1–359 aggregates (Fig. 3C).

Figure 3. Truncated FUS 1–359 is not recruited into stress granules in naïve SH-SY5Y cells. Representative confocal images of cells transfected with plasmids expressing various GFP-tagged FUS variants and stained with antibodies that detect stress granules. Foci spontaneously formed by FUS R522G or FUS 1–513 in a fraction of transfected cells are positive for TIAR (A) or G3BP1 (B), markers for stress granules, whereas neither large juxtanuclear, nor small dispersed aggregates formed by FUS 1–359 contain these proteins. (C) Emetine treatment disperses foci formed by FUS R522G, but has no effect on FUS 1–359 aggregates. Scale bars: 15 µm for all panels.
To assess whether FUS variants can be recruited to induced SGs, we treated transfected cells with sodium arsenite or applied a heat shock and visualized SGs by either anti-TIAR or anti-G3BP1 staining. With the exception of FUS 1–359, all other studied FUS proteins, including variants lacking RRM or bearing the largest known ALS-associated truncation (FUS 1–466) were readily recruited to SGs (Fig. 4A; Fig. S2A). In contrast, FUS 1–359 was completely excluded from induced SGs (Fig. 4A; Fig. S2A). As in the case of “constitutive” SGs formed in response to FUS overexpression, additional treatment of stressed cells with emetine or cycloheximide abolished SG formation and FUS recruitment in FUS 1–513-expressing cells but did not affect the integrity of either small precursors or aggresomes formed by FUS 1–359 (Fig. 4B; Fig. S2B).
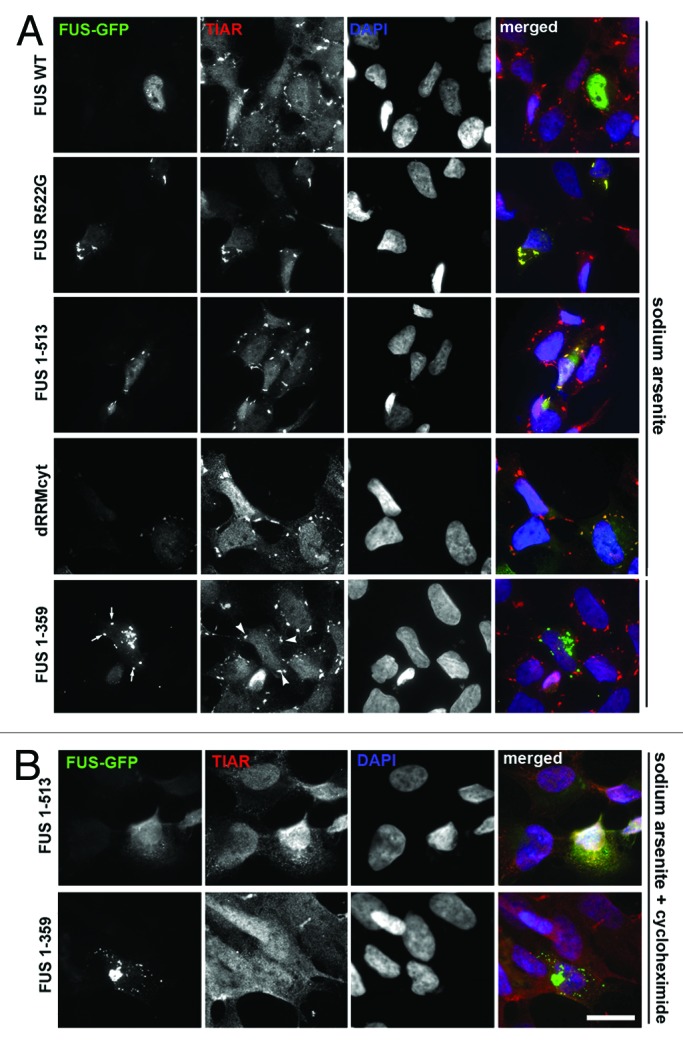
Figure 4. Truncated FUS 1–359 is not recruited into stress granules upon oxidative insult. (A) Cells transfected with GFP-tagged FUS expressing plasmids were treated with 0.5 mM sodium arsenite for 1 h and stained with anti-TIAR antibody. Foci formed by FUS R522G and FUS 1–513 are positive for TIAR, while FUS 1–359 aggregates (arrows) do not co-localize with TIAR-positive stress granules (arrowheads). (B) Cycloheximide treatment blocks formation of stress granules including those containing FUS 1–513, but does not affect FUS 1–359 aggregates. Scale bar: 15 µm for all panels.
Aggregation of C-terminally truncated FUS can be prevented by its fusion with RNA-binding domains of TDP-43
If the impaired ability for RNA binding and/or SG recruitment seen in FUS variants with deletions of RNA binding domains indeed results in their irreversible aggregation in the cytoplasm, restoration of this ability should impede or prevent such aggregation. To test this idea, we constructed a chimeric protein that was structurally different, yet possessing high RNA-binding capacity and SG affinity motifs, namely RRM1 and RRM2 from TDP-43, fused to the C terminus of FUS 1–359 (Fig. 5A). In transfected cells, this chimeric protein either displayed diffuse cytoplasmic staining or formed multiple irregular shape cytoplasmic foci (Fig. 5C, left panel), i.e., behaved similarly to FUS variants with disturbed NLS but intact RNA-binding domains (compare Fig. 5C left panel with R522G, 1–513 and 1–466 panels in Fig. 1D). Also as predicted, this protein was readily sequestered to SGs induced by sodium arsenite treatment (Fig. 5D). Furthermore, introduction of 3 single amino acid substitutions in the RRM1 domain (L106D, V108D, L111D) that abolish RNA binding by TDP-4336 resulted in the reversal to the aggregation properties typical to the parental FUS protein: accumulation of the chimeric protein in large juxtanuclear aggregates similar to those formed by FUS 1–359 (compare Fig. 5C right panel with FUS 1–359 panels in Fig. 1D) and inability to enter SG (Fig. 5E). These differences in aggregation patterns were not caused by differences in amounts of accumulating proteins, as very similar levels of FUS 1–359 and both FUS-TDP-43 chimeras were detected in cells transfected with corresponding plasmids (Fig. 5B). Results of these rescue experiments strongly suggest that FUS with lost ability to bind target RNA, and, consequently, impaired affinity to SGs, is prone to irreversible aggregation in the cell cytoplasm.

Figure 5. Aggregation of truncated FUS 1–359 in cultured cells can be prevented by fusion with functional RNA-binding domains of TDP-43. (A) Scheme of FUS-TDP-43 chimeric proteins. (B) Western blot analysis of FUS 1–359 and FUS-TDP-43 fusions using anti-GFP antibody demonstrates their equal levels upon expression in neuroblastoma SH-SY5Y cells. The size of proteins in kDa is shown. (C) Intracellular distribution of GFP-tagged chimeric proteins in transfected SH-SY5Y cells reveals granule-like aggregates with diffuse distribution of FUS-TDP-43 with intact RRMs and formation of aggresomes when RNA-binding capacity of RRM1 is abolished by 3 amino acid substitutions, L106D, V108D, L111D (3D mutant). Scale bar: 15 µm for both panels. (D and E) FUS-TDP-43 with intact RRMs (D) but not FUS-TDP-43 bearing substitutions in RRM1 disrupting RNA binding (E) is able to enter stress granules induced by arsenite treatment. Scale bar: 15 µm for all panels.
FUS 1–359 is able to seed aggregation
The N terminus of FUS contains a strong prion-like domain,37 and therefore, it is plausible that FUS 1–359 would seed aggregation of structurally normal FUS molecules. To test this, we employed antibodies specifically recognizing C terminus of FUS, which strongly labeled both endogenous FUS and all variants we used with the exception of FUS 1–359 (Fig. S3). We performed co-transfection of FUS 1–359 with either wild-type or cytoplasmically mislocalized mutants and observed that FUS 1–359 is able to recruit and sequester within aggregates other variants of FUS expressed in the same cell as well as endogenous FUS (Fig. 6). However, this recruitment was dependent upon retention of these proteins in the cytoplasm, as only a fraction of the predominantly nuclear wild-type FUS could be found in cytoplasmic aggregates formed by FUS 1–359, in contrast to nearly complete co-localization seen for cytoplasmic FUS R522G or FUS 1–513 variants (Fig. 6).
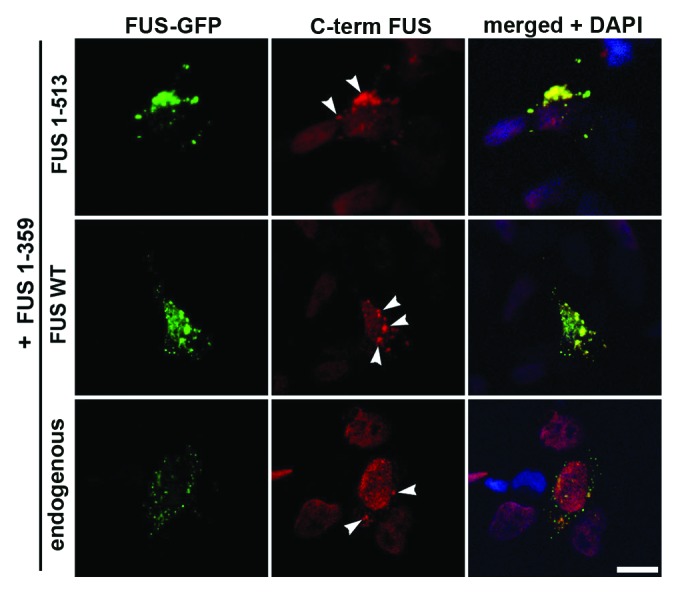
Figure 6. Truncated FUS 1–359 protein is able to sequester both normal and mutated FUS variants in aggregates. Cells were co-transfected with shown GFP-tagged FUS variants, and stained with antibody against C terminus of FUS. Cytoplasmic variants FUS R522G and FUS 1–513 are recruited into morphologically distinct aggregates formed by FUS 1–359 (arrowheads). In some, relatively rare cells FUS 1–359 recruits into these aggregates even a fraction of co-expressed wild-type FUS or endogenous wild-type FUS retained in the cytoplasm (lower panel). Scale bar: 10 µm for all panels.
Discussion
Our studies of human FUS protein variants expressed in cultured neuroblastoma cells demonstrated that disease-associated point mutations or deletions at the C-terminal region that change normal, predominantly nuclear localization of FUS to predominantly cytoplasmic do not trigger aggregation of modified proteins with formation of stable inclusions. In transfected cells, these FUS variants are diffusely distributed throughout the cytoplasm, and when a cell becomes stressed, either because of the ectopic protein accumulation, or by external factor(s), they appear to be sequestered into SGs. However, when inactivation of the C-terminal NLS is accompanied by compromised ability to bind RNA due to deletion of relevant domains, FUS becomes excluded from SGs, which is consistent with previously demonstrated importance of major RNA binding domains for SG recruitment,16 and rapidly aggregates with the formation of typical aggresomes. These results suggest that sequestering of cytoplasmic FUS in SGs prevents its aggregation in stressed cells. Thus, a temporary failure to form SGs might be a “second hit” that triggers aggregation of cytoplasmic forms of FUS under conditions of stress.
There is a growing body of evidence that in human diseases the presence of a mutation that modifies FUS compartmentalization from predominantly nuclear to predominantly cytoplasmic is not per se sufficient for the development of pathological changes in neurons, and additional genetic or environmental events are required for triggering this process.38-41 It is feasible that alterations in cytoplasmic RNP metabolism caused by such external factors may contribute to FUS irreversible aggregation by preventing its sequestration. Even when these alterations are only transient, aggregation of cytosolic FUS, once triggered, might gradually progress via seeding mechanism, resulting in formation of large inclusions and/or cell death due to toxicity of certain aggregation products and secondary effects on RNA metabolism (Fig. 7). Interestingly, a C-terminal caspase-cleaved 25 kD fragment of TDP-43 lacking abilities to bind target RNA and to be efficiently sequestered by stress granules16 is abundant in pathological inclusions in TDP-43 proteinopathies and plays an important role in pathogenesis of these diseases.25,28,29,42-45 Moreover, it has been demonstrated that interaction with target RNA can ameliorate aggregation of binding-competent TDP-43 variants in vitro,31 while acute RNA depletion might stimulate aggregation of TDP-43.32

Figure 7. A mechanism of pathological aggregation of FUS protein in the cell cytoplasm. A proposed sequence of internal and external events triggering the development of FUSopathy. FUS-containing stress granules are shown in yellow, and FUS aggregates in green.
The requirement of additional factors for efficient aggregation of FUS protein in vivo might explain difficulties with achieving FUS aggregation and therefore establishing adequate models of FUSopathy in short-living laboratory animals.46-49 The presence of prion-like domains within the C-terminal region of TDP-43 (amino acids 277–414) and the N-terminal region of FUS (amino acids 1–239) has been recently reported.37 Our observations that endogenous FUS proteins become recruited to aggregates formed by FUS 1–359 in cultured cells suggest that certain products of FUS aggregation are capable of seeding aggregation of other, including structurally normal, FUS proteins. Consistent with this idea, in transgenic mice expressing relatively low levels of FUS 1–359 in their neurons and abruptly developing fatal motor neuron pathology, endogenous mouse FUS is present in pathological inclusions formed by transgenically expressed truncated protein.50 It is feasible that a similar mechanism might be involved in the formation of inclusions and even spreading of pathology in human FUSopathies.
In conclusion, our results suggest that sequestering in cytoplasmic RNP particles protects mislocalized FUS from aggregation, while certain structural modifications of the protein or alterations of cellular RNA metabolism might compromise this process, leading to irreversible aggregation and further pathological changes in the affected cells.
Materials and Methods
Expression plasmids and transfection of eukaryotic cells
Human FUS or TDP-43 cDNA fragments carrying point mutations or deletion were produced by PCR amplification from the full-length cDNA using designed primers cloned into pTOPO-Blunt vector (Invitrogen) and, after verification of the insert sequence, subcloned into the pEGFP-C1 vector (Clontech) downstream and in-frame with the GFP coding region. SH-SY5Y human neuroblastoma cells were maintained in Dulbecco modified Eagle medium (Invitrogen), supplemented with 10% fetal bovine serum. For immunofluorescence, cells were grown on poly-L-lysine-coated coverslips. Cells were transfected with the expression plasmids using Lipofectamine2000 reagent (Invitrogen) according to the manufacturer's instructions. For co-transfection, equal amounts of plasmid DNA for each construct were taken to the reaction. For disruption of microtubules, cells were treated with nocodazole (Sigma-Aldrich) at a final concentration of 0.5 or 1 µM for 4 h. In heat shock experiments, cells were subjected to 43 °C for 1 h. For induction of oxidative stress, cells were exposed to 0.5 mM sodium arsenite (Sigma-Aldrich) for 1 h. Emetine and cycloheximide (Sigma-Aldrich) were used at final concentrations of 20 and 10 µg/ml, respectively.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde on ice for 15 min, followed by washes with PBS, and 5 min permeabilization in cold methanol. Immunostaining and detection was performed as described previously.50 After 3 washes with PBS and blocking in 5% goat serum/PBS/0.1% Triton X100 for 1 h at room temperature, samples were incubated with primary antibodies (see below) diluted in blocking solution. Alexa Fluor-conjugated anti-mouse or anti-rabbit immunoglobulins (Molecular Probes) were used as secondary antibodies (1:1000 in PBS/0.1% Triton X100), and cell nuclei were visualized with DAPI. Epifluorescent images were taken using either BX61 microscope (Olympus) or confocal Leica DM6000B microscope (Leica Microsystems) and processed using Cell-F software or Leica Application Suite AF software.
Live-cell imaging
Time-lapse images were obtained using a Leica TCS SP2 MP confocal microscope, equipped with an on-scope incubator with temperature control (Leica Microsystems). SH-SY5Y or COS7 cells were plated on glass-bottomed tissue culture dishes and transfected with FUS 1–359 construct as described above. Eight hours post-transfection, regular culture media was replaced with HEPES-buffered media (10 mM HEPES-KOH, pH7.5) for maintaining physiological pH outside a CO2 incubator. Cells were visualized under Fluotar L 63 × 1.4 oil objective. A sequence of images taken with 8 min intervals was further transformed into video using Leica Application Suite AF software.
Antibodies
Primary antibodies against the following antigens were used: C terminus of FUS protein (mouse monoclonal, clone 4H11, Santa Cruz Biotechnology); N terminus of FUS protein (rabbit polyclonal, Abcam; mouse polyclonal, BD Biosciences); GFP (mouse monoclonal, clone 3A9, Protein Synthesis); TIAR (mouse monoclonal, clone 6, BD Biosciences); G3BP1(mouse monoclonal, BD Biosciences); vimentin (mouse monoclonal, clone rv202, BD Biosciences); gamma-tubulin (mouse monoclonal, clone GTU-88, Sigma-Aldrich); HSP27 (rabbit polyclonal, Cell Signaling); LAMP-1 (mouse monoclonal, clone H4A3, BD Biosciences); LAMP-2 (mouse monoclonal, clone H4B4, BD Biosciences); β-actin (mouse monoclonal, clone AC-15, Sigma-Aldrich). All primary antibodies were used in 1:1000 dilution for both immunofluorescence and western blotting.
Western blotting
Cells or tissues were homogenized directly in SDS-PAGE loading buffer and denatured at 100 °C for 10 min. Equal amounts of total protein were run on SDS-PAGE, transferred to PVDF membrane by semi-dry blotting followed by blocking, incubation with primary and HRP-conjugated secondary (GE Healthcare) antibodies, and ECL detection. Equal loading was confirmed by reprobing membranes with antibodies against β-actin.
Supplementary Material
Acknowledgments
This work was supported by a Research Grant from the Welcome Trust (075615/Z/04/z) to VLB. TAS was supported by the Short-Term EMBO Fellowship (ASTF 351–2011) and Russian Foundation for Basic Research_mol_a Grant (12-04-31791), and HR by the Cardiff NMHRI 4-y PhD Studentship Programme. We are grateful for Natalia Ninkina for critical reading of the manuscript.
Glossary
Abbreviations:
- FUS
fused in sarcoma
- TDP-43
TAR DNA-binding protein of 43 kDa
- ALS
amyotrophic lateral sclerosis
- NLS
nuclear localization signal
- SGs
stress granules
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/26241
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26241
References
- 1.Kwiatkowski TJ, Jr., Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–8. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 2.Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–11. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deng HX, Zhai H, Bigio EH, Yan J, Fecto F, Ajroud K, Mishra M, Ajroud-Driss S, Heller S, Sufit R, et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann Neurol. 2010;67:739–48. doi: 10.1002/ana.22051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009;132:2922–31. doi: 10.1093/brain/awp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neumann M, Roeber S, Kretzschmar HA, Rademakers R, Baker M, Mackenzie IR. Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol. 2009;118:605–16. doi: 10.1007/s00401-009-0581-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munoz DG, Neumann M, Kusaka H, Yokota O, Ishihara K, Terada S, Kuroda S, Mackenzie IR. FUS pathology in basophilic inclusion body disease. Acta Neuropathol. 2009;118:617–27. doi: 10.1007/s00401-009-0598-9. [DOI] [PubMed] [Google Scholar]
- 7.Cohen NR, Hammans SR, Macpherson J, Nicoll JA. New neuropathological findings in Unverricht-Lundborg disease: neuronal intranuclear and cytoplasmic inclusions. Acta Neuropathol. 2011;121:421–7. doi: 10.1007/s00401-010-0738-2. [DOI] [PubMed] [Google Scholar]
- 8.Da Cruz S, Cleveland DW. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr Opin Neurobiol. 2011;21:904–19. doi: 10.1016/j.conb.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lattante S, Rouleau GA, Kabashi E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum Mutat. 2013;34:812–26. doi: 10.1002/humu.22319. [DOI] [PubMed] [Google Scholar]
- 10.Dormann D, Madl T, Valori CF, Bentmann E, Tahirovic S, Abou-Ajram C, Kremmer E, Ansorge O, Mackenzie IR, Neumann M, et al. Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J. 2012;31:4258–75. doi: 10.1038/emboj.2012.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kedersha N, Anderson P. Mammalian stress granules and processing bodies. Methods Enzymol. 2007;431:61–81. doi: 10.1016/S0076-6879(07)31005-7. [DOI] [PubMed] [Google Scholar]
- 12.Bosco DA, Lemay N, Ko HK, Zhou H, Burke C, Kwiatkowski TJ, Jr., Sapp P, McKenna-Yasek D, Brown RH, Jr., Hayward LJ. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet. 2010;19:4160–75. doi: 10.1093/hmg/ddq335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Schmid B, et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010;29:2841–57. doi: 10.1038/emboj.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gal J, Zhang J, Kwinter DM, Zhai J, Jia H, Jia J, Zhu H. Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol Aging. 2011;32:2323.e27–40. doi: 10.1016/j.neurobiolaging.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ito D, Seki M, Tsunoda Y, Uchiyama H, Suzuki N. Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann Neurol. 2011;69:152–62. doi: 10.1002/ana.22246. [DOI] [PubMed] [Google Scholar]
- 16.Bentmann E, Neumann M, Tahirovic S, Rodde R, Dormann D, Haass C. Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43) J Biol Chem. 2012;287:23079–94. doi: 10.1074/jbc.M111.328757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cléry A, Blatter M, Allain FH. RNA recognition motifs: boring? Not quite. Curr Opin Struct Biol. 2008;18:290–8. doi: 10.1016/j.sbi.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 18.Kuo PH, Doudeva LG, Wang YT, Shen CK, Yuan HS. Structural insights into TDP-43 in nucleic-acid binding and domain interactions. Nucleic Acids Res. 2009;37:1799–808. doi: 10.1093/nar/gkp013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lerga A, Hallier M, Delva L, Orvain C, Gallais I, Marie J, Moreau-Gachelin F. Identification of an RNA binding specificity for the potential splicing factor TLS. J Biol Chem. 2001;276:6807–16. doi: 10.1074/jbc.M008304200. [DOI] [PubMed] [Google Scholar]
- 20.Liu X, Niu C, Ren J, Zhang J, Xie X, Zhu H, Feng W, Gong W. The RRM domain of human fused in sarcoma protein reveals a non-canonical nucleic acid binding site. Biochim Biophys Acta. 2013;1832:375–85. doi: 10.1016/j.bbadis.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li YR, King OD, Shorter J, Gitler AD. Stress granules as crucibles of ALS pathogenesis. J Cell Biol. 2013;201:361–72. doi: 10.1083/jcb.201302044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolozin B. Regulated protein aggregation: stress granules and neurodegeneration. Mol Neurodegener. 2012;7:56. doi: 10.1186/1750-1326-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kedersha N, Cho MR, Li W, Yacono PW, Chen S, Gilks N, Golan DE, Anderson P. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J Cell Biol. 2000;151:1257–68. doi: 10.1083/jcb.151.6.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J, Okabe K, Tani T, Funatsu T. Dynamic association-dissociation and harboring of endogenous mRNAs in stress granules. J Cell Sci. 2011;124:4087–95. doi: 10.1242/jcs.090951. [DOI] [PubMed] [Google Scholar]
- 25.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 26.Baloh RH. How do the RNA-binding proteins TDP-43 and FUS relate to amyotrophic lateral sclerosis and frontotemporal degeneration, and to each other? Curr Opin Neurol. 2012;25:701–7. doi: 10.1097/WCO.0b013e32835a269b. [DOI] [PubMed] [Google Scholar]
- 27.Zhang YJ, Xu YF, Dickey CA, Buratti E, Baralle F, Bailey R, Pickering-Brown S, Dickson D, Petrucelli L. Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J Neurosci. 2007;27:10530–4. doi: 10.1523/JNEUROSCI.3421-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rutherford NJ, Zhang YJ, Baker M, Gass JM, Finch NA, Xu YF, Stewart H, Kelley BJ, Kuntz K, Crook RJ, et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008;4:e1000193. doi: 10.1371/journal.pgen.1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Igaz LM, Kwong LK, Xu Y, Truax AC, Uryu K, Neumann M, Clark CM, Elman LB, Miller BL, Grossman M, et al. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am J Pathol. 2008;173:182–94. doi: 10.2353/ajpath.2008.080003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furukawa Y, Kaneko K, Nukina N. Molecular properties of TAR DNA binding protein-43 fragments are dependent upon its cleavage site. Biochim Biophys Acta. 2011;1812:1577–83. doi: 10.1016/j.bbadis.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 31.Pesiridis GS, Tripathy K, Tanik S, Trojanowski JQ, Lee VMA. A “two-hit” hypothesis for inclusion formation by carboxyl-terminal fragments of TDP-43 protein linked to RNA depletion and impaired microtubule-dependent transport. J Biol Chem. 2011;286:18845–55. doi: 10.1074/jbc.M111.231118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang YC, Lin KF, He RY, Tu PH, Koubek J, Hsu YC, Huang JJ. Inhibition of TDP-43 aggregation by nucleic acid binding. PLoS One. 2013;8:e64002. doi: 10.1371/journal.pone.0064002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kino Y, Washizu C, Aquilanti E, Okuno M, Kurosawa M, Yamada M, Doi H, Nukina N. Intracellular localization and splicing regulation of FUS/TLS are variably affected by amyotrophic lateral sclerosis-linked mutations. Nucleic Acids Res. 2011;39:2781–98. doi: 10.1093/nar/gkq1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shelkovnikova TA, Ustyugov AA, Smirnov AP, Skvortsova VI, Buchman VL, Bachurin SO, Ninkina NN. FUS gene mutations associated with familiar forms of amyotrophic lateral sclerosis affect cellular localization and aggregation properties of the encoded protein. Dokl Biochem Biophys. 2011;438:123–6. doi: 10.1134/S1607672911030045. [DOI] [PubMed] [Google Scholar]
- 35.Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–30. doi: 10.1016/S0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- 36.Buratti E, Baralle FE. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem. 2001;276:36337–43. doi: 10.1074/jbc.M104236200. [DOI] [PubMed] [Google Scholar]
- 37.Cushman M, Johnson BS, King OD, Gitler AD, Shorter J. Prion-like disorders: blurring the divide between transmissibility and infectivity. J Cell Sci. 2010;123:1191–201. doi: 10.1242/jcs.051672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Blitterswijk M, van Es MA, Hennekam EA, Dooijes D, van Rheenen W, Medic J, Bourque PR, Schelhaas HJ, van der Kooi AJ, de Visser M, et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum Mol Genet. 2012;21:3776–84. doi: 10.1093/hmg/dds199. [DOI] [PubMed] [Google Scholar]
- 39.Millecamps S, Salachas F, Cazeneuve C, Gordon P, Bricka B, Camuzat A, Guillot-Noël L, Russaouen O, Bruneteau G, Pradat PF, et al. SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: genotype-phenotype correlations. J Med Genet. 2010;47:554–60. doi: 10.1136/jmg.2010.077180. [DOI] [PubMed] [Google Scholar]
- 40.Groen EJ, van Es MA, van Vught PW, Spliet WG, van Engelen-Lee J, de Visser M, Wokke JH, Schelhaas HJ, Ophoff RA, Fumoto K, et al. FUS mutations in familial amyotrophic lateral sclerosis in the Netherlands. Arch Neurol. 2010;67:224–30. doi: 10.1001/archneurol.2009.329. [DOI] [PubMed] [Google Scholar]
- 41.Blair IP, Williams KL, Warraich ST, Durnall JC, Thoeng AD, Manavis J, Blumbergs PC, Vucic S, Kiernan MC, Nicholson GA. FUS mutations in amyotrophic lateral sclerosis: clinical, pathological, neurophysiological and genetic analysis. J Neurol Neurosurg Psychiatry. 2010;81:639–45. doi: 10.1136/jnnp.2009.194399. [DOI] [PubMed] [Google Scholar]
- 42.Caccamo A, Majumder S, Oddo S. Cognitive decline typical of frontotemporal lobar degeneration in transgenic mice expressing the 25-kDa C-terminal fragment of TDP-43. Am J Pathol. 2012;180:293–302. doi: 10.1016/j.ajpath.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cuijt I, Smits V, Ceuterick-de Groote C, Van Broeckhoven C, Kumar-Singh S. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2010;107:3858–63. doi: 10.1073/pnas.0912417107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang YJ, Xu YF, Cook C, Gendron TF, Roettges P, Link CD, Lin WL, Tong J, Castanedes-Casey M, Ash P, et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci U S A. 2009;106:7607–12. doi: 10.1073/pnas.0900688106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dormann D, Capell A, Carlson AM, Shankaran SS, Rodde R, Neumann M, Kremmer E, Matsuwaki T, Yamanouchi K, Nishihara M, et al. Proteolytic processing of TAR DNA binding protein-43 by caspases produces C-terminal fragments with disease defining properties independent of progranulin. J Neurochem. 2009;110:1082–94. doi: 10.1111/j.1471-4159.2009.06211.x. [DOI] [PubMed] [Google Scholar]
- 46.Chen Y, Yang M, Deng J, Chen X, Ye Y, Zhu L, Liu J, Ye H, Shen Y, Li Y, et al. Expression of human FUS protein in Drosophila leads to progressive neurodegeneration. Protein Cell. 2011;2:477–86. doi: 10.1007/s13238-011-1065-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miguel L, Avequin T, Delarue M, Feuillette S, Frébourg T, Campion D, Lecourtois M. Accumulation of insoluble forms of FUS protein correlates with toxicity in Drosophila. Neurobiol Aging. 2012;33:1008.e1–15. doi: 10.1016/j.neurobiolaging.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 48.Murakami T, Yang SP, Xie L, Kawano T, Fu D, Mukai A, Bohm C, Chen F, Robertson J, Suzuki H, et al. ALS mutations in FUS cause neuronal dysfunction and death in Caenorhabditis elegans by a dominant gain-of-function mechanism. Hum Mol Genet. 2012;21:1–9. doi: 10.1093/hmg/ddr417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang C, Zhou H, Tong J, Chen H, Liu YJ, Wang D, Wei X, Xia XG. FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genet. 2011;7:e1002011. doi: 10.1371/journal.pgen.1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shelkovnikova TA, Peters OM, Deykin AV, Connor-Robson N, Robinson H, Ustyugov AA, Bachurin SO, Ermolkevich TG, Goldman IL, Sadchikova ER, et al. Fused in sarcoma (FUS) protein lacking nuclear localization signal (NLS) and major RNA binding motifs triggers proteinopathy and severe motor phenotype in transgenic mice. J Biol Chem. 2013;288:25266–74. doi: 10.1074/jbc.M113.492017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.