

Abstract
Here, we developed a model system to evaluate the metabolic effects of oncogene(s) on the host microenvironment. A matched set of “normal” and oncogenically transformed epithelial cell lines were co-cultured with human fibroblasts, to determine the “bystander” effects of oncogenes on stromal cells. ROS production and glucose uptake were measured by FACS analysis. In addition, expression of a panel of metabolic protein biomarkers (Caveolin-1, MCT1, and MCT4) was analyzed in parallel. Interestingly, oncogene activation in cancer cells was sufficient to induce the metabolic reprogramming of cancer-associated fibroblasts toward glycolysis, via oxidative stress. Evidence for “metabolic symbiosis” between oxidative cancer cells and glycolytic fibroblasts was provided by MCT1/4 immunostaining. As such, oncogenes drive the establishment of a stromal-epithelial “lactate-shuttle”, to fuel the anabolic growth of cancer cells. Similar results were obtained with two divergent oncogenes (RAS and NFκB), indicating that ROS production and inflammation metabolically converge on the tumor stroma, driving glycolysis and upregulation of MCT4. These findings make stromal MCT4 an attractive target for new drug discovery, as MCT4 is a shared endpoint for the metabolic effects of many oncogenic stimuli. Thus, diverse oncogenes stimulate a common metabolic response in the tumor stroma. Conversely, we also show that fibroblasts protect cancer cells against oncogenic stress and senescence by reducing ROS production in tumor cells. Ras-transformed cells were also able to metabolically reprogram normal adjacent epithelia, indicating that cancer cells can use either fibroblasts or epithelial cells as “partners” for metabolic symbiosis. The antioxidant N-acetyl-cysteine (NAC) selectively halted mitochondrial biogenesis in Ras-transformed cells, but not in normal epithelia. NAC also blocked stromal induction of MCT4, indicating that NAC effectively functions as an “MCT4 inhibitor”. Taken together, our data provide new strategies for achieving more effective anticancer therapy. We conclude that oncogenes enable cancer cells to behave as selfish “metabolic parasites”, like foreign organisms (bacteria, fungi, viruses). Thus, we should consider treating cancer like an infectious disease, with new classes of metabolically targeted “antibiotics” to selectively starve cancer cells. Our results provide new support for the “seed and soil” hypothesis, which was first proposed in 1889 by the English surgeon, Stephen Paget.
Keywords: oncogene, oxidative stress, glycolysis, reverse Warburg effect, RAS, inflammation, NFkB, cancer associated fibroblast, tumor microenvironment, HaCaT, MCT1, MCT4, caveolin-1, TOMM20, mitochondrial metabolism, wound healing, response to injury, field cancerization, metabolic parasite, autophagy, senescence, oncogenic stress, stromal biomarkers
Introduction
Previous studies have shown that mutational activation and/or overexpression of diverse oncogenes is indeed sufficient to confer cell transformation and to drive tumor growth and metastasis in whole-animal models.1,2 This has led to the over-simplified notion that cancer is a cell-autonomous disease. However, this view is inherently incomplete, as it does not explain the critical role of the tumor microenvironment or the host organism in tumor initiation, progression, and metastasis.3-7 As such, a more global or metabolic view of cancer will be necessary for us to begin to design and implement new more effective anticancer therapies.8-11
Also, little is known regarding the process of tumor cell engraftment. We believe that understanding the metabolic requirements for tumor cell engraftment should allow us to develop new strategies for cancer prevention and therapy. Tumor cell engraftment most likely requires that the cancer cells gain the ability to metabolically reprogram their microenvironment, essentially a “nesting” type of cell behavior. In support of this notion, many human epithelial tumor types consist of “cancer cell nests”, surrounded by cancer-associated fibroblasts, which were originally thought to play mainly a structural role.
However, more recent studies indicate that epithelial cancer cells also derive many of their nutrients directly from cancer-associated fibroblasts and other stromal cells, such as adipocytes.12-54 This would be especially critical during the early phases of tumor initiation and the later stages of metastasis, when a neo-angiogenic blood supply is clearly lacking. We have termed this type of symbiotic relationship between cancer cells and stromal cells “two-compartment tumor metabolism.”31,33,40,42-45 In this scenario, glycolytic fibroblasts directly provide mitochondrial fuels (such as L-lactate and ketone bodies) for oxidative cancer cells to burn via the TCA cycle and OXPHOS. This energy transfer scheme also requires the establishment of a stromal-epithelial “lactate shuttle”, for the efficient transfer of these high-energy nutrients from fibroblasts to cancer cells. To accomplish this metabolic coupling, fibroblasts export L-lactate and ketone bodies via MCT4 transporters.55,56 In turn, epithelial cancer cells import and recycle these fuels via the MCT1 transporter to produce ATP efficiently.
So, how do we integrate the “oncogene theory” of cancer with this new form of cancer metabolism or “metabolic symbiosis”? One simple idea is that oncogenes give cancer cells the ability to manipulate and/or sculpt their microenvironment for their own personal benefit at a significant cost to the host organism.31 This would also mechanistically explain tumor cell engraftment. Thus, oncogenes may give predatory cancer cells the ability to function as “metabolic parasites”. This would mean that oncogenes in epithelial cancer cells must also have long-distance or bystander effects on the tumor microenvironment in order for cancer cells to make a proper nest.
Here, we have begun to test this new hypothesis using a matched set of 3 well-defined epithelial cell lines.57 The parental cell line (HaCaT cells) is immortalized, but not transformed, and does not form tumors in nude mice, so it is incapable of cancer cell engraftment.58 The two other HaCaT cells lines we used harbor activated oncogenes (H-Ras [G12V] or NFkB [p65]), are transformed, and form tumors in nude mice, so they efficiently undergo cancer cell engraftment. Interestingly, we show that only HaCaT cells that harbor activated oncogenes gain the ability to glycolytically reprogram the tumor microenvironment, via the induction of oxidative stress in cancer-associated fibroblasts. Thus, it appears that oncogenes also act at a distance, via ROS production and inflammation, to induce metabolic symbiosis between cancer cells and the tumor stroma. As a consequence, diverse oncogenes (Ras and NFkB) act via a common convergent mechanism (oxidative stress) to upregulate MCT4 in the tumor stroma. As MCT4 controls the “fuel supply” for cancer cells, this makes MCT4 an extremely attractive druggable target for new therapeutic interventions aimed at “starving” oncogenically activated tumor cells. As such, diverse oncogenes stimulate a common metabolic response to “injury” in the tumor microenvironment. This idea is consistent with the hypothesis that cancer behaves as a wound that does not heal.
N-acetyl-cysteine (NAC) is among the smallest FDA-approved drug molecules currently in use, and it has both antioxidant and anti-inflammatory properties. Surprisingly, however, it is not used clinically for the treatment of human cancers. As we show here that NAC inhibits the induction of stromal MCT4 by preventing oxidative stress, this may also explain why NAC so effectively prevents tumor initiation, progression, and metastasis, in numerous pre-clinical animal models.59-70 As such, new clinical trials with NAC should be considered, to alleviate oxidative stress and inflammation in the tumor microenvironment. In addition, since NAC effectively functions as an “MCT4 inhibitor”, the development of more targeted and selective MCT4 inhibitors may be warranted.
Results
HaCaT cells: An experimental model for understanding the requirements of tumor cell engraftment
HaCaT cells are considered to be a relatively “normal”, non-transformed, spontaneously immortalized human epithelial keratinocyte cell line.58 In accordance with this idea, they fail to form tumors in immunodeficient mouse animal models.58 Thus, they are an ideal cell line to study the factors that are required for successful tumor cell engraftment. Indeed, several laboratories have now shown that the recombinant expression of activated oncogenes in HaCaT cells is sufficient to confer effective cell transformation and tumor formation in nude mice. In this regard, expression of either activated H-Ras (G12V) or the p65 subunit of NFkB in HaCaT cells greatly facilitates successful tumor cell engraftment.57
Thus, this isogenic matched series of HaCaT cell lines (control, H-Ras [G12V], and NFkB [p65]) provides a unique, well-characterized model system to begin to dissect the possible metabolic requirements for tumor formation and efficient epithelial tumor cell engraftment (Fig. 1).
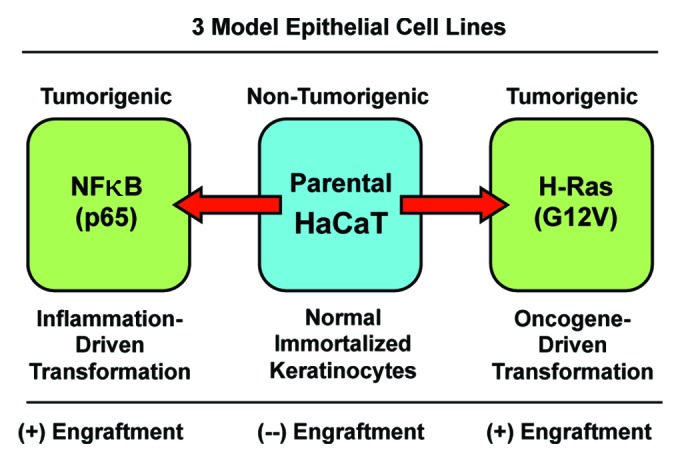
Figure 1. The HaCaT cell system: Immortalized and transformed with oncogenes. HaCaT cells are immortalized epidermal keratinocytes, but they are not transformed, and they do not form tumors in immunodeficient mice. However, overexpression of activated H-Ras (G12V) or NFkB (p65 subunit) drives cell transformation, and confers the capacity for tumor cell engraftment, leading to tumor formation in nude mice. Thus, phenotypic comparisons of these 3 matched cell lines should allow one to better understand the metabolic requirements for successful tumor cell engraftment within the naïve host microenvironment.
For this purpose, we co-cultured HaCaT cells with hTERT-immortalized fibroblasts, to mimic and monitor their reciprocal interactions with the host tumor microenvironment.
Ras activation and inflammation in epithelial cancer cells metabolically alters the tumor microenvironment, driving stromal oxidative stress and glycolysis
To model and dissect the metabolic basis of tumor-stromal interactions, epithelial HaCaT cells (control, H-Ras [G12V], and NFkB [p65]) were co-cultured with immortalized fibroblasts and then subjected to FACS analysis with a series of fluorescent metabolic probes. These small reporter molecules allowed us to quantitatively monitor ROS production (a measure of oxidative stress) and glucose uptake (a measure of glycolysis or glycolytic power) in a compartment-specific fashion. Thus, we compared ROS production and glucose uptake in epithelial cancer cells and adjacent normal fibroblasts to determine how epithelial oncogene activation metabolically reprograms the tumor stromal microenvironment.
Figure 2A and B show the status of these metabolic parameters in HaCaT cells co-cultured with fibroblasts. Interestingly, Ras activation leads to a 1.5-fold increase in ROS production, as expected based on the literature. However, NFkB activation did not increase ROS production significantly. Moreover, oncogene-activation (Ras or NFkB) did not result in increased glucose uptake. Thus, the metabolic effects of oncogene activation in epithelial cancer cells appeared to be relatively minimal.
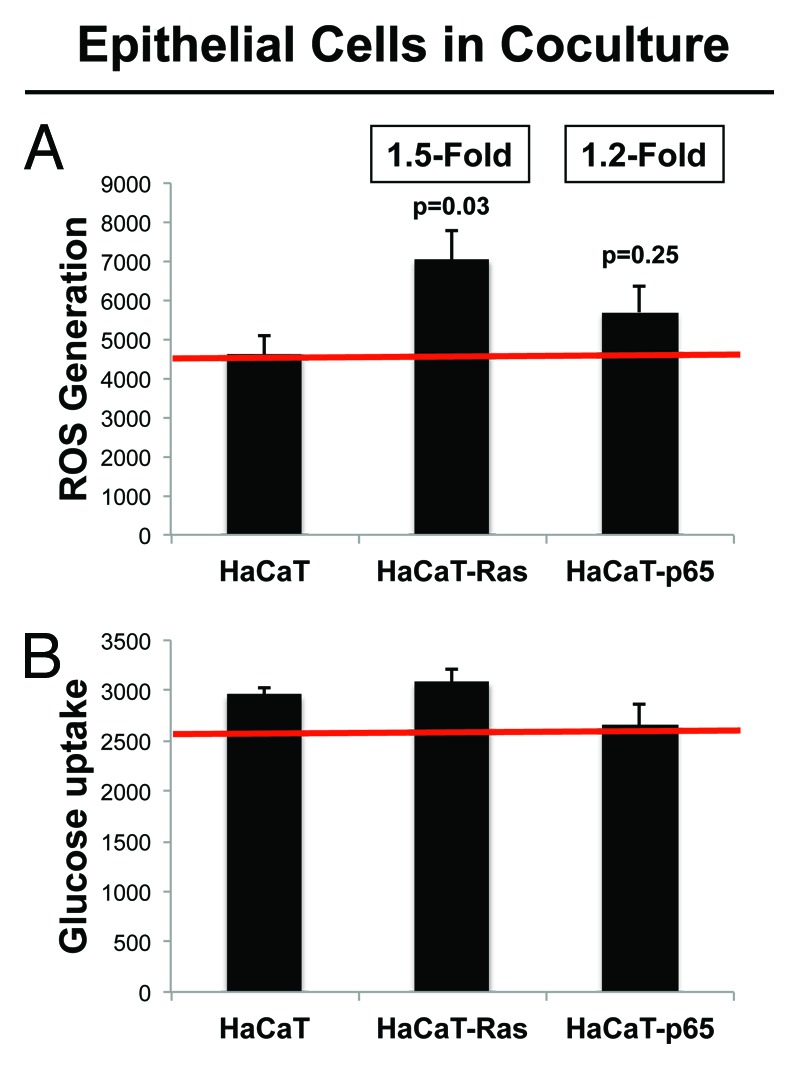
Figure 2. HaCaT-fibroblast co-cultures: Transformed HaCaT cells show a modest increase in ROS production, without any significant increases in glucose uptake. (A) ROS-production. (B) Glucose uptake. HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were co-cultured for 4 days with hTERT-immortalized fibroblasts (RFP[+]). Then, ROS production (a measure of oxidative stress) and glucose uptake (a measure of glycolytic activity) in HaCaT cells were quantitatively determined by FACS sorting. Note that only HaCaT-Ras cells show a significant increase in ROS production (1.5-fold; P = 0.03), without any detectable increases in glucose uptake.
Next, we examined ROS production and glucose uptake in the population of hTERT fibroblasts co-cultured with HaCaT cells (Fig. 3A and B). Large metabolic changes were observed in both ROS production and glucose uptake. More specifically, hTERT-fibroblasts co-cultured with HaCaT-Ras cells showed a significant increase in both ROS production (2.5-fold; P = 0.008) and glucose uptake (2.2-fold; P = 0.03). Similarly, hTERT-fibroblasts co-cultured with HaCaT-p65 cells showed a significant increase in ROS production (1.9-fold; P = 0.01) and glucose uptake (1.7-fold; P = 0.02). Thus, oncogene-transformed epithelial cancer cells metabolically reprogram adjacent normal fibroblasts.
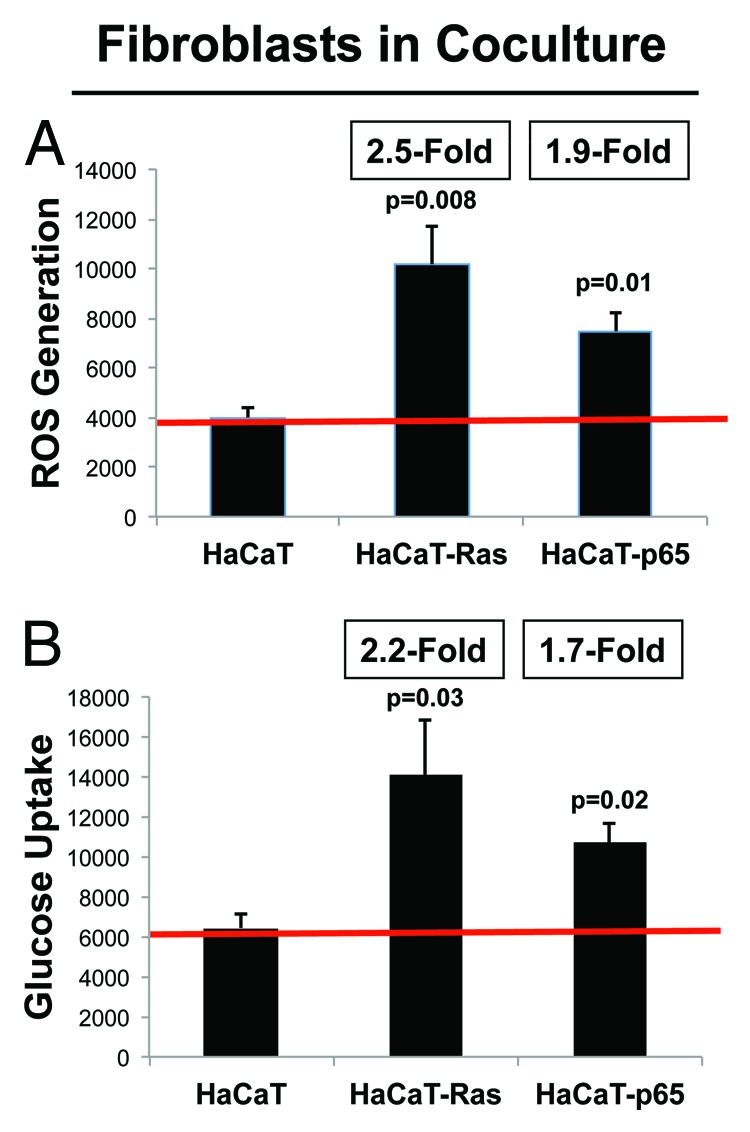
Figure 3. HaCaT-fibroblast co-cultures: Cancer-associated fibroblasts show dramatic increases in both ROS production and glucose uptake. (A) ROS-production. (B) Glucose uptake. HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were co-cultured for 4 days with hTERT-immortalized fibroblasts (RFP[+]). Then, ROS production (a measure of oxidative stress) and glucose uptake (a measure of glycolytic activity) in hTERT-fibroblasts were quantitatively determined by FACS sorting. Note that hTERT-fibroblasts co-cultured with HaCaT-Ras cells show a significant increase in both ROS production (2.5-fold; P = 0.008) and glucose uptake (2.2-fold; P = 0.03). Similarly, hTERT-fibroblasts co-cultured with HaCaT-p65 cells show a significant increase in ROS production (1.9-fold; P = 0.01) and glucose uptake (1.7-fold; P = 0.02). Thus, oncogene-transformed epithelial cancer cells metabolically reprogram adjacent normal fibroblasts.
Cancer-associated fibroblasts produce more ROS and are more glycolytic, as directly compared with epithelial cancer cells
Figure 4 shows the absolute magnitude of ROS production and glucose uptake in epithelial cancer cells and fibroblasts. This direct, side-by-side comparison allows one to appreciate that although the epithelial cancer cells harbor the activated oncogenes, their largest effects on cellular metabolism are actually occurring in neighboring normal fibroblasts.
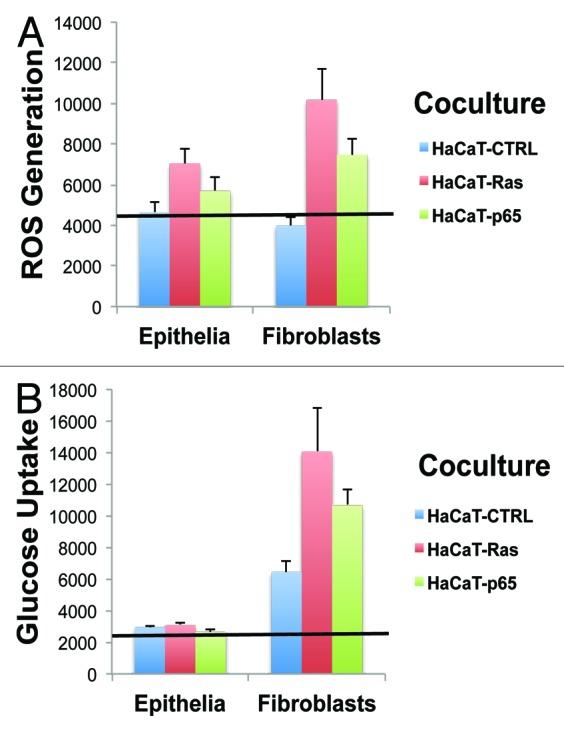
Figure 4. Cancer-associated fibroblasts Show the largest increases in ROS production and glucose uptake, as directly compared with adjacent epithelial cancer cells. (A) ROS-production. (B) Glucose uptake. Data originally presented in Figures 2 and 3 is presented again in Figure 4, in another format. In this case, the absolute magnitude of ROS production and glucose uptake in epithelial cancer cells and fibroblasts is directly compared, side-by-side, on the same graphs. This allows one to better appreciate that although the epithelial cancer cells harbor the activated oncogenes, their largest effects on cellular metabolism actually occur in neighboring normal fibroblasts. Thus, the “bystander” effect of oncogenes on the tumor microenvironment is one of the most significant metabolic effects, in terms of metabolic reprogramming.
Thus, the “bystander” effect of oncogenes on the tumor microenvironment appears to be one of the most significant metabolic effects, in terms of metabolic reprogramming.
Ras oncogene activation and inflammation drive a loss of stromal Cav-1 expression in adjacent cancer-associated fibroblasts
Loss of stromal caveolin-1 (Cav-1) is a biomarker of poor clinical outcome in several distinct types of human epithelial cancers, including breast, prostate, and gastric carcinomas, as well as in metastatic melanoma.71-82 In human breast cancers, reductions in stromal Cav-1 are clinically associated with early tumor recurrence, lymph-node metastasis, tamoxifen-resistance, and premature death. Mechanistically, loss of Cav-1 occurs via autophagic/lysosomal degradation due to oxidative stress in cancer-associated fibroblasts.30 Thus, loss of stromal Cav-1 is a functional biosensor of oxidative stress, autophagy, and glycolysis in the tumor microenvironment.22,34
As a consequence, we next examined the ability of HaCaT cells to downregulate Cav-1 expression in normal adjacent fibroblasts, during co-culture. For this purpose, HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were co-cultured with hTERT-immortalized fibroblasts (GFP[+]). Then, the cells were fixed and immunostained with specific antibody probes. Figure 5A shows that loss of stromal Cav-1 occurs only in fibroblasts co-cultured with HaCaT-Ras and HaCaT-p65 cells. However, HaCaT control cells did not induce a loss of Cav-1 in stromal fibroblasts. Image quantitation regarding the oncogene-induced loss of stromal Cav-1 is presented in Figure 5B. Note that there is a significant 2–3-fold reduction in Cav-1 expression.
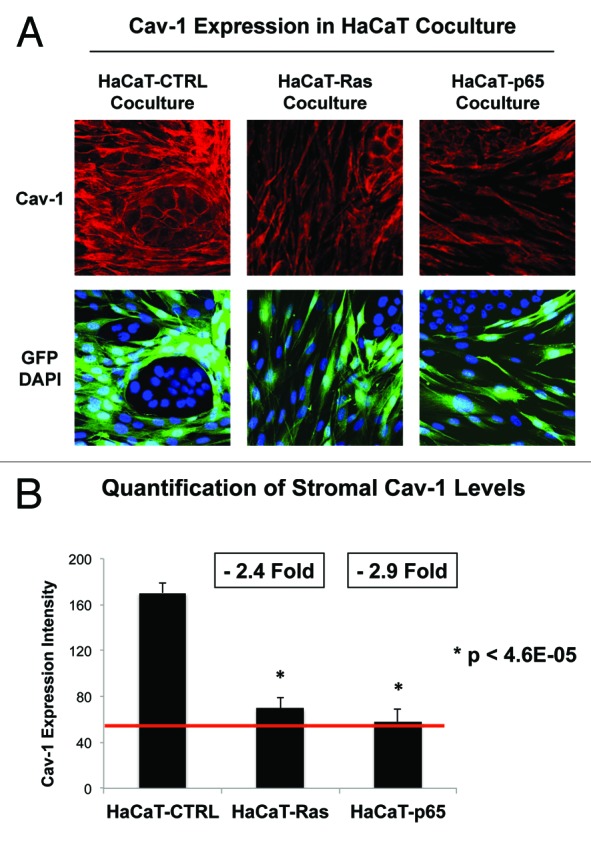
Figure 5. Ras oncogene activation and inflammation drive a loss of Cav-1 expression in adjacent cancer-associated fibroblasts. (A) HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were co-cultured for 4 days with hTERT-immortalized fibroblasts (GFP+). Then, the cells were fixed and immunostained with specific antibody probes. Note that a loss of stromal Cav-1 occurs only in fibroblasts co-cultured with HaCaT-Ras and HaCaT-p65 cells. HaCaT control cells did not induce a loss of Cav-1 in stromal fibroblasts. DAPI (blue nuclear staining) is also shown for reference. (B) Image quantitation regarding the oncogene-induced loss of stromal Cav-1 is presented; note that there is a significant 2–3-fold reduction in Cav-1 expression.
Thus, a loss of stromal Cav-1 is essentially a “mirror” or “reporter” of oncogenic transformation in adjacent epithelial cancer cells during the transition to malignancy.
Ras oncogene activation and inflammation drive the upregulation of MCT4 expression in adjacent cancer-associated fibroblasts, via oxidative stress
Like Cav-1, MCT4 is another biomarker of oxidative stress in cancer-associated fibroblasts.55,56 In head and neck cancers, MCT4 is a highly specific marker of cancer-associated fibroblasts, as compared with normal fibroblasts.83 In addition, stromal MCT4 expression in triple-negative breast cancers is a single independent powerful predictor of poor clinical outcome.84 In this context, a loss of stromal Cav-1 is strictly correlated with stromal MCT4 upregulation and lethality in triple-negative breast cancer patients.84 Physiologically, MCT4 allows the efficient export of L-lactate and ketone bodies from glycolytic cells. Mechanistically, MCT4 is a HIF1-α target gene that is upregulated under conditions of hypoxia and/or oxidative stress (a.k.a., pseudo-hypoxia). Hence, MCT4 is a functional biomarker of oxidative stress, hypoxia, and glycolysis, as well as mitochondrial dysfunction.
As such, we next examined the ability of HaCaT cells to upregulate MCT4 expression in normal adjacent fibroblasts during co-culture. HaCaT epithelial cells (control, H-Ras [G12V] or NFkB [p65]) were co-cultured with hTERT-immortalized fibroblasts (GFP+). Then, the cells were fixed and immunostained with specific antibody probes. Figure 6A shows that MCT4 expression is increased most significantly in fibroblasts co-cultured with HaCaT-Ras and HaCaT-p65 cells. In contrast, HaCaT control cells only induce a mild or modest increase in stromal MCT4 expression, close to the “border” or epithelial-stromal interface. Image quantitation regarding the oncogene-induced upregulation of stromal MCT4 is presented in Figure 6B and C. Note that stromal MCT4 expression is increased by up to > 4-fold during fibroblast co-culture with HaCaT-Ras and HaCaT-p65 cells.
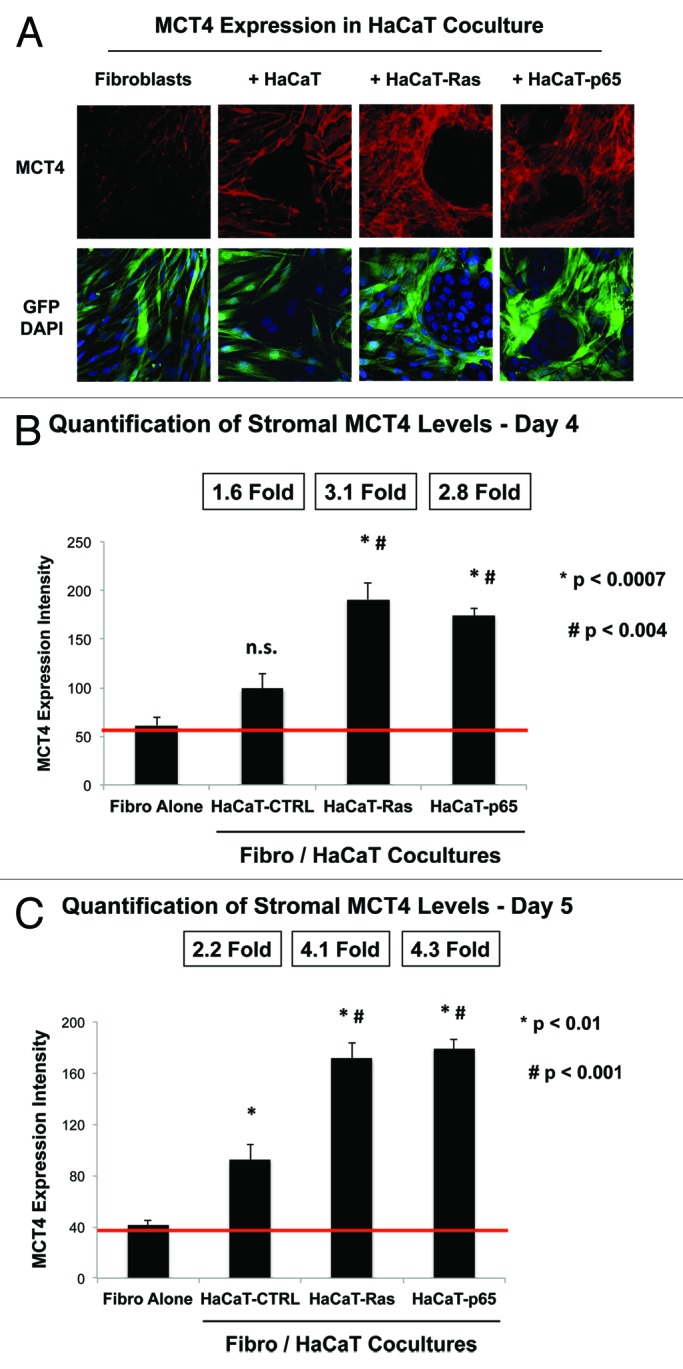
Figure 6. Ras oncogene activation and inflammation drive the upregulation of MCT4 expression in adjacent cancer-associated fibroblasts. (A) HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were co-cultured for 4 days with hTERT-immortalized fibroblasts (GFP+). Then, the cells were fixed and immunostained with specific antibody probes. Note that MCT4 expression is increased most significantly in fibroblasts co-cultured with HaCaT-Ras and HaCaT-p65 cells. In contrast, HaCaT control cells only induced a mild or modest increase in stromal MCT4 expression. DAPI (blue nuclear staining) is also shown for reference. (B) Image quantitation regarding the oncogene-induced upregulation of stromal MCT4 at day 4 is presented; note that there is a significant 3-fold upregulation of MCT4 expression. P values vs. fibroblasts alone (P < 0.0007) and vs. HaCaT-CTRL co-cultures (P < 0.004) are both shown. (C) Image quantitation regarding the oncogene-induced upregulation of stromal MCT4 at day 5 is presented; note that there is a significant 4-fold upregulation of MCT4 expression. P values vs. fibroblasts alone (P < 0.01) and vs. HaCaT-CTRL co-cultures (P < 0.001) are both shown.
To directly validate the idea that increased MCT4 expression represents oxidative stress in cancer-associated fibroblasts, we explored the functional effects of N-acetyl cysteine (NAC) [10 mM], a powerful antioxidant. Indeed, Figure 7 shows that NAC treatment reversed or prevented the induction of MCT4 in the stromal microenvironment in Ras and NFkB co-cultures only, directly implicating oxidative stress. Quantitation of these findings is presented in Figure 8A and B. Note that NAC treatment normalizes MCT4 expression in HaCaT-Ras and HaCaT-p65 co-cultures. Thus, NAC effectively functions as an “MCT4 inhibitor”.
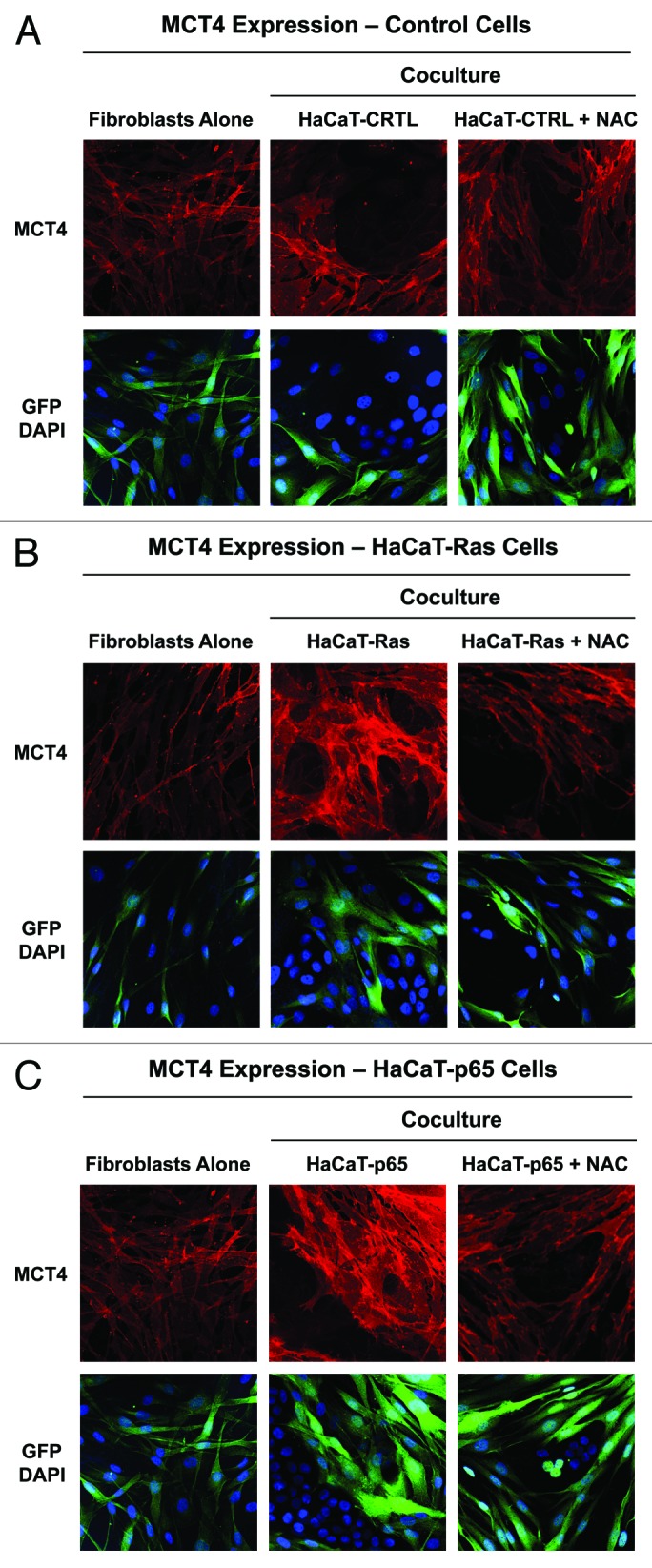
Figure 7. Oxidative stress mediates the upregulation of MCT4 in cancer-associated fibroblasts: rescue with NAC, a powerful antioxidant. HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were co-cultured for 5 days with hTERT-immortalized fibroblasts (GFP+). Then, the cells were fixed and immunostained with specific antibody probes. Note that MCT4 expression is increased most significantly in fibroblasts co-cultured with HaCaT-Ras cells and HaCaT-p65 cells, and that this can be reversed or prevented by the addition of the powerful antioxidant, N-acetyl cysteine (NAC) [10 mM], to the culture media. (A) HaCaT control co-cultures; (B) HaCaT-Ras co-cultures; (C) HaCaT-p65 co-cultures. DAPI (blue nuclear staining) is also shown for reference.
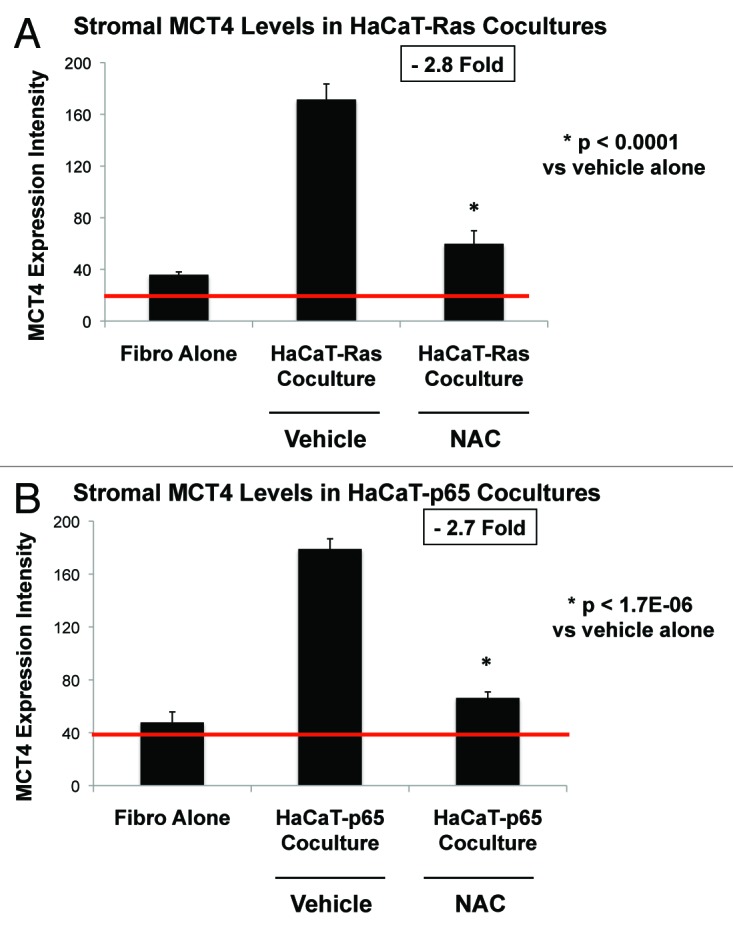
Figure 8. NAC quantitatively reduces oncogene-induced expression of MCT4 in cancer associated fibroblasts. (A) HaCaT-Ras co-cultures; (B) HaCaT-p65 co-cultures. MCT4 expression, illustrated in Figure 7, was subjected to image quantitation, as detailed under “Materials and Methods”. Note that HaCaT-Ras and HaCaT-p65 cells significantly induce MCT4 expression in adjacent stromal fibroblasts, during co-culture. However, treatment with NAC [10 mM] reduced stromal MCT4 expression levels by nearly 3-fold, approaching baseline levels. Thus, NAC effectively functions as an “MCT4 inhibitor”.
Co-culture with fibroblasts upregulates the expression of MCT1 in Ras-transformed and NFkB-expressing epithelial cancer cells: Establishment of metabolic symbiosis
We have previously provided evidence that a “lactate shuttle” exists in human tumors.45,55,56,83 In this scenario, cancer-associated fibroblasts express MCT4 and export mitochondrial fuels (such as L-lactate and ketone bodies) into the tumor microenvironment. Conversely, epithelial cancer cells express MCT1, which allows them to efficiently import these mitochondrial fuels to “burn” as an energy source for oxidative mitochondrial metabolism. This metabolic symbiosis mechanism could represent how tumor cells achieve efficient engraftment by metabolically co-operating with and rewiring their host microenvironment. To further test this hypothesis, we next examined the expression of MCT1 in HaCaT cells, either cultured alone or in the presence of stromal fibroblasts.
More specifically, HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were co-cultured with hTERT-immortalized fibroblasts (GFP[+]). Alternatively, HaCaT cells were cultured alone (without fibroblasts) for the same amount of time. Then, the cells were fixed and immunostained with specific antibody probes.
Figure 9 shows that MCT1 expression and plasma membrane localization is increased most significantly in HaCaT-Ras cells and HaCaT-p65 cells co-cultured with fibroblasts, relative to the same HaCaT cells cultured alone. Thus, induction of MCT1 in epithelial cancer cells is a functional biomarker of “metabolic symbiosis”, which occurs during co-culture with stromal fibroblasts.
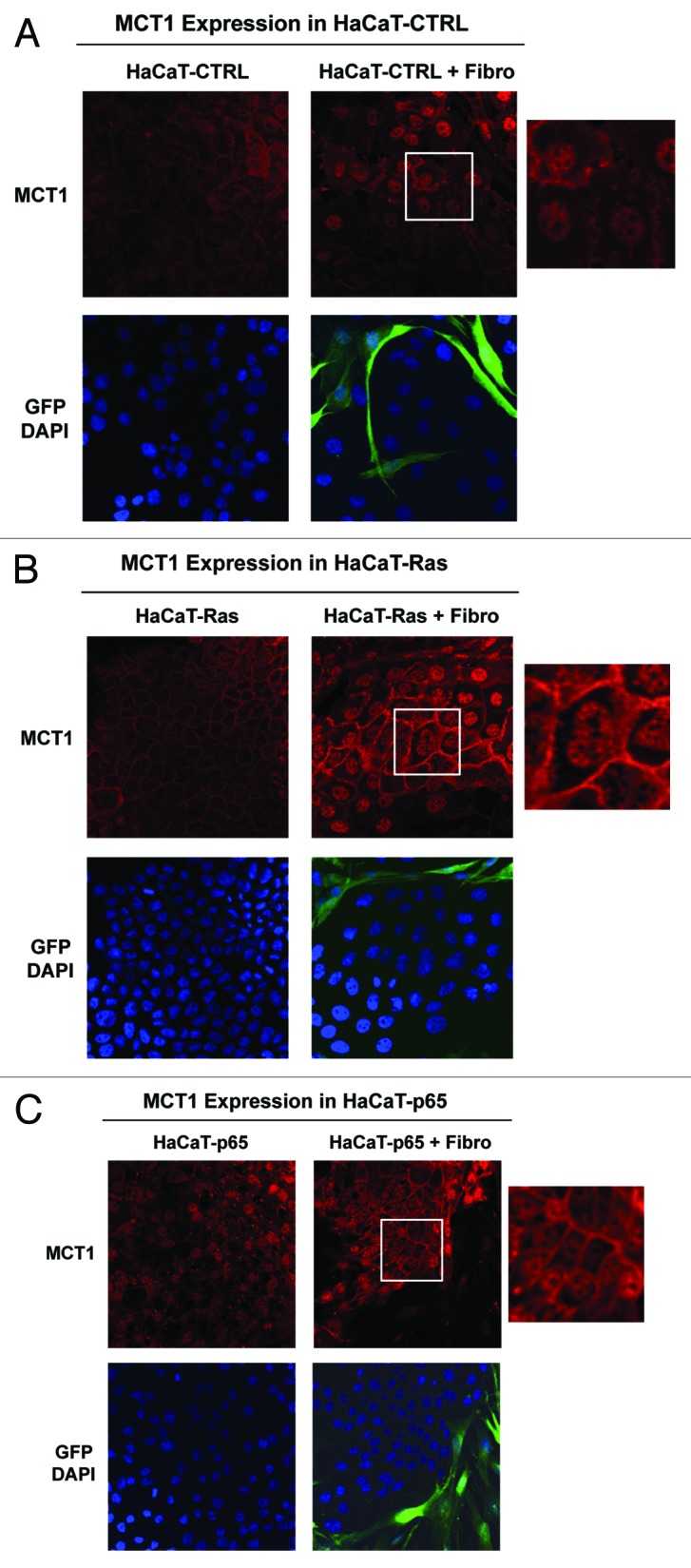
Figure 9. Co-culture with fibroblasts upregulates the expression of MCT1 in Ras-transformed and NFkB-expressing epithelial cells. HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were co-cultured for 5 days with hTERT-immortalized fibroblasts (GFP+). Alternatively, HaCaT cells were cultured alone (without fibroblasts) for the same amount of time. Then, the cells were fixed and immunostained with specific antibody probes. Note that MCT1 expression and plasma membrane localization is increased most significantly in HaCaT-Ras cells and HaCaT-p65 cells co-cultured with fibroblasts, relative to the same HaCaT cells cultured alone. Insets at higher magnification are shown to highlight the plasma membrane staining of MCT1. DAPI (blue nuclear staining) is also shown for reference.
Interestingly, we have previously shown that MCT1 is a biomarker of increased mitochondrial mass and activity, as well as high proliferation rates in vivo.83 For example, MCT1 expression is tightly correlated with Ki-67 immunostaining in head and neck cancers and in normal mucosal tissues, especially within the basal stem cell compartment.83 Thus, MCT1 may also be a marker of increased stem cell activity.
Homotypic cultures of epithelial cancer cells show that Ras transformation increases ROS production and MCT4 expression
For comparison purposes, we also examined some of the metabolic properties of HaCaT cells cultured alone under homotypic culture conditions. For this purpose, HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were cultured alone and then subjected to either FACS analysis to determine ROS production, or immunostaining with antibodies directed against MCT4, a marker of oxidative stress.
Interestingly, Figure 10 shows that Ras-activation in HaCaT cells significantly increases ROS production (Fig. 10A; 1.5-fold; P = 0.02) and MCT4 expression at the plasma membrane (Fig. 10B). Thus, HaCaT-Ras cells cultured alone also show an increase in oxidative stress. In contrast, HaCaT-p65 cells did not show significant evidence of oxidative stress when cultured alone. Thus, they may induce oxidative stress in fibroblasts via cytokine production.
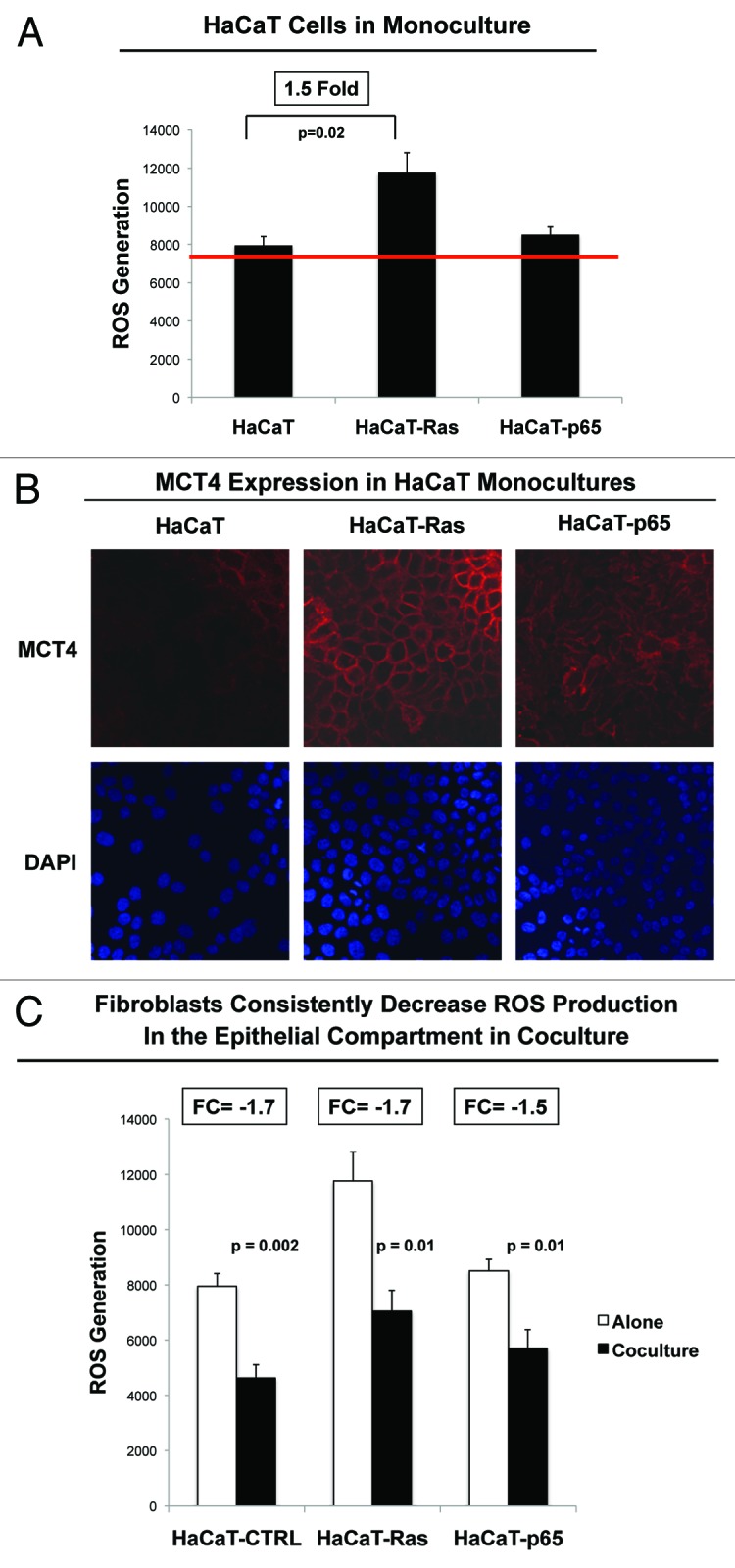
Figure 10. Homotypic cultures of epithelial cancer cells show that Ras-transformation increases ROS production and MCT4 expression. HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were cultured alone and then subjected to either FACS analysis to determine ROS production (A), or immunostaining with antibodies directed against MCT4, a marker of oxidative stress (B). Note that Ras-activation in HaCaT cells significantly increases ROS production (1.5-fold; P = 0.02) and MCT4 expression plasma membrane staining. DAPI (blue nuclear staining) is also shown for reference. (C) shows ROS production in HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) alone or during co-culture with fibroblasts, by comparing data presented in Figure 2 and Figure 10A; these data are derived from the same experiments. Note that co-culture of normal and epithelial cancer cells with fibroblasts reduced ROS-production in all cases, indicating that fibroblasts may help induce an antioxidant response in adjacent epithelial cells and protect these epithelial cells from oxidative stress.
Importantly, co-culture of normal and epithelial cancer cells with fibroblasts reduced ROS-production in all cases, indicating that fibroblasts may help induce an antioxidant response in adjacent epithelial cells and protect epithelial cells from oxidative stress (Fig. 10C). These findings are consistent with recent studies showing that bone marrow-derived stromal cells provide cysteine (cys), which is transferred to cancer cells (in chronic lymphocytic leukemia [CLL]) and is then converted to glutathione (gamma-glut-cys-gly), fueling an antioxidant response.85
Oncogene activation can drive the upregulation of mitochondrial activity in epithelial cancer cells
We also examined the functional effects of oncogenic transformation on the status of mitochondria in epithelial cancer cells. Briefly, HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were cultured alone and then subjected to FACS analysis to determine mitochondrial activity, via MitoTracker staining.
Figure 11 shows that HaCaT-Ras cells (1.5-fold; P = 0.009) and HaCaT-p65 cells (1.6-fold; P = 0.006) both show a significant increase in MitoTracker activity staining. Thus, oncogenic tranformation can also promote increases in mitochondrial mass and/or activity in epithelial cancer cells, so that they are “primed” to undergo metabolic symbiosis with a glycolytic stromal microenvironment.
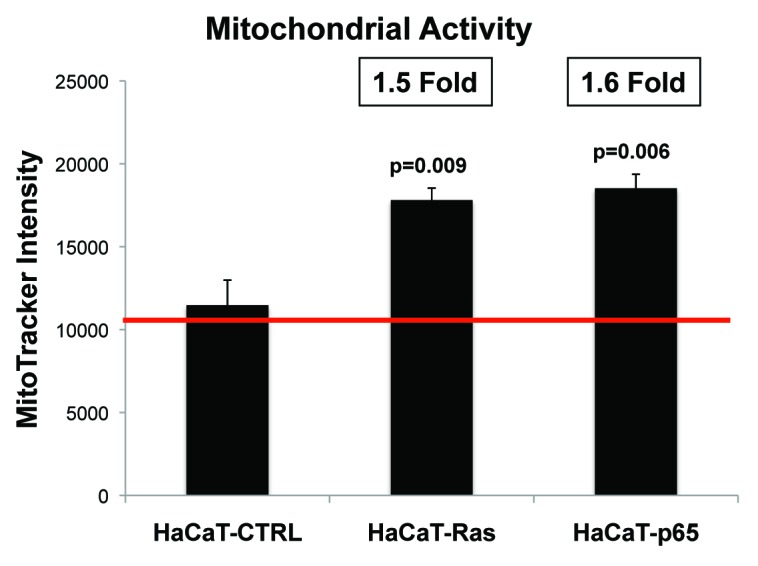
Figure 11. Ras oncogene activation and inflammation both drive the upregulation of mitochondrial activity in epithelial cancer cells. HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were cultured alone and then subjected to FACS analysis to determine mitochondrial activity, via MitoTracker staining. Note that HaCaT-Ras cells (1.5-fold; P = 0.009) and HaCaT-p65 cells (1.6-fold; P = 0.006) both show a significant increase in MitoTracker activity staining.
Antioxidants inhibit mitochondrial biogenesis in Ras-transformed cancer cells
We also followed mitochondrial mass in HaCaT using a specific mitochondrial marker, namely TOMM20, which also serves as a marker of mitochondrial biogenesis. In these studies, HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were cultured alone (in the absence or presence of NAC [10 mM]) and then subjected to immunostaining with TOMM20.
Figure 12 illustrates that HaCaT-Ras cells show the most significant increases in mitochondrial mass (consistent with the MitoTracker data), and that this is strictly dependent on oxidative stress. Note that NAC treatment almost completely eliminated mitochondrial staining in HaCaT-Ras, but had little or no effect on mitochondrial mass in HaCaT-p65 cells and control HaCaT cells. As such, it appears that NAC selectively halts mitochondrial biogenesis in Ras-transformed cells, possibly explaining how antioxidants could be used to “starve” cancer cells. Image quantitation revealed that the expression of TOMM20 was reduced by >5-fold in NAC-treated HaCaT-Ras cells (data not shown).

Figure 12. Antioxidant treatment selectively inhibits mitochondrial biogenesis in Ras-transformed epithelial cancer cells. HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were cultured alone (in the absence or presence of NAC [10 mM]) and then subjected to immunostaining with TOMM20, a marker of mitochondrial mass. Note that HaCaT-Ras cells show the most significant increase in mitochondrial mass, and that this is strictly dependent on oxidative stress. DAPI (blue nuclear staining) is also shown for reference.
Thus, ROS production in HaCaT-Ras cells apparently drives mitochondrial biogenesis in epithelial cancer cells, and then almost simultaneously “fertilizes” the tumor microenvironment, via “bystander” oxidative stress in cancer-associated fibroblasts.
Fibroblasts protect cancer cells against programmed cell death
As fibroblasts appeared to protect HaCaT-Ras and HaCaT-p65 cells against oxidative stress (Fig. 10C), we also examined their effects on programmed cell death (apoptosis), which was quantitated by FACS analysis using Annexin-V and PI staining.
Figure 13A shows that fibroblasts did not affect cell death in normal HaCaT control cells but significantly rescued HaCaT-Ras and HaCaT-p65 cells from apoptosis. However, the most significant fibroblast-mediated reductions in programmed cell death were observed with HaCaT-Ras cells, which showed a 2.4-fold reduction in apoptosis.

Figure 13. Fibroblasts protect cancer cells against apoptosis, autophagy, and sensecence: quantitation via FACS. (A) Apoptosis. Cell death was quantified by flow cytometry using propidium iodide and Annexin-V-APC. After a total of 4 days in culture, the cells were collected by centrifugation. Then, the annexin V-APC conjugate and propidium iodide was added. Cells were then analyzed by flow cytometry. Note that fibroblasts (GFP[+]) did not affect cell death in HaCaT control cells, but significantly rescued HaCaT-Ras and HaCaT-p65 cells from apoptosis. However, the most significant fibroblast-mediated reductions in cell death were observed with HaCaT-Ras cells, which showed a 2.4-fold reduction in apoptosis. (B and C) Autophagy and senescence. Cells were treated using the FluoReporter lacZ Flow Cytometry Kit (Molecular probes). Assay results were evaluated by flow-cytometry analysis. Note that HaCaT-Ras cells cultured alone showed very dramatic increases in β-GAL activity, as compared directly with HaCaT control cells. However, β-GAL levels were progressively reduced (by >2-fold) in HaCaT-Ras cells during co-culture with hTERT-immortalized fibroblasts, over a 2–4 d period. Thus, fibroblasts induce autophagy-resistance and reverse senescence in HaCaT-Ras cancer cells. (B) Day 2 co-culture; (C) day 4 co-culture.
Thus, fibroblasts functionally rescue HaCaT-Ras and HaCaT-p65 cells from oxidative stress and programmed cell death, likely by reducing oncogene-induced stress.
Fibroblasts confer resistance to autophagy and senescence during Ras activation in cancer cells
Previously, we and others have shown that β-galactosidase (β-GAL) is a metabolic marker of both autophagy and senescence, as it is a key lysosomal enzyme which accumulates in lysosomes and autophagosomes in senescent cells.49-51,54
Since fibroblasts effectively reduced oxidative stress and protected cancer cells against apoptosis, we also speculated that these reductions in oxidative stress may also protect HaCaT-Ras cells against autophagy and senescence. HaCaT-Ras cells should be particularly susceptible to autophagy and senescence, as H-Ras (G12V) expression is known to confer significant oncogenic stress. Thus, we quantitatively measured β-galactosidase (β-GAL) activity levels by FACS analysis.
First, we examined β-GAL activity in HaCaT cells (CTRL vs. Ras). These results are shown in Figure 13B and C. Note that HaCaT-Ras cells cultured alone showed very dramatic increases in β-GAL activity, as compared directly with HaCaT control cells. However, β-GAL levels were progressively reduced (by >2-fold) in HaCaT-Ras cells during co-culture with hTERT-immortalized fibroblasts over a 2–4 day period.
This provides independent support for the idea that cancer cells use fibroblasts to rescue themselves from oncogene-induced stress and to confer autophagy resistance and suppress senescence in cancer cells.
Ras activation in cancer cells significantly alters metabolism in normal adjacent epithelial cells, driving oxidative stress and glycolysis
To determine if Ras activation in epithelial cancer cells also affects metabolism in normal adjacent epithelial cells, HaCaT-Ras (RFP[+]) cells were co-cultured with normal HaCaT control cells, which are not transformed. Then, these epithelial–epithelial co-cultures were subjected to FACS analysis with a series of fluorescent metabolic probes. These small reporter molecules allowed us to quantitatively monitor ROS production (a measure of oxidative stress) and glucose uptake (a measure of glycolysis or glycolytic power) in a compartment-specific fashion. Thus, we compared ROS production and glucose uptake in Ras-transformed epithelial cancer cells and adjacent “normal” epithelial cells to determine how epithelial oncogene-activation metabolically reprograms adjacent normal epithelial cells.
Figure 14 shows the status of these metabolic parameters in normal HaCaT control cells co-cultured with HaCaT-Ras (RFP[+]) cells. More specifically, we examined ROS production and glucose uptake in the population of normal HaCaT control cells co-cultured with HaCaT-Ras cells. Significant changes were observed in both ROS production and glucose uptake. More specifically, normal HaCaT control cells that were co-cultured with HaCaT-Ras cells showed a significant increase in both ROS production (1.7-fold; P = 0.03) and glucose uptake (1.3-fold; P = 6 × 10−5). Thus, oncogene-transformed epithelial cancer cells can metabolically reprogram normal adjacent epithelial cells as well as fibroblasts.

Figure 14. HaCaT epithelial-cancer cell co-cultures: Oncogene-transformed cancer cells can metabolically reprogram normal adjacent epithelial cells. (A) ROS-production. (B) Glucose uptake. HaCaT-Ras (RFP[+]) epithelial cancer cells were co-cultured for 4 days with normal HaCaT control cells. Then, ROS production (a measure of oxidative stress) and glucose uptake (a measure of glycolytic activity) in both cell types were quantitatively determined by FACS sorting. Note that normal HaCaT control cells co-cultured with HaCaT-Ras cells show a significant increase in both ROS production (1.7-fold; P = 0.03) and glucose uptake (1.3-fold; P = 6 × 10−5). However, normal HaCaT control cells did not change ROS production or glucose uptake in HaCaT-Ras cells, indicating that HaCaT-Ras cells exert a significant dominant effect over normal epithelial cells. Thus, oncogene-transformed epithelial cancer cells can metabolically reprogram adjacent normal epithelial cells, to establish “metabolic symbiosis”.
In contrast, normal HaCaT control cells did not change ROS production or glucose uptake in HaCaT-Ras cells, indicating that HaCaT-Ras cells exert a significant dominant effect over normal epithelial cells. Thus, unlike fibroblasts, normal epithelial cells do not relieve oxidative stress in epithelial cancer cells. This is an important distinction, which suggests that fibroblasts also make better partners for metabolic symbiosis.
We conclude that epithelial cancer cells can use either adjacent fibroblasts, or even adjacent normal epithelial cells, as “partners” to engage in a form of “metabolic symbiosis”.
Thus, as previously proposed by Sonveaux, Feron, and Dewhirst, a “lactate shuttle” may also exist between two populations of oxidative and glycolytic epithelial cells.86-88 This occurs clinically in head and neck cancers, where the oxidative cancer cells with functional mitochondrial are hyper-proliferative and are MCT1(+)-positive.83 In contrast, the glycolytic cancer cells are mitochondria-deficient, non-proliferative, and are MCT4(+)-positive.83 As such, this glycolytic cancer cell population may actually represent relatively “normal” epithelial cells, which are helping to fuel their neighbors via metabolic symbiosis. In fact, these glycolytic MCT4(+) epithelial tumor cells appeared to be well differentiated in head and neck cancers.83
Discussion
Cancer is a systemic metabolic disease, fueled by oxidative stress and inflammation
Here, we provide provocative new evidence that cancer is not a cell autonomous disease, but rather it is a systemic disease, of the host stromal microenvironment (Fig. 15A). More specifically, we show that RAS oncogene-induced ROS production is transmitted from epithelial cancer cells to neighboring normal fibroblasts. As a consequence, epithelial ROS production drives the onset of the cancer-associated fibroblast phenotype, resulting in an amplification of ROS production, literally creating a “field of oxidative stress”. This has important functional consequences, as ROS production in cancer-associated fibroblasts causes a shift toward aerobic glycolysis, initiating the “reverse Warburg effect.”33,40,41,84,89 This fibroblastic shift toward glycolysis was quantitatively followed by FACS analysis and NBD–glucose uptake.
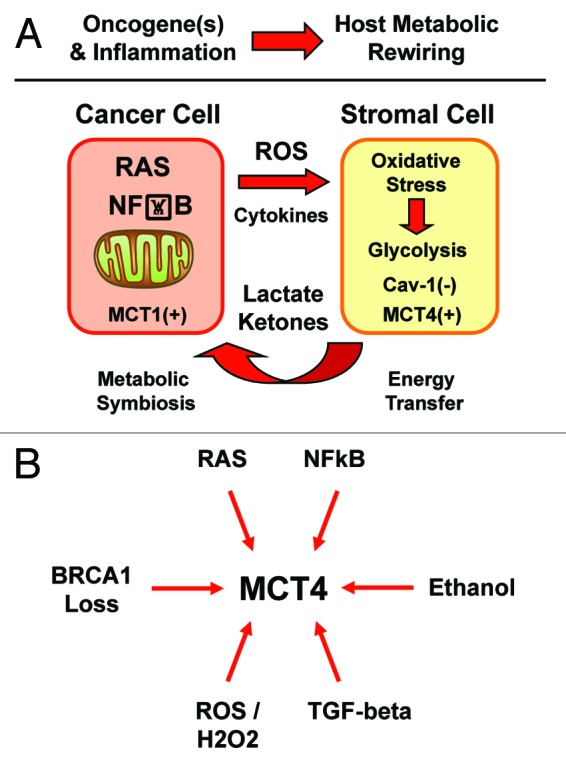
Figure 15. Diverse oncogenic stimuli metabolically rewire the microenvironment, via a common mechanism. (A)Oxidative stress and inflammation reprogram the microenvironment. Here, we show that activation of Ras or the innate immune response in epithelial cancer cells are both sufficient to actively induce metabolic reprogramming of the tumor microenvironment. ROS production and/or cytokine release drives oxidative stress in adjacent cancer-associated fibroblasts. Oxidative stress in myofibroblasts then mediates the induction of stromal aerobic glycolysis, i.e., “metabolic symbiosis” or the “reverse Warburg effect”. Thus, diverse epithelial oncogenes (H-Ras [G12V] and NFkB [p65]) induce oxidative stress in the tumor stroma. MCT1 and MCT4 are markers of metabolic symbiosis. (B) Oncogenic stimuli converge on stromal MCT4. HaCaT cells which harbor activated oncogenes gain the ability to glycolytically reprogram the tumor microenvironment, via the induction of oxidative stress in cancer-associated fibroblasts. Thus, diverse oncogenes (Ras and NFkB) act via a common mechanism (oxidative stress), to upregulate MCT4 in the tumor stroma. As MCT4 controls the “energy” supply for cancer cells, this makes MCT4 an attractive target, for new therapeutic interventions aimed at “starving” tumor cells. Similarly, other pro-oncogenic stimuli, such as loss of BRCA1, TGF-β secretion, and ethanol, as well as ROS and hydrogen peroxide production, all converge on MCT4 upregulation in the tumor stromal compartment. In addition, all these pro-oncogenic stimuli (RAS, NFkB, BRCA1 loss, TGF-β, ROS/H2O2, and ethanol) also drive a loss of stromal Cav-1 expression in cancer associated fibroblasts. Thus, Cav-1 and MCT4 are stromal biosensors of the transition to malignancy.
Virtually identical results were obtained using epithelial cancer cells that were genetically reprogrammed toward a pro-inflammatory phenotype, via NFkB activation. As such, oncogene-induced ROS production and/or inflammation have similar negative metabolic effects on the host microenvironment, allowing for effective tumor cell engraftment.
Our results explain how activated oncogenes, inflammation, and ROS production “collaborate”, resulting in tumor initiation and “fertilization” of the host microenvironment.18,19,35,38 Thus, activated oncogenes and chronic inflammation have local and systemic metabolic effect(s), which establish metabolic symbiosis between epithelial cancer cells and cancer-associated fibroblasts.17-19 This metabolic reprogramming results in the paracrine establishment of “two-compartment tumor metabolism”90-92.
Our findings have important practical applications for changing the existing paradigm(s) of cancer diagnosis, FDG-PET-imaging, and anticancer therapy toward more holistic and non-toxic approaches.18,25 They also directly implicate the use of antioxidants and anti-inflammatory therapies in cancer prevention and personalized medicine.42-45,93
Hence, cancer may be viewed as a host-based disease of chronic oxidative stress and inflammation that starts locally as a point source in the cancer cell, which is then amplified and spreads systemically in the host, ultimately driving whole-body catabolism and “metabolic catastrophe” or “metabolic collapse”, especially during metastasis.
Diverse oncogenic stimuli converge on a common druggable target: MCT4 in the tumor microenvironment
Cancer cells use oncogene-driven oxidative stress as a “weapon” to induce a specific metabolic response to injury in the tumor microenvironment, allowing them to establish metabolic symbiosis and successful cancer cell engraftment within the host organism. In many ways, our current results are consistent with the “seed and soil” hypothesis, which was originally proposed in 1889, by Dr Stephen Paget in England, more than 100 y ago.94-97
We conclude that oncogenes can also act at a distance, via ROS production and inflammation, to induce metabolic symbiosis between cancer cells and the tumor stroma. Diverse oncogenes (Ras and NFkB) may act via a common convergent mechanism (oxidative stress), to upregulate MCT4 in the tumor stroma (Fig. 15B). In addition, we have previously shown that several other pro-oncogenic stimuli, such as loss of BRCA1,20,21 TGF-β secretion,98 and ethanol treatment,99 as well as ROS and hydrogen peroxide (H2O2) production,56 all converge on Cav-1 downregulation and MCT4 upregulation in the tumor stromal compartment. As such, changes in the expression of stromal Cav-1/MCT4 can be used to monitor the transition to malignancy during tumor initiation and progression. Taken together, these data make stromal MCT4 an attractive new druggable target for novel therapeutic interventions, as MCT4 controls the “food” supply for cancer cells.56,83,84
In summary, oncogenic stress stimulates a common metabolic response to “injury” in the tumor microenvironment. This idea is consistent with the hypothesis that cancer behaves as a wound that does not heal (Fig. 16).

Figure 16. Understanding why cancer behaves as a wound that does not heal. Oncogenic stress stimulates a common metabolic response to “injury” in the tumor microenvironment. Four steps or events are outlined, leading to “metabolic symbiosis”. This idea is consistent with the hypothesis that cancer behaves as a wound that does not heal.
Cancer cells behave as “metabolic parasites”: Can we treat cancer like an infectious disease with “metabolic” antibiotics?
The parallels with infectious disease are now clear. Cancer cells behave as predatory “metabolic parasites”, just like foreign microorganisms (bacteria, fungi, and viruses).31,36,40,44 Thus, we should consider treating cancer like an infectious disease, with new classes of “metabolic” antibiotics that can starve cancer cells to death.
Some of these “metabolic” antibiotics may already be available. For example, tigecycline, a member of the tetracycline family of antibiotics, selectively kills cancer cells (but not normal cells), by preventing mitochondrial biogenesis in cancer cells.100 Furthermore, doxycyline, another tetracycline family member, inhibits breast cancer metastasis by >70% in pre-clinical animal models,101 likely by targeting mitochondrial protein synthesis in cancer cells.102
In accordance with these findings, we have previously shown that 15 markers of mitochondrial biogenesis are increased in human breast cancer cells in vivo (e.g., TOMM20, MCT1, MRP [mitochondrial ribosomal proteins]),93 and that the overexpression of genes that functionally drive mitochondrial biogenesis (GOLPH3, PGC1a/b, MitoNEET, or POLRMT)91,92 in breast cancer cells is indeed sufficient to increase tumor growth rates in xenografted mice.
Interestingly, we also show here that NAC halts mitochondrial biogenesis in Ras-transformed epithelial cancer cells, most likely by eliminating oxidative stress. Thus, NAC targets both epithelial cancer cells and the tumor microenvironment, “hitting” multiple genetic and spatial targets at the same time.
Inflammation is a critical mediator of the transition to malignancy: Implications for chemoprevention with anti-inflammatory drugs
It has long been suggested that chronic inflammation (due to injury or infections), may be a critical trigger for tumor initiation.103,104 In accordance with this notion, the most common global causes of cancer are due to infections in the cervix (HPV), liver (HBV), lung (TB), nasopharyngeal tissues (EBV), and stomach (H. pylori). In addition, other chronic inflammatory conditions, such as ulcerative colitis, Crohn disease, and scleroderma, all lead to an increased risk of developing cancer. Moreover, in the GI tract, ROS-producing bacteria have been implicated in the initiation of colon cancer. Finally, H. pylori also increases ROS (hydrogen peroxide) and RNS (nitric oxide) in the stomach.
Notably, in DCIS patients, loss of stromal Cav-1 is directly associated with inflammation in the tumor stroma.71 More specifically, in this patient cohort, either loss of stromal Cav-1 or inflammation were both individually sufficient to predict DCIS recurrence and progression to invasive breast cancer.71 Importantly, loss of stromal Cav-1 was a better predictor (as compared with inflammation) of DCIS recurrence and/or progression, indicating that glycolytic stromal metabolism is the most critical event for tumor progression.71
Similarly, we have shown here that oncogenes and inflammation induce a loss of Cav-1, as well as increased oxidative stress, and glycolysis in the host microenvironment. This leads to metabolic symbiosis between oxidative cancer cells and glycolytic stromal fibroblasts. Thus, inflammation is a critical mediator of tumor initiation and progression, suggesting that we should consider new anti-inflammatory strategies for chemoprevention, and as anticancer therapies.
Understanding the desmoplastic reaction, during the transition to malignancy: Implications for cancer therapy
The “desmoplastic reaction” refers to the growth of connective tissue fibroblasts or stroma in and around an area, usually in response to an injury or the presence of a focus of cancer cells.15,105-107 Thus, the desmoplastic reaction is part of the normal wound response and is consistent with the notion that a tumor is a wound that does not heal.105
In tumors, the desmoplastic reaction is most often associated with myo-fibroblastic differentiation and the increased secretion of extracellular matrix proteins, such as collagen I and tenascin C, among others.108 This can also lead to scar formation. Interestingly, tumors with a “central scar” or “fibrotic focus” are associated with a worse clinical outcome, such as recurrence, metastasis, and poor overall survival.109-114
Given our current findings, a new function for the desmoplastic reaction, which is a common feature of many different types of malignancies, may be to help cancer cells overcome oncogenic stress. More specifically, we show here that glycolytic stromal fibroblasts reduce oxidative stress in cancer cells, which effectively protects these cancer cells against apoptosis, autophagy, and even senescence.
Indeed, over the years, it has proved quite difficult to generate new cancer cells lines, in addition to the NCI-60 panel of tumor cells. One possible explanation is that primary cancer cells suffer from oncogene-induced senescence, and that this becomes most apparent when we isolate cancer cells away from stromal cells in an attempt to have pure cultures of cancer cells. Thus, the best way to kill primary cancer cells may be to separate them from stromal fibroblasts.
As such, new therapies should be developed to target the tumor stroma. Once stromal cells are effectively killed, an expected consequence would be the onset of oncogene-induced senescence in the primary cancer cells, resulting in tumor regression.
Materials and Methods
Materials
Antibodies were as follows: MCT4 and MCT1 (generous gifts of Dr Nancy Philp, which are isoform-specific rabbit polyclonal antibody against 18-mer synthetic oligo-peptides corresponding to the C-terminal amino acids of human MCT4 or MCT1115); TOMM20 (sc-17764, Santa Cruz Biotechnologies). Secondary antibodies for immunofluorescence were Alexa Orange-Red 546 nm and Alexa far red 633 nm (Invitrogen). Other reagents were as follows: N-acetyl cysteine (NAC) was from Sigma, 2-(N-[7-nitrobenz-2-oxa-1,3-diazol-4-yl]amino)-2-deoxy-D-glucose (2-NBDG), 4,6-diamidino-2-phenylindole (DAPI) were from Invitrogen. The CellROX assay for measuring reactive oxygen species was from Invitrogen.
Cell cultures
The HaCaT cancer cell lines were provided by Dr Ulrich Rodeck and experiments were performed in the recommended complete growth medium (DMEM with 10% FBS and Penicillin 100 units/mL-Streptomycin 100 µg/mL). Human skin fibroblasts (BJ-1) immortalized with the telomerase reverse transcriptase catalytic domain (hTERT) were originally purchased from Clontech, Inc, and clones were generated with either GFP or RFP overexpression. BJ-1-GFP or BJ-1-RFP fibroblasts were cultured in HaCaT complete growth media. After 24 h, the media was changed to DMEM with 10% nuserum and penicillin 100 units/mL-streptomycin 100 µg/mL.
Co-cultures of HaCaT cells and fibroblasts
BJ-1 fibroblasts overexpressing GFP or RFP and one of the HaCaT cell lines (HaCaT-CTRL, HaCaT-Ras(G12V), HaCaT-p65) were plated on glass coverslips in 12-well plates in 1 ml of complete media. Epithelial cells were plated within 2 h of fibroblast plating. The total number of cells per well was 1 × 105. Experiments were performed at a 5:1 fibroblast-to-epithelial cell ratio. As controls, homotypic mono-cultures of fibroblasts and epithelial cells were seeded using the same number of cells as the total corresponding co-cultures. Cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2.
Co-cultures of HaCaT-control cells and HaCaT-Ras cells
HaCaT-Ras cells overexpressing RFP and HaCaT-Control cell lines were plated onto glass coverslips in 12-well plates in 1 ml of complete media. Both cell lines were plated within 2 h of the one being plated. The total number of cells per well was 1 × 105. Experiments were performed at a 5:1 HaCaT-Control -to-HaCaT-Ras cell ratio. As controls, homotypic mono-cultures of HaCaT-Control and HaCaT-Ras cells were seeded using the same number of cells as the total corresponding co-cultures. Cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2.
Flow cytometric analysis
HaCaT cells were plated in co-culture with BJ-1 fibroblasts (which were either GFP [+] or RFP[+]) or plated in mono-culture. Cells were grown for a total culture duration of 4 d. Then, to isolate the GFP(+) or RFP(+) BJ-1 cell population, co-cultured cells were sorted using a 488 nm and a 543 nm laser. As a critical control, mono-cultures of Hacat cells, which are GFP(−), and BJ-1 fibroblast cells, which are GFP (+) or RFP (+), were sorted in parallel.
Measurement of reactive oxygen species
CellROX (cat #C10422, Invitrogen) is a cell-permeable dye used to measure intracellular ROS. CellROX is non-fluorescent in a reduced state. When oxidized by reactive oxygen species witihin cells it has high fluorescence, with absorption/emission maxima of ~644–665 nm. The fluorescence method was used according to the manufacturer’s instructions. Briefly, BJ-1 fibroblasts which were RFP(+) were cultured homotypically or in co-culture with HaCaT cells. Cells were then incubated with 5 μM CellROX in DMEM with 10% FBS for 30 min at 37 °C. Cells were then washed in PBS ×3, harvested, and re-suspended in 500 μL of PBS. Cells were analyzed by flow cytometry using an RFP signal detector to distinguish fibroblasts from HaCaT cells (fibroblasts are the RFP[+] population) and an APC signal detector to measure levels of intracellular reactive oxygen species.
Measurement of glucose uptake
Glucose uptake was performed using 2-NBDG. HaCaT cells in homo-typic culture or co-cultured with BJ-1 fibroblasts (RFP[+]) were cultured for 4 d. Then, the cells were incubated with 2-NBDG solution (diluted in phenol red free DMEM with 10% FBS to a final concentration of 250 μM) for 30 min at 37 °C. All subsequent steps were performed in the dark. Cells were then washed in PBS, harvested, and re-suspended in 500 μL of PBS. Cells were then analyzed by flow cytometry using a GFP signal detector with excitation wavelength of 488 nm and emission of 530 nm (to detect 2-NBDG), and an RFP signal detector with excitation wavelength of 496 nm and emission of 615 (to detect RFP-positive BJ-1 cells). Thus, HaCaT cells in co-culture were the RFP-negative population.
Mitochondrial activity
To measure mitochondrial activity, cells were stained with MitoTracker Orange (CMTMRos cat #M7510, Invitrogen), whose accumulation in mitochondria is dependent upon membrane potential. Briefly, HaCaT cells were cultured alone for 3 days in DMEM with 10% NuSerum. Cells were then incubated with pre-warmed MitoTracker staining solution (diluted in serum free DMEM to a final concentration of 10 nM) for 10 min at 37 °C. All subsequent steps were performed in the dark. Cells were washed in PBS, harvested, and re-suspended in 500 μL of PBS. Cells were then analyzed by flow cytometry using a GFP signal detector with excitation wavelength of 488 nm and emission of 530 nm (to detect GFP-positive fibroblasts), and a PE Texas Red signal detector with excitation wavelength of 496 nm and emission of 615 (to detect MitoTracker). Data analysis was performed using FlowJo 8.8 software.
Immunocytochemistry
Cells were fixed after 4 days of culture. Then, the ICC protocol was performed as previously described, with minor modifications.56 Briefly, cells were fixed for 30 min at room temperature in 2% para-formaldehyde diluted in PBS, after which they were permeabilized with cold methanol at −20 °C for 5 min. The cells were rinsed with PBS with 0.1 mM calcium chloride and 1 mM magnesium chloride (PBS/CM). Then, cells were incubated with NH4Cl in PBS to quench free aldehyde groups. Rinsing with PBS/CM was followed by blocking with immunofluorescence (IF) buffer (PBS, 1% BSA, 0.1% Tween 20) for 1 h at room temperature. Primary anti-MCT4 and TOMM20 antibodies were incubated in IF buffer for 1 h at room temperature. After washing with IF buffer (3×, 10 min each), cells were incubated for 30 min at room temperature with fluorochrome-conjugated anti-rabbit and anti-mouse secondary antibodies diluted in IF buffer. Finally, slides were washed at room temperature with IF buffer (3×, 10 min each), rinsed with PBS/CM and counter-stained with DAPI (1 μg/ml) in PBS and mounted with Prolong Gold anti-fade reagent.
Confocal microscopy
Images were collected with a Zeiss LSM510 meta confocal system using a 405 nm diode excitation laser with a band pass filter of 420–480 nm, a 488 nm Argon excitation laser with a band pass filter of 505–550 nm, a 543 nm HeNe1 excitation laser with a 561–604 nm filter and a 633 nM HeNe2 excitation laser with a 657–754 nm filter. Images were acquired with a 40× objective. Immunofluorescence staining image quantitation was performed using ImageJ 1.46 (National Institutes of Health,). Briefly, the channel was split into 8-bit single color images. Polygon selection was used to delineate the cells so as to measure cell fluorescence intensity and area. Quantification was expressed as intensity per unit area intensity/area(pixel2). Different conditions were compared by Student t test.
Apoptosis measurement
Cell death was quantified by flow cytometry using propidium iodide and Annexin-V-APC, as previously described, with minor modifications.23 Briefly, HaCaT cells and BJ-1 cells were plated in 12-well plates, with the BJ-1 cells being GFP(+). The day after, media was changed to DMEM with 10% NuSerum. After a total of 4 d in culture, cells were collected by centrifugation and re-suspended in 500 μL of Annexin-V binding buffer. Then, the annexin V-APC conjugate (BD Biosciences 550474) (4 μL) and propidium iodide (1 μL) was added and incubated in the dark at room temperature for 5 min. Cells were then analyzed by flow cytometry using a GFP signal detector (to detect BJ-1-GFP cells), a PE Texas Red signal detector and an APC signal detector.
Beta-galactosidase assay
Assays were performed as previously described.50 Briefly, 240 000 cells were seeded per well in 6-well plates in DMEM with 10% FBS and 1% P/S. The next day, the media was changed to DMEM with 10% Nu serum. Cells were then incubated at 37 °C with 5% CO2, under normal conditions. Then, the cells were trypsinized, centrifuged, and counted to obtain 106 cells. Afterwards, cells were treated according the manufacturer’s instructions, using the FluoReporter lacZ Flow Cytometry Kit (Molecular probes, #F-1930). Assay results were evaluated by flow-cytometry analysis.
Acknowledgments
We are extremely grateful to Dr Nancy Philp, who provided isoform-specific rabbit antibodies directed against MCT1 and MCT4, and Dr Ulrich Rodeck, who provided the 3 well-characterized HaCaT cell lines.
Drs Michael P Lisanti and Federica Sotgia were supported by the resources of Thomas Jefferson University in Philadelphia, USA. Dr Ubaldo E Martinez-Outschoorn was supported by a Young Investigator Award from the Margaret Q Landenberger Research Foundation. Funds were also contributed by the Margaret Q Landenberger Research Foundation (to MPL).
Also, Dr Lisanti’s and Dr Sotgia’s current affiliation is the University of Manchester (United Kingdom), where they receive funding from the Manchester Cancer Research Centre (MCRC), Breakthrough Breast Cancer (BBC), and The European Research Council (ERC).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25510
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–22. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 4.Bissell MJ, Labarge MA. Context, tissue plasticity, and cancer: are tumor stem cells also regulated by the microenvironment? Cancer Cell. 2005;7:17–23. doi: 10.1016/j.ccr.2004.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kenny PA, Lee GY, Bissell MJ. Targeting the tumor microenvironment. Front Biosci. 2007;12:3468–74. doi: 10.2741/2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kenny PA, Nelson CM, Bissell MJ. The Ecology of Tumors: By perturbing the microenvironment, wounds and infection may be key to tumor development. Scientist. 2006;20:30. [PMC free article] [PubMed] [Google Scholar]
- 7.Rønnov-Jessen L, Bissell MJ. Breast cancer by proxy: can the microenvironment be both the cause and consequence? Trends Mol Med. 2009;15:5–13. doi: 10.1016/j.molmed.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12:685–98. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491:364–73. doi: 10.1038/nature11706. [DOI] [PubMed] [Google Scholar]
- 10.Maes H, Rubio N, Garg AD, Agostinis P. Autophagy: shaping the tumor microenvironment and therapeutic response. Trends Mol Med. 2013;19:428–46. doi: 10.1016/j.molmed.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 11.Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10:417–27. doi: 10.1038/nrd3455. [DOI] [PubMed] [Google Scholar]
- 12.Brauer HA, Makowski L, Hoadley KA, Casbas-Hernandez P, Lang LJ, Romàn-Pèrez E, et al. Impact of tumor microenvironment and epithelial phenotypes on metabolism in breast cancer. Clin Cancer Res. 2013;19:571–85. doi: 10.1158/1078-0432.CCR-12-2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17:1498–503. doi: 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De Donatis A, et al. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012;72:5130–40. doi: 10.1158/0008-5472.CAN-12-1949. [DOI] [PubMed] [Google Scholar]
- 15.Fordyce CA, Patten KT, Fessenden TB, Defilippis R, Hwang ES, Zhao J, et al. Cell-extrinsic consequences of epithelial stress: activation of protumorigenic tissue phenotypes. Breast Cancer Res. 2012;14:R155. doi: 10.1186/bcr3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chaudhri VK, Salzler GG, Dick SA, Buckman MS, Sordella R, Karoly ED, et al. Metabolic alterations in lung cancer-associated fibroblasts correlated with increased glycolytic metabolism of the tumor. Mol Cancer Res. 2013;11:579–92. doi: 10.1158/1541-7786.MCR-12-0437-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lisanti MP, Martinez-Outschoorn UE, Chiavarina B, Pavlides S, Whitaker-Menezes D, Tsirigos A, et al. Understanding the “lethal” drivers of tumor-stroma co-evolution: emerging role(s) for hypoxia, oxidative stress and autophagy/mitophagy in the tumor microenvironment. Cancer Biol Ther. 2010;10:537–42. doi: 10.4161/cbt.10.6.13370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lisanti MP, Martinez-Outschoorn UE, Lin Z, Pavlides S, Whitaker-Menezes D, Pestell RG, et al. Hydrogen peroxide fuels aging, inflammation, cancer metabolism and metastasis: the seed and soil also needs “fertilizer”. Cell Cycle. 2011;10:2440–9. doi: 10.4161/cc.10.15.16870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lisanti MP, Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Pestell RG, Howell A, et al. Accelerated aging in the tumor microenvironment: connecting aging, inflammation and cancer metabolism with personalized medicine. Cell Cycle. 2011;10:2059–63. doi: 10.4161/cc.10.13.16233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinez-Outschoorn UE, Balliet R, Lin Z, Whitaker-Menezes D, Birbe RC, Bombonati A, et al. BRCA1 mutations drive oxidative stress and glycolysis in the tumor microenvironment: implications for breast cancer prevention with antioxidant therapies. Cell Cycle. 2012;11:4402–13. doi: 10.4161/cc.22776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez-Outschoorn UE, Balliet RM, Lin Z, Whitaker-Menezes D, Howell A, Sotgia F, et al. Hereditary ovarian cancer and two-compartment tumor metabolism: epithelial loss of BRCA1 induces hydrogen peroxide production, driving oxidative stress and NFκB activation in the tumor stroma. Cell Cycle. 2012;11:4152–66. doi: 10.4161/cc.22226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle. 2010;9:3256–76. doi: 10.4161/cc.9.16.12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez-Outschoorn UE, Goldberg A, Lin Z, Ko YH, Flomenberg N, Wang C, et al. Anti-estrogen resistance in breast cancer is induced by the tumor microenvironment and can be overcome by inhibiting mitochondrial function in epithelial cancer cells. Cancer Biol Ther. 2011;12:924–38. doi: 10.4161/cbt.12.10.17780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez-Outschoorn UE, Lin Z, Ko YH, Goldberg AF, Flomenberg N, Wang C, et al. Understanding the metabolic basis of drug resistance: therapeutic induction of the Warburg effect kills cancer cells. Cell Cycle. 2011;10:2521–8. doi: 10.4161/cc.10.15.16584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinez-Outschoorn UE, Lin Z, Trimmer C, Flomenberg N, Wang C, Pavlides S, et al. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: implications for PET imaging of human tumors. Cell Cycle. 2011;10:2504–20. doi: 10.4161/cc.10.15.16585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinez-Outschoorn UE, Lin Z, Whitaker-Menezes D, Howell A, Lisanti MP, Sotgia F. Ketone bodies and two-compartment tumor metabolism: stromal ketone production fuels mitochondrial biogenesis in epithelial cancer cells. Cell Cycle. 2012;11:3956–63. doi: 10.4161/cc.22136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinez-Outschoorn UE, Lin Z, Whitaker-Menezes D, Howell A, Sotgia F, Lisanti MP. Ketone body utilization drives tumor growth and metastasis. Cell Cycle. 2012;11:3964–71. doi: 10.4161/cc.22137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Tanowitz HB, Sotgia F, et al. Stromal-epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment. Int J Biochem Cell Biol. 2011;43:1045–51. doi: 10.1016/j.biocel.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez-Outschoorn UE, Pavlides S, Sotgia F, Lisanti MP. Mitochondrial biogenesis drives tumor cell proliferation. Am J Pathol. 2011;178:1949–52. doi: 10.1016/j.ajpath.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN, Chiavarina B, et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle. 2010;9:2423–33. doi: 10.4161/cc.9.12.12048. [DOI] [PubMed] [Google Scholar]
- 31.Martinez-Outschoorn UE, Pestell RG, Howell A, Tykocinski ML, Nagajyothi F, Machado FS, et al. Energy transfer in “parasitic” cancer metabolism: mitochondria are the powerhouse and Achilles’ heel of tumor cells. Cell Cycle. 2011;10:4208–16. doi: 10.4161/cc.10.24.18487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinez-Outschoorn UE, Prisco M, Ertel A, Tsirigos A, Lin Z, Pavlides S, et al. Ketones and lactate increase cancer cell “stemness”, driving recurrence, metastasis and poor clinical outcome in breast cancer: achieving personalized medicine via Metabolo-Genomics. Cell Cycle. 2011;10:1271–86. doi: 10.4161/cc.10.8.15330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Power surge: supporting cells “fuel” cancer cell mitochondria. Cell Metab. 2012;15:4–5. doi: 10.1016/j.cmet.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 34.Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment. Cell Cycle. 2010;9:3515–33. doi: 10.4161/cc.9.17.12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martinez-Outschoorn UE, Whitaker-Menezes D, Lin Z, Flomenberg N, Howell A, Pestell RG, et al. Cytokine production and inflammation drive autophagy in the tumor microenvironment: role of stromal caveolin-1 as a key regulator. Cell Cycle. 2011;10:1784–93. doi: 10.4161/cc.10.11.15674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinez-Outschoorn UE, Whitaker-Menezes D, Pavlides S, Chiavarina B, Bonuccelli G, Casey T, et al. The autophagic tumor stroma model of cancer or “battery-operated tumor growth”: A simple solution to the autophagy paradox. Cell Cycle. 2010;9:4297–306. doi: 10.4161/cc.9.21.13817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle. 2010;9:3485–505. doi: 10.4161/cc.9.17.12721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Loss of stromal caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the “reverse Warburg effect”: a transcriptional informatics analysis with validation. Cell Cycle. 2010;9:2201–19. doi: 10.4161/cc.9.11.11848. [DOI] [PubMed] [Google Scholar]
- 39.Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Transcriptional evidence for the “Reverse Warburg Effect” in human breast cancer tumor stroma and metastasis: similarities with oxidative stress, inflammation, Alzheimer’s disease, and “Neuron-Glia Metabolic Coupling”. Aging (Albany NY) 2010;2:185–99. doi: 10.18632/aging.100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pavlides S, Vera I, Gandara R, Sneddon S, Pestell RG, Mercier I, et al. Warburg meets autophagy: cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid Redox Signal. 2012;16:1264–84. doi: 10.1089/ars.2011.4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8:3984–4001. doi: 10.4161/cc.8.23.10238. [DOI] [PubMed] [Google Scholar]
- 42.Sotgia F, Martinez-Outschoorn UE, Howell A, Pestell RG, Pavlides S, Lisanti MP. Caveolin-1 and cancer metabolism in the tumor microenvironment: markers, models, and mechanisms. Annu Rev Pathol. 2012;7:423–67. doi: 10.1146/annurev-pathol-011811-120856. [DOI] [PubMed] [Google Scholar]
- 43.Sotgia F, Martinez-Outschoorn UE, Lisanti MP. Mitochondrial oxidative stress drives tumor progression and metastasis: should we use antioxidants as a key component of cancer treatment and prevention? BMC Med. 2011;9:62. doi: 10.1186/1741-7015-9-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sotgia F, Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Lisanti MP. Understanding the Warburg effect and the prognostic value of stromal caveolin-1 as a marker of a lethal tumor microenvironment. Breast Cancer Res. 2011;13:213. doi: 10.1186/bcr2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sotgia F, Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, et al. Mitochondrial metabolism in cancer metastasis: visualizing tumor cell mitochondria and the “reverse Warburg effect” in positive lymph node tissue. Cell Cycle. 2012;11:1445–54. doi: 10.4161/cc.19841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chiavarina B, Martinez-Outschoorn UE, Whitaker-Menezes D, Howell A, Tanowitz HB, Pestell RG, et al. Metabolic reprogramming and two-compartment tumor metabolism: opposing role(s) of HIF1α and HIF2α in tumor-associated fibroblasts and human breast cancer cells. Cell Cycle. 2012;11:3280–9. doi: 10.4161/cc.21643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chiavarina B, Whitaker-Menezes D, Martinez-Outschoorn UE, Witkiewicz AK, Birbe RC, Howell A, et al. Pyruvate kinase expression (PKM1 and PKM2) in cancer-associated fibroblasts drives stromal nutrient production and tumor growth. Cancer Biol Ther. 2011;12:1101–13. doi: 10.4161/cbt.12.12.18703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiavarina B, Whitaker-Menezes D, Migneco G, Martinez-Outschoorn UE, Pavlides S, Howell A, et al. HIF1-alpha functions as a tumor promoter in cancer associated fibroblasts, and as a tumor suppressor in breast cancer cells: Autophagy drives compartment-specific oncogenesis. Cell Cycle. 2010;9:3534–51. doi: 10.4161/cc.9.17.12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Capparelli C, Chiavarina B, Whitaker-Menezes D, Pestell TG, Pestell RG, Hulit J, et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle. 2012;11:3599–610. doi: 10.4161/cc.21884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Capparelli C, Guido C, Whitaker-Menezes D, Bonuccelli G, Balliet R, Pestell TG, et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production. Cell Cycle. 2012;11:2285–302. doi: 10.4161/cc.20718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Capparelli C, Whitaker-Menezes D, Guido C, Balliet R, Pestell TG, Howell A, et al. CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle. 2012;11:2272–84. doi: 10.4161/cc.20717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carito V, Bonuccelli G, Martinez-Outschoorn UE, Whitaker-Menezes D, Caroleo MC, Cione E, et al. Metabolic remodeling of the tumor microenvironment: migration stimulating factor (MSF) reprograms myofibroblasts toward lactate production, fueling anabolic tumor growth. Cell Cycle. 2012;11:3403–14. doi: 10.4161/cc.21701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Migneco G, Whitaker-Menezes D, Chiavarina B, Castello-Cros R, Pavlides S, Pestell RG, et al. Glycolytic cancer associated fibroblasts promote breast cancer tumor growth, without a measurable increase in angiogenesis: evidence for stromal-epithelial metabolic coupling. Cell Cycle. 2010;9:2412–22. doi: 10.4161/cc.9.12.11989. [DOI] [PubMed] [Google Scholar]
- 54.Avena P, Anselmo W, Whitaker-Menezes D, Wang C, Pestell RG, Lamb RS, et al. Compartment-specific activation of PPARγ governs breast cancer tumor growth, via metabolic reprogramming and symbiosis. Cell Cycle. 2013;12:1360–70. doi: 10.4161/cc.24289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle. 2011;10:4047–64. doi: 10.4161/cc.10.23.18151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle. 2011;10:1772–83. doi: 10.4161/cc.10.11.15659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ren Q, Kari C, Quadros MR, Burd R, McCue P, Dicker AP, et al. Malignant transformation of immortalized HaCaT keratinocytes through deregulated nuclear factor kappaB signaling. Cancer Res. 2006;66:5209–15. doi: 10.1158/0008-5472.CAN-05-4158. [DOI] [PubMed] [Google Scholar]
- 58.Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988;106:761–71. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mates JM, Segura JA, Alonso FJ, Marquez J. Sulphur-containing non enzymatic antioxidants: therapeutic tools against cancer. Front Biosci (Schol Ed) 2012;4:722–48. doi: 10.2741/S296. [Schol Ed] [DOI] [PubMed] [Google Scholar]
- 60.Shimamoto K, Hayashi H, Taniai E, Morita R, Imaoka M, Ishii Y, et al. Antioxidant N-acetyl-L-cysteine (NAC) supplementation reduces reactive oxygen species (ROS)-mediated hepatocellular tumor promotion of indole-3-carbinol (I3C) in rats. J Toxicol Sci. 2011;36:775–86. doi: 10.2131/jts.36.775. [DOI] [PubMed] [Google Scholar]
- 61.Miller MS, Moore JE, Walb MC, Kock ND, Attia A, Isom S, et al. Chemoprevention by N-acetylcysteine of low-dose CT-induced murine lung tumorigenesis. Carcinogenesis. 2013;34:319–24. doi: 10.1093/carcin/bgs332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sathish P, Paramasivan V, Palani V, Sivanesan K. N-acetylcysteine attenuates dimethylnitrosamine induced oxidative stress in rats. Eur J Pharmacol. 2011;654:181–6. doi: 10.1016/j.ejphar.2010.10.080. [DOI] [PubMed] [Google Scholar]
- 63.Izzotti A, Calin GA, Steele VE, Cartiglia C, Longobardi M, Croce CM, et al. Chemoprevention of cigarette smoke-induced alterations of MicroRNA expression in rat lungs. Cancer Prev Res (Phila) 2010;3:62–72. doi: 10.1158/1940-6207.CAPR-09-0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Balansky R, Ganchev G, Iltcheva M, Steele VE, De Flora S. Prevention of cigarette smoke-induced lung tumors in mice by budesonide, phenethyl isothiocyanate, and N-acetylcysteine. Int J Cancer. 2010;126:1047–54. doi: 10.1002/ijc.24942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Balansky R, Ganchev G, Iltcheva M, Steele VE, De Flora S. Prenatal N-acetylcysteine prevents cigarette smoke-induced lung cancer in neonatal mice. Carcinogenesis. 2009;30:1398–401. doi: 10.1093/carcin/bgp128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.De Flora S, Scarfì S, Izzotti A, D’Agostini F, Chang CC, Bagnasco M, et al. Induction by 7,12-dimethylbenz(a)anthracene of molecular and biochemical alterations in transformed human mammary epithelial stem cells, and protection by N-acetylcysteine. Int J Oncol. 2006;29:521–9. [PubMed] [Google Scholar]
- 67.Gao P, Zhang H, Dinavahi R, Li F, Xiang Y, Raman V, et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell. 2007;12:230–8. doi: 10.1016/j.ccr.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wilpart M, Mainguet P, Geeroms D, Roberfroid M. Desmutagenic effects of N-acetylcysteine on direct and indirect mutagens. Mutat Res. 1985;142:169–77. doi: 10.1016/0165-7992(85)90018-1. [DOI] [PubMed] [Google Scholar]
- 69.De Flora S, Rossi GA, De Flora A. Metabolic, desmutagenic and anticarcinogenic effects of N-acetylcysteine. Respiration. 1986;50(Suppl 1):43–9. doi: 10.1159/000195087. [DOI] [PubMed] [Google Scholar]
- 70.van Zandwijk N. N-acetylcysteine (NAC) and glutathione (GSH): antioxidant and chemopreventive properties, with special reference to lung cancer. J Cell Biochem Suppl. 1995;22:24–32. doi: 10.1002/jcb.240590805. [DOI] [PubMed] [Google Scholar]
- 71.Witkiewicz AK, Dasgupta A, Nguyen KH, Liu C, Kovatich AJ, Schwartz GF, et al. Stromal caveolin-1 levels predict early DCIS progression to invasive breast cancer. Cancer Biol Ther. 2009;8:1071–9. doi: 10.4161/cbt.8.11.8874. [DOI] [PubMed] [Google Scholar]
- 72.Witkiewicz AK, Dasgupta A, Sammons S, Er O, Potoczek MB, Guiles F, et al. Loss of stromal caveolin-1 expression predicts poor clinical outcome in triple negative and basal-like breast cancers. Cancer Biol Ther. 2010;10:135–43. doi: 10.4161/cbt.10.2.11983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Witkiewicz AK, Dasgupta A, Sotgia F, Mercier I, Pestell RG, Sabel M, et al. An absence of stromal caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breast cancers. Am J Pathol. 2009;174:2023–34. doi: 10.2353/ajpath.2009.080873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Witkiewicz AK, Kline J, Queenan M, Brody JR, Tsirigos A, Bilal E, et al. Molecular profiling of a lethal tumor microenvironment, as defined by stromal caveolin-1 status in breast cancers. Cell Cycle. 2011;10:1794–809. doi: 10.4161/cc.10.11.15675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu KN, Queenan M, Brody JR, Potoczek M, Sotgia F, Lisanti MP, et al. Loss of stromal caveolin-1 expression in malignant melanoma metastases predicts poor survival. Cell Cycle. 2011;10:4250–5. doi: 10.4161/cc.10.24.18551. [DOI] [PubMed] [Google Scholar]
- 76.Di Vizio D, Morello M, Sotgia F, Pestell RG, Freeman MR, Lisanti MP. An absence of stromal caveolin-1 is associated with advanced prostate cancer, metastatic disease and epithelial Akt activation. Cell Cycle. 2009;8:2420–4. doi: 10.4161/cc.8.15.9116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Koo JS, Park S, Kim SI, Lee S, Park BW. The impact of caveolin protein expression in tumor stroma on prognosis of breast cancer. Tumour Biol. 2011;32:787–99. doi: 10.1007/s13277-011-0181-6. [DOI] [PubMed] [Google Scholar]
- 78.Qian N, Ueno T, Kawaguchi-Sakita N, Kawashima M, Yoshida N, Mikami Y, et al. Prognostic significance of tumor/stromal caveolin-1 expression in breast cancer patients. Cancer Sci. 2011;102:1590–6. doi: 10.1111/j.1349-7006.2011.01985.x. [DOI] [PubMed] [Google Scholar]
- 79.Simpkins SA, Hanby AM, Holliday DL, Speirs V. Clinical and functional significance of loss of caveolin-1 expression in breast cancer-associated fibroblasts. J Pathol. 2012;227:490–8. doi: 10.1002/path.4034. [DOI] [PubMed] [Google Scholar]
- 80.Sloan EK, Ciocca DR, Pouliot N, Natoli A, Restall C, Henderson MA, et al. Stromal cell expression of caveolin-1 predicts outcome in breast cancer. Am J Pathol. 2009;174:2035–43. doi: 10.2353/ajpath.2009.080924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.El-Gendi SM, Mostafa MF, El-Gendi AM. Stromal caveolin-1 expression in breast carcinoma. Correlation with early tumor recurrence and clinical outcome. Pathol Oncol Res. 2012;18:459–69. doi: 10.1007/s12253-011-9469-5. [DOI] [PubMed] [Google Scholar]
- 82.Ayala G, Morello M, Frolov A, You S, Li R, Rosati F, et al. Loss of Caveolin-1 in Prostate Cancer Stroma Correlates with Reduced Relapse-Free Survival and is Functionally Relevant to Tumor Progression. J Pathol. 2013 doi: 10.1002/path.4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Curry JM, Tuluc M, Whitaker-Menezes D, Ames JA, Anantharaman A, Butera A, et al. Cancer metabolism, stemness and tumor recurrence: MCT1 and MCT4 are functional biomarkers of metabolic symbiosis in head and neck cancer. Cell Cycle. 2013;12:1371–84. doi: 10.4161/cc.24092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Witkiewicz AK, Whitaker-Menezes D, Dasgupta A, Philp NJ, Lin Z, Gandara R, et al. Using the “reverse Warburg effect” to identify high-risk breast cancer patients: stromal MCT4 predicts poor clinical outcome in triple-negative breast cancers. Cell Cycle. 2012;11:1108–17. doi: 10.4161/cc.11.6.19530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang W, Trachootham D, Liu J, Chen G, Pelicano H, Garcia-Prieto C, et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol. 2012;14:276–86. doi: 10.1038/ncb2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118:3930–42. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Semenza GL. Tumor metabolism: cancer cells give and take lactate. J Clin Invest. 2008;118:3835–7. doi: 10.1172/JCI37373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dhup S, Dadhich RK, Porporato PE, Sonveaux P. Multiple biological activities of lactic acid in cancer: influences on tumor growth, angiogenesis and metastasis. Curr Pharm Des. 2012;18:1319–30. doi: 10.2174/138161212799504902. [DOI] [PubMed] [Google Scholar]
- 89.Martinez-Outschoorn UE, Lin Z, Trimmer C, Flomenberg N, Wang C, Pavlides S, et al. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: implications for PET imaging of human tumors. Cell Cycle. 2011;10:2504–20. doi: 10.4161/cc.10.15.16585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Salem AF, Howell A, Sartini M, Sotgia F, Lisanti MP. Downregulation of stromal BRCA1 drives breast cancer tumor growth via upregulation of HIF-1α, autophagy and ketone body production. Cell Cycle. 2012;11:4167–73. doi: 10.4161/cc.22316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Salem AF, Whitaker-Menezes D, Howell A, Sotgia F, Lisanti MP. Mitochondrial biogenesis in epithelial cancer cells promotes breast cancer tumor growth and confers autophagy resistance. Cell Cycle. 2012;11:4174–80. doi: 10.4161/cc.22376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Salem AF, Whitaker-Menezes D, Lin Z, Martinez-Outschoorn UE, Tanowitz HB, Al-Zoubi MS, et al. Two-compartment tumor metabolism: autophagy in the tumor microenvironment and oxidative mitochondrial metabolism (OXPHOS) in cancer cells. Cell Cycle. 2012;11:2545–56. doi: 10.4161/cc.20920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sotgia F, Whitaker-Menezes D, Martinez-Outschoorn UE, Salem AF, Tsirigos A, Lamb R, et al. Mitochondria “fuel” breast cancer metabolism: fifteen markers of mitochondrial biogenesis label epithelial cancer cells, but are excluded from adjacent stromal cells. Cell Cycle. 2012;11:4390–401. doi: 10.4161/cc.22777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Langley RR, Fidler IJ. The seed and soil hypothesis revisited--the role of tumor-stroma interactions in metastasis to different organs. Int J Cancer. 2011;128:2527–35. doi: 10.1002/ijc.26031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3:453–8. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 96.Fidler IJ, Poste G. The “seed and soil” hypothesis revisited. Lancet Oncol. 2008;9:808. doi: 10.1016/S1470-2045(08)70201-8. [DOI] [PubMed] [Google Scholar]
- 97.Fidler IJ, Yano S, Zhang RD, Fujimaki T, Bucana CD. The seed and soil hypothesis: vascularisation and brain metastases. Lancet Oncol. 2002;3:53–7. doi: 10.1016/S1470-2045(01)00622-2. [DOI] [PubMed] [Google Scholar]
- 98.Guido C, Whitaker-Menezes D, Capparelli C, Balliet R, Lin Z, Pestell RG, et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: connecting TGF-β signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell Cycle. 2012;11:3019–35. doi: 10.4161/cc.21384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sanchez-Alvarez R, Martinez-Outschoorn UE, Lin Z, Lamb R, Hulit J, Howell A, et al. Ethanol exposure induces the cancer-associated fibroblast phenotype and lethal tumor metabolism: implications for breast cancer prevention. Cell Cycle. 2013;12:289–301. doi: 10.4161/cc.23109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Skrtić M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2011;20:674–88. doi: 10.1016/j.ccr.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Duivenvoorden WC, Popović SV, Lhoták S, Seidlitz E, Hirte HW, Tozer RG, et al. Doxycycline decreases tumor burden in a bone metastasis model of human breast cancer. Cancer Res. 2002;62:1588–91. [PubMed] [Google Scholar]
- 102.van den Bogert C, Dontje BH, Holtrop M, Melis TE, Romijn JC, van Dongen JW, et al. Arrest of the proliferation of renal and prostate carcinomas of human origin by inhibition of mitochondrial protein synthesis. Cancer Res. 1986;46:3283–9. [PubMed] [Google Scholar]
- 103.Oikonomopoulou K, Brinc D, Kyriacou K, Diamandis EP. Infection and cancer: revaluation of the hygiene hypothesis. Clin Cancer Res. 2013;19:2834–41. doi: 10.1158/1078-0432.CCR-12-3661. [DOI] [PubMed] [Google Scholar]
- 104.Maeda H. The link between infection and cancer: Tumor vasculature, free radicals, and drug delivery to tumors via the EPR effect. Cancer Sci 2013; http://www.ncbi.nlm.nih.gov/pubmed/23495730 [DOI] [PMC free article] [PubMed]
- 105.Sarrazy V, Billet F, Micallef L, Coulomb B, Desmoulière A. Mechanisms of pathological scarring: role of myofibroblasts and current developments. Wound Repair Regen. 2011;19(Suppl 1):s10–5. doi: 10.1111/j.1524-475X.2011.00708.x. [DOI] [PubMed] [Google Scholar]
- 106.Angeli F, Koumakis G, Chen MC, Kumar S, Delinassios JG. Role of stromal fibroblasts in cancer: promoting or impeding? Tumour Biol. 2009;30:109–20. doi: 10.1159/000218708. [DOI] [PubMed] [Google Scholar]
- 107.Zalatnai A. Molecular aspects of stromal-parenchymal interactions in malignant neoplasms. Curr Mol Med. 2006;6:685–93. doi: 10.2174/156652406778195053. [DOI] [PubMed] [Google Scholar]
- 108.Linder S, Castaños-Velez E, von Rosen A, Biberfeld P. Immunohistochemical expression of extracellular matrix proteins and adhesion molecules in pancreatic carcinoma. Hepatogastroenterology. 2001;48:1321–7. [PubMed] [Google Scholar]
- 109.Hasebe T, Tsuda H, Hirohashi S, Shimosato Y, Iwai M, Imoto S, et al. Fibrotic focus in invasive ductal carcinoma: an indicator of high tumor aggressiveness. Jpn J Cancer Res. 1996;87:385–94. doi: 10.1111/j.1349-7006.1996.tb00234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hasebe T, Imoto S, Ogura T, Mukai K. Significance of basic fibroblast growth factor and fibroblast growth factor receptor protein expression in the formation of fibrotic focus in invasive ductal carcinoma of the breast. Jpn J Cancer Res. 1997;88:877–85. doi: 10.1111/j.1349-7006.1997.tb00464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hasebe T, Mukai K, Tsuda H, Ochiai A. New prognostic histological parameter of invasive ductal carcinoma of the breast: clinicopathological significance of fibrotic focus. Pathol Int. 2000;50:263–72. doi: 10.1046/j.1440-1827.2000.01035.x. [DOI] [PubMed] [Google Scholar]
- 112.Hasebe T, Sasaki S, Imoto S, Mukai K, Yokose T, Ochiai A. Prognostic significance of fibrotic focus in invasive ductal carcinoma of the breast: a prospective observational study. Mod Pathol. 2002;15:502–16. doi: 10.1038/modpathol.3880555. [DOI] [PubMed] [Google Scholar]
- 113.Hasebe T, Sasaki S, Imoto S, Ochiai A. Highly proliferative fibroblasts forming fibrotic focus govern metastasis of invasive ductal carcinoma of the breast. Mod Pathol. 2001;14:325–37. doi: 10.1038/modpathol.3880310. [DOI] [PubMed] [Google Scholar]
- 114.Hasebe T, Tsuda H, Tsubono Y, Imoto S, Mukai K. Fibrotic focus in invasive ductal carcinoma of the breast: a histopathological prognostic parameter for tumor recurrence and tumor death within three years after the initial operation. Jpn J Cancer Res. 1997;88:590–9. doi: 10.1111/j.1349-7006.1997.tb00423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gallagher SM, Castorino JJ, Wang D, Philp NJ. Monocarboxylate transporter 4 regulates maturation and trafficking of CD147 to the plasma membrane in the metastatic breast cancer cell line MDA-MB-231. Cancer Res. 2007;67:4182–9. doi: 10.1158/0008-5472.CAN-06-3184. [DOI] [PubMed] [Google Scholar]