

The Ras-related C3 botulinum toxin substrate 1 (Rac1) belongs to the Ras-homologous (Rho) family of small GTPases, which transduce signals from the outside to the inside of a cell. Rac1 becomes activated upon ligand binding of a variety of receptors, including receptor tyrosine kinases and heterotrimeric G-protein-coupled receptors. After GTP loading by guanine exchange factors (GEFs), GTP-bound Rac1 engages numerous effector proteins, thereby eventually regulating cell motility and adhesion, cell cycle progression through G1, mitosis and meiosis, as well as cell death and metastasis.1 Besides, Rac1 adjusts cellular responses to genotoxic agents, such as UV light and alkylating agents, by rapid activation of stress kinases, including stress-activated protein kinases (SAPK/JNK) and p38 kinase, as well as transcription factors (e.g., AP-1, NFκB).
Apart from regulating stress responses originating from the outer membrane, Rac1 may have additional nuclear functions. This assumption rests on the fact that Rac1 is not only present in the cytoplasm, but also in the nucleus, where it interacts with topoisomerase II (topo II),2 which is involved in the regulation of replication and transcription. Topo II isoforms are the major target of potent and widely used anticancer drugs, such as anthracyclines (e.g., doxorubicin) and podophyllotoxins (e.g., etoposide). Binding of these drugs to topo II causes inhibition of its DNA religation function, resulting in the formation of DNA double-strand breaks (DSBs). Pharmacological targeting of Rac1 by both HMG-CoA-reductase inhibitors (statins), small-molecule inhibitors, or clostridial toxins mitigates the DNA damage response (DDR) induced by topo II poisons.3 As the DDR is decision-making regarding activation of checkpoint control mechanisms, DNA repair, and/or apoptosis,4 it is conceivable that Rac1 holds a so-far-unappreciated nuclear function in response to genomic DNA damage. Since the Ras-homologous GTPase RhoA is also present in the nucleus, where it can be activated by ionizing radiation, it is possible that RhoA and Rac1 are integrated into the complex network of cellular responses to at least a subset of genotoxins by affecting both DNA damage-independent and DNA damage-dependent stress responses.
Inhibition of Rac1 is a major mechanism of the well-known beneficial effect of statins in cardiovascular disease. Notably, statins also protect from cardiotoxicity evoked by anthracyclines in therapeutic relevant settings in vivo.5,6 Using Cre-based cardiac-specific knockout, inhibition of topo IIβ was recently suggested as the major cause of cardiotoxicity following anthracycline treatment.7 Hence, it is tempting to speculate that cardioprotection by statins rests on the inhibition of the Rac1-topo IIβ axis. This hypothesis, however, remains to be scrutinized by the analysis of cardiomyocyte-specific genetic knockouts of rac1. Analysis of the anthracycline response of mice lacking hepatic rac1 points to a double-edged function of Rac1 in acute and subacute settings.8 While rac1-deficient hepatocytes were protected from DNA damage induction and acute inflammatory and fibrotic responses following doxorubicin treatment, lack of rac1 promoted the subacute hepatotoxicity. Apparently, the influence of Rac1 signaling on the outcome of anthracycline exposure changes with time, illustrating the complexity of Rac1-regulated functions. Rac1 signaling enhances the interaction of topo II with its respective poisons, putatively by regulating the activity of protein kinases that phosphorylate topo II. In consequence, DSBs are formed, leading to the activation of the DDR. Rac1 also regulates inflammatory and fibrotic mechanisms by controlling the mRNA expression of cytokines, likely by regulating the activity of relevant transcription factors (e.g., AP-1, NFκB, STAT, Smad). Correspondingly, pharmacological or genetic abrogation of Rac1-regulated mechanisms disrupts stress responses stimulated by topo II poisons, resulting in beneficial effects in the short-term. On the other hand, Rac1 is required to alleviate the delayed harmful consequences of doxorubicin-induced injury by mitigating late inflammatory and fibrotic effects that emanate from anthracycline-induced cytotoxicity.
The mechanisms underlying the dual biological activity of Rac1 in the context of anthracycline-induced stress responses, which comprise both beneficial and adverse effects, are still obscure. Furthermore, the influence of Rac1 on genotoxin-induced response seems to be agent-specific, as DNA damage and DDR evoked by ionizing radiation are not affected by Rac1.5,8 Apart from anticancer therapy-related aspects, there is another highly important issue that remains to be addressed. In vitro and in vivo data have demonstrated a key role of Rac1 in epidermal to mesenchymal transition (EMT) as well as Ras-mediated lung carcinogenesis. Whether Rac1 affects the acute and subacute response to chemical mutagens and, most importantly, also impacts chemical carcinogenesis is unknown. Thus, it would be of interest to elucidate, whether the mechanisms involved in tumor initiation and promotion are differently affected by Rac1 signaling. Moreover, Rac1 seems to interfere with aging as it regulates target of rapamycin (TOR), insulin secretion, and signaling, and also suppresses the expression of the senescence-associated cyclin-dependent kinase inhibitor p16.8 Taken together, targeting of Rac1 is proposed as a promising approach to interfere with manifold pathophysiological stress responses that favor DDR, inflammation, carcinogenesis, cell death, and aging. (Fig. 1)
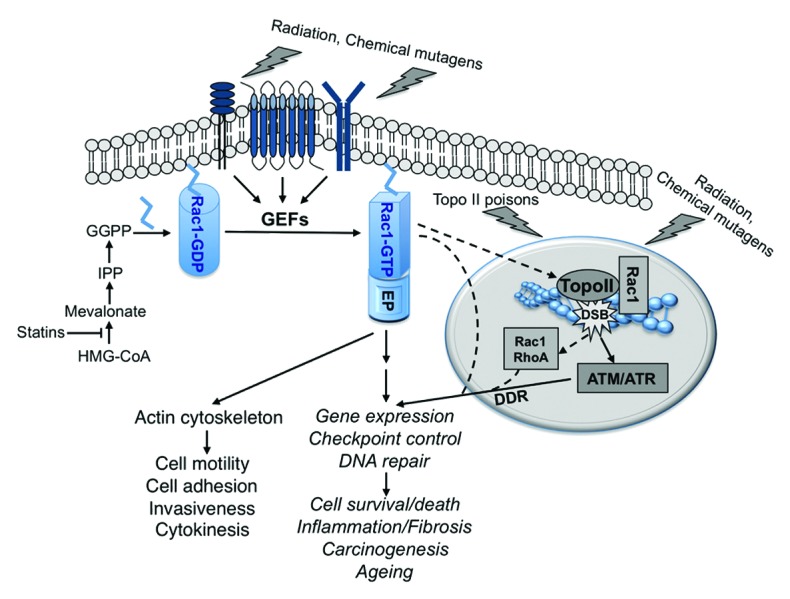
Figure 1.Model of Rac1-regulated response to genotoxic stress. Exposure to genotoxic agents rapidly stimulates membrane-bound receptors. In turn, guanine exchange factors (GEFs) for Rac1 become activated. GTP-bound active Rac1 couples to different effector proteins (EP), eventually leading to pleiotropic biological effects, which are either closely related to the actin cytoskeleton, or are independent of it. Genotoxins also damage DNA, thereby triggering the DNA damage response (DDR). Rac1-regulated mechanisms are required to provoke the DDR following treatment with topoisomerase II (topo II) poisons. Membrane-bound Rac1 might be involved in this process. Bearing in mind that Rac1 is present in the nucleus associated with topo II, it is also conceivable that a so-far-unknown nuclear function of Rac1 affects the DDR. We hypothesize that Rac1, together with RhoA, which was also found in the nucleus, ameliorates early cellular responses to genotoxins resulting from damage to membrane and nuclear structures. Yet, while transducing signals originating from both membrane structures and damaged DNA, Rac1 appears to differently influence early and delayed responses to genotoxins. Nevertheless, targeting of Rac1 signaling is suggested as a promising strategy to interfere with the complex stress responses that are evoked by DNA damaging anticancer drugs and chemical mutagens.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25807
References
- 1.Hall A. The cytoskeleton and cancer. Cancer Metastasis Rev. 2009;28:5–14. doi: 10.1007/s10555-008-9166-3. [DOI] [PubMed] [Google Scholar]
- 2.Sandrock K, Bielek H, Schradi K, Schmidt G, Klugbauer N. The nuclear import of the small GTPase Rac1 is mediated by the direct interaction with karyopherin alpha2. Traffic. 2010;11:198–209. doi: 10.1111/j.1600-0854.2009.01015.x. [DOI] [PubMed] [Google Scholar]
- 3.Huelsenbeck SC, Schorr A, Roos WP, Huelsenbeck J, Henninger C, Kaina B, et al. Rac1 protein signaling is required for DNA damage response stimulated by topoisomerase II poisons. J Biol Chem. 2012;287:38590–9. doi: 10.1074/jbc.M112.377903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–45. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 5.Huelsenbeck J, Henninger C, Schad A, Lackner KJ, Kaina B, Fritz G. Inhibition of Rac1 signaling by lovastatin protects against anthracycline-induced cardiac toxicity. Cell Death Dis. 2011;2:e190. doi: 10.1038/cddis.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma J, Wang Y, Zheng D, Wei M, Xu H, Peng T. Rac1 signalling mediates doxorubicin-induced cardiotoxicity through both reactive oxygen species-dependent and -independent pathways. Cardiovasc Res. 2013;97:77–87. doi: 10.1093/cvr/cvs309. [DOI] [PubMed] [Google Scholar]
- 7.Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012;18:1639–42. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]
- 8.Bopp A, Wartlick F, Henninger C, Kaina B, Fritz G. Rac1 modulates acute and subacute genotoxin-induced hepatic stress responses, fibrosis and liver aging. Cell Death Dis. 2013;4:e558. doi: 10.1038/cddis.2013.57. [DOI] [PMC free article] [PubMed] [Google Scholar]