

Abstract
Background
In dilated cardiomyopathy (DCM), the clinical and prognostic implications of rare variants in sarcomeric genes remain poorly understood. To address this question, we analyzed the outcome of rare sarcomeric gene variants in patients enrolled in our Familial Cardiomyopathy Registry.
Methods
DCM families harboring rare sarcomeric variants in MYH6, MYH7, MYBPC3, TNNT2, and TTN were identified. Genotype–phenotype association analysis was performed, and long‐term survival‐free from death or heart transplant was compared between carriers and noncarriers.
Results
We found 24 rare variants (3 in MYH6, 3 in MYH7, 3 in MYBPC3, 2 in TNNT2, and 13 in TTN) affecting 52 subjects in 25 families. The phenotypes of variant carriers were severe (3 sudden deaths, 6 heart failure deaths, 8 heart transplants, 2 ventricular fibrillations). There was no difference in the overall long‐term survival between carriers and the 33 noncarriers (p = 0.322). However after 50 years of age, the combined endpoint of death or transplant was decreased in carriers as compared to noncarriers (p = 0.026).
Conclusions
Patients with DCM carrying rare variants in sarcomeric genes manifest a poorer prognosis as compared to noncarriers after the age of 50 years. These data further support the role of genetic testing in DCM for risk stratification.
Keywords: cardiomyopathy, genes, prognosis, molecular genetics, genetics, phenotyping
Introduction
Dilated cardiomyopathy (DCM) is a heart muscle disease characterized by dilatation and systolic dysfunction of the left or both ventricles,1 and represents a major cause of heart failure, sudden death and heart transplantation. At least 30–50% of DCM cases are familial, suggesting the involvement of a defective gene.2, 3 Currently, mutations in over 30 genes across a wide variety of cellular components and pathways have been associated with DCM4, 5; among these are sarcomeric genes, which when mutated also cause hypertrophic cardiomyopathy (HCM) and restrictive cardiomyopathy.
In DCM, the most common sarcomeric mutations are reported in cardiac beta‐myosin heavy chain (MYH7), in cardiac troponin T (TNNT2), and in myosin binding protein C (MYBPC3).6, 7 We previously reported alpha‐myosin heavy chain (MYH6) mutations in DCM families.8 Hershberger et al. also found rare variants in genes of the sarcomeric complex “likely” or “possibly” causing the disease in their population.9, 10 Recently, Herman et al. reported high frequency of “deleterious variants” in the titin (TTN) gene in a large multicenter DCM cohort.11 Finally, Lackdawala et al. reported that subclinical DCM carriers of sarcomeric gene mutations have subtle abnormalities in systolic function, despite normal left ventricular size and ejection fraction, in contrast with HCM where early manifestations appear to affect the diastolic function.12 In spite of these genetic data, longitudinal clinical prognostic data of the impact of sarcomeric variants are lacking in DCM as compared to HCM.13
Here, using longitudinal clinical data, we performed a genotype–phenotype analysis to estimate the effect of rare sarcomeric variants in MYH6, MYH7, MYBPC3, TNNT2, and TTN, which were suspected to be pathogenic, on the natural history of a large cohort of DCM patients and their families enrolled in the International Familial Cardiomyopathy Registry.
Methods
Patient population
Our study population comprised 179 families, studied longitudinally at the University of Colorado Cardiovascular Institute and the Cardiovascular Department of the University Hospital of Trieste, Italy, and enrolled in the International Familial Cardiomyopathy Registry from 1988 (Table 1). Study subjects underwent physical examination, electrocardiogram, echocardiogram, and laboratory investigations according to the current familial DCM guidelines.13, 14 When clinically indicated, additional studies were performed, including right and left heart catheterization, ventriculography, coronary angiography, endomyocardial biopsy, and neuromuscular evaluation. Genetic screening was systematically performed in the proband and any available affected individual from each family. The Registry has collected clinical and genetic data of study subject for over 20 years (1988–2013), and as new families are continuously added to the Registry, the number of families screened for each sequentially tested gene has increased over time (Table 1).
Table 1.
Demographic data of the study population
| MYH6 | MYH7 | MYBPC3 | TNNT2 | TTN | ||
|---|---|---|---|---|---|---|
| Total number of families | 69 | 67 | 168 | 68 | 147 | |
| FDC (n) | 48 | 43 | 120 | 48 | 122 | |
| Sporadic (n) | 21 | 24 | 48 | 20 | 25 | |
| Males (%) | 65 | 65 | 63 | 64 | 94 | |
| Age of diagnosis (years) | 43 ± 15 | 43 ± 15 | 43 ± 16 | 42 ± 15 | 42 ± 13 |
FDC = familial dilated cardiomyopathy.
Criteria for the diagnosis of DCM were the presence of left ventricular fractional shortening <25% and/or an ejection fraction <45%, and left ventricular end‐diastolic diameter >117% of the predicted value by the Henry formula.13, 14 Exclusion criteria included any of the following conditions: blood pressure >160/110 mmHg, obstruction >50% of a major coronary artery branch, alcohol intake >100 g/day, persistent high‐rate supraventricular arrhythmia, systemic diseases, pericardial diseases, congenital heart diseases, cor pulmonale, and myocarditis. Familial DCM was defined by the presence of two or more affected subjects in the same family with DCM meeting the published criteria.13, 14 Informed consent was obtained from all enrolled subjects, and the study was approved by the respective institutional review committees.
Molecular genetic screening
Blood samples were collected for DNA analysis and previously studied for rare variants in MYH6 (GenBank accession no. NM_002471 / P13533), MYH7 (GenBank accession no. NM_000257.2 / NP_000248.2), MYBPC3 (NM_000256.3 / NP_000247.2), TNNT2 (NM_001001430.1 / NP_001001430.1) and TTN (NM_133378 / NM_00319 / NM_133379 / Q8WZ42) by denaturing high performance liquid chromatography (DHPLC) or by Sanger sequencing. In families in which we found a putative disease‐causing mutation, all available relatives were screened for segregation of the genetic variant. Criteria for classifying variants as putative disease‐causing mutations included changes in predicted amino acid sequences, segregation within the family, conservation across different species (http://www.ncbi.nlm.nih.gov/BLAST/), absence in a control population of 150 subjects and >5,000 healthy ethnically similar subjects from public databases. Putative mutations were filtered by PolyPhen2, BDGP splice site detection software.15, 16 and 1,000 Genome Project (http://www.1000genomes.org/home). As the combination of testing healthy controls and bioinformatics analysis can suggest but not prove pathogenicity, the “putative mutations” are referred to hereafter as “rare variants.”
Effects of sarcomeric gene rare variants on prognosis
To estimate the effect of sarcomeric gene rare variants on the natural history of DCM, we compared the long‐term death/heart transplantation event‐free survival of subjects carrying rare variants to a population of 33 Registry patients in whom MYH6, MYH7, MYBPC3, TNNT2, and TTN genes were all tested and resulted negative for mutations. In the control group, there was one known LMNA A/C and one known SCN5A mutation carrier. The same comparison has been made between probands carrying and not carrying sarcomere gene rare variants. Finally, to replicate our analysis in an independent cohort, we compared the long‐term survival between our population of sarcomeric gene variant carriers with a cohort of sarcomeric mutation carriers from seven other studies with similar diagnostic criteria and available outcome data8, 17, 18, 19, 20, 21, 22 (92 variant carriers).
Statistical analysis
Summary statistics of clinical and instrumental variables at enrollment were expressed as mean and standard deviation or count and percentage, as appropriate. Comparison between rare variant carriers and noncarriers was made by the Anova Test on continuous variables and the Chi‐square test for discrete variables. Event‐free survival curves for death/heart transplantation were estimated and plotted using the Kaplan–Meier method and the log‐rank test was applied in order to investigate for differences in long‐term survival. To take into account the clustered failure times (i.e., relatives within families cannot be taken as entirely independent) a survival regression Cox model was also estimated, with “group” (with levels: carriers or noncarriers) as the unique covariate and the “family index” as a cluster indicator. Statistical analyses were performed with IBM SPSS Statistical Package 19.0 and the R statistical package version 2.14.1.
Results
Molecular genetics of sarcomeric genes in DCM
The cohorts screened for sarcomeric variants were not of equal size as they were tested at different times; rare variant frequencies are reported by each screened cohort. We found 24 sarcomeric missense rare variants, accounting for 4.4% of tested families in MYH6 (3/69; c.2489 C>T, p.P830L; c.3010 G>T, p.A1004S; 4369 G>A, p.E1457K), 4.4% in MYH7 (3/67; c.2945 T>C, p.M982T; c.4300 C>T, p.R1434C; c.4498 C>T, p.R1500W), 1.8% in MYBPC3 (3/168; c.649 A>G, p.S217G; c.1373 G>A, p.R458H; c.2870 C>G, p.T957S), 4.4% in TNNT2 (3/68; c.391 C>T, p.R131W, and c.517 C>T, p.R173W the latter found in two nuclear families) and 8.8% in TTN (13/147; c.6247delG, p.R2883fs; c.91043delA, p.N30348fs; c.49077G>A, p.W16359X; c.51883G>A, p.R17295X; c.52408C>T, p.R17470X; c.53347G>T, p.E17783X; c.56953C>T, p.R18985X; c.79896G>A, p.W26632X; c.81046A>T, p.K27016X; c.87953G>A, p.w29318X; c.88242C>T, p.R29415X; c.50346+3A>G, p.K16782; c.53145_53146insG, p.E17715fs). All rare variants were novel and suspected of being pathogenic or had previously been reported as mutations in DCM or HCM10, 21, 22, 23, 24, 25, 26, 27, 29) and were absent in control samples. The characteristics of the rare variants are shown in Table S1 and the pedigrees of familial DCM cases in Figures S1–S3.
Genotype–phenotype correlation, natural history, and prognosis of sarcomeric genes in DCM
Table S2 reports the phenotypic characteristics of the 52 patients (probands and affected relatives) carrying a rare sarcomeric variant. The phenotypes of probands and first‐degree relatives were frequently severe with 19 patients (36.5%) having ventricular arrhythmias, including two case of ventricular fibrillation. Furthermore outcomes were notably severe: 3 patients experienced sudden death, 6 pump failure death, and 8 required cardiac transplantation for refractory heart failure out of 21 major events. Supraventricular arrhythmias were also present in carriers, and could have influenced the development and progression of heart failure. Major hypokinetic arrhythmias were not detected, only three pacemaker were required (one was implanted in a patient with sick sinus syndrome; the second in a patient with 1st degree atrioventricular block, left bundle branch block and need of beta‐blockers, later requiring heart transplant; the third in a patient with complete left bundle branch block).
Compared with noncarriers, carriers had higher left ventricular ejection fraction (LVEF) (37 ± 15% vs. 29 ± 12%, p = 0.03), no other significant differences were found concerning age of disease onset, gender, heart failure symptoms, or echocardiographic features (Table 2). Although no differences in the overall long‐term survival were found between carriers and noncarriers (p = 0.322), in sarcomeric carriers death/HTx‐free survival dramatically decreased after 50 years of age compared to noncarriers (83–38% vs. 81–66% at 50 and 75 years of age in carriers and noncarriers, respectively, p = 0.026) (Figure 1). Likewise, the hazard ratio estimated by the clustered Cox model was globally not significant (HR = 1.35, 95% CI: 0.28–5.93), but become highly significant after 50 years of age (HR = 3.74, 95% CI: 1.15–9.8). The same results were found while repeating the survival analysis on carrier and noncarrier probands only (Figure 2). Furthermore we performed a gene‐centric analysis for TTN truncating rare variants without finding survival differences between the 26 carriers and the 133 noncarriers (p = 0.98) (Figure S4); for the other four genes the gene‐centric analysis was not possible for the exiguous number of carriers. Finally no significant differences in survival trends were found when we compared our carriers with those from seven other studies on sarcomere genes with similar available prognostic data present in literature.8, 17, 18, 19, 20, 21, 22 (Figure S5.)
Table 2.
Comparison of clinical and echocardiographic characteristics at enrollment between sarcomeric gene rare variant carriers and noncarriers
| Carriers (52 patients) | Noncarriers (33 patients) | p value | |
|---|---|---|---|
| Male gender (%) | 71 | 70 | 0.886 |
| Age, years (mean ± SD) | 41 ± 17 | 41 ± 13 | 0.901 |
| New York Heart Association classes III–IV (%) | 23 | 30 | 0.543 |
| Left ventricular ejection fraction, % (mean ± SD) | 37 ± 15 | 29 ± 12 | 0.03 |
| Left ventricular end diastolic diameter, mm (mean ± SD) | 62 ± 13 | 67 ± 10 | 0.08 |
| Interventricular septum, mm (mean ± SD) | 9 ± 2 | 8 ± 2 | 0.623 |
| Left ventricular fractional shortening, % (mean ± SD) | 18 ± 8 | 18 ± 9 | 0.957 |
| Moderate to severe mitral regurgitation (%) | 26 | 27 | 0.974 |
Figure 1.
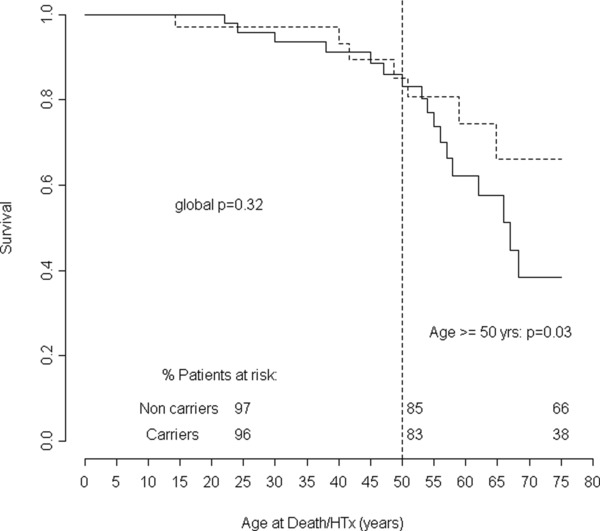
Comparison of long‐term natural history between 52 sarcomeric genes (MYH6, MYH7, MYBPC3, TNNT2, and TTN) rare variant carriers and noncarriers population (33 patients). Follow‐up from birth to end‐point/last follow‐up evaluation. Survival rates (as percentages) are provided at ages 25, 50, and 75 years. D/HTx: death or heart transplant.
Figure 2.
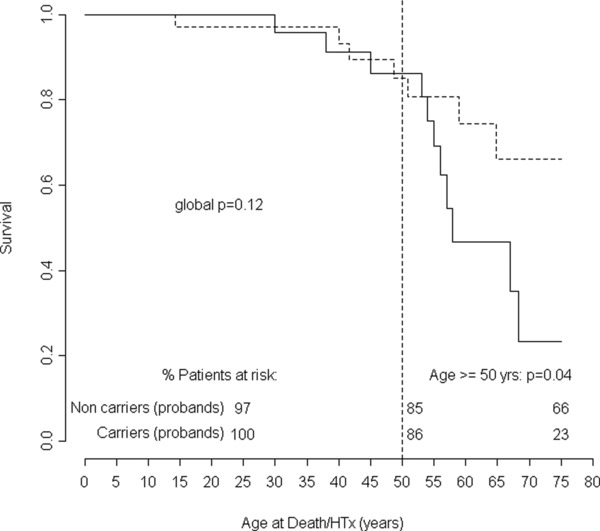
Comparison of long‐term natural history between 25 probands carrying sarcomeric genes (MYH6, MYH7, MYBPC3, TNNT2, and TTN) rare variant carriers and noncarriers population (33 probands). Follow‐up from birth to end‐point/last follow‐up evaluation. Survival rates (as percentages) are provided at ages 25, 50, and 75 years. D/HTx = death or heart transplant.
Discussion
Frequency and characteristics of sarcomeric gene rare variants in DCM
In the present study, we evaluated a well‐characterized cohort of DCM patients from the International Familial Cardiomyopathy Registry, extensively investigated from the clinical and genetic perspectives and followed longitudinally for over 20 years. We detected a frequency of rare variants suspected to be pathogenic between 1.5% and 8.8% in 5 sarcomeric genes (MYH6, MYH7, MYBPC3, TNNT2, and TTN), in agreement with an overall frequency of sarcomeric gene mutations between 4% and 8% as reported in the literature.4, 9, 10, 12, 14
Sarcomeric gene mutations are characterized by phenotypic heterogeneity, leading to a range of phenotypes including DCM, HCM, left ventricular noncompaction and restrictive cardiomyopathy, variable expressivity and overlapping phenotypes.4, 28 In our study, a MYBPC3 variant (p.R458H) was present in a family with DCM and unexplained muscular dystrophy, and the same mutation was previously found in HCM.29 Furthermore, the TNNT2 variant (p.R131W) was found in our study in a family with a complex phenotype including DCM and left ventricular noncompaction and was previously reported as a pathogenic mutation causing DCM and causing altered protein interactions in a functional mammalian two‐hybrid assay.21 This variant is reported as pathogenic in dbSNP (rs74315380; OMIM 191045.0007‐8; no frequency data available: absent in the NHLBI Exome Sequencing Project). Interestingly, this variant was not present in our proband's affected mother and was presumably inherited from his father, who was healthy by history but had no available family history data. The R131W variant may therefore represent a rare variant with reduced penetrance or a variant of unknown significance.
Genotype–phenotype correlation
The availability of extensive longitudinal clinical follow‐up data in our patient cohort allowed us to provide the first assessment of the effect of sarcomeric mutations on the natural history of DCM. We found that carriers of sarcomeric gene rare variants represented a subgroup of DCM patients with a particularly severe phenotype characterized by a high frequency of ventricular arrhythmias and high incidence of cardiovascular events, major arrhythmia event and pump failure. In spite of a higher LVEF at enrollment, carriers showed a more rapid progression towards death or heart transplantation compared to the noncarriers between 50 and 75 years of age (rates of death or transplantation: 17% and 62% vs. 19% and 34% at 50 and 75 years in carriers and noncarriers, respectively). Furthermore, strengthening our results, the long‐term survival of our carriers was not different when compared with carriers form the other studies in literature with available prognostic data on sarcomeric genes rare variants.8, 17, 18, 19, 20, 21, 22
Study limitations
A limitation of our study is the retrospective approach; however, the cohort studied includes extensive longitudinal data relying on data from over two decades. Due to continuous ascertainment of new families and the sequential nature of cardiomyopathy genes studied over the years, the number of subjects screened for each gene was not the same and the distribution of nonsarcomeric DCM gene mutations was not known for all samples. When used, DHPLC may have had lower sensitivity compared to direct sequencing. Furthermore, our DCM cohort, enrolled in tertiary referral heart failure centers could represent a more severe group of DCM patients than the general DCM population due to selection bias. As discussed above, it should be noted that the causal role of some of these variants has not been established. In particular, variant p.R131W, which had previously been reported in DCM,21 did not segregate with the disease in the family. However, it should be emphasized that Bick et al. showed that in the general population of the Framingham Heart Study, sarcomere variants were associated with an increased risk for adverse cardiovascular events (hazard ratio: 2.3), suggesting that cardiovascular risk assessment in the general population can benefit from rare variant analysis.30 Overall these findings strongly suggest that the sarcomere plays a primary pathogenic role in DCM. It should finally be noted that the statistical significance in outcome was identified in the subgroup analysis, requiring further validation. Future studies are also necessary to analyze the prognostic role and the genotype–phenotype correlation also of other emerging sarcomeric genes, to determine if the malignant prognosis we identified in our study is a common feature of the sarcomere in DCM, as previously shown for genes of the nuclear lamina.31, 32, 33
Conclusions
The extensive period of enrollment and follow‐up represent unique features of this study, providing valuable insight into the clinical impact of sarcomeric gene rare variants on the natural history of DCM. A poorer clinical outcome after the fifth decade of life was evident in sarcomeric rare variant carriers. These findings have important clinical implications for the management of DCM, risk stratification, and prognostic assessment. Indeed, systematic genetic testing, which has recently been endorsed for cardiomyopathies, can add prognostic information and improve the management of patients who are found mutation carriers toward more aggressive therapeutic and follow‐up strategies in order to improve their natural history.4, 14
Conflicts of Interest
None.
Supporting information
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Figure S1. Pedigrees of the families “MYH7 p.M982T,” “MYH7 p.R1434C,” “MYBPC3 p.R458H,” “MYBPC3 p.T957S,” and “TNNT2 p.R131W” with familial DCM carrying a sarcomeric gene rare variant. Males and females indicated by squares and circles, respectively. Individuals with DCM and/or who are deceased are indicated by black shading and diagonal lines, respectively. Variant status is indicated by “+” and “‐” for mutation carriers and noncarriers, respectively. Five variants (MYH6 p.P830L, p.A1004S, p.E1457K; MYH7 p.R1500W; MYBPC3 p.S217G) occurred in nonfamilial DCM cases.
Figure S2. Pedigree of 2 families (A and B) with familial DCM carrying the sarcomeric gene rare variant TNNT2 p.R173W. Males and females indicated by squares and circles, respectively. Individuals with DCM and/or who are deceased are indicated by black shading and diagonal lines, respectively. Variant status is indicated by “+” and “−” for mutation carriers and noncarriers, respectively.
Figure S3. Pedigrees of 7 families with DCM carrying the sarcomeric gene variants TTN (p.N30348fs, p.R17470X, p.E17783X, p.W26632X, p.K27016X, p.R29415X, p.E17715fs). Males and females indicated by squares and circles, respectively. Individuals with DCM and/or who are deceased are indicated by black shading and diagonal lines, respectively. Variant status is indicated by “+” and “−” for mutation carriers and noncarriers, respectively.
Figure S4. Comparison of long‐term natural history between 26 TTN gene carriers and noncarriers population (133 patients). Follow‐up from birth to end‐point/last follow‐up evaluation. Survival rates (as percentages) are provided at ages 25, 50, and 75 years. D/HTx = death or heart transplant.
Figure S5. Comparison of long‐term natural history between 52 gene variant carriers and carriers from all the other previous 7 studies on sarcomere genes with similar available prognostic data (92 patients). Follow‐up from birth to end‐point/last follow‐up evaluation. Survival rates (as percentages) are provided at ages 25, 50, and 75 years. D/HTx = death or heart transplant.
Table S1. Characteristics of identified rare variants in MYH6, MYH7, MYBPC3, TNNT2, and TTN.
Table S2. Phenotype at enrollment and outcome of the sarcomeric genes rare variant carriers.
Acknowledgments
The authors thank the family members for their participation in these studies. This study was supported by the NIH grants UL1 RR025780 and RO1 HL69071 to LM, and K23 JL067915 and 1R01HL109209‐01A1 to MRGT.
References
- 1. Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O‘Connell J, Olsen E, Thiene G, Goodwin J, Gyarfas I, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 1996; 93: 841–842. [DOI] [PubMed] [Google Scholar]
- 2. Baig MK, Goldman JH, Caforio ALP, Coonar AS, Keeling PJ, McKenna WJ. Familial dilated cardiomyopathy: cardiac abnormalities are common in asymptomatic relatives and may represent early disease. J Am Coll Cardiol. 1998; 31: 195–201. [DOI] [PubMed] [Google Scholar]
- 3. Mestroni L, Rocco C, Gregori D, Sinagra G, Di Lenarda A, Miocic S, Vatta M, Pinamonti B, Muntoni F, Caforio AL, et al. Familial dilated cardiomyopathy: evidence for genetic and phenotypic heterogeneity. J Am Coll Cardiol. 1999; 34: 181–190. [DOI] [PubMed] [Google Scholar]
- 4. Hershberger RE, Morales A, Siegfried JD. Clinical and genetic issues in dilated cardiomyopathy: a review for genetics professionals. Genet Med. 2011; 12: 655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Watkins H, Ashrafian H, Redwood C. Inherited cardiomyopathies. N Engl J Med. 2011; 364: 1643–1656. [DOI] [PubMed] [Google Scholar]
- 6. Chang AN, Potter JD. Sarcomeric protein mutations in dilated cardiomyopathy. Heart Fail Rev. 2005; 10: 225–235. [DOI] [PubMed] [Google Scholar]
- 7. Moller DV, Andersen PS, Hedley P, Ersbøll MK, Bundgaard H, Moolman‐Smook J, Christiansen M, Køber L. The role of sarcomere gene mutations in patients with idiopathic dilated cardiomyopathy. Eur J Hum Genet. 2009; 17: 1241–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carniel E, Taylor MR, Sinagra G, Di Lenarda A, Ku L, Fain PR, Boucek MM, Cavanaugh J, Miocic S, Slavov D, et al. Alpha‐myosin heavy chain: a sarcomeric gene associated with dilated and hypertrophic phenotypes of cardiomyopathy. Circulation 2005; 112: 54–59. [DOI] [PubMed] [Google Scholar]
- 9. Hershberger RE, Norton N, Morales A, Li D, Siegfried JD, Gonzalez‐Quintana J. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ Cardiovasc Genet. 2010; 3: 155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hershberger RE, Parks SB, Kushner JD, Li D, Ludwigsen S, Jakobs P, Nauman D, Burgess D, Partain J, Litt M. Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin Transl Sci. 2008; 1: 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012; 366: 619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lakdawala NK, Thune JJ, Colan SD, Cirino AL, Farrohi F, Rivero J, McDonough B, Sparks E, Orav EJ, Seidman JG, et al. Subtle abnormalities in contractile function are an early manifestation of sarcomere mutations in dilated cardiomyopathy. Circ Cardiovasc Genet. 2012; 5: 503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mestroni L, Maisch B, McKenna WJ, Schwartz K, Charron P, Rocco C, Tesson F, Richter A, Wilke A, Komajda M. Guidelines for the study of familial dilated cardiomyopathies. Eur Heart J. 1999; 20: 93–102. [DOI] [PubMed] [Google Scholar]
- 14. Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, Towbin JA. Heart Failure Society of America. Genetic evaluation of cardiomyopathy–a Heart Failure Society of America practice guideline. J Card Fail. 2009; 15: 83–97. [DOI] [PubMed] [Google Scholar]
- 15. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. 1997; 4: 311–323. [DOI] [PubMed] [Google Scholar]
- 17. Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, Smoot L, Mullen MP, Woolf PK, Wigle ED, et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med 2000; 343: 1688–1696. [DOI] [PubMed] [Google Scholar]
- 18. Villard E, Duboscq‐Bidot L, Charron P, Benaiche A, Conraads V, Sylvius N, Komajda M. Mutation screening in dilated cardiomyopathy: prominent role of the beta myosin heavy chain gene. Eur Heart J 2005; 26: 794–803. [DOI] [PubMed] [Google Scholar]
- 19. Daehmlow S, Erdmann J, Knueppel T, Gille C, Froemmel C, Hummel M, Hetzer R, Regitz‐Zagrosek V. Novel mutations in sarcomeric protein genes in dilated cardiomyopathy. Biochem Biophys Res Commun. 2002; 298: 116–120. [DOI] [PubMed] [Google Scholar]
- 20. Lakdawala NK, Dellefave L, Redwood CS, Sparks E, Cirino AL, Depalma S, Colan SD, Funke B, Zimmerman RS, Robinson P, et al. Familial dilated cardiomyopathy caused by an alpha‐ tropomyosin mutation: the distinctive natural history of sarcomeric dilated cardiomyopathy. J Am Coll Cardiol. 2010; 55: 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mogensen J, Murphy RT, Shaw T, Bahl A, Redwood C, Watkins H, Burke M, Elliott PM, McKenna WJ. Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2004; 44: 2033–2040. [DOI] [PubMed] [Google Scholar]
- 22. Karkkainen S, Helio T, Jaaskelainen P, Miettinen R, Tuomainen P, Ylitalo K, Kaartinen M, Reissell E, Toivonen L, Nieminen MS, et al. Two novel mutations in the beta‐myosin heavy chain gene associated with dilated cardiomyopathy. Eur J Heart Fail. 2004; 6: 861–868. [DOI] [PubMed] [Google Scholar]
- 23. Blair E, Redwood C, de Jesus Oliveira M, Moolman‐Smook JC, Brink P, Corfield VA, Ostman‐Smith I, Watkins H. Mutations of the light meromyosin domain of the beta‐myosin heavy chain rod in hypertrophic cardiomyopathy. Circ Res. 2002; 90: 263–269. [DOI] [PubMed] [Google Scholar]
- 24. Ehlermann P, Weichenhan D, Zehelein J, Steen H, Pribe R, Zeller R, Lehrke S, Zugck C, Ivandic BT, Katus HA. Adverse events in families with hypertrophic or dilated cardiomyopathy and mutations in the MYBPC3 gene. BMC Med Genet 2008; 9: 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harris SP, Lyons RG, Bezold KL. In the thick of it: HCM‐causing mutations in myosin binding proteins of the thick filament. Circ Res 2011; 108: 751–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roncarati R, Latronico MV, Musumeci B, Aurino S, Torella A, Bang ML, Jotti GS, Puca AA, Volpe M, Nigro V, et al. Unexpectedly low mutation rates in beta‐myosin heavy chain and cardiac myosin binding protein genes in italian patients with hypertrophic cardiomyopathy. J Cell Physiol 2011; 226: 2894–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sun N, Yazawa M, Liu J, Han L, Sanchez‐Freire V, Abilez OJ, Navarrete EG, Hu S, Wang L, Lee A, et al. Patient‐specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci Transl Med 2012; 4: 130ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Towbin JA, Lorts A. Arrhythmias and dilated cardiomyopathy common pathogenetic pathways? J Am Coll Cardiol 2011; 57: 2169–2171. [DOI] [PubMed] [Google Scholar]
- 29. Van Driest SL, Vasile VC, Ommen SR, Will ML, Tajik AJ, Gersh BJ, Ackerman MJ. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol 2004; 44: 1903–1910. [DOI] [PubMed] [Google Scholar]
- 30. Bick AG, Flannick J, Ito K, Cheng S, Vasan RS, Parfenov MG, Herman DS, DePalma SR, Gupta N, Gabriel SB, et al. Burden of rare sarcomere gene variants in the Framingham and Jackson heart study cohorts. Am J Hum Genet 2012; 91: 513–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Taylor MR, Fain PR, Sinagra G, Robinson ML, Robertson AD, Carniel E, Di Lenarda A, Bohlmeyer TJ, Ferguson DA, Brodsky GL. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol 2003; 41: 771–780. [DOI] [PubMed] [Google Scholar]
- 32. Taylor MR, Slavov D, Gajewski A, Vlcek S, Ku L, Fain PR, Carniel E, Di Lenarda A, Sinagra G, Boucek MM, et al. Thymopoietin (lamina‐associated polypeptide 2) gene mutation associated with dilated cardiomyopathy. Hum Mutat 2005; 26: 566–574. [DOI] [PubMed] [Google Scholar]
- 33. van Rijsingen IA, Arbustini E, Elliott PM, Mogensen J, Hermans‐van Ast JF, van der Kooi AJ, van Tintelen JP, van den Berg MP, Pilotto A, Pasotti M, et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J Am Coll Cardiol 2012; 59: 493–500. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Figure S1. Pedigrees of the families “MYH7 p.M982T,” “MYH7 p.R1434C,” “MYBPC3 p.R458H,” “MYBPC3 p.T957S,” and “TNNT2 p.R131W” with familial DCM carrying a sarcomeric gene rare variant. Males and females indicated by squares and circles, respectively. Individuals with DCM and/or who are deceased are indicated by black shading and diagonal lines, respectively. Variant status is indicated by “+” and “‐” for mutation carriers and noncarriers, respectively. Five variants (MYH6 p.P830L, p.A1004S, p.E1457K; MYH7 p.R1500W; MYBPC3 p.S217G) occurred in nonfamilial DCM cases.
Figure S2. Pedigree of 2 families (A and B) with familial DCM carrying the sarcomeric gene rare variant TNNT2 p.R173W. Males and females indicated by squares and circles, respectively. Individuals with DCM and/or who are deceased are indicated by black shading and diagonal lines, respectively. Variant status is indicated by “+” and “−” for mutation carriers and noncarriers, respectively.
Figure S3. Pedigrees of 7 families with DCM carrying the sarcomeric gene variants TTN (p.N30348fs, p.R17470X, p.E17783X, p.W26632X, p.K27016X, p.R29415X, p.E17715fs). Males and females indicated by squares and circles, respectively. Individuals with DCM and/or who are deceased are indicated by black shading and diagonal lines, respectively. Variant status is indicated by “+” and “−” for mutation carriers and noncarriers, respectively.
Figure S4. Comparison of long‐term natural history between 26 TTN gene carriers and noncarriers population (133 patients). Follow‐up from birth to end‐point/last follow‐up evaluation. Survival rates (as percentages) are provided at ages 25, 50, and 75 years. D/HTx = death or heart transplant.
Figure S5. Comparison of long‐term natural history between 52 gene variant carriers and carriers from all the other previous 7 studies on sarcomere genes with similar available prognostic data (92 patients). Follow‐up from birth to end‐point/last follow‐up evaluation. Survival rates (as percentages) are provided at ages 25, 50, and 75 years. D/HTx = death or heart transplant.
Table S1. Characteristics of identified rare variants in MYH6, MYH7, MYBPC3, TNNT2, and TTN.
Table S2. Phenotype at enrollment and outcome of the sarcomeric genes rare variant carriers.