

Abstract
The crystal structure of Rad4/Rad23, the yeast homolog of the human nucleotide excision repair (NER) lesion recognition factor XPC-RAD23B (Min and Pavletich, (2007) Nature 449:570–575) reveals that the lesion-partner base is flipped out of the helix and binds the protein. This suggests the hypothesis that flipping of this partner base must overcome a free energy barrier, which constitutes one element contributing to changes in the thermodynamic properties induced by the DNA damage and sensed by the recognition protein. We explored this hypothesis by computing complete flipping free energy profiles for two lesions derived from the procarcinogenic polycyclic aromatic hydrocarbons (PAHs), dibenzo[a,l]pyrene (DB[a,l]P) and benzo[a]pyrene (B[a]P), R-trans-anti-DB[a,l]P-N6-dA (R-DB[a,l]P-dA) and R-trans-anti-B[a]P-N6-dA (R-B[a]P-dA), and the corresponding unmodified duplex. The DB[a,l]P and B[a]P adducts differ in number and organization of their aromatic rings. We integrate these results with prior profiles for the R-trans-anti-DB[a,l]P-dG adduct (Zheng et al., (2010) Chem. Res. Toxicol. 23:1868–1870). All adopt conformational themes involving intercalation of the PAH aromatic ring system into the DNA duplex; however, R-DB[a,l]P-dA and R-B[a]P-dA intercalate from the major groove, while R-DB[a,l]P-dG intercalates from the minor groove. These structural differences produce different computed van der Waals stacking interaction energies between the flipping partner base with the lesion aromatic ring system and adjacent bases; we find that the better the stacking, the higher the relative flipping free energy barrier and hence lower flipping probability. The better relative NER susceptibilities correlate with greater ease of flipping in these three differently intercalated lesions. In addition to partner base-flipping, the Rad4/Rad23 crystal structure shows that a protein-β-hairpin, BHD3, intrudes from the major groove side between the DNA strands at the lesion site. We present a molecular modeling study for the R-DB[a,l]P-dG lesion in Rad4/Rad23 showing BHD3 β-hairpin intrusion with lesion eviction, and we hypothesize that lesion steric effects play a role in the recognition of intercalated adducts.
Keywords: base-flipping, nucleotide excision repair, free energy profile
INTRODUCTION
DNA duplexes covalently damaged by metabolically activated polycyclic aromatic hydrocarbons (PAH) have provided a rich model system for investigating the nucleotide excision repair (NER) susceptibility of lesion-containing DNAs.1–5 These bulky adducts are repaired by the NER machinery with remarkably different susceptibilities.6,7 However, the underlying origins of these differences remain unclear.6,8 Lesion recognition in NER is performed by XPC-RAD23B in an initial step9 that is rate limiting for NER,10 and therefore governs the success of subsequent downstream stages of the process. The crystal structure of Rad4/Rad23, the S. cerevisiae homologue of XPC-RAD23B shows that a β-hairpin loop, BHD3, of this recognition factor inserts from the major groove side between the two strands of the DNA duplex at the site of a cis-syn thymine dimer lesion, and two opposite bases that are mismatched thymines are flipped into the protein.11 These structural studies suggest that insertion of this BHD3 β-hairpin through the double helix occurs cooperatively with binding to the two nucleotides which flip, and that local thermodynamic destabilization of the damaged duplex4,12–14 facilitates the cooperative flipping and β-hairpin intrusion. Accordingly, flipping free energy profiles of partner bases to the damage site may yield clues to differential NER susceptibilities of bulky lesions.
In the present study we are focusing on lesions derived from dibenzo[a,l]pyrene (DB[a,l]P) and benzo[a]pyrene (B[a]P). DB[a,l]P is the most potent polycylic aromatic hydrocarbon (PAH) tumorigen yet identified,15–19 and the International Agency for Research on Cancer (IARC)20 has classified B[a]P as a human carcinogen. DB[a,l]P is a member of the “fjord region” (defined in Figure 1A) PAH family; in this family there is steric hinderance (between carbon 1 and 14 in the case of DB[a,l]P), producing non-planarity with a twist across the fjord region.21,22 B[a]P is a member of the “bay region” (defined in Figure 1A) family and is planar. Metabolic activation through the well-studied diol epoxide pathway18,23–25 of both DB[a,l]P and B[a]P yields stereoisomeric adducts to adenine and guanine that are mutagenic.18,26,27 Structural studies involving high resolution NMR and molecular modeling with molecular dynamics (MD) simulations have been employed to obtain structures for various members of these adduct families in duplex DNA.28–33
Figure 1.
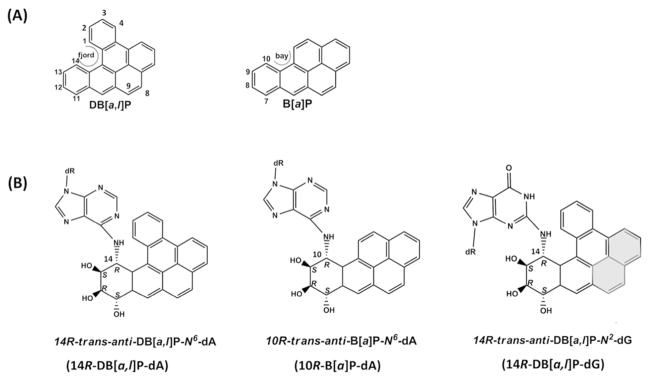
(A) The chemical structures of DB[a,l]P and B[a]P with definitions of the fjord region and the bay region, respectively. (B) The chemical structures for the 14R-DB[a,l]P-dA, 10R-B[a]P-dA and 14R-DB[a,l]P-dG adducts. The distal aromatic rings highlighted in grey cause steric crowding in the 14R-DB[a,l]P-dG structure.
We have investigated the fjord 14R-DB[a,l]P-dA and the bay 10R-B[a]P-dA adducts (Figure 1B), with the former containing five aromatic rings and the latter possessing four. These both adopt classical intercalation structures in which the aromatic ring system is intercalated from the major groove, on the 5′-side of the damaged base, through duplex stretching and unwinding (Figure 2A).33–37 Watson-Crick base pairing is distorted but not ruptured. However, the adaptable fjord region twist of the 14R-DB[a,l]P-dA34 adduct allows better accommodation in the intercalation pocket than the planar, rigid bay region of the 10R-B[a]P-dA adduct. NER in human HeLa cell extracts has shown that the 14R-DB[a,l]P-dA adduct is repair-resistant38 while the 10R-B[a]P-dA adduct is modestly repaired. 33, 38–40
Figure 2.
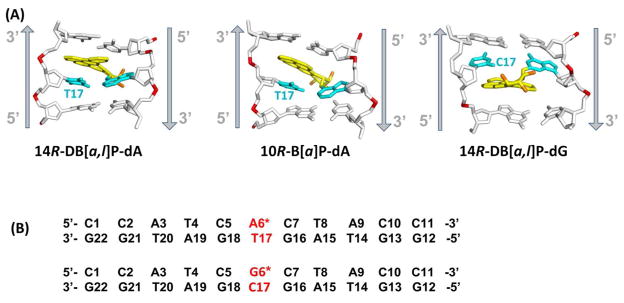
(A) Best representative structures49 from MD simulations for the 14R-DB[a,l]P-dA, and 10R-B[a]P-dA adducts. The NMR solution structure for the 14R-DB[a,l]P-dG adduct is also shown.42 Only the central 3-mers are presented. The view is looking into the minor groove. For the DB[a,l]P and B[a]P moieties, the carbon atoms are colored yellow and the oxygen atoms orange. The damaged bases are colored cyan and the DNA duplexes are colored white, except for the phosphorus atoms, which are red. Hydrogen atoms are not displayed for clarity. The arrows indicate 5′ to 3′ direction. Movies S1 – S3, showing these structures rotating are provided in Supporting Information. (B) The base sequence contexts for the 14R-DB[a,l]P-dA and the 14R-DB[a,l]P-dG 11-mer duplexes. A* and G* designate the damaged bases.
We have obtained complete free energy profiles for partner base flipping in damaged DNA duplexes for these two adducts as well as their corresponding unmodified duplexes (Figure 2B). Previously, we had obtained flipping free energy profiles41 for the well-repaired 14R-DB[a,l]P-dG adduct38 and its corresponding unmodified duplex (Figure 2B). This adduct also adopts an intercalative conformation, but it is intercalated from the minor groove, and on the 3′-side of the damaged base (Figure 2A); as recently shown by Tang et al,42 the Watson-Crick partner bases at the lesion site are stretched apart so that they are not in hydrogen bonding range, but they remain stacked with the PAH aromatic ring system. The previous work41 together with our current studies provide a set of data for lesions that are members of intercalative conformational families, but with differing local structural and energetic properties. These data provide an opportunity to examine the hypothesis that flipping barriers for the partner base are one component of the energetic properties that are sensed by the recognition protein. More broadly, an opportunity for further elucidating structural and energetic elements that contribute to determining relative NER susceptibilities is afforded, including consideration of the adduct structures in relation to that of the Rad4/Rad23 yeast homolog of XPC.
MATERIALS AND METHODS
Adduct structural models
For the 14R-DB[a,l]P-dA adduct, a duplex 11-mer model had previously been constructed;34, 43 it was based on the high resolution NMR solution structure of the benzo[c]phenanthrene (B[c]Ph)-derived fjord region analogue with the same stereochemistry at the linkage site, the 1R (+)-trans-anti-B[c]Ph-N6-dA adduct,35 by adding two aromatic rings, followed by 30.0 ns of MD simulations.34 As discussed previously,34 an intercalative conformation for the 14R-DB[a,l]P-dA adduct is supported by a red-shifted UV absorbance maximum.44 For the 10R-B[a]P-dA adduct, an initial structure that was based on the NMR solution structure of this adduct37 with remodeling of the sequence33 (Figure 2B) had been previously subjected to MD simulations, as had the corresponding unmodified duplex.32 We note that a number of 10R-B[a]P-dA adduct NMR solution structures have all revealed similar 5′-intercalated structures in a number of sequence contexts.33, 36, 37, 45 We utilized 30 ns from the MD ensembles for analyses of helix structural properties using MD Toolchest,46, 47 for van der Waals stacking energy analyses using the ANAL module of the AMBER 11 simulation package,48 as well as to obtain the best representative structures49 (shown in Figure 2A), which were used to initiate the flipping studies. The computed Rise and Twist values at the lesion site were between base pairs comprising the intercalation pocket: for the 5′-intercalated dA adducts and their corresponding unmodified duplex, between C5:G18 and A6:T17, and for the 3′-intercalated dG adduct and its corresponding unmodified duplex, between G6:C17 and C7:G16. For the van der Waals stacking interaction energies of the flipping partner base, we computed interactions between the flipping base T17/C17 with the bases C5, C7, G16, G18, A6/G6 and the lesion aromatic rings. Analyses for the 14R-DB[a,l]P-dG adduct42 given in the present work are based on the ensemble of five structures, derived from the 1 ns NMR restrained MD simulations, that were selected to best represent the NMR data42 [PDB50 ID: 2LZK].
Flipping free energy profiles
The best representative structures from the 30 ns MD simulations of the 14R-DB[a,l]P-dA and 10R-B[a]P-dA adducts (shown in Figure 2) and the corresponding unmodified duplex, were the initial models for the flipping studies. These modified and unmodified 11-mer duplexes were subjected to 2 ns of unrestrained MD simulations for the subsequent free energy profile calculations. Next, utilizing a restraint force constant of 1 × 104 kcal/(mol×radian2), 0.5 ps simulations were performed serially for 72 windows at 5 degree intervals of the reaction coordinate (Figure 3) as the partner nucleotide dT17 (Figure 2A) is extruded, employing the last structure of the previous window to initialize the current window. This protocol provided the structures to begin the 3 ns MD simulations for sampling each umbrella window. The AMBER 10 software package51 modified for base flipping along the pseudo-dihedral angle reaction coordinate52–54 (definition shown in Figure 3A) was employed. Random snapshots from the MD simulations of the 14R-DB[a,l]P-dA duplex along different reaction coordinate values (−180°, −90°, 0°, 90°) are depicted in Figure 3B. The free energy profiles were computed with data collected between 1.0 and 3.0 ns with a 1.0 fs time step. Thus, a total of 2 × 106 structures were utilized to compute the free energy profile for each window; for the 72 windows, the total number of structures was 1.44 × 108. The statistical errors were calculated based on the differences among different time periods (1–2 ns versus 1 – 3 ns, 2 –3 ns versus 1 – 3 ns) (Figure S2, Supporting Information). The force constants for the 72 windows were in the range of 20 to 200 kcal/(mol×radian2) to ensure sufficient overlap between the windows. The free energy profiles along the reaction coordinate were obtained with the weighted histogram analysis method (WHAM).55
Figure 3.

(A) The pseudo-dihedral angle is defined by the center of mass of the circled groups. (B) Random snapshots of the 14R-DB[a,l]P-dA duplex 11-mer along different reaction coordinate values. (a) Pseudo-dihedral = −180°. (b) Pseudo-dihedral = −90°. (c) Pseudo-dihedral = 0°. (d) Pseudo-dihedral = 90°. Movie S4, Supporting Information, shows random snapshots along the pseudo-dihedral at 5 ° intervals from 0° to 355°. Views are into the minor groove.
Modeling Rad4/Rad23 in complex with DNA containing the 14R-DB[a,l]P-dG adduct
Loop filling
Residues 518 to 525 were missing in a β-hairpin loop (BH2) of the crystal structure of Rad4/Rad23 (PDB50 ID: 2QSG11 ) (Figure S1, Supporting Information). We used BLAST (http://blast.ncbi.nlm.nih.gov/) to homology model the sequence of residues 511 to 532. Using the BLOSUM45 matrix for scoring, the PDB structure that provided the best alignment, with an e-value of 1.8, was residues 61 to 70 of phosphoglycerate kinase from B. anthracis with PDB ID:3UWD.56 The missing loop (amino acids: grpkgeae) of Rad4 was modeled based on this homology sequence (amino acids: grpkgqav) from the phosphoglycerate kinase structure. The loop structure of the homology sequence was modeled to Rad4 using Discovery Studio 2. The terminus of the loop was adjusted to covalently bond to the terminus of the broken chain, and analogous residues were mutated to those in the missing loop of Rad4. Side chains and ϕ/ψ torsion angles were adjusted to avoid clashing with other residues in Rad4 and the DNA duplex.
Starting model for lesion-containing DNA in Rad4/Rad23
In the Rad4/Rad23-DNA complex structure (PDB ID: 2QSG11 ), the cyclobutane pyrimidine dimer (CPD) lesion is missing, resulting in a gap of two nucleotides in the damaged strand (Figure S1, Supporting Information). We modeled to this gap the dinucleotide containing the 14R-DB[a,l]P-dG adduct with its 3′-side neighboring base dC; it was cut from the NMR refined structure of the 14R-DB[a,l]P-dG adduct.42 There is very limited space to fit the lesion and its neighboring nucleotide into the gap. Utilizing Insight II from Accelrys Inc, we placed the excised dinucleotide containing the lesion so that it was as close as possible to forming the missing covalent bonds in the gapped crystal structure, while having minimum collisions. The bond distances at this stage were about 3 Å too long. We used steepest descent energy minimization with the AMBER 11 simulation package48 to form the bond, followed by 10 ns of MD, to further equilibrate the model.
Force field
The Cornell et al. force field,57 the parm99 parameter set,58 the parmbsc0 DNA parameters,59 and force field parameters developed for the 14R-DB[a,l]P-dA and 10R-B[a]P-dA adducts were utilized, together with explicit solvent and counterions. These partial charges and other added parameters for the 14R-DB[a,l]P-dA34 and 10R-B[a]P-dA60 adducts on the nucleoside level were reported previously.
Minimization and MD protocols
The AMBER 11 simulation package48 was utilized to carry out all minimization and MD simulations. The LEaP module was used to add hydrogen atoms and neutralize the system with Na+ counterions. A periodic rectangular box of TIP3P water61 with 10.0 Å buffer was created around the Rad4/Rad23-DNA-14R-DB[a,l]P-dG complex with the LEaP module. The box dimensions were approximately 127 Å × 103 Å × 97 Å, with a total of ~32205 water molecules. The following minimization, heating, MD equilibration and production protocols were utilized: to begin with, the counterions and water molecules were minimized for 500 steps of steepest descent and 500 steps of conjugate gradient, with 50 kcal/mol restraints on the solute atoms. Then, 30 ps initial MD at 10 K with 25 kcal/mol restraints on solute were performed to allow the solvent to relax. Next, the system was heated from 10 K to 300 K at constant volume over 30 ps with 10 kcal/mol restraint on the protein-DNA complex. The restraints on the solute were relaxed from 10 kcal/mol (for 30 ps) to 1 kcal/mol (for 40 ps) to 0.1 kcal/mol (for 50 ps). Subsequently, production MD was conducted at 1 atm, 300 K for 10.0 ns, with 1 ps coupling constant for both pressure and temperature. In the MD simulations, the Particle-Mesh Ewald62,63 method with 9.0 Å cutoff for the non-bonded interactions was used. A 2.0 fs time step and the SHAKE algorithm64 were applied in the MD simulations. All other parameters were default values in the AMBER 11 simulation package.48
RESULTS AND DISCUSSION
Relative flipping free energy barriers of lesion partner bases follow the trend of relative NER susceptibilities in investigated intercalated lesions
We have determined the complete flipping free energy profiles for the partner base dT17 (Figure 2A) to the damaged adenine in the 14R-DB[a,l]P-dA and the 10R-B[a]P-dA adducts, and the corresponding unmodified duplex utilizing the Weighted Histogram Analysis Method (WHAM)55 with a pseudo-dihedral angle reaction coordinate (Figure 3).52,54 We wished to explore the hypothesis that the flipping free energy profiles reflect a component in the local thermodynamic stability of the lesion-containing duplexes that is considered to play an important role in their relative NER excision efficiencies.2,4,38,65 These profiles are determined from structural models based on NMR solution data, molecular modeling and MD simulations as detailed in Methods.
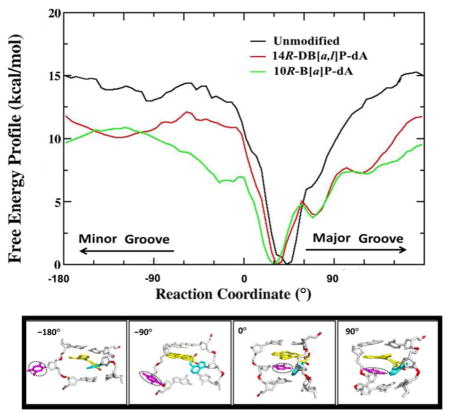
The calculated free energy profiles for the 14R-DB[a,l]P-dA and the 10R-B[a]P-dA adducts, and the unmodified duplex 11-mers are shown in Figure 4. They have converged reasonably well as shown in Figure S2, Supporting Information. For the unmodified duplex 11-mer, the calculated maximal free energy barrier for flipping dT17 is 15.0 ± 0.3 kcal/mol (Figure 4A), in a similar range to base flipping barriers computed for unmodified duplexes in previous studies (~ 15 to 20 kcal/mol);41, 53, 54, 66, 67 barriers for pyrimidines are very similar through major or minor groove pathways, as also seen in our results (Figure 4). For the 14R-DB[a,l]P-dA adduct, the calculated maximal free energy barrier is 11.8 ± 0.6 kcal/mol (Figure 4A), with minor and major groove routes nearly the same. For the 10R-B[a]P-dA adduct, the calculated maximal free energy barrier is 10.9 ± 0.2 kcal/mol (Figure 4A), and it is via the minor groove. In prior work,41 we obtained flipping free energy profiles for the 14R-DB[a,l]P-dG adduct and the corresponding unmodified duplex in the otherwise identical sequence context (Figure 2B). For comparison we show this profile in Figure 4B. In this case the maximal barrier for flipping the dC17 base in the unmodified duplex is 18.1 ± 0.8 kcal/mol, with major and minor groove paths essentially the same, while in the 14R-DB[a,l]P-dG adduct, it is 10.4 ± 0.6 kcal/mol (Figure 4B). In a recent study of flipping free energy profiles in several aromatic amine-derived DNA adducts, groove-dependence of maximum barriers differ in the various lesions and sequences.68
Figure 4.

(A) The calculated free energy profiles for the 14R-DB[a,l]P-dA duplex 11-mer (red), the 10R-B[a]P-dA duplex 11-mer (green) and their corresponding unmodified duplex 11-mer (black). (B) The calculated free energy profiles for the 14R-DB[a,l]P-dG duplex 11-mer (red) and its corresponding unmodified duplex 11-mer (black). Maximal barrier values are given.
To provide insights on the relative difficulty of flipping the base that is the partner to the damaged base, we are interested in the maximal barrier differences between the corresponding damaged and undamaged duplexes, since dC and dT bases must have intrinsically different barriers in the same duplex sequence context.53 We hence define ΔΔGFlip = ΔGFlip,mod − ΔGFlip,unmod, where ΔGFlip,mod is the maximal flipping free energy barrier for the partner base to the lesion in the modified duplex, and ΔGFlip,unmod is the maximal flipping free energy barrier for the corresponding base in the unmodified duplex. Table 1 presents barriers for the modified and corresponding unmodified duplexes we have studied, and their differences, ΔΔGFlip. The base flips more easily the lower its relative flipping free energy barrier, ΔΔGFlip. The table also presents relative NER efficiencies. We observe that ΔΔGFlip for the well repaired 14R-DB[a,l]P-dG adduct is lowest in energy, followed by the modestly repaired 10R-B[a]P-dA, and then the repair-resistant 14R-DB[a,l]P-dA.
Table 1.
Relative flipping free energy barriers are correlated with lesion-base stacking energies and relative NER susceptibilities in investigated intercalated adducts. Standard deviations are given in parentheses.
| PAH-DNA adduct | Structural characteristics | Ensemble average vdW stacking energy of partner (kcal/mol)a | Relative flipping barrier ΔΔGFlipping (kcal/mol)b,c | NERd |
|---|---|---|---|---|
| R-DB[a,l]P-dA | Fjord region, 5′-intercalation from major groove, 5 aromatic rings |
−16.6 (1.6) | −3.2 (0.7) | Resistant |
| R-B[a]P-dA | Bay region, 5′-intercalation from major groove, 4 aromatic rings |
−15.2 (1.4) | −4.1 (0.6) | Modest |
| R-DB[a,l]P-dG | Fjord region, 3′-intercalation from minor groove, 5 aromatic rings |
−12.0 (0.7) | −7.7 (1.0) | High |
Table S1, Supporting Information, provides breakdowns of van der Waals stacking interaction energies between the flipping base and all nearby bases and the PAH aromatic ring system (See Methods).
Flipping data for the 14R-DB[a,l]P-dG adduct and its corresponding unmodified duplex was published previously.41
The maximal base flipping barrier energy ΔG is 11.8 ± 0.6 kcal/mol for the 14R-DB[a,l]P-dA adduct, 10.9 ± 0.2 kcal/mol for the 10R-B[a]P-dA adduct, and 15.0 ± 0.3 kcal/mol for the corresponding dA unmodified, 10.4 ± 0.6 kcal/mol for the 14R-DB[a,l]P-dG adduct and 18.1± 0.8 kcal/mol for the corresponding dG unmodified. See Figure 4. ΔΔG is the difference between the ΔG for the unmodified and the corresponding modified duplex.
Distinct structural features of the intercalated lesions explain differences in partner base van der Waals stacking interaction energies and ease of flipping, which correlate with relative NER susceptibilities
Structural and energetic lesion properties can explain the relative flipping barriers. NMR solution data, molecular modeling and MD simulations were utilized to obtain our structural models, as described in Methods. The lesion structures are shown in Figure 2A and Movies S1–3, Supporting Information. Figure 5 shows stacking patterns and predominant van der Waals interactions involving the flipping partner base for each lesion. Figure S4, Supporting Information shows stereo views. The repair-resistant38 14R-DB[a,l]P-dA and the modestly repaired33,39,40 10R-B[a]P-dA adducts are intercalated from the spacious major groove on the 5′-side of the damaged adenine; however, the 14R-DB[a,l]P-dG adduct is intercalated from the narrow minor groove on the 3′-side of the damaged guanine. In each case, the position of the modified amino group in B-DNA determines the groove from which intercalation occurs (Figure 7A). In the well-repaired38 14R-DB[a,l]P-dG adduct, intercalation of the bulky aromatic rings from the minor groove causes the lesion-modified base pair to stretch so that these bases are no longer within Watson-Crick hydrogen bonding range,42 due to steric crowding caused by the distal aromatic rings (highlighted in grey in Figure 1B). This stretching is accompanied by unusually enlarged Rise of 9.1 Å, compared to 7.6 Å in the 14R-DB[a,l]P-dA adduct intercalation pocket (Figure S3, Supporting Information) and severe unwinding accompanied by minor groove opening (Figure S3, Supporting Information).42 However, the damaged base pair dG6*:dC17 retains stacking with the DB[a,l]P aromatic ring system: the C17 base stacks mostly with the 8,9 ring (Figure 1) of the DB[a,l]P moiety, and G6* stacks with the 2,3,4 ring (Figures 1). The flipping C17 also stacks with G18. By contrast, for the 14R-DB[a,l]P-dA adduct, the DB[a,l]P ring system is well intercalated from the spacious major groove on the 5′-side, without being sterically obstructing; Watson-Crick pairs are maintained, and the dT17 partner base remains well stacked both with the DB[a,l]P rings and the G16 base. These structural properties explain the ΔΔGFlip values for dT17 in 14R-DB[a,l]P-dA vs dC17 in 14R-DB[a,l]P-dG of −3.2 kcal/mol and −7.7 kcal/mol, respectively (Figure 6A); thus dT17, with higher barrier, flips with greater difficulty than dC17. Consistent with the ΔΔGFlip values, the van der Waals stacking interaction energy between dT17 and surrounding bases and the DB[a,l]P lesion aromatic rings is −16.6 kcal/mol, while for the dC17 it is only −12.0 kcal/mol (Figure 6B).
Figure 5.

Stick (a) and CPK (b) renderings of views looking down the helix axis of the (dA6*:dT17) • (dC5:dG18) segment in the 14R-DB[a,l]P-dA (A) and 10R-B[a]P-dA (B) 11-mer duplexes, and the (dC7:dG16) • (dG6*:dC17) segment in the 14R-DB[a,l]P-dG (C) 11-mer duplex. The black dot circles designate the partner bases to be flipped. Note that PAH ring overlap with an adjacent base does not properly project stacking when the two ring systems are not parallel, as is particularly the case for the dT17 in 10R-B[a]P-dA (See Figure 2). (c) Views looking into the minor groove that show predominant stacking interactions (indicated by arrows) involving the flipping base. The color code is the same as in Figure 2A. Stereo views are given in Figure S4, Supporting Information. Table S1, Supporting Information gives ensemble average breakdowns of van der Waals stacking interaction energies between the flipping base and all adjacent bases and the PAH aromatic ring system (See Methods).
Figure 7.

(A) Cartoons illustrating the groove location and BHD3 β-hairpin insertion direction. (B) Best representative structure for the Rad4/Rad23-DNA-14R-DB[a,l]P-dG complex from 10.0 ns of MD simulations. (C) Close-up view of the BHD3 β-hairpin insertion and lesion site. See Movie S5, Supporting Information.
Figure 6.

(A) Summary of ΔΔGFlip, the maximal flipping free energy barrier differences between the modified and the corresponding unmodified duplexes for the 10R-B[a]P-dA, 14R-DB[a,l]P-dA and the 14R-DB[a,l]P-dG adducts. (B) Ensemble average values of the van der Waals stacking interaction energy between the partner base and nearby bases and the lesion aromatic ring system. (C) Nucleotide excision repair susceptibilities relative to the well-repaired standard R(+)-cis-anti-B[a]P-N2-dG lesion. Data are from references.33,38–40
On the other hand, the modestly repaired 10R-B[a]P-dA adduct differs from the repair resistant 14R-DB[a,l]P-dA adduct in topology: it has a planar and rigid bay region geometry and contains four aromatic rings, while the five-ring 14R-DB[a,l]P-dA adduct has a flexible and twisted fjord region, which accommodates to optimize stacking interactions.34 The rigidity of the bay region causes the intercalation pocket to have a large rise of 8.9 Å, compared to 7.6 Å for the 14R-DB[a,l]P-dA adduct and the duplex is locally more unwound (Figure S3, Supporting Information and Figure 2). Accordingly, the van der Waals stacking interaction energy of the dT17 partner base for the 10R-B[a]P-dA adduct is ~1.5 kcal/mol less stabilizing than for the 14R-DB[a,l]P-dA adduct (Table 1). This is consistent with easier flipping, with the ΔΔGFlip being ~1 kcal/mol lower than for the 14R-DB[a,l]P-dA adduct (Table 1). However, the intercalation from the major groove in the B[a]P case is still better than for the minor groove intercalated 14R-DB[a,l]P-dG adduct, and the dC17 in the 14R-DB[a,l]P-dG case is less well stacked than the dT17 in the 10R-B[a]P-dA adduct. Stacking and other lesion properties are summarized in Table 1. See Figures 2 and 5 for structures.
Figure 6 shows how relative flipping free energy barriers ΔΔGFlip, that reflect ease of flipping, relate to van der Waals stacking interaction energies involving the flipping base, and to relative NER susceptibilities. From this figure, it is clear that ΔΔGFlip (Figure 6A) parallels the stacking interaction energies (Figure 6B), and that these both correlate inversely with relative NER susceptibilities (Figure 6C). These results support the hypothesis that ease of partner base flipping is a component of the local thermodynamic factors that contribute to relative NER susceptibilities in the current series of intercalated adducts.
Resistance to NER has been correlated with minimal decrease or even more striking increases in the thermal stabilities of damaged DNA duplexes compared to the analogous unmodified sequences. 38, 65, 69 In our current series, there is a remarkable stabilization of thermal melting by ~ 10 °C for the repair resistant 14R-DB[a,l]P-dA adduct, while the repaired ones are both destabilized by ~10 °C to 12 °C.42 While thermal melting does not quantitatively correlate with NER susceptibility, for reasons that have been discussed,38 thermal stabilization has provided strong correlation with repair resistance, and structural connections with lesion-induced van der Waals stacking stabilization have been presented.38,65,69,70
Lesion recognition through β-hairpin insertion from the major groove and partner base flipping: combined effects of local lesion-induced thermodynamic impact and steric properties in intercalated adducts
The crystal structure of the XPC-RAD23B yeast homologue, Rad4/Rad23, provides an opportunity to gain some further understanding of how the lesion recognition process may function with intercalated lesions whose repair susceptibility varies. This structure shows that the β-hairpin BHD3 is inserted between the two DNA strands from the major groove side (Figure 7), extending through the width of the Watson-Crick double helix and causing the two mismatched dT bases opposite to the cis-syn thymine dimer lesion to flip into the protein. With the approach of the BHD3 β-hairpin from the major groove side, it appears that partner flipping must take the minor groove route to avoid steric clashes with the incoming hairpin. This route appears to be sterically unavoidable for this NER system. For comparison, a computational investigation of 8-oxo-guanine eversion free energy paths in the prokaryotic base excision repair Fpg enzyme, where the damaged base itself is flipped into a specific pocket and then excised, showed that a major groove route provided the lowest free energy path.71
Our structural and energetic studies for the 14R-DB[a,l]P-dA adduct, combined with the crystal structure, provide further understanding for the repair resistance of this adduct. The ΔΔGFlip is the highest, with flipping most difficult, due to the excellent stacking of the partner base dT17 with the five ring twisted fjord DB[a,l]P (Table 1 and Figure 5B (a)). This factor, combined with the intercalation of the adduct from the major groove side, could obstruct the BHD3 β-hairpin insertion and concomitant partner flipping. The 10R-B[a]P-dA adduct’s modest repair, also plausibly related to its major groove intercalation and associated steric hinderance to hairpin insertion, is somewhat better than that of the repair-resistant 14R-DB[a,l]P-dA adduct. Its ΔΔGFlip is modestly more favorable due to weaker stacking interactions of the dT17 with the rigid, planar, bay region B[a]P -- which causes enlarged Rise in the intercalation pocket (Figure S3, Supporting Information) -- and with its adjacent G16 base (Figure 5B (c)).
By contrast, the well-repaired 14R-DB[a,l]P-dG adduct has the most flipping-favorable ΔΔGFlip, because the partner dC17 is stacked least well (Figures 5C (a)), stemming from the intercalation of its bulky ring system from the minor groove side (Figures 2A and 5A). One could envision that the BHD3 β-hairpin -- coming from the major groove side -- could evict the intercalated DB[a,l]P-dG adduct together with its 3′-side base dC7, with which the DB[a,l]P aromatic rings are partly stacked (Figures 2A and 5C(a)), into the minor groove. The large untwisting (Figure S3, Supporting Information) would facilitate this process, since the DNA contains this distortion in the crystal structure11 (Figure S1, Supporting Information).
To illustrate such an extruded structure, we have used molecular modeling and a brief 10 ns MD simulation for equilibration to propose such a model (see Methods) for the 14R-DB[a,l]P-dG adduct, based on the Rad4/Rad23 structure. This model essentially preserves the local NMR solution structure42 stacking pattern of the damaged strand (Figure 2A) on the dinucleotide level, with the 14R-DB[a,l]P-dG aromatic ring system and its 3′-neighbor dC base stacked, except that this region is extruded into the minor groove (Figure 7). The BHD3 β-hairpin and the two partner dT bases remain as in the crystal structure; the crystal Phe 599 also remains stacked with the thymine below as in the crystal (Figure 7 and Movie S5, Supporting Information). In essence, the lesioned region is placed approximately in the space occupied by the thymine dimer in the crystal (although coordinates for the dimer could not be resolved). The BH2 residue Arg 519 (coordinates were missing in the crystal and hence were modeled, as detailed in Methods), forms a hydrogen bond with the DNA backbone. We emphasize, however, that the brief MD only served the purpose of equilibrating the model, but is far too short for analyses of the constellation of protein-DNA interactions that stabilize the structure. Our model is intended to illustrate how a well-repaired destabilizing lesion,38 which is intercalated from the minor groove and hence can be readily extruded by the major groove-intruding BHD3 β-hairpin, may be accommodated in Rad4/Rad23.
CONCLUSION
The yeast Rad4/Rad23 crystal structure11 shows that lesion recognition for NER requires partner base flipping and insertion of a protein β-hairpin between the DNA strands at the damage site, from the major groove side, and requires the concerted action of these two processes. Our studies of flipping free energy barriers of major and minor groove intercalated PAHs, as well as molecular modeling studies of the yeast Rad4/Rad23 homolog of XPC-RAD23B, suggest that both energetic and structural lesion properties may impact NER susceptibility. Relative flipping barriers follow the trend of relative NER susceptibilities, and the lesion partner base flipping free energy barrier is related to how well the partner base is stacked with the lesion aromatic rings and adjacent bases for the three differently intercalated lesions investigated (Figure 6). However, steric effects of intercalated lesions that occlude the major groove should also be considered. Lesion-governed modulation of base flipping and/or hairpin insertion should affect the rate-determining lesion recognition step10 and the subsequent rate of excision. Of course, downstream effects including the subsequent steps of lesion verification are also key regulators of NER efficiencies.7, 8, 72–74
Supplementary Material
Acknowledgments
Funding Sources
This research was supported by NIH Grants CA-28038 to S.B. and CA-099194 to N.E.G.. Computational infrastructure and systems management was partially supported by CA-75449 to S.B.. We appreciate the computational resources and support provided by NYU-ITS. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation (NSF) grant MCB060037. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
ABBREVIATIONS
- NER
nucleotide excision repair
- PAHs
polycyclic aromatic hydrocarbons
- DB[a,l]P
dibenzo[a,l]pyrene
- B[a]P
benzo[a]pyrene
- 14R-DB[a,l]P-dG
14R(+)-trans-anti-DB[a,l]P-N2-dG
- 14R-DB[a,l]P-dA
14R (+)-trans-anti-DB[a,l]P-N6-dA
- R-cis-B[a]P-N2-dG
10R (+)-cis-anti-B[a]P-N2-dG
- 10R-B[a]P-dA
10R (−)-trans-anti-B[a]P-N6-dA
- MD
molecular dynamics
Footnotes
ASSOCIATED CONTENT
Supporting Information. Supporting Movies S1–3 show the 14R-DB[a,l]P-dA, 14R-DB[a,l]P-dG and 10R-B[a]P-dA structures rotating. Supporting Movie S4 shows the flipping process for the 14R-DB[a,l]P-dA adduct. Supporting Movie S5 shows the 14R-DB[a,l]P-dG-containing Rad4/Rad23 structure. Figure S1 showing crystal structure of Rad4/Rad23 with PDB ID: 2QSG; Figure S2 showing free energy profiles for the 14R-DB[a,l]P-dA, 10R-B[a]P-dA adducts and the corresponding unmodified duplex 11-mer; Figure S3 showing ensemble average Rise and Twist values between base pairs comprising the intercalation pocket; Figure S4 showing stereo views for Figure 5; Table S1 supplies breakdowns of van der Waals stacking interaction energies for the flipping base. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cai Y, Kropachev K, Xu R, Tang Y, Kolbanovskii M, Kolbanovskii A, Amin S, Patel DJ, Broyde S, Geacintov NE. Distant neighbor base sequence context effects in human nucleotide excision repair of a benzo[a]pyrene-derived DNA lesion. J Mol Biol. 2010;399:397–409. doi: 10.1016/j.jmb.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cai Y, Patel DJ, Broyde S, Geacintov NE. Base sequence context effects on nucleotide excision repair. J Nucleic Acids. 2010;2010 doi: 10.4061/2010/174252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cai Y, Patel DJ, Geacintov NE, Broyde S. Differential nucleotide excision repair susceptibility of bulky DNA adducts in different sequence contexts: hierarchies of recognition signals. J Mol Biol. 2009;385:30–44. doi: 10.1016/j.jmb.2008.09.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geacintov NE, Broyde S, Buterin T, Naegeli H, Wu M, Yan S, Patel DJ. Thermodynamic and structural factors in the removal of bulky DNA adducts by the nucleotide excision repair machinery. Biopolymers. 2002;65:202–210. doi: 10.1002/bip.10239. [DOI] [PubMed] [Google Scholar]
- 5.Kropachev K, Kolbanovskii M, Cai Y, Rodriguez F, Kolbanovskii A, Liu Y, Zhang L, Amin S, Patel D, Broyde S, Geacintov NE. The sequence dependence of human nucleotide excision repair efficiencies of benzo[a]pyrene-derived DNA lesions: insights into the structural factors that favor dual incisions. J Mol Biol. 2009;386:1193–1203. doi: 10.1016/j.jmb.2008.12.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gillet LC, Scharer OD. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem Rev. 2006;106:253–276. doi: 10.1021/cr040483f. [DOI] [PubMed] [Google Scholar]
- 7.Peng Y, Wang H, Santana dos Santos L, Kisker C, Van Houten B. Nucleotide excision repair from bacteria to humans: Structure–function studies. In: Penning TM, editor. Chemical Carcinogenesis, Current Cancer Research. Springer Science+Business Media, LLC; 2011. pp. 267–296. [Google Scholar]
- 8.Schärer OD, Campbell AJ. Mechanisms of base excision repair and nucleotide excision repair. In: Geacintov NE, Broyde S, editors. The Chemical Biology of DNA Damage. Wiley-VCH; Weinheim, Germany: 2010. pp. 239–260. [Google Scholar]
- 9.Sugasawa K, Ng JM, Masutani C, Iwai S, van der Spek PJ, Eker AP, Hanaoka F, Bootsma D, Hoeijmakers JH. Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol Cell. 1998;2:223–232. doi: 10.1016/s1097-2765(00)80132-x. [DOI] [PubMed] [Google Scholar]
- 10.Luijsterburg MS, von Bornstaedt G, Gourdin AM, Politi AZ, Mone MJ, Warmerdam DO, Goedhart J, Vermeulen W, van Driel R, Hofer T. Stochastic and reversible assembly of a multiprotein DNA repair complex ensures accurate target site recognition and efficient repair. JCell Biol. 2010;189:445–463. doi: 10.1083/jcb.200909175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Min JH, Pavletich NP. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature. 2007;449:570–575. doi: 10.1038/nature06155. [DOI] [PubMed] [Google Scholar]
- 12.Schärer OD. Achieving broad substrate specificity in damage recognition by binding accessible nondamaged DNA. Mol Cell. 2007;28:184–186. doi: 10.1016/j.molcel.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 13.Schärer OD. Hot topics in DNA repair: the molecular basis for different disease states caused by mutations in TFIIH and XPG. DNA Repair (Amst) 2008;7:339–344. doi: 10.1016/j.dnarep.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gunz D, Hess MT, Naegeli H. Recognition of DNA adducts by human nucleotide excision repair. Evidence for a thermodynamic probing mechanism. J Biol Chem. 1996;271:25089–25098. doi: 10.1074/jbc.271.41.25089. [DOI] [PubMed] [Google Scholar]
- 15.Cavalieri EL, Higginbotham S, RamaKrishna NV, Devanesan PD, Todorovic R, Rogan EG, Salmasi S. Comparative dose-response tumorigenicity studies of dibenzo[alpha,l]pyrene versus 7,12-dimethylbenz[alpha]anthracene, benzo[alpha]pyrene and two dibenzo[alpha,l]pyrene dihydrodiols in mouse skin and rat mammary gland. Carcinogenesis. 1991;12:1939–1944. doi: 10.1093/carcin/12.10.1939. [DOI] [PubMed] [Google Scholar]
- 16.Cavalieri EL, Higginbotham S, Rogan EG. Dibenzo[a,l]pyrene: The most potent carcinogenic aromatic hydrocarbon. Polycyclic Aromat Compd. 1994;6:177–183. [Google Scholar]
- 17.Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91:1194–1210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- 18.Luch A. On the impact of the molecule structure in chemical carcinogenesis. EXS. 2009;99:151–179. doi: 10.1007/978-3-7643-8336-7_6. [DOI] [PubMed] [Google Scholar]
- 19.Phillips DH. Smoking-related DNA and protein adducts in human tissues. Carcinogenesis. 2002;23:1979–2004. doi: 10.1093/carcin/23.12.1979. [DOI] [PubMed] [Google Scholar]
- 20.IARC. IARC Monogr Eval Carcinog Risks Hum. International Agency for Research on Cancer; Lyon, France: 2010. Some non-heterocyclic polycyclic aromatic hydrocarbons and some related exposures; pp. 1–853. [PMC free article] [PubMed] [Google Scholar]
- 21.Hirshfeld FL. The structure of overcrowded aromatic compounds. Part VII. Out-of-plane deformation in benzo[c]-phenanthrene and 1,12-dimethyl-benzo[c]phenanthrene. J Chem Soc. 1963;1963:2108–2125. [Google Scholar]
- 22.Katz AK, Carrell HL, Glusker JP. Dibenzo[a,l]pyrene (dibenzo[def,p]chrysene): fjord-region distortions. Carcinogenesis. 1998;19:1641–1648. doi: 10.1093/carcin/19.9.1641. [DOI] [PubMed] [Google Scholar]
- 23.Conney AH. Induction of microsomal enzymes by foreign chemicals and carcinogenesis by polycyclic aromatic hydrocarbons: G. H. A. Clowes Memorial Lecture. Cancer Res. 1982;42:4875–4917. [PubMed] [Google Scholar]
- 24.Xue W, Warshawsky D. Metabolic activation of polycyclic and heterocyclic aromatic hydrocarbons and DNA damage: a review. Toxicol Appl Pharmacol. 2005;206:73–93. doi: 10.1016/j.taap.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 25.Penning TM. Polycyclic aromatic hydrocarbons: Multiple metabolic pathways and the DNA lesions formed. In: Geacintov NE, Broyde S, editors. The Chemical Biology of DNA Damage. Chapter 6. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2010. pp. 131–155. [Google Scholar]
- 26.Henkler F, Stolpmann K, Luch A. Exposure to polycyclic aromatic hydrocarbons: bulky DNA adducts and cellular responses. EXS. 2012;101:107–131. doi: 10.1007/978-3-7643-8340-4_5. [DOI] [PubMed] [Google Scholar]
- 27.Hanrahan CJ, Bacolod MD, Vyas RR, Liu T, Geacintov NE, Loechler EL, Basu AK. Sequence specific mutagenesis of the major (+)-anti-benzo[a]pyrene diol epoxide-DNA adduct at a mutational hot spot in vitro and in Escherichia coli cells. Chem Res Toxicol. 1997;10:369–377. doi: 10.1021/tx9601925. [DOI] [PubMed] [Google Scholar]
- 28.Geacintov NE, Cosman M, Hingerty BE, Amin S, Broyde S, Patel DJ. NMR solution structures of stereoisometric covalent polycyclic aromatic carcinogen–DNA adduct: principles, patterns, and diversity. Chem Res Toxicol. 1997;10:111–146. doi: 10.1021/tx9601418. [DOI] [PubMed] [Google Scholar]
- 29.Lukin M, de Los Santos C. NMR structures of damaged DNA. Chem Rev. 2006;106:607–686. doi: 10.1021/cr0404646. [DOI] [PubMed] [Google Scholar]
- 30.Cho BP. Dynamic conformational heterogeneities of carcinogen-DNA adducts and their mutagenic relevance. J Environ Sci Health Pt C Environ Carcinogen Ecotoxi Rev. 2004;C22:57–90. doi: 10.1081/LESC-200038217. [DOI] [PubMed] [Google Scholar]
- 31.Stone MP, Huang H, Brown KL, Shanmugam G. Chemistry and structural biology of DNA damage and biological consequences. Chem Biodivers. 2011;8:1571–1615. doi: 10.1002/cbdv.201100033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cai Y, Geacintov NE, Broyde S. Nucleotide excision repair efficiencies of bulky carcinogen-DNA adducts are governed by a balance between stabilizing and destabilizing interactions. Biochemistry. 2012;51:1486–1499. doi: 10.1021/bi201794x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan S, Wu M, Buterin T, Naegeli H, Geacintov NE, Broyde S. Role of base sequence context in conformational equilibria and nucleotide excision repair of benzo[a]pyrene diol epoxide-adenine adducts. Biochemistry. 2003;42:2339–2354. doi: 10.1021/bi0270081. [DOI] [PubMed] [Google Scholar]
- 34.Cai Y, Ding S, Geacintov NE, Broyde S. Intercalative conformations of the 14R (+)- and 14S (−)-trans-anti-DB[a,l]P-N6-dA adducts: molecular modeling and MD simulations. Chem Res Toxicol. 2011;24:522–531. doi: 10.1021/tx1004002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cosman M, Fiala R, Hingerty BE, Laryea A, Lee H, Harvey RG, Amin S, Geacintov NE, Broyde S, Patel D. Solution conformation of the (+)-trans-anti-[BPh]dA adduct opposite dT in a DNA duplex: intercalation of the covalently attached benzo[c]phenanthrene to the 5′-side of the adduct site without disruption of the modified base pair. Biochemistry. 1993;32:12488–12497. doi: 10.1021/bi00097a029. [DOI] [PubMed] [Google Scholar]
- 36.Zegar IS, Chary P, Jabil RJ, Tamura PJ, Johansen TN, Lloyd RS, Harris CM, Harris TM, Stone MP. Multiple conformations of an intercalated (−)-(7S,8R,9S,10R)-N6-[10-(7,8,9,10-tetrahydrobenzo[a]pyrenyl)]-2′-deoxyadenosyl adduct in the N-ras codon 61 sequence. Biochemistry. 1998;37:16516–16528. doi: 10.1021/bi9817616. [DOI] [PubMed] [Google Scholar]
- 37.Zegar IS, Kim SJ, Johansen TN, Horton PJ, Harris CM, Harris TM, Stone MP. Adduction of the human N-ras codon 61 sequence with (−)-(7S,8R,9R,10S)-7,8-dihydroxy-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene: structural refinement of the intercalated SRSR(61,2) (−)-(7S,8R,9S,10R)-N6-[10-(7,8,9,10-tetrahydrobenzo[a]pyrenyl)]-2′-deoxyadenosyl adduct from 1H NMR. Biochemistry. 1996;35:6212–6224. doi: 10.1021/bi9524732. [DOI] [PubMed] [Google Scholar]
- 38.Kropachev K, Kolbanovskiy M, Liu Z, Cai Y, Zhang L, Schwaid AG, Kolbanovskiy A, Ding S, Amin S, Broyde S, Geacintov NE. Adenine-DNA adducts derived from the highly tumorigenic dibenzo[a,l]pyrene are resistant to nucleotide excision repair while guanine adducts are not. Chem Res Toxicol. 2013;26:783–793. doi: 10.1021/tx400080k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Geacintov N, Naegeli H, Patel DJ, Broyde S. Structural aspects of polycyclic aromatic carcinogen-damaged DNA and Its recognition by NER proteins. In: Siede W, Kow Y, Doetsch P, editors. DNA Damage Recognition. Taylor and Francis; New York, N.Y: 2005. pp. 263–296. [Google Scholar]
- 40.Buterin T, Hess MT, Luneva N, Geacintov NE, Amin S, Kroth H, Seidel A, Naegeli H. Unrepaired fjord region polycyclic aromatic hydrocarbon–DNA adducts in ras codon 61 mutational hot spots. Cancer Res. 2000;60:1849–1856. [PubMed] [Google Scholar]
- 41.Zheng H, Cai Y, Ding S, Tang Y, Kropachev K, Zhou Y, Wang L, Wang S, Geacintov NE, Zhang Y, Broyde S. Base flipping free energy profiles for damaged and undamaged DNA. Chem Res Toxicol. 2010;23:1868–1870. doi: 10.1021/tx1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang Y, Liu Z, Ding S, Rodriguez FA, Sayer JM, Jerina DM, Cai Y, Amin S, Broyde S, Geacintov NE. NMR solution structure of an adduct derived from the potent tumorigen dibenzo[a,l]pyrene linked to N2-guanine: intercalation from the B-DNA minor groove with ruptured Watson-Crick pairing at the lesion site. Biochemistry. 2012;51:9751–9762. doi: 10.1021/bi3013577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cai Y, Wang L, Ding S, Schwaid A, Geacintov NE, Broyde S. A bulky DNA lesion derived from a highly potent polycyclic aromatic tumorigen stabilizes nucleosome core particle structure. Biochemistry. 2010;49:9943–9945. doi: 10.1021/bi101560y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwaid A, Liu Z, Tang Y, Rodriguez F, Kolbanovskii A, Cai Y, Ding S, Amin S, Broyde S, Geacintov NE. Correlations between the characteristics of fjord region dibenzo[a,l]pyrene diol epoxide-guanine and -adenine adducts in double-stranded DNA and nucleotide excision repair. Chem Res Toxicol. 2010;23:281–282. [Google Scholar]
- 45.Mao B, Gu Z, Gorin A, Chen J, Hingerty BE, Amin S, Broyde S, Geacintov NE, Patel DJ. Solution structure of the (+)-cis-anti-benzo[a]pyrene-dA ([BP]dA) adduct opposite dT in a DNA duplex. Biochemistry. 1999;38:10831–10842. doi: 10.1021/bi991212f. [DOI] [PubMed] [Google Scholar]
- 46.Ravishanker G, Swaminathan S, Beveridge DL, Lavery R, Sklenar H. Conformational and helicoidal analysis of 30 PS of molecular dynamics on the d(CGCGAATTCGCG) double helix: “curves”, dials and windows. J Biomol Struct Dyn. 1989;6:669–699. doi: 10.1080/07391102.1989.10507729. [DOI] [PubMed] [Google Scholar]
- 47.Ravishanker G, Wang W, Beveridge DL. MD Toolchest. Wesleyan University; Middletown, CT: 1993. [Google Scholar]
- 48.Case DA, Darden TA, Cheatham TE, 3rd, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Wang B, Hayik S, Roitberg A, Seabra G, Kolossváry I, Wong KF, Paesani F, Vanicek J, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh MJ, Cui G, Roe DR, Mathews DH, Seetin MG, Sagui C, Babin V, Gusarov S, Kovalenko A, Kollman PA. AMBER. 11. University of California, San Francisco; San Francisco, CA: 2010. [Google Scholar]
- 49.Simmerling C, Elber R, Zhang J. Modelling of biomolecular structure and mechanisms. Netherlands: Kluwer; 1995. pp. 241–265. [Google Scholar]
- 50.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Case DA, Darden TA, Cheatham TE, 3rd, Simmerling CL, Wang J, Duke RE, Luo R, Crowley M, Walker RC, Zhang W, Merz KM, Wang B, Hayik S, Roitberg A, Seabra G, Kolossváry I, Wong KF, Paesani F, Vanicek J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Mathews DH, Seetin MG, Sagui C, Babin V, Kollman PA. AMBER. 10. University of California San Francisco; San Francisco, CA: 2008. [Google Scholar]
- 52.Huang N, Banavali NK, MacKerell AD., Jr Protein-facilitated base flipping in DNA by cytosine-5-methyltransferase. Proc Natl Acad Sci U S A. 2003;100:68–73. doi: 10.1073/pnas.0135427100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Priyakumar UD, MacKerell AD., Jr Computational approaches for investigating base flipping in oligonucleotides. Chem Rev. 2006;106:489–505. doi: 10.1021/cr040475z. [DOI] [PubMed] [Google Scholar]
- 54.Priyakumar UD, MacKerell AD. Base flipping in a GCGC containing DNA dodecamer: A comparative study of the performance of the nucleic acid force fields, CHARMM, AMBER, and BMS. J Chem Theory Comput. 2006;2:187–200. doi: 10.1021/ct0501957. [DOI] [PubMed] [Google Scholar]
- 55.Roux B. The Calculation of the Potential of Mean Force Using Computer-Simulations. Comput Phys Commun. 1995;91:275–282. [Google Scholar]
- 56.Zheng H, Filippova EV, Tkaczuk KL, Dworzynski P, Chruszcz M, Porebski PJ, Wawrzak Z, Onopriyenko O, Kudritska M, Grimshaw S, Savchenko A, Anderson WF, Minor W. Crystal structures of putative phosphoglycerate kinases from B. anthracis and C. jejuni. J Struct Funct Genomics. 2012;13:15–26. doi: 10.1007/s10969-012-9131-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 58.Cheatham TE, Cieplak P, Kollman PA. A modified version of the Cornell et al. force field with improved sugar pucker phases and helical repeat. J Biomol Struct Dyn. 1999;16:845–862. doi: 10.1080/07391102.1999.10508297. [DOI] [PubMed] [Google Scholar]
- 59.Pérez A, Marchán I, Svozil D, Sponer J, Cheatham TE, 3rd, Laughton CA, Orozco M. Refinement of the AMBER force field for nucleic acids: improving the description of α/γ conformers. Biophys J. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yan S, Shapiro R, Geacintov NE, Broyde S. Stereochemical, structural, and thermodynamic origins of stability differences between stereoisomeric benzo[a]pyrene diol epoxide deoxyadenosine adducts in a DNA mutational hot spot sequence. J Am Chem Soc. 2001;123:7054–7066. doi: 10.1021/ja0043035. [DOI] [PubMed] [Google Scholar]
- 61.Jorgensen WL, Chandreskhar J, Madura JD, Imprey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 62.Darden T, York D, Pedersen L. Particle mesh Ewald: an N log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 63.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pederson LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 64.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comp Phys. 1977;23:327–341. [Google Scholar]
- 65.Lukin M, Zaliznyak T, Johnson F, de Los Santos C. Structure and stability of DNA containing an aristolactam II-dA lesion: implications for the NER recognition of bulky adducts. Nucleic Acids Res. 2012;40:2759–2770. doi: 10.1093/nar/gkr1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Song K, Campbell AJ, Bergonzo C, de los Santos C, Grollman AP, Simmerling C. An improved reaction coordinate for nucleic acid base flipping studies. J Chem Theory Comput. 2009;5:3105–3113. doi: 10.1021/ct9001575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Varnai P, Lavery R. Base flipping in DNA: pathways and energetics studied with molecular dynamic simulations. J Am Chem Soc. 2002;124:7272–7273. doi: 10.1021/ja025980x. [DOI] [PubMed] [Google Scholar]
- 68.Jain V, Hilton B, Lin B, Patnaik S, Liang F, Darian E, Zou Y, Mackerell AD, Jr, Cho BP. Unusual sequence effects on nucleotide excision repair of arylamine lesions: DNA bending/distortion as a primary recognition factor. Nucleic Acids Res. 2013;41:869–880. doi: 10.1093/nar/gks1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu Y, Reeves D, Kropachev K, Cai Y, Ding S, Kolbanovskiy M, Kolbanovskiy A, Bolton JL, Broyde S, Van Houten B, Geacintov NE. Probing for DNA damage with beta-hairpins: similarities in incision efficiencies of bulky DNA adducts by prokaryotic and human nucleotide excision repair systems in vitro. DNA Repair (Amst) 2011;10:684–696. doi: 10.1016/j.dnarep.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Reeves DA, Mu H, Kropachev K, Cai Y, Ding S, Kolbanovskiy A, Kolbanovskiy M, Chen Y, Krzeminski J, Amin S, Patel DJ, Broyde S, Geacintov NE. Resistance of bulky DNA lesions to nucleotide excision repair can result from extensive aromatic lesion-base stacking interactions. Nucleic Acids Res. 2011;39:8752–8764. doi: 10.1093/nar/gkr537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bergonzo C, Campbell AJ, de los Santos C, Grollman AP, Simmerling C. Energetic preference of 8-oxoG eversion pathways in a DNA glycosylase. J Am Chem Soc. 2011;133:14504–14506. doi: 10.1021/ja205142d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kuper J, Kisker C. Damage recognition in nucleotide excision DNA repair. Curr Opin Struct Biol. 2012;22:88–93. doi: 10.1016/j.sbi.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 73.Naegeli H, Sugasawa K. The xeroderma pigmentosum pathway: decision tree analysis of DNA quality. DNA Repair (Amst) 2011;10:673–683. doi: 10.1016/j.dnarep.2011.04.019. [DOI] [PubMed] [Google Scholar]
- 74.Sugasawa K, Akagi J, Nishi R, Iwai S, Hanaoka F. Two-step recognition of DNA damage for mammalian nucleotide excision repair: Directional binding of the XPC complex and DNA strand scanning. Mol Cell. 2009;36:642–653. doi: 10.1016/j.molcel.2009.09.035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.