

Abstract
The Wip1 phosphatase is an oncogene that is overexpressed in a variety of primary human cancers. We were interested in identifying genetic variants that could change Wip1 activity. We identified 3 missense SNPs of the human Wip1 phosphatase, L120F, P322Q, and I496V confer a dominant-negative phenotype. On the other hand, in primary human cancers, PPM1D mutations commonly result in a gain-of-function phenotype, leading us to identify a hot-spot truncating mutation at position 525. Surprisingly, we also found a significant number of loss-of-function mutations of PPM1D in primary human cancers, both in the phosphatase domain and in the C terminus. Thus, PPM1D has evolved to generate genetic variants with lower activity, potentially providing a better fitness for the organism through suppression of multiple diseases. In cancer, however, the situation is more complex, and the presence of both activating and inhibiting mutations requires further investigation to understand their contribution to tumorigenesis.
Keywords: Wip1, PPM1D, genetic variants, mutations, human cancer, ATM, Chk2
Introduction
The wild-type p53-induced phosphatase 1, Wip1 (encoded by the gene PPM1D), is a serine/threonine phosphatase that is amplified and overexpressed in a variety of human cancers.1-6 Wip1 is a regulator of the DNA damage response and has been shown to dephosphorylate key members of the checkpoint pathway including ATM,7 p53,8 Chk2,9,10 and Chk1.11 In mice, Wip1 deficiency has been shown to alleviate many cancer phenotypes, including mammary tumorigenesis driven by H-ras or Neu,12 Eµ-myc-induced B-cell lymphomas,13 APC-induced intestinal polyposis,14 as well as spontaneous tumorigenesis.15 More recently we found that deletion of Ppm1d in mice results in suppression of body fat accumulation and atherosclerosis through regulation of ATM-dependent suppression of the mTor pathway.16 Given the importance of ATM in regulating a variety of pathological conditions, it is therefore conceivable that the existence of natural variants of human PPM1D could be linked to risk of several diseases, including cancer and cardiovascular pathologies.
The most common natural sequence variation in the human genome is the stable substitution of a single base, the single nucleotide polymorphism (SNP). By definition, a SNP has a minor allele frequency of greater than 1% in at least one population. In turn, sequence variations with allelic frequency of less than 1% are referred to as genetic variants. SNPs and genetic variants can change the activity or expression of a protein and are associated with risk for diseases.17 In addition, a recent study identified several C-terminal mutations in Wip1 that are associated with predisposition to breast and ovarian cancer.18
Here we show that several variants of Wip1 can attenuate its phosphatase activity and modulate the response to DNA damage. Moreover, we identified numerous point mutations in primary human cancers, including hot spot-truncating mutations at positions E525 and R552. Surprisingly, these mutations resulted both in gain- and loss-of-function in Wip1. Thus, the role of Wip1 in primary human cancer is complex and most likely context-dependent by both promoting tumorigenesis in some cases while suppressing in others.
Results
Missense mutations account for approximately half of all DNA mutations that are known to cause genetic disease according to the Human Gene Mutation Database.19 Some of these mutations are SNPs and/or genetic variants that have been shown to affect protein activity and expression levels in the cell. Therefore we focused our attention on 4 published missense SNPs/genetic variants of Wip1 affecting amino acids A82S, L120F, P322Q, and I496V, as shown in Table 1. Three of these genetic variants, A82S, L120F, and P322Q, have mutations which lie in the phosphatase domain of Wip1 (amino acids G67-T368), thus potentially affecting its activity, whereas I496V is localized to the C-terminal domain (amino acids S369-C605). The predicted protein structure model using modeling software (Fig. 1A) shows the di-magnesium active site and the residues A82S, L120F, and P332Q (I496V is not in the catalytic domain and therefore could not be modeled). The protein is shown as a transparent surface, which depicts the active site cleft and the relative position of the three residues. Although A82 is nearest to the active site, it is exposed on the other side of the protein. Mutation to Ser most likely would have little effect on the activity due to its hydrophilicity and thus should not apply any distorting force on the active-site geometry (Fig. 1B). However, L120 is buried in the domain that makes the cleft-wall to the right of the active site (Fig. 1B). Due to tight packing of that area, the mutation to Phe could distort the conformation of this domain. This should cause a reduction in activity by reducing access to the active site and/or distort the geometry around D105 and the Mg2+-binding. Similarly, Figure 1C shows how P322 is also structurally linked to Mg2+ binding through D314 in the active site. P322 also initiates an α helix, which is common for proline residues due to their stabilized backbone geometry. Mutation to Gln may destabilize this helix and the link to the active site, compromising the activity. Additionally, although P322 is exposed to the solvent, it is located at the center of a hydrophobic patch. Thus the change to polar Gln may also lead to distortion of the active site and reduction in accessibility. These results suggest that these variants may affect Wip1 phosphatase activity.
Table 1.PPM1D genetic variants.
| Region | Contig position | mRNA position | rs# | Heterozygosity | mutation type | nucleotide change | amino acid position | amino acid change |
|---|---|---|---|---|---|---|---|---|
| exon1 |
17330926 |
312 |
rs16944543 |
0.075 |
synonymous |
G→A |
30 |
E→E |
| |
17331080 |
466 |
rs34632861 |
0.043 |
missense |
G→T |
82 |
A→S |
| |
17331196 |
582 |
rs17853242 |
ND |
missense |
G→T |
120 |
L→F |
| exon 2 |
17354080 |
832 |
rs35595160 |
ND |
frame shift |
A→- |
204 |
K→X |
| exon 4 |
17378452 |
1187 |
rs17855093 |
ND |
missense |
C→A |
322 |
P→Q |
| exon 6 | 17393642 | 1708 | rs35491690 | 0.028 | missense | A→G | 496 | I→V |
Genetic variants within PPM1D were selected from the National Center for Biotechnology Information database (http://www.ncbi.nlm.nih.gov/SNP). ND, not determined.
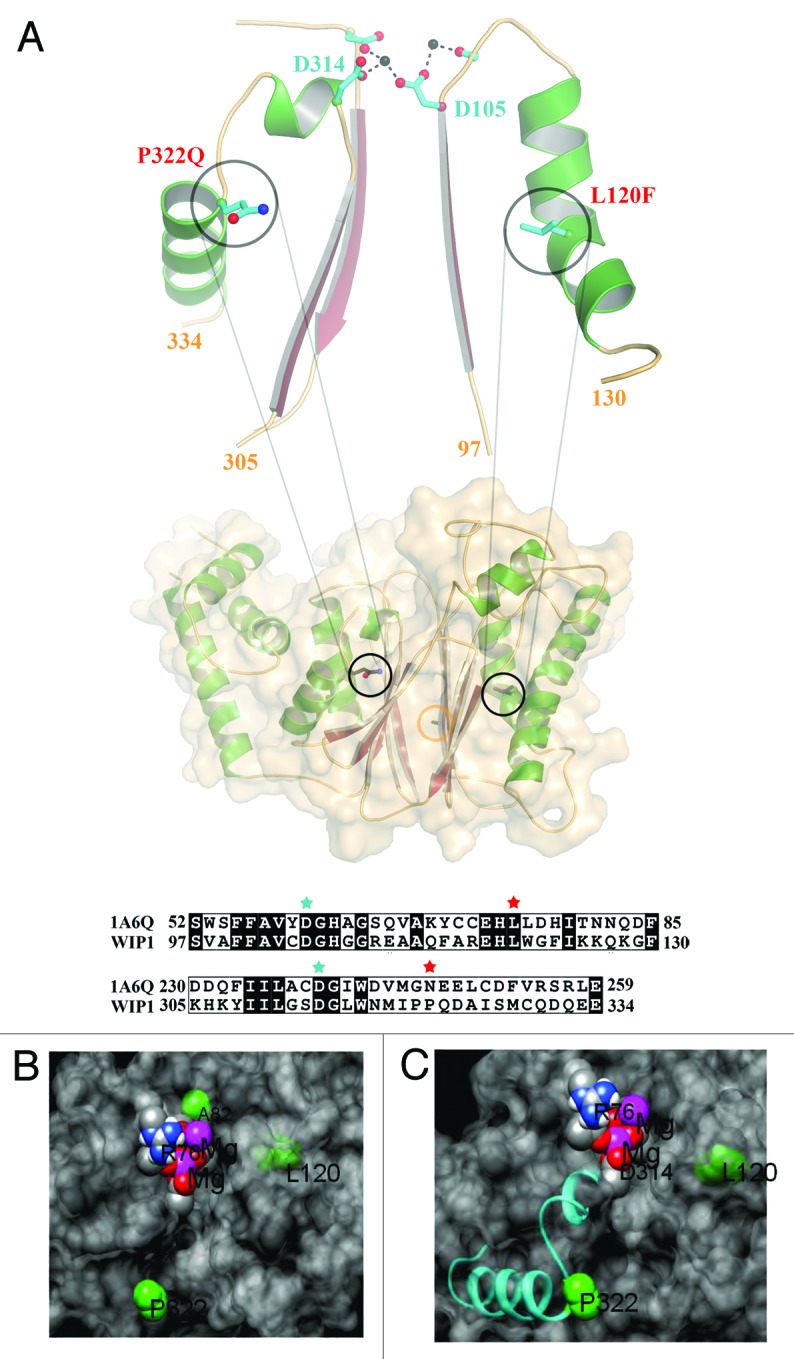
Figure 1. The structural model for Wip1 genetic variants. (A) The model of Wip1 was generated based on the structure of phosphatase 2C (1A6G). L120F and P322Q point towards β-sheets in this α-β-β-α sandwich fold. Both mutations are located in α-helices that follow loops, which contain metal ion coordinating aspartic acids in the active site, Asp105 and Asp314, respectively. The L120F (L is shown) mutation may be expected to alter the conformation of the preceding loop and Asp105 due to its increased size being accommodated in the α-β packing. P322Q (Q is shown) defines the end of the α-helix. The restricted geometry of helix-breaking proline and the increased bulk of glutamine will similarly affect the path of the preceding loop and Asp314. Sequence alignments of these regions of Wip1 with phosphatase 2C (1A6G) are shown in the lower panel. L120 and P322 are highlighted by red stars. D105 and D314 are denoted by cyan stars. The position of Ala82 is demarked by an orange circle. The model was generated in the program COOT. (B and C) Close views of the variants (green) in relation to the surface (gray), magnesium ions (purple), oxygen atoms of D105 and D314 (red) and the P332 containing helix (cyan).
To determine the activity of the genetic variants located in the phosphatase domain, we expressed and purified the mutant proteins. For these experiments, we had to use a truncated form of Wip1 (missing exon 6), as the full-length Wip1 is poorly soluble. The purified proteins were then used to measure the phospho-ATM peptide concentration dependence of Wip1 phosphatase activity. Both Km and Vmax values for the A82S mutant were almost the same as WT (Fig. 2A). These data indicated that the single amino acid substitution Ser for Ala in position 82 has no effect on the catalytic activity of Wip1. The Km value for the L120F mutant was 9 times higher than the value obtained for the WT, indicating that this mutant is less efficient in recognizing the phospho-ATM peptide as a substrate. The Km value for the P322Q mutant was only 2 times higher than the value for WT, whereas the Vmax value was 4 times lower than the WT (Fig. 2A). These data suggest that the phospho-ATM peptide binds to the P322Q mutant, but dephosphorylation by Wip1 is compromised. Taken together, A82S most likely behaves like WT Wip1, whereas L120F and P322Q could have reduced phosphatase activity and potentially have differences in biological activity in vivo.

Figure 2. Wip1 variants A82S, L120F, and P322Q affect the DNA damage response pathway and biological response to IR. (A) Phosphatase activity was measured by a malachite green/molybdate-based assay as described in “Materials and Methods”. Wip1 L120F(green triangle) and P322Q (blue square) both have a reduced phosphatase activity, while Wip1 A82S (orange triangle) has a phosphatase activity similar to Wip1 WT (red circle). (B) HeLa cells were transfected with Wip1 WT and variants, then irradiated the next day for 30 min with 5 Gy IR. Western blotting was completed using antibodies specific to p-ATM (Chemicon), ATM, p-Chk2, Chk2, GFP, and p38 (loading control). (C)After transfection of a 1:1 ratio of Wip1 WT and L120F (left) or P322Q (right), cells were treated as in (B) and probed with the same antibodies.(D)Colony-formation assay was performed using HeLa cells transfected with empty vector, pCAG-GFP Wip1 WT, or L120F or P322Q. Following irradiation, cells were plated for colony formation as described in “Materials and Methods”. Colonies were imaged (left) and counted (right).
In order to analyze the significance of the mutant alleles in vivo, we transiently transfected HeLa cells with either the Wip1 WT or genetic variants to determine the response of the cells to irradiation-induced DNA damage. Consistent with Figure 2A and previous reports, overexpression of WT or A82S allele attenuated the ATM-mediated DNA damage response pathway, as shown by decreased phosphorylation of ATM and Chk2. However, the results were different in cells expressing the L120F and P322Q alleles (Fig. 2B), behaving similar to controls without Wip1 overexpression, suggesting that these two variants have reduced phosphatase activity in vivo.
Since the phenotype of A82S allele mirrored that of WT, we decided to investigate only L120F and P322Q in detail. To understand whether these two variants could function as a dominant negative, we overexpressed the WT and L120F (Fig. 2C, left panel) or P322Q (Fig. 2C, right panel) allele in a 1:1 ratio in HeLa cells. A subtle but clear difference in the levels of phospho-ATM and phospho-Chk2 were seen, suggesting that L120F and P322Q can function to suppress the ability of WT Wip1 to reduce ATM-dependent signaling.
To understand the biological significance of these differences, we next transfected cells with either GFP-tagged Wip1 WT, L120F, or P322Q. ATM activity is required for efficient suppression of colony formation,20 and suppression of ATM with WT Wip1 results in increased number of colonies after irradiation of cells with 6 Gy of IR (Fig. 2D). In contrast, cells expressing L120F and P322Q formed fewer colonies compared with that of the WT allele, confirming that the ATM-mediated DNA damage pathway was not downregulated efficiently in these cells. Furthermore, when L120F or P322Q were expressed together with the WT allele in a 1:1 ratio, the colony-forming ability was also significantly diminished, demonstrating that the mutant alleles can function as a dominant negative. These results suggest that the presence of a single allele of either genetic variant could have a dominant-negative effect on the remaining wild-type allele.
We repeated the same analysis used for A82S, L120F, and P322Q on I496V, which is a missense mutation located in the C terminus before the two putative localization signals (KRTLEESNSGPLMKKHRR (amino acids 535–552) and RRRLRGQKK (amino acids 581–589 aa).10 We could not express a full-length protein containing I496V mutation in bacteria, as it is poorly soluble, so we analyzed its activity after transfection into HeLa cells. Analysis of phosphatase activity based on dephosphorylation of a phospho-ATM peptide demonstrated that I496V has phosphatase activity, although more of the protein is required for that activity (Fig. 3A). To determine the significance of I496V in vivo, we transiently transfected HeLa cells as before and irradiated them to ascertain the ATM-mediated DNA damage response. I496V behaved similarly to Wip1 WT when transfected solo, causing a decrease in p-ATM and p-Chk2 after IR (Fig. 3B), although surprisingly it behaved more like a dominant negative when mixed with WT at a ratio of 1:1. To confirm this activity, colony formation was performed following 6 Gy irradiation. There was a slight decrease in colony formation of I496V compared with WT (P = 0.04), whereas the combination of I496V with WT gave a significant reduction in colony formation (P < 0.001) (Fig. 3C). To understand the mechanism of these results, the localization of I496V was ascertained to see if the mutant remained nuclear, as is required for Wip1’s phosphatase activity on ATM, H2AX, and Chk2. As shown in Figure 3D, I496V was localized to both the cytoplasm and nucleus with a higher ratio than WT (A82S, L120F, and P322Q had no change in localization, data no shown). Taken together, these results suggest that I496V has diminished phosphatase activity, partly due to its localization in vivo, but has a weaker dominant-negative phenotype than L120F and P322Q.

Figure 3. Wip1 I496V affects the response to the DNA damage pathway. (A) 293T cells were transfected with the plasmid encoding either Wip1-WT-Flag (10 µg) or Wip1-I496V-Flag (12.5, 15, 17.5, and 20 µg). Phosphatase activity of Wip1 was measured in vitro with immunoprecipitated Flag-Wip1 WT or I496V protein in the presence of 100 µM ATM- pS1981 peptide (left). Immunoprecipitated Wip1 was also blotted for Wip1 (Santa Cruz, H300) to check protein amounts. (B) HeLa cells were transfected with a Flag-Wip1 WT or I496V and irradiated as before. Protein extracts were blotted for p-ATM, ATM, p-Chk2, Chk2, Flag, and actin as a loading control. (C) HeLa cells were transfected as in (B) and treated as before in Figure 2D.(D)Wip1 WT and I496V cellular localization was determined by nuclear/cytoplasmic extraction (left) and immunofluorescence (right). Nuclear and cytoplasmic extract was blotted using Flag (Wip1), H3 as a nuclear extract control, and GAPDH as a cytoplasmic extract control (left). Flag antibody was used to detect Wip1 WT and I496V, with DAPI as a DNA marker.
We next turned to the analysis of Wip1 mutations in primary human cancers using available cancer data sets (http://www.cbioportal.org). We found that Wip1 mutations were most frequent in uterine corpus endometrioid carcinoma (4.4%) followed by lung adenocarcinoma (2.2%), renal carcinoma (1.8%), colon and rectum adenocarcinoma (1.8%), stomach adenocarcinoma (1.7%), and thyroid carcinoma (1.5%), and some other types of cancer with the frequency less than 1% (Fig. 4A). No PPM1D mutations were found in acute myeloid leukemia, cancers of the bladder, prostate, and ovaries. Analysis of mutation positions revealed that two-thirds of them were located in the C terminus of Wip1, although one-quarter was found within the phosphatase domain (Table 2). All the mutations were present on different amino acids, with the exception of only 2 sites, amino acid E525 (4 out of 41) and R552 (3 out of 41).
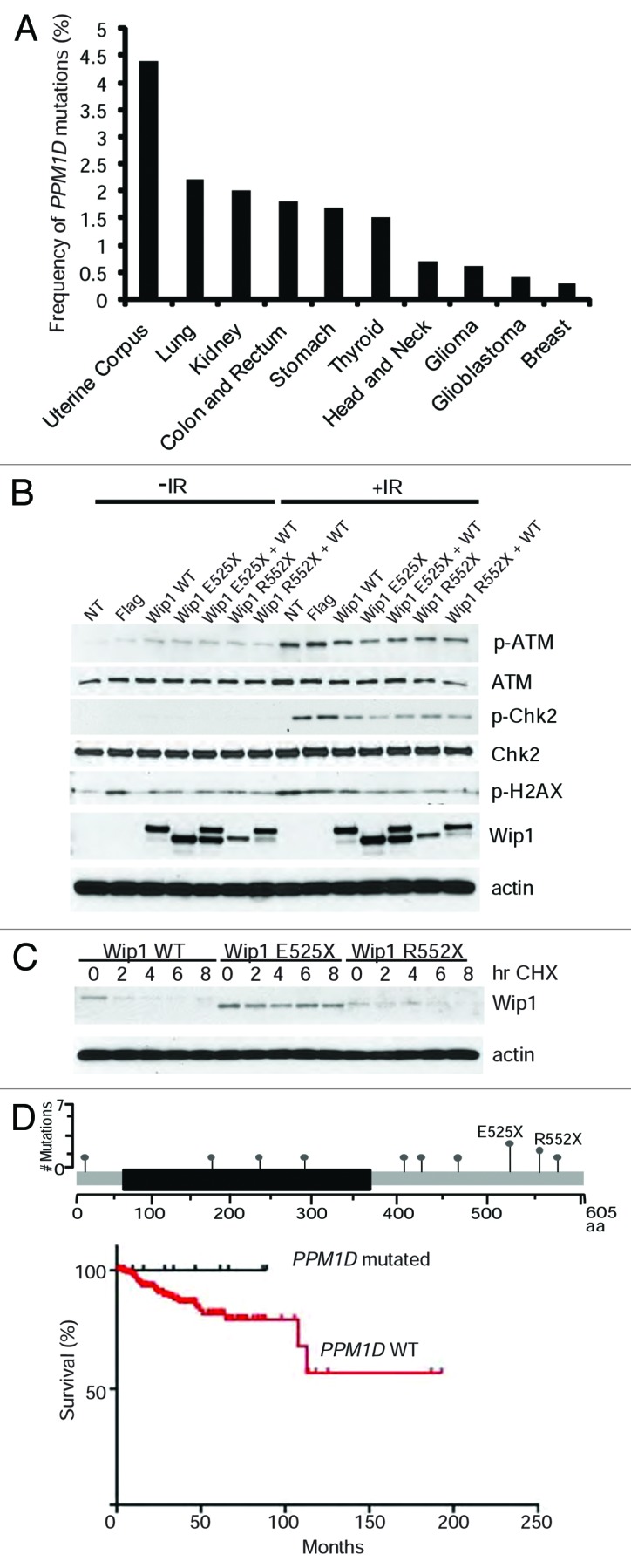
Figure 4. Loss- and gain-of-function mutations in primary human cancers. (A) Analysis different primary human tumors for the presence of PPM1D mutations. The frequency of mutations is shown. (B) Wip1 truncation mutants, E525X and R552X, have varying biochemical behaviors. HeLa cells were transfected with plasmids containing Wip1 WT, Wip1 E525X, Wip1 R552X, and a combination of WT with the truncation mutants. The cells were irradiated 24 h post-transfection with 0 or 5 Gy and protein harvested after 1 h. Antibodies used for western blotting are p-ATM, ATM, p-CHK2, CHK2, Wip1 (H300), and actin as a loading control. The experiment was repeated in triplicate. (C)Wip1 truncation mutants display variances in their protein stability. HeLa cells were transfected for 24 h with 25 ng of plasmid containing either Wip1 WT, Wip1 E525X, or Wip1 R552X. Cycloheximide (50 μg/ml) was added and protein harvested at the indicated time points. Western blots were performed using Wip1 (H300) to visualize all forms of Wip1 and actin as a loading control. (D)Top: positions of PPM1D mutations identified from patients with uterine corpus endometroid carcinoma. Bottom: survival of patients with and without PPM1D mutations (P = 0.2395).
Table 2.PPM1D mutations in human cancers.
| Cancer type | PPM1D mutation | TP53 mutation |
|---|---|---|
| Uterine corpus endometrioid carcinoma |
E525X |
R342X |
|
R429M / R552Q |
R213X |
|
| R572Q |
|
|
| F288C/E525X |
|
|
|
R552X |
|
|
|
E229K |
|
|
|
I180T |
|
|
| Q462X |
|
|
| C407R |
|
|
| E525X |
|
|
| Y5C |
|
|
| Lung adenocarcinoma |
P321A |
E286G |
|
A140S |
R110L |
|
|
D282fs / L301I |
G187_splice |
|
|
T261S |
|
|
|
S133F |
|
|
| Lung squamous cell carcinoma |
R243C |
K164E |
| Kidney renal papillary cell carcinoma |
R581Q |
|
| E451X |
|
|
| Colon and rectum adenocarcinoma |
N448S |
M237I |
| V491I |
|
|
| Q510fs |
|
|
| A481V |
|
|
| Stomach adenocarcinoma |
R583I / G586S |
G245R |
|
R552X |
K382fs / S215G |
|
| A370V |
R273C / R175C / E171K |
|
| Thyroid carcinoma |
S468fs |
|
| N574fs |
|
|
| T406fs |
|
|
| D470N |
|
|
| V430fs |
|
|
| Head and neck squamous cell carcinoma |
G122V |
L194P |
| R458X |
|
|
| Brain lower grade glioma |
E229K |
R175C |
| Glioblastoma Mmultiforme |
E525X |
R213X |
| Breast invasive carcinoma |
V289M |
R213X |
| C478X |
Mutations within the PPM1D gene were selected from the TCGA database (http://www.cbioportal.org). Loss-of-function PPM1D mutations are underlined.
To further investigate the functions of these hot-spot mutations, we generated appropriate point mutants in Wip1. We found that overexpression of Wip1 E525X was more efficient in downregulating the phosphorylation of ATM, Chk2, and H2AX after IR in comparison with wild-type Wip1 (Fig. 4B). In this assay, however, we were unable to see significant differences in properties of the Wip1 R552X mutant. As it has been recently reported that mutations in Wip1 could result in changes in protein stability,21 we looked at the stability of both proteins in the presence of cycloheximide. Consistent with the previous report,21 we found that one of the truncating mutations, E525X, resulted in an increase in protein stability of Wip1 (Fig. 4C). In contrast, the R552X truncation mutant was expressed at a significantly lower level than wild-type Wip1 and was highly unstable, suggesting that truncation at this position results in loss-of-function. We next analyzed patient survival based on PPM1D mutations in uterine corpus endometriod cancer (Fig. 4D), since it had the highest percentage of PPM1D mutations, including sites E525 and R552. Although the data did not reach statistical significance (P = 0.2), it did show a clear trend toward better overall survival for patients having a PPM1D mutation regardless of type of mutations. Truncating mutations in the C terminus of Wip1 have been reported as gain-of-function mutations.18,21 In contrast, mutations in the phosphatase domain of Wip1 contribute to loss of function and, as such, should result in upregulation of p53 activity.12 In addition, we found that some mutations in the C terminus (around amino acids 322, 496, and 552) of Wip1 could also lead to loss of function (Figs. 2–4). In this scenario, for a tumor to develop, p53 must be inactivated. Thus, one should expect that tumor samples with PPM1D loss-of-function mutations should have a high frequency of mutation in TP53. On the contrary, gain-of-function mutations, similar to PPM1D amplification,22 should remove selective pressure to simultaneously inactivate p53. Our analysis of tumor samples with PPM1D mutations revealed that 11 out of 15 samples with loss-of-function mutations in PPM1D had simultaneous mutations in TP53 (Table 2). In contrast, only 4 out of 21 samples with potential gain-of-function mutations had TP53 mutations. Thus, primary human tumors contain PPM1D with both loss- and gain-of-function mutations; however, if Wip1 is inactivated, it would most likely require an additional mutation in TP53 for tumor development.
Discussion
In order to evaluate the role of any SNP/genetic variant in human disease, one must have sufficient epidemiological studies and adequate statistical power. This is demonstrated by meta-analysis of large-scale samples.23,24 Our own preliminary findings with I496V suggested a reduction of breast cancer risk (normal subjects, n = 200, 1% frequency; breast cancer subjects, n = 620, 0% frequency). The frequency in the normal population for L120F and P322Q was much lower (around 0.1%). However, the data set was limited and could not achieve a sufficiently high statistical power to be significant. A larger data set of normal subjects (n > 5000) would be needed to achieve the correct statistical power.
While most SNPs that are found to have any biological activity are linked with a predisposition or increase in a disease state, the Wip1 genetic variants L120F, P322Q, and I496V found here may display the opposite effect. Having these dominant-negative variants would be advantageous to the organism under certain conditions; they could alleviate or prevent several types of cancer as well as reduce obesity and atherosclerosis. In fact, Wip1-null mice have a decreased cancer risk, as demonstrated by the great reduction in mammary,12 intestinal,14 and B-cell cancers,13 as well as reduced spontaneous tumorigenesis.15 Recently, we also found that Wip1-deficient mice are resistant to obesity and atherosclerosis,16 providing another potential direction for epidemiological studies to look at the frequencies of identified SNPs.
The situation in human cancers is more complex, as we found PPM1D mutations with both gain- and loss-of-function properties. Recent reports suggested that the majority of C-terminal truncating mutations found in human cancers are gain-of-function.18,21 However, here we identified the second most frequent mutation at position R552 as a loss-of-function mutation (Fig. 4C). It therefore needs to be determined whether inactivation of Wip1 at any stage of tumorigenesis favors cancer progression. Unfortunately, our own analysis of patients survival based on PPM1D mutations did not reach statistical significance, although it did show a clear trend toward better overall survival in the careers of PPM1D mutations regardless of type of mutations (Fig. 4D). It is also noteworthy that PPM1D inhibitory mutations probably could enhance tumorigenesis under certain conditions. In one such scenario, inactivation of Wip1 could create additional selective pressure to mutate p53 and as such to progress more rapidly to advanced stages of tumorigenesis. Further analysis of PPM1D mutations at different stages of cancer would resolve this issue. In conclusion, we suggest that several Wip1 genetic variants and SNP alleles can function as biomarkers for decreased risk of cancer, and potentially obesity and atherosclerosis. In turn, the role of Wip1 in human cancer evolution is more complex and most likely context-dependent, promoting tumorigenesis in some tumors while suppressing in others.
Materials and Methods
Structure modeling and phosphatase assays
The model of Wip1 was generated in the program COOT25 based on the structure of phosphatase 2C (1A6G).26 Phosphatase activity for Wip1 A82S, L120F, P322Q was measured by a malachite green/molybdate-based assay as described.27 The amount of phosphate released was calculated from a phosphate standard curve. All assays were performed in phosphatase buffer (50 mM TRIS-HCl [pH 7.5], 0.1 mM EGTA, and 0.02% 2-mercaptoethanol) with 60 mM MgCl2 and incubation with phosphopeptide for 5–10 min at 30 °C. To determine the kinetic parameters Km and kcat, the initial velocities (v) were measured at various peptide concentrations ([S]), and data were fitted to the Michaelis-Menten equation, v = kcat [S] / (Km + [S]). The phosphatase activity of Wip1 I496V was measured by transfecting 293T cells with the plasmid encoding either Wip1-WT-Flag (10 µg) or Wip1-I496V-Flag (12.5, 15, 17.5, and 20 µg). Following cell lysis, anti-Flag antibody conjugated beads were added to immunoprecipitate the Flag-tagged Wip1 protein. Phosphatase activity of Wip1 was measured in vitro with immunoprecipitated Flag-Wip1 WT or I496V protein in the presence of 100 µM ATM- pS1981 peptide. After the reaction, the solution was divided into 2 fractions for colorimetry to estimate the released Pi (O.D.620 nm) and western blotted for Wip1 (Santa Cruz, H300) to check protein amounts.
Transfections, site-directed mutagenesis, and treatments
HeLa cells were purchased from ATCC and maintained in a 37 °C incubator at 5% CO2. Site-directed mutagenesis was performed using the site-directed mutagenesis kit (Stratagene) following the manufacturer’s protocol. Primers used for mutation of Wip1WT to A82S, L120F, P322Q, I496V, E525X, and R552X are available upon request. Wip1 WT, L120F, P322Q, I496V, E525X, and R552X were transfected using lipofectamine 2000 into HeLa cells, then irradiated for 30 min or 1 h with 5 Gy IR the following day. To study the dominant-negative effect of variants on WT activity, a 1:1 ratio of WT:mutant was used, keeping the total amount of DNA the same as the single WT/mutant transfection. For the cycloheximide study, cycloheximide (50 μg/ml) was added the next day following transfection of Wip1WT, E525X, and R552X and protein harvested at the indicated time points.
Western blotting and antibodies
Cells were treated as described and protein extracted using RIPA buffer, blotted, and probed with antibodies specific to p-ATM (S1981), ATM, p-Chk2, Chk2, p-H2AX (Cell Signaling), Wip1 (H300), gapdh, actin, p38, H3, (Santa Cruz), GFP (Roche), and Flag (Sigma).
Colony formation assay
Colony formation assay was performed using HeLa cells transfected with empty vector, pCAG-GFP Wip1 WT, L120F, P322Q, or pIRES2neoFlag Wip1 I496V, E525X, or R552X as before. Two days post-transfection, cells were sorted and cells were either mock irradiated or irradiated at 5 Gy and plated at 1000 cells/well of a 6-well plate in triplicates. After 2–3 wk, colonies were fixed and stained with crystal violet solution, images taken using a scanner and colonies counted. To investigate the dominant-negative effect of Wip1 mutants in colony-formation assay, pCAG-Wip1 WT was mixed with the variants as before.
Nuclear extraction and immunofluorescence
For nuclear extraction, HeLa cells were plated on 6-well dishes and transfected the next day with Flag-Wip1 WT or I496V as before. The next day, protein was harvested using the NE-PER kit (Pierce) following the manufacturer’s procedure. Nuclear and cytoplasmic extracts were blotted using Flag (to detect Wip1), H3 as a nuclear extract control, and GAPDH as a cytoplasmic extract control. For immunofluorescence, cells were fixed with 3.7% paraformaldehyde and permeabilized with 0.25% Triton X100, then subjected to immunofluorescence staining for Flag to identify cellular localization.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25694
References
- 1.Hirasawa A, Saito-Ohara F, Inoue J, Aoki D, Susumu N, Yokoyama T, et al. Association of 17q21-q24 gain in ovarian clear cell adenocarcinomas with poor prognosis and identification of PPM1D and APPBP2 as likely amplification targets. Clin Cancer Res. 2003;9:1995–2004. [PubMed] [Google Scholar]
- 2.Saito-Ohara F, Imoto I, Inoue J, Hosoi H, Nakagawara A, Sugimoto T, et al. PPM1D is a potential target for 17q gain in neuroblastoma. Cancer Res. 2003;63:1876–83. [PubMed] [Google Scholar]
- 3.Loukopoulos P, Shibata T, Katoh H, Kokubu A, Sakamoto M, Yamazaki K, et al. Genome-wide array-based comparative genomic hybridization analysis of pancreatic adenocarcinoma: identification of genetic indicators that predict patient outcome. Cancer Sci. 2007;98:392–400. doi: 10.1111/j.1349-7006.2007.00395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castellino RC, De Bortoli M, Lu X, Moon SH, Nguyen TA, Shepard MA, et al. Medulloblastomas overexpress the p53-inactivating oncogene WIP1/PPM1D. J Neurooncol. 2008;86:245–56. doi: 10.1007/s11060-007-9470-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Natrajan R, Lambros MB, Rodríguez-Pinilla SM, Moreno-Bueno G, Tan DS, Marchió C, et al. Tiling path genomic profiling of grade 3 invasive ductal breast cancers. Clin Cancer Res. 2009;15:2711–22. doi: 10.1158/1078-0432.CCR-08-1878. [DOI] [PubMed] [Google Scholar]
- 6.Tan DS, Lambros MB, Rayter S, Natrajan R, Vatcheva R, Gao Q, et al. PPM1D is a potential therapeutic target in ovarian clear cell carcinomas. Clin Cancer Res. 2009;15:2269–80. doi: 10.1158/1078-0432.CCR-08-2403. [DOI] [PubMed] [Google Scholar]
- 7.Shreeram S, Demidov ON, Hee WK, Yamaguchi H, Onishi N, Kek C, et al. Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol Cell. 2006;23:757–64. doi: 10.1016/j.molcel.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 8.Lu X, Ma O, Nguyen TA, Jones SN, Oren M, Donehower LA. The Wip1 Phosphatase acts as a gatekeeper in the p53-Mdm2 autoregulatory loop. Cancer Cell. 2007;12:342–54. doi: 10.1016/j.ccr.2007.08.033. [DOI] [PubMed] [Google Scholar]
- 9.Fujimoto H, Onishi N, Kato N, Takekawa M, Xu XZ, Kosugi A, et al. Regulation of the antioncogenic Chk2 kinase by the oncogenic Wip1 phosphatase. Cell Death Differ. 2006;13:1170–80. doi: 10.1038/sj.cdd.4401801. [DOI] [PubMed] [Google Scholar]
- 10.Yoda A, Xu XZ, Onishi N, Toyoshima K, Fujimoto H, Kato N, et al. Intrinsic kinase activity and SQ/TQ domain of Chk2 kinase as well as N-terminal domain of Wip1 phosphatase are required for regulation of Chk2 by Wip1. J Biol Chem. 2006;281:24847–62. doi: 10.1074/jbc.M600403200. [DOI] [PubMed] [Google Scholar]
- 11.Lu X, Nannenga B, Donehower LA. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 2005;19:1162–74. doi: 10.1101/gad.1291305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bulavin DV, Phillips C, Nannenga B, Timofeev O, Donehower LA, Anderson CW, et al. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat Genet. 2004;36:343–50. doi: 10.1038/ng1317. [DOI] [PubMed] [Google Scholar]
- 13.Shreeram S, Hee WK, Demidov ON, Kek C, Yamaguchi H, Fornace AJ, Jr., et al. Regulation of ATM/p53-dependent suppression of myc-induced lymphomas by Wip1 phosphatase. J Exp Med. 2006;203:2793–9. doi: 10.1084/jem.20061563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demidov ON, Timofeev O, Lwin HN, Kek C, Appella E, Bulavin DV. Wip1 phosphatase regulates p53-dependent apoptosis of stem cells and tumorigenesis in the mouse intestine. Cell Stem Cell. 2007;1:180–90. doi: 10.1016/j.stem.2007.05.020. [DOI] [PubMed] [Google Scholar]
- 15.Nannenga B, Lu X, Dumble M, Van Maanen M, Nguyen TA, Sutton R, et al. Augmented cancer resistance and DNA damage response phenotypes in PPM1D null mice. Mol Carcinog. 2006;45:594–604. doi: 10.1002/mc.20195. [DOI] [PubMed] [Google Scholar]
- 16.Le Guezennec X, Brichkina A, Huang YF, Kostromina E, Han W, Bulavin DV. Wip1-dependent regulation of autophagy, obesity, and atherosclerosis. Cell Metab. 2012;16:68–80. doi: 10.1016/j.cmet.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 17.Schork NJ, Fallin D, Lanchbury JS. Single nucleotide polymorphisms and the future of genetic epidemiology. Clin Genet. 2000;58:250–64. doi: 10.1034/j.1399-0004.2000.580402.x. [DOI] [PubMed] [Google Scholar]
- 18.Ruark E, Snape K, Humburg P, Loveday C, Bajrami I, Brough R, et al. Breast and Ovarian Cancer Susceptibility Collaboration. Wellcome Trust Case Control Consortium Mosaic PPM1D mutations are associated with predisposition to breast and ovarian cancer. Nature. 2013;493:406–10. doi: 10.1038/nature11725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krawczak M, Ball EV, Fenton I, Stenson PD, Abeysinghe S, Thomas N, et al. Human gene mutation database-a biomedical information and research resource. Hum Mutat. 2000;15:45–51. doi: 10.1002/(SICI)1098-1004(200001)15:1<45::AID-HUMU10>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 20.Becker-Catania SG, Chen G, Hwang MJ, Wang Z, Sun X, Sanal O, et al. Ataxia-telangiectasia: phenotype/genotype studies of ATM protein expression, mutations, and radiosensitivity. Mol Genet Metab. 2000;70:122–33. doi: 10.1006/mgme.2000.2998. [DOI] [PubMed] [Google Scholar]
- 21.Kleiblova P, Shaltiel IA, Benada J, Ševčík J, Pecháčková S, Pohlreich P, et al. Gain-of-function mutations of PPM1D/Wip1 impair the p53-dependent G1 checkpoint. J Cell Biol. 2013;201:511–21. doi: 10.1083/jcb.201210031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bulavin DV, Demidov ON, Saito S, Kauraniemi P, Phillips C, Amundson SA, et al. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat Genet. 2002;31:210–5. doi: 10.1038/ng894. [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Zhao H, Sun L, Huang L, Yang Q, Kong B. MDM2 SNP309 is associated with endometrial cancer susceptibility: a meta-analysis. Hum Cell. 2011;24:57–64. doi: 10.1007/s13577-011-0013-4. [DOI] [PubMed] [Google Scholar]
- 24.Palanca Suela S, Esteban Cardeñosa E, Barragán González E, de Juan Jiménez I, Chirivella González I, Segura Huerta A, et al. Group for Assessment of Hereditary Cancer of Valencia Community CASP8 D302H polymorphism delays the age of onset of breast cancer in BRCA1 and BRCA2 carriers. Breast Cancer Res Treat. 2010;119:87–93. doi: 10.1007/s10549-009-0316-2. [DOI] [PubMed] [Google Scholar]
- 25.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Das AK, Helps NR, Cohen PT, Barford D. Crystal structure of the protein serine/threonine phosphatase 2C at 2.0 A resolution. EMBO J. 1996;15:6798–809. [PMC free article] [PubMed] [Google Scholar]
- 27.Yamaguchi H, Minopoli G, Demidov ON, Chatterjee DK, Anderson CW, Durell SR, et al. Substrate specificity of the human protein phosphatase 2Cdelta, Wip1. Biochemistry. 2005;44:5285–94. doi: 10.1021/bi0476634. [DOI] [PubMed] [Google Scholar]