

Abstract
In eukaryotic cells, genomic DNA is organized into a chromatin structure, which not only serves as the template for DNA-based nuclear processes, but also as a platform integrating intracellular and extracellular signals. Although much effort has been spent to characterize chromatin modifying/remodeling activities, little is known about cell signaling pathways targeting these chromatin modulators. Here, we report that cyclin-dependent kinase 1 (CDK1) phosphorylates the histone H2A deubiquitinase Ubp-M at serine 552 (S552P), and, importantly, this phosphorylation is required for cell cycle progression. Mass spectrometry analysis confirmed Ubp-M is phosphorylated at serine 552, and in vitro and in vivo assays demonstrated that CDK1/cyclin B kinase is responsible for Ubp-M S552P. Interestingly, Ubp-M S552P is not required for Ubp-M tetramer formation, deubiquitination activity, substrate specificity, or regulation of gene expression. However, Ubp-M S552P is required for cell proliferation and cell cycle G2/M phase progression. Ubp-M S552P reduces Ubp-M interaction with nuclear export protein CRM1 and facilitates Ubp-M nuclear localization. Therefore, these studies confirm that Ubp-M is phosphorylated at S552 and identify CDK1 as the enzyme responsible for the phosphorylation. Importantly, this study specifically links Ubp-M S552P to cell cycle G2/M phase progression.
Keywords: CDK1, CRM1, G2/M phase, H2A deubiquitination, Ubp-M, cell cycle, gene expression, phosphorylation
Introduction
In eukaryotic cells, genomic DNA is organized into a chromatin structure by association with histone and non-histone proteins.1 This organization not only allows eukaryotic cells to store their genetic information efficiently, but also provides an elegant means to regulate DNA-based nuclear processes including transcription, DNA damage repair, replication, and recombination.2-4 Two major mechanisms, ATP-dependent chromatin remodeling and covalent modifications of chromatin, have been shown to modulate the structure of chromatin and facilitate chromatin-based nuclear processes. While ATP-dependent chromatin remodeling activity transiently alters the interaction between histones and DNA,5,6 covalent modifications of chromatin components are involved in the stable alteration of chromatin structure.7,8 Covalent chromatin modifications include DNA methylation and posttranslational histone modifications, such as acetylation, methylation, phosphorylation, ubiquitination, and ADP ribosylation. These mechanisms function coordinately to precisely control the conformation of chromatin structure and facilitate nuclear processes on the chromatin template.
Although a great variety of chromatin remodeling/modifying enzymes have been described in past years,5,7 much less is known about how these enzymes are regulated by extrinsic or intrinsic cell signal transduction pathways.9-11 Recent studies uncover tight links between cell signaling and epigenetic regulation. MAPK, Wnt, Notch, JAK-STAT, JNK, NFκB, and PKA signaling pathways all have intrinsic links to epigenetic regulation.12,13 Current evidence indicates that such regulation may be critical. EZH2, the primary histone methyltransferase for H3K27, is phosphorylated by AKT and CDK1/CDK2 at serine 21, threonine 345 (threonine 350 in humans), and threonine 487 (threonine 492 in humans).14-18 Serine 21 phosphorylation impedes the binding of PRC2 to chromatin and reduces H3K27 methylation.14 Phosphorylation at threonine 345 is important for the recruitment of EZH2 and maintenance of H3K27 methylation at target genes, possibly by affecting EZH2 binding to HOTAIR.16,17 Phosphorylation of threonine 487 disrupts EZH2 interaction with other PRC2 components.15 Phosphorylation of EZH2 may also affect its ubiquitination and protein stability.18 In addition, Mel18 requires phosphorylation to direct H2A ubiquitination activity.19 Therefore, it is apparent that cell signaling pathways play important roles in regulating the activity, stability, and recruitment of chromatin modifiers.
In previous studies, we identified Ubp-M as a histone H2A deubiquitinase that regulates cell cycle progression and gene expression.20 Pioneer studies conducted by Dr Marchesi’s group revealed that Ubp-M is phosphorylated at the onset of mitosis and dephosphorylated during the metaphase/anaphase transition.21 Further studies revealed that the purified cdc-2/cyclin B complexes or mitotic cell extracts could phosphorylate Ubp-M in vitro. Although the phosphorylation site(s) was not identified in Dr Marchesi’s study, several recent studies identify serine 552 as a major phosphorylation site for Ubp-M during mitosis.22,23 Based on sequence analysis, the enzyme responsible for S552P is likely cdc-2/cyclin B; however, there is no direct experimental evidence for this. In addition, how Ubp-M S552P regulates the function of Ubp-M is not known.
To answer these questions, we investigated the role of phosphorylation in regulating the histone H2A deubiquitinase Ubp-M. Consistent with previous studies, we also identified 552 serine residue as a major Ubp-M phosphorylation site in addition to several other sites.22,23 Our studies provide direct experimental evidence for CDK1/cyclin B kinase in Ubp-M S552P. Importantly, our study revealed that Ubp-M S552P is not required for Ubp-M in vitro deubiquitination activity and in vivo gene expression regulation; instead, our studies specifically link Ubp-M S552P to cell proliferation and cell cycle G2/M phase progression.
Results
UBP-M is phosphorylated at serine 552
Ubp-M purified from sf9 cells appears as a doublet on SDS-PAGE gels, similar to Ubp-M purified from HeLa cells (Fig. 1A, lane 2, and data not shown). Since both bands are recognized by the Ubp-M antibody, we determined whether the doublet is due to phosphorylation. For this purpose, we treated Ubp-M with Calf Intestinal Alkaline Phosphatase (CIP, NEB). After CIP treatment, the intensity of the top band decreased, with a concomitant increase of the intensity of the bottom band (Fig. 1A, compare lane 3 with 2), suggesting that Ubp-M is phosphorylated.
Figure 1.
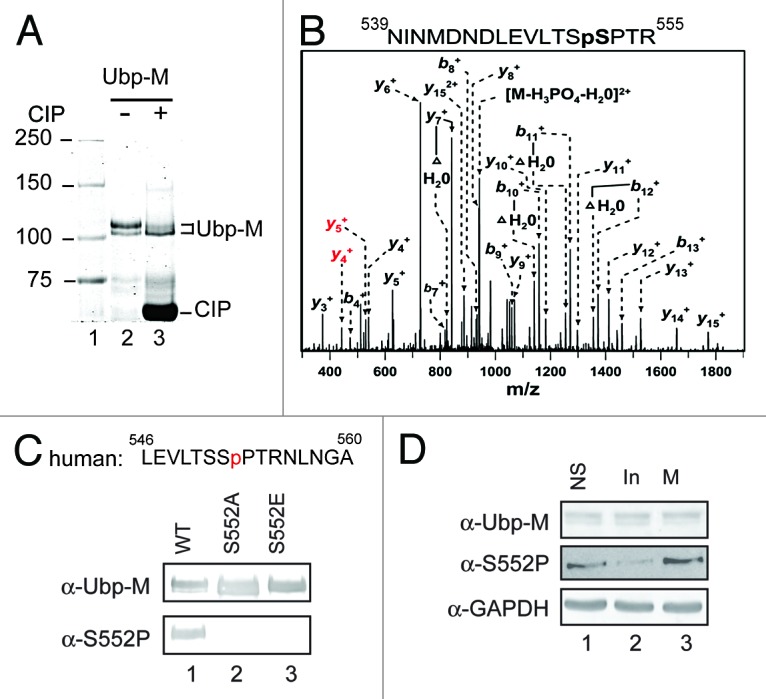
Ubp-M is phosphorylated at the serine 552 residue. (A) Ubp-M is phosphorylated in vivo. Coomassie blue staining of affinity purified Ubp-M from sf9 cells incubated with (lane 3) or without (lane 2) Calf intestinal alkaline phosphatase (CIP). CIP treatment decreases the intensity of the top band and increases the intensity of the bottom band. (B) The serine 552 residue of Ubp-M is phosphorylated. Mass spectrometry analysis indicated that Ubp-M is phosphorylation at serine 552. (C) Generation of the Ubp-M phosphorylated serine 552 (S552P) antibody. Top: peptide sequence used to generate the Ubp-M S552P antibody. Bottom: characterization of the Ubp-M S552P antibody. Antibodies used are labeled on the left side of the panel. (D) Ubp-M S552P occurs in cell cycle M phase. Western blot analysis of extracts from unsynchronized, interphase, and M phase cells. Antibodies used are labeled on the left side of the panels.
To determine the phosphorylation site(s), we analyzed Ubp-M using high-resolution mass spectrometry. Four phosphorylation sites were identified by accurate mass and LTQ collision-induced dissociation tandem mass spectrometry: serine 552, 530, 414, and 329 (Fig. 1B and data not shown). Both serine 552 and 414 phosphorylation were also reported in 2 recent studies.22,23 In this study, we specifically focused on Ubp-M serine 552 phosphorylation (S552P). To confirm the mass spectrometry results and investigate the function of Ubp-M S552P, we generated an antibody against phosphorylated serine 552 Ubp-M (Fig. 1C). Since Ubp-M S552 is not conserved in mice and rats (Fig. S1), we used a phosphorylated peptide corresponding to human Ubp-M for antibody production. Characterization of the antibody revealed that it specifically recognizes wild-type but not S552A and S552E mutants (Fig. 1C, bottom panels). In concordance with our CIP studies, this antibody specifically recognized the upper band of the Ubp-M doublet. After confirming the specificity of the antibody, we determined whether Ubp-M S552P is regulated in a cell cycle-dependent manner. As shown in Figure 1D, Ubp-M S552P is detected in unsynchronized cells and cells in M phase, but is less abundant in interphase cells. These studies suggest that S552P might regulate Ubp-M function during cell cycle.
Cyclin-dependent kinase 1 is responsible for Ubp-M S552P
Visual and computer-assisted inspection of the Ubp-M sequence revealed that the 552 serine residue is adjacent to a fully consensus SPTR CDK recognition motif (Fig. 2A, top panel). Since Ubp-M is phosphorylated in M phase, when CDK1-cyclin B kinase is active, we tested whether Ubp-M S552 is phosphorylated by CDK1–cyclin B kinase. For this purpose, we incubated full-length wild-type Ubp-M and the Ubp-M S552A and S552E mutants with purified CDK1/cyclin B kinase in the presence of γ-ATP. As shown in Figure S2, Ubp-M was phosphorylated by CDK1/cyclin B kinase; however, Ubp-M S552A and S552E were also phosphorylated to a similar level. This result indicates that Ubp-M contains multiple CDK1 phosphorylation sites. To overcome this problem, we repeated this assay using an Ubp-M fragment (529–599) that does not contain other potential CDK phosphorylation sites. As shown in Figure 2A, the wild-type Ubp-M fragment could be phosphorylated by CDK1, while the S552A and S552E mutant fragments were not (compare lanes 2 and 3 with 1). These data suggest that the 552 serine residue is a target site for CDK1/cycle B kinase in vitro.
Figure 2.
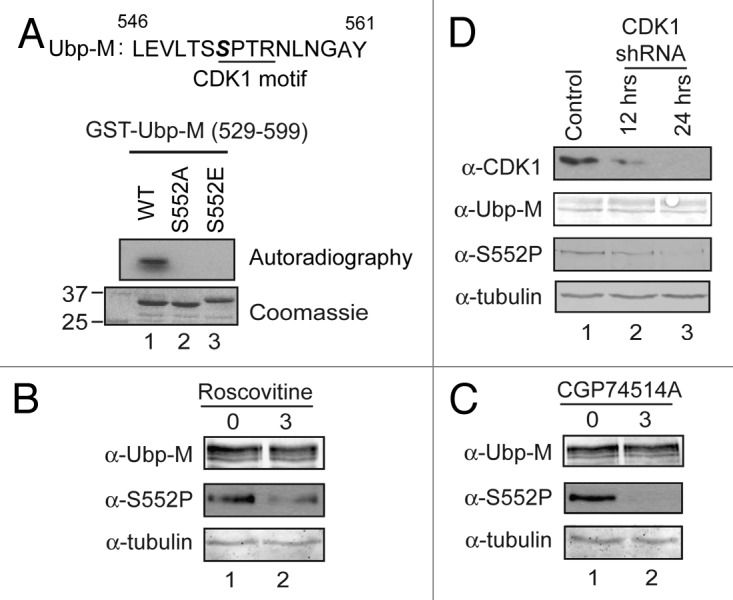
Cyclin-dependent kinase 1 is responsible for Ubp-M serine 552 phosphorylation. (A) Cyclin-dependent kinase 1 (CDK1) phosphorylates Ubp-M serine 552 in vitro. Top: peptide sequence flanking serine 552 contains a fully conserved CDK1 binding motif. Bottom: in vitro kinase assay using Ubp-M fragments (529–599) as substrates. (B) Roscovitine treatment reduces Ubp-M S552P (middle panel) without affecting total Ubp-M levels (top panel). (C) CGP74514A treatment reduces Ubp-M S552P (middle panel) without affecting total Ubp-M levels (top panel). (D) CDK1 knockdown abolishes Ubp-M S552P. Top panel indicates that the level of CDK1 is significantly reduced by shRNA treatment. Middle two panels indicate that knockdown of CDK1 reduces Ubp-M S552P levels (third panel) without affecting total Ubp-M levels (second panel).
To determine whether CDK1/cyclin B kinase phosphorylates Ubp-M in vivo, we first treated cells with roscovitine, a general inhibitor for CDK kinases. As shown in Figure 2B, roscovitine treatment substantially reduced the levels of Ubp-M S552P without affecting the total levels of Ubp-M (top 2 panels). To further confirm this result, we treated cells with the specific CDK1 inhibitor CGP74514A at 2 μM concentration. As shown in Figure 2C, CGP74514A treatment dramatically reduced Ubp-M S552P without affecting total Ubp-M levels (compare lane 2 with 1). These data suggest that Ubp-M S552P is likely mediated by the CDK1/cycle B kinase. To further confirm the role of CDK1 in Ubp-M S552P, we knocked down CDK1 kinase by specific shRNA and examined the effects on Ubp-M S552P. As shown in Figure 2D, compared with control shRNA knockdown, the levels of CDK1 were significantly reduced (top panel, compare lanes 2 and 3 with 1). Importantly, when CDK1 was knocked down, we observed a significant reduction of Ubp-M S552P without apparent effects on total Ubp-M levels (Fig. 2D, second and third panels; compare lanes 2 and 3 with 1). Together, these data reveal that CDK1 is responsible for Ubp-M S552P.
S552P is not required for Ubp-M deubiquitination activity in vitro and gene regulation in vivo
To determine how S552P regulates the function of Ubp-M, we purified wild-type Ubp-M and Ubp-M S552A and S552E mutants from sf9 cells. As shown in Figure 3A, compared with wild-type Ubp-M, the Ubp-M S552A mutation completely abolished the mobility shift, while the S552E mutation partially mimicked the mobility shift. These data suggest that Ubp-M S552 phosphorylation might account for at least some of the observed mobility shift of Ubp-M in SDS-PAGE gels.
Figure 3.

S552P does not affect Ubp-M deubiquitination activity in vitro or in vivo. (A) Purification of wild-type and mutant (S552A and S552E) Ubp-M. S552P accounts for the majority of the Ubp-M mobility shift observed by western blot. Mutation of serine 552 to alanine caused a significant reduction in mobility shifted Ubp-M, while mutation of serine 552 to glutamic acid partially mimics the phosphorylation-mediated mobility shift. (B) S552P does not affect Ubp-M tetramer formation. Superose 6 analysis revealed that wild-type and mutant (S552A and S552E) elute at a molecular weight of 443 kDa. (C) Ubp-M S552A and S552E mutations do not affect Ubp-M H2A deubiquitination activity in vitro. Histone H2A deubiquitination assay with mononuclosomes (second and third panels) and core histone (fourth panel) as substrates. Wild-type and mutant (S552A and S552E) Ubp-M display similar uH2A deubiquitination activity in vitro. The amount of Ubp-M used and the H2A levels were similar in core histone (20 min), and mononucleosome (10 min) reaction. His. indicates histone substrates and Nucl. indicates nucleosome substrates. (D) Ubp-M S552A and S552E mutations do not affect the substrate specificity of Ubp-M. Histone deubiquitination assay with uH2B-containing mononucleosomes and core histones as substrates. His. indicates histone substrates and Nucl. indicates nucleosome substrates. (E) Ubp-M S552P is not required for regulation of global H2A ubiquitination levels. The indicated constructs were transfected into HeLa cell lines stably expressing Flag-H2A and HA-ubiquitin and the effects on global H2A ubiquitination levels were analyzed by western blot assay. (F) Ubp-M S552 phosphorylation is not required for Ubp-M regulation of gene expression. The constructs indicated on top of the panel were transfected into control and Ubp-M knockdown cells and the effects on the expression of HoxD10 were analyzed by RT-PCR assay.
Our previous studies reveal that Ubp-M functions as a homotetramer.20 To determine whether S552P affects the formation of Ubp-M homotetramer, we measured the elution profile of wild-type and mutant (S552A and S552E) Ubp-M by gel filtration Superose 6 column. As shown in Figure 3B, wild-type Ubp-M was eluted at peak fraction 62, corresponding to a molecular weight of 443 kDa (top panel). S552A and S552E mutants were also eluted at peak fraction 62, equivalent to a molecular weight of 443 kDa (Fig. 3B, bottom 2 panels). These data suggest that S552P does not affect the homotetramer formation of Ubp-M.
To determine whether S552P affects Ubp-M’s H2A deubiquitination and substrate specificity, we first performed histone H2A deubiquitination assays with uH2A containing nucleosomes as substrates. As shown in Figure 3C, the Ubp-M S552A and S552E mutants deubiquitinated uH2A as efficiently as wild-type Ubp-M (second and third panels, compare lanes 3 and 4 with 1 and 2; see the time-dependent decrease of uH2A signals). These data suggest that S552P does not affect Ubp-M deubiquitination activity. To determine whether S552P affects the substrate specificity of Ubp-M, we performed in vitro deubiquitination assays with uH2A-containing core histones and uH2B-containing nucleosomes and core histones as substrates. As shown in Figure 3C (third panel) and Figure 3D (second and fourth panels), Ubp-M S552A and S552E mutants also do not exhibit activities toward these substrates (compare lanes 2–4 with 1 and 5). Together, these data suggest that S552P does not affect Ubp-M’s deubiquitination activity and substrate specificity.
To determine whether S552P regulates Ubp-M-mediated H2A deubiquitination activity in vivo, we transfected vectors encoding wild-type Ubp-M, Ubp-M S552A, or Ubp-M S552E into Hela cells stably expressing Flag-H2A and HA-ubiquitin. As shown in Figure 3E, overexpression of wild-type Ubp-M results in a significant decrease in the levels of H2A ubiquitination, as compared with cells transfected with control empty vectors (compare lane 2 with 1). Overexpression of Ubp-M S552A and S552E mutant reduced the levels of H2A ubiquitination to a similar extent (Fig. 3E, compare lanes 3 and 4 with 1 and 2). These data suggest that S552P is not required for Ubp-M-mediated global H2A deubiquitination in vivo.
In previous studies, we demonstrated that Ubp-M regulates both cell cycle progression and gene expression.20 To determine whether Ubp-M S552P regulates gene expression, we investigated whether Ubp-M S552A and S552E mutants could rescue HoxD10 gene expression in Ubp-M-knockdown cells. As shown in Figure 3F, wild-type Ubp-M was able to rescue the expression of the HoxD10 gene in knockdown cells, consistent with our previous studies (middle panel, compare lane 3 with 1 and 2). Interestingly, when expressed at similar levels (Fig. 3F, top panel, compare lanes 3–5 with 1), the expression of HoxD10 could also be rescued by S552A and S552E mutant Ubp-M to a similar extent (Fig. 3F, middle panel, compare 4 and 5 with 1–3). These studies suggest that S552P is not required for the regulation of HoxD10 gene expression. We next investigated whether Ubp-M S552P is required for the regulation of other gene expression. As shown in Figure S3, both S552A and S552E could rescue the expression of p19 and p16, similar to wild-type Ubp-M. Together, these studies reveal that Ubp-M S552P is not required for the regulation of gene expression.
Ubp-M S552P is required for cell proliferation and cell cycle progression
Since S552P occurs in cell cycle M phase, we investigated whether S552P regulates cell cycle progression. Consistent with previous observations, Ubp-M S552P occurs as cells enter G2/M phase and is correlated with H2A deubiquitination during cell cycle progression (Fig. 4A, lanes 5–7). FACS analysis also revealed that S552P is present in M phase cells (Fig. 4B). These data confirm that the S552P modification appears during M phase of the cell cycle.
Figure 4.

Ubp-M S552P regulates cell cycle G2/M phase progression. (A) Ubp-M S552P occurs in cell cycle M phase. Western blot assay of extracts from cells at the indicated time points following release from double thymidine block. Antibodies used are labeled in the left side of the panel. (B) Ubp-M S552P stains M phase cells. Hela cells were stained with S552P antibody and PI, and cell cycle was analyzed by FACS. (C) Characterization of Ubp-M stable knockdown cell lines expressing Flag tagged wild-type, S552A, or S552E Ubp-M. Antibodies used are labeled in the left side of the panel. (D) Growth curve of control, Ubp-M knockdown, and Ubp-M knockdown cells expressing wild-type or mutant (S552A and S552E) Ubp-M. Wild-type Ubp-M, but not the S552A and S552E mutants, rescued the growth defects in Ubp-M knockdown cells. (E) Expression of wild-type, but not mutant (S552A and S552E) mutant Ubp-M, rescues the decrease in G2/M phase cell population caused by Ubp-M knockdown.
To determine the functional significance of Ubp-M S552P, we investigated whether Ubp-M S552P affects cell proliferation. For this purpose, we established stable Ubp-M-knockdown cell lines also expressing wild-type, S552A, or S552E Ubp-M. As shown in Figure 4C, wild-type, S552A, and S552E Ubp-M were expressed at levels similar to endogenous Ubp-M (top panel, compare lanes 3–5 with 1). S552P is restored in cells expressing wild-type Ubp-M but not in Ubp-M S552A and S552E mutants (Fig. 4C, third panel, compare lanes 3–5 with 1 and 2). Proliferation rates of knockdown cells expressing wild-type Ubp-M was largely restored to control levels (Fig. 4D, blue line; Fig. S4 for other independent cell lines). The slight difference between the proliferation rate of these cells and control cells was possibly due to the presence of several antibiotics or the incorporation of Ubp-M at specific sites. In contrast, the growth of cells expressing S552A and S552E is similar to that of Ubp-M-knockdown cells (Fig. 4D; Fig. S4, compare red line with purple and tan lines). These data reveal that S552P specifically regulates cell proliferation.
In previous studies, we demonstrated that Ubp-M knockdown results in a significant decrease of the G2/M phase cell population. To determine whether the rescue of cell proliferation rate observed in knockdown cells expressing wild-type Ubp-M is related to changes in G2/M phase cell populations, we measured the H3Ser10P-positive cells in control, knockdown cells, and knockdown cells expressing different Ubp-M constructs. As shown in Figure 4E, expression of wild-type Ubp-M largely restored the G2/M cell numbers, but expression of S552A and S552E mutant did not (Fig. 4E). These data suggest that S552P regulates cell growth through regulation of cell cycle G2/M progression.
S552P regulates the interaction between Ubp-M and CRM1
Although Ubp-M is a histone H2A-specific deubiquitinase, it is predominately localized in the cytoplasm during cell cycle interphase (Fig. 5A, top panels). To determine the mechanism for this subcellular localization, we tested whether the cytoplasmic localization of Ubp-M was controlled by active nuclear export mechanisms. For this purpose, we treated cells with leptomycin B (LMB), an inhibitor of the nuclear export receptor CRM1.24 As shown in Figure 5A (top panels), without LMB treatment, Ubp-M was predominately localized to the cytoplasm during interphase. After LMB treatment, we found that a significant number of cells retained Ubp-M in the nucleus (Fig. 5A, bottom panels). However, LMB treatment has no effects on the nuclear localization of Ubp-M S552A and S552E mutant (Fig. 5B). In addition, overexpression of CRM1 caused a significant reduction of Ubp-M nuclear localization but had no effects on the nuclear localization of Ubp-M S552A and S552E mutants (Fig. 5C; Fig. S5). Together, these data reveal that Ubp-M is actively exported from the nucleus.
Figure 5.

Ubp-M S552P regulates the interaction between Ubp-M and CRM1. (A) Ubp-M subcellular localization is regulated by CRM1 mediated nuclear export. LMB treatment increased the nuclear localization of RFP-Ubp-M. Two hundred RFP-positive cells were examined and representative images shown. (B) Fold change in the number of cells with nuclear wild-type or mutant (S552A or S552E) RFP-Ubp-M in control and LMB treated cells. 200 RFP-positive cells were examined. The percentage of cells with nuclear wild-type, S552A or S552E RFP-Ubp-M without LMB treatment was set to 1. (C) Fold change of nuclear wild-type and mutant (S552A and S552E) RFP-Ubp-M in control and CRM1-overexpressing cells. 200 RFP-positive cells were examined. The percentage of nuclear wild-type and mutant (S552A and S552E) Ubp-M in control cells was set to 1. (D) Ubp-M S552P regulates the interaction between Ubp-M and CRM1. CRM1 was only detected in anti-Flag immunoprecipitates of wild-type, but not S552A and S552E mutant, Ubp-M. (E) CRM1 interacts with non-phosphorylated Ubp-M. CRM1 specifically pulled down non-phosphorylated Ubp-M but not phosphorylated Ubp-M, as confirmed by anti-S552P antibody. (F) A proposed model for the function of Ubp-M S552P. Ubp-M subcellular localization is determined by the balance between export and import processes. As cells prepare to enter M phase, cytoplasmic cycle B/CDK1 kinase is activated and phosphorylates Ubp-M S552. S552P disrupts Ubp-M interaction with CRM1, allowing Ubp-M to be retained in the nucleus and deubiquitinate nucleosomal uH2A, which is required for chromosome condensation and cell cycle progression.
To determine whether S552P affects Ubp-M interaction with the nuclear export receptor CRM1, we determined the interaction between Ubp-M and CRM1 by co-immunoprecipitation. As shown in Figure 5D, wild-type Ubp-M co-immunoprecipitated with CRM1. However, Ubp-M S552 mutations disrupt the interaction between Ubp-M and CRM1 (bottom panel), suggesting that Ubp-M interaction with CRM1 requires serine 552. The failure of both Ubp-M S552A and S552E mutants to interact with CRM1 suggests that both substantive amino acids may not be able to mimic in vivo Ubp-M phosphorylation. To further determine the effect of Ubp-M S552P on the interaction between CRM1 and Ubp-M, we performed reciprocal co-immunoprecipitation experiments. As shown in Figure 5E, CRM1 specifically pulled down the bottom band of Ubp-M doublet, corresponding to non-phosphoryated Ubp-M (middle panel, compare lane 2 with 3). Western blot with Ubp-M S552P antibody further confirmed the non-phosphorylated form of Ubp-M (Fig. 5E, bottom panel, compare lane 2 with 3). Based on these data, we propose a model for Ubp-M S552P function (Fig. 5F). In interphase cells, Ubp-M is predominately located in the cytoplasm, as determined by a balance between export and import processes. As cells prepare to enter M phase, CDK1 kinase is activated in the cytoplasm and phosphorylates Ubp-M S552. S552P disrupts Ubp-M interaction with CRM1, allowing Ubp-M to be retained in the nucleus and deubiquitinate nucleosomal uH2A, which is required for chromosome condensation and cell cycle progression.
Discussion
The onset of mitosis requires increased activity of CDK1 associated with cyclin A and cyclin B. The CDK1/cyclin B complex is the major regulator of the G2/M transition and has maximal activity during metaphase.25 Several studies based on large-scale screening methods and complementary computational approaches have provided lists of potential CDK targets in S. cerevisiase and, to a limited extent, in higher organisms.25,26 These substrates regulate a diverse spectrum of cellular processes including transcription, translation, chromatin remodeling, apoptosis, and telomere regulation. Recent studies indicate that CDK1 also targets epigenetic regulators for phosphorylation. EZH2 phosphorylation by CDK1 at different sites regulates its interaction with other PRC2 subunits and chromatin targeting, indicating a precise coordination of gene repression and cell proliferation.14-18 In this study we identified the H2A deubiquitinase Ubp-M as a substrate for CDK1, and, importantly, our studies revealed that phosphorylation of Ubp-M by CDK1 is specifically required for cell cycle progression. Mutation of S552 impairs cell cycle G2/M phase progression, indicating that Ubp-M is part of the coordinated, phosphorylation-mediated reorganization of cellular machineries required for cell cycle progression. Therefore, these studies identified Ubp-M as a novel and essential substrate for CDK1 during cell cycle progression.
Interestingly, S552 is not conserved during evolution and is present in primates but not in mice and rats (Fig. S1). Our unpublished results also reveal that Ubp-M knockout does not affect mouse embryonic stem cell cycle progression, suggesting that S552P regulation is limited to certain organisms. The shift of CDK1 phosphorylation sites during evolution is consistent with the notion that regulation of protein function by phosphorylation depends on simple nonspecific mechanism that disrupt and enhance protein–protein interactions.26 Specifically, S552P disrupts the interaction between Ubp-M and nuclear export protein CRM1, increasing Ubp-M nuclear retention as cells progress into M phase, revealing a critical function for Ubp-M S552P.
As a histone H2A-specific deubiquitinase, Ubp-M is primarily located in the cytoplasm during interphase (Fig. 5A). The separation of a deubiquitinase from its physiological substrates is likely a general mechanism for regulation of deubiquitinase function and might be vital for Ubp-M function.27 The cytoplasmic localization of Ubp-M is controlled by at least one active export mechanism. It should be noted that other mechanisms also contribute to Ubp-M subcellular localization, as even after prolonged LMB treatment, only a small fraction of Ubp-M is in the nucleus. In an independent experiment to define the subcellular localization of Ubp-M, we made serial N-terminal Ubp-M deletions. When these deletions were transfected into Cos-7 cells, multiple regions were found to play a role in determining Ubp-M subcellular localization (Fig. S6). S552P alone does not affect the subcellular localization of Ubp-M in cell cycle interphase (data not shown); however, when cells enter G2/M phase, CDK1-dependent Ubp-M phosphorylation indeed plays a role in cell cycle G2/M phase progression, as mutation of serine 552 to alanine delays cell entry into G2/M phase, as compared with wild-type Ubp-M (Fig. 4D and E). Therefore, this study specifically links Ubp-M S552P to cell cycle progression. Moreover, Ubp-M function might also be regulated by protein–protein interaction,28 and these interactions might be affected by phosphorylation
Interestingly, phosphorylation of Ubp-M by CDK1 is not required for transcriptional regulation of HoxD10, a gene previously shown to be regulated by Ubp-M20 and several other genes we have examined. Therefore, this study identified 2 independent functions of Ubp-M: gene regulation, which is not dependent on S552P, and cell cycle progression, which depends on S552P. Since Ubp-M is also phosphorylated at other sites by distinct kinases, phosphorylation at different sites might also be involved in priming Ubp-M for distinct functions. Identification and elucidation of other phosphorylation events will be needed to demonstrate how the cell cycle progression and gene expression regulatory functions of Ubp-M are coordinated.
Materials and Methods
Protein expression and purification
Baculovirus expressing wild-type Flag-tagged Ubp-M has been described.20 Flag-tagged mutant forms of Ubp-M (S552A and S552E) were constructed by PCR-based mutagenesis and confirmed by DNA sequencing. Wild-type and mutated Ubp-M were purified by anti-Flag M2 immunoprecipitation and gel filtration as described.20 Ubp-M fragments containing the phosphorylation site were generated by PCR and cloned into pGEX-KG with a GST tag at the N-terminus. GST fusion proteins were purified as described.29 A synthetic peptide corresponding to human Ubp-M amino acid 546–561 containing phosphorylated serine 552 residue was used for antibody production. Phosphorylation-specific antibody was purified by S552 phosphorylated peptides. For subcellular localization, Ubp-M and its mutant forms were cloned into the pEGFP-C1 vector (Clontech) by PCR, confirmed by sequencing, transfected into HeLa cells, and examined by microscopy.
Mass spectrometry analysis of Ubp-M phosphorylation
To identify the phosphorylation sites on Ubp-M, gel bands containing phosphorylated forms of Ubp-M were excised and subjected to mass spectrometry analysis by use of a high-resolution linear quadruple ion trap Fourier transform ion cyclotron resonance mass spectrometer (LTQ FT-ICR MS, ICR, Thermo Fisher Scientific). Briefly, slices were destained with 5:8 ratio of potassium ferricyanide:sodium thiosulfate, reduced with 10 mM DTT at 37 °C for 45 min, alkylated with 50 mM iodoacetamide at 37 °C for 45 min, and digested with trypsin overnight at 37 °C. Peptides were extracted from the gel using 50% acetonitrile and concentrated in a speed vacuum. Tryptic digests were loaded onto a 100 μm diameter, 11 cm pulled tip packed column with Jupiter 5 μm C18 reversed-phase beads (Phenomenex) using a Micro AS autosampler and LC nanopump (Eksigent). An acetonitrile gradient in 0.1% formic acid was run from 5% to 40% over 50 min at a flow rate of 650 nl/min. The eluting peptides were analyzed by CID fragmentation on a LTQ FT-ICR. The LTQ FT-ICR parameters were set as described previously.30 Fully tryptic human peptides were identified using TurboSEQUEST v.27 (rev.12, Thermo Fisher Scientific),31 MASCOT 2.2 (Matrix Biosciences),32 and Protein Prospector v.5.2.2 (University of California, San Francisco)33 algorithms with a parent ion mass accuracy of 10.0 ppm from the Uniref 100 database (06/2009) and the inclusion of phosphorylation as a possible variable modification. SEQUEST and MASCOT results were further refined through the trans-proteomic pipeline using the Peptide Prophet34 and Protein Prophet35 models. Phosphorylated peptide hits were manually sequenced to validate the sites of attachment.
Kinase assay, histone deubiquitination assay, and chemical treatment
Full-length and Ubp-M fragments were incubated with CDK1–cyclin B kinase (NEB, P6020) at 37° for 15 min. The reaction mixtures were then resolved by a 10% SDS-PAGE. After Coomassie blue staining and destaining, gels were dried and exposed to X-ray film. In vitro and in vivo histone deubiquitination reactions were performed as described.20,36 For dephosphorylation assays, purified Ubp-M (1 μg) was incubated with calf intestinal alkaline phosphatase (CIP, NEB) at 37 ° for 30 min, and the reaction mixture was then separated on a 8% SDS-PAGE gel and visualized by Coomassie blue staining. For kinase activity inhibition, cells were treated with 2 μM CGP74514A (Sigma, C3352) or 50 μM roscovitine (Sigma, R7772) for indicated time, and cells were then collected for western blot assay. For Ubp-M localization assay, cells were treated with 10 ng/ml leptomycin B (LMB, Sigma, L2913) for indicated times, and cells were then examined by microscopy. Two hundred RFP-positive cells were examined, and the percentage of cells with nuclear localization was calculated. For cell synchronization, HeLa cells were treated with thymidine (2.5 mM) for 18 h, released for 10 h, and then treated with thymidine for another 17 h. Cells were collected at different time points and used for western blot assay.
Stable cell line establishment and proliferation assay
HeLa cells were co-transfected with vectors expressing hairpin RNAs targeting Ubp-M20 and expression constructs for wild-type and mutant Ubp-M. Transfected cells were selected in the presence of 2 μg/ml puromycin and 4 μg/ml G418. Selected clones were amplified, and the efficiency of endogenous Ubp-M knockdown as well as the expression of epitope tagged proteins were analyzed by western blot.20 For proliferation assay, 5 × 104 cells were seeded in 6-well plates, and at different time points, viable cells were counted by trypan blue exclusion.
Lentivirus infection, transfection, RT-PCR, immunofluorescent, and co-immunoprecipitation assay
Lentivirus targeting CDK1 was packaged and used to infect HeLa cells as described.15 Transfection was performed with Effectene (Qiagen) following the manufacturer’s instructions. RT-PCR procedure and primers used were described in a previous publication.20 For quantitative PCR, 0.5 μl cDNA was used as template in a 25 μl reaction containing 1 μg Oligo dT 15 primers, 0.5 mM dNTPs, RNase inhibitor, and M-MLV and 5X M-MLV buffer. All quantitative PCR reactions were performed on a Roche LightCycler 480 thermocycler (Roche). Fold change of gene expression levels were calculated according to the 2-△△Ct method as described previously37 and normalized to GAPDH. Primers sequences for p16: F:5′-CGCACCGAAT AGTTACGGTC G-3′, R:5′-GTGCAGCACC ACCAGCGTGT C-3′; and for p19: F:5′-CTCAACCGCT TCGGCAAGAC-3′, R:5′-GCCATCAGGC ACGTTGACAT C-3′.
Fluorescence study was performed as described.38 For immunoprecipitation assay, cells were transfected with wild-type or mutant Ubp-M or Flag-CRM1, cross-linked with 1% formaldehyde, dissolved in denature buffer by sonication, and subjected to anti-Flag immunoprecipitation.
Supplementary Material
Acknowledgment
We thank Dr. Mien-Chie Hung for control and shRNA against CDK1, Dr. Mary Dasso and Ms Maia Ouspenskaia for CRM1 construct, Drs Peter H King and Natalia Kedishvile for microscopy, and UAB Rheumatic Diseases Core Center (RDCC) Analytic and Preparative Cytometry Facility (APCF) for FACS analysis. We thank Ms Amanda Jones for critical reading of the manuscript. HW is a Leukemia and Lymphoma Scholar and is supported by NIH grant (GM081489). UAB RDCC APCF is supported by NIH grant (P30 AR48311).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/26278
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26278
References
- 1.Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–94. doi: 10.1016/S0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- 2.Kornberg RD. The molecular basis of eukaryotic transcription. Proc Natl Acad Sci U S A. 2007;104:12955–61. doi: 10.1073/pnas.0704138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–12. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 4.Campos EI, Reinberg D. Histones: annotating chromatin. Annu Rev Genet. 2009;43:559–99. doi: 10.1146/annurev.genet.032608.103928. [DOI] [PubMed] [Google Scholar]
- 5.Hargreaves DC, Crabtree GR. ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 2011;21:396–420. doi: 10.1038/cr.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vignali M, Hassan AH, Neely KE, Workman JL. ATP-dependent chromatin-remodeling complexes. Mol Cell Biol. 2000;20:1899–910. doi: 10.1128/MCB.20.6.1899-1910.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 8.Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem. 2006;75:243–69. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- 9.Suganuma T, Workman JL. Signals and combinatorial functions of histone modifications. Annu Rev Biochem. 2011;80:473–99. doi: 10.1146/annurev-biochem-061809-175347. [DOI] [PubMed] [Google Scholar]
- 10.Suganuma T, Workman JL. Chromatin and signaling. Curr Opin Cell Biol. 2013;25:322–6. doi: 10.1016/j.ceb.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 11.Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science. 2010;330:612–6. doi: 10.1126/science.1191078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arzate-Mejía RG, Valle-García D, Recillas-Targa F. Signaling epigenetics: novel insights on cell signaling and epigenetic regulation. IUBMB Life. 2011;63:881–95. doi: 10.1002/iub.557. [DOI] [PubMed] [Google Scholar]
- 13.Mohammad HP, Baylin SB. Linking cell signaling and the epigenetic machinery. Nat Biotechnol. 2010;28:1033–8. doi: 10.1038/nbt1010-1033. [DOI] [PubMed] [Google Scholar]
- 14.Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, Ping B, Otte AP, Hung MC. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science. 2005;310:306–10. doi: 10.1126/science.1118947. [DOI] [PubMed] [Google Scholar]
- 15.Wei Y, Chen YH, Li LY, Lang J, Yeh SP, Shi B, Yang CC, Yang JY, Lin CY, Lai CC, et al. CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nat Cell Biol. 2011;13:87–94. doi: 10.1038/ncb2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen S, Bohrer LR, Rai AN, Pan Y, Gan L, Zhou X, Bagchi A, Simon JA, Huang H. Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2. Nat Cell Biol. 2010;12:1108–14. doi: 10.1038/ncb2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaneko S, Li G, Son J, Xu CF, Margueron R, Neubert TA, Reinberg D. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev. 2010;24:2615–20. doi: 10.1101/gad.1983810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu SC, Zhang Y. Cyclin-dependent kinase 1 (CDK1)-mediated phosphorylation of enhancer of zeste 2 (Ezh2) regulates its stability. J Biol Chem. 2011;286:28511–9. doi: 10.1074/jbc.M111.240515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elderkin S, Maertens GN, Endoh M, Mallery DL, Morrice N, Koseki H, Peters G, Brockdorff N, Hiom K. A phosphorylated form of Mel-18 targets the Ring1B histone H2A ubiquitin ligase to chromatin. Mol Cell. 2007;28:107–20. doi: 10.1016/j.molcel.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 20.Joo HY, Zhai L, Yang C, Nie S, Erdjument-Bromage H, Tempst P, Chang C, Wang H. Regulation of cell cycle progression and gene expression by H2A deubiquitination. Nature. 2007;449:1068–72. doi: 10.1038/nature06256. [DOI] [PubMed] [Google Scholar]
- 21.Cai SY, Babbitt RW, Marchesi VT. A mutant deubiquitinating enzyme (Ubp-M) associates with mitotic chromosomes and blocks cell division. Proc Natl Acad Sci U S A. 1999;96:2828–33. doi: 10.1073/pnas.96.6.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dephoure N, Zhou C, Villén J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A. 2008;105:10762–7. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beausoleil SA, Jedrychowski M, Schwartz D, Elias JE, Villén J, Li J, Cohn MA, Cantley LC, Gygi SP. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc Natl Acad Sci U S A. 2004;101:12130–5. doi: 10.1073/pnas.0404720101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gademann K. Controlling protein transport by small molecules. Curr Drug Targets. 2011;12:1574–80. doi: 10.2174/138945011798109446. [DOI] [PubMed] [Google Scholar]
- 25.Errico A, Deshmukh K, Tanaka Y, Pozniakovsky A, Hunt T. Identification of substrates for cyclin dependent kinases. Adv Enzyme Regul. 2010;50:375–99. doi: 10.1016/j.advenzreg.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 26.Holt LJ, Tuch BB, Villén J, Johnson AD, Gygi SP, Morgan DO. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science. 2009;325:1682–6. doi: 10.1126/science.1172867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem. 2009;78:363–97. doi: 10.1146/annurev.biochem.78.082307.091526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, Zhang Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004;431:873–8. doi: 10.1038/nature02985. [DOI] [PubMed] [Google Scholar]
- 30.Renfrow MB, Mackay CL, Chalmers MJ, Julian BA, Mestecky J, Kilian M, Poulsen K, Emmett MR, Marshall AG, Novak J. Analysis of O-glycan heterogeneity in IgA1 myeloma proteins by Fourier transform ion cyclotron resonance mass spectrometry: implications for IgA nephropathy. Anal Bioanal Chem. 2007;389:1397–407. doi: 10.1007/s00216-007-1500-z. [DOI] [PubMed] [Google Scholar]
- 31.Yates JR, 3rd, Eng JK, McCormack AL, Schieltz D. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal Chem. 1995;67:1426–36. doi: 10.1021/ac00104a020. [DOI] [PubMed] [Google Scholar]
- 32.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–67. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 33.Clauser KR, Baker P, Burlingame AL. Role of accurate mass measurement (+/- 10 ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal Chem. 1999;71:2871–82. doi: 10.1021/ac9810516. [DOI] [PubMed] [Google Scholar]
- 34.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–92. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 35.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–58. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 36.Joo HY, Jones A, Yang C, Zhai L, Smith AD, 4th, Zhang Z, Chandrasekharan MB, Sun ZW, Renfrow MB, Wang Y, et al. Regulation of histone H2A and H2B deubiquitination and Xenopus development by USP12 and USP46. J Biol Chem. 2011;286:7190–201. doi: 10.1074/jbc.M110.158311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 38.Yu JH, Lin BY, Deng W, Broker TR, Chow LT. Mitogen-activated protein kinases activate the nuclear localization sequence of human papillomavirus type 11 E1 DNA helicase to promote efficient nuclear import. J Virol. 2007;81:5066–78. doi: 10.1128/JVI.02480-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.