

Abstract
Objective
Diabetic subjects are at high risk for developing atherosclerosis through a variety of mechanisms. As the metabolism of glucose results in production of activators of protein kinase C (PKC)β, it was logical to investigate the role of PKCβ in modulation of atherosclerosis in diabetes.
Approach and Results
ApoE−/− and PKCβ −/−/ApoE−/− mice were rendered diabetic with streptozotocin. Quantification of atherosclerosis, gene expression profiling or analysis of signaling molecules was performed on aortic sinus or aortas from diabetic mice. Diabetes-accelerated atherosclerosis increased the level of phosphorylated ERK1/2 and JNK mitogen activated protein (MAP) kinases and augmented vascular expression of inflammatory mediators, as well as increased monocyte/macrophage infiltration and CD11c+ cells accumulation in diabetic ApoE−/− mice; processes which were diminished in diabetic PKCβ −/−/ApoE−/− mice. In addition, pharmacological inhibition of PKCβ reduced atherosclerotic lesion size in diabetic ApoE−/− mice. In vitro, the inhibitors of PKCβ and ERK1/2, as well as small interfering RNA (siRNA) to Egr-1 significantly decreased high glucose-induced expression of CD11c (Itgax), chemokine (C-C motif) ligand 2 (CCL2) and interleukin (IL)-1β in U937 macrophages.
Conclusions
These data link enhanced activation of PKCβ to accelerated diabetic atherosclerosis via a mechanism that includes modulation of gene transcription and signal transduction in the vascular wall; processes that contribute to acceleration of vascular inflammation and atherosclerosis in diabetes. Our results uncover a novel role for PKCβ in modulating CD11c expression and inflammatory response of macrophages in the development of diabetic atherosclerosis. These findings support PKCβ activation as a potential therapeutic target for prevention and treatment of diabetic atherosclerosis.
Keywords: atherosclerosis, diabetes, PKCβ, CD11c, inflammation
Introduction
Diabetic subjects are at two-to six fold greater risk of developing atherosclerosis than nondiabetic individuals,1, 2 constituting the main cause of morbidity and mortality.3, 4 In diabetes, an emerging view is that events leading to accelerated atherosclerosis in affected subjects are underway prior to the diagnosis of diabetes.5 Thus, an intensive search for new therapeutic targets in diabetic atherosclerosis is needed. These considerations led us to investigate molecular mechanisms by which diabetes accelerates atherosclerosis.
The ubiquitous enzyme Protein Kinase C (PKC), a family of at least 12 isoforms of serine and threonine kinases, has been linked to the pathogenesis of vascular injury. Although several PKC isoforms are expressed in vascular tissue, in the rodent model of diabetes there is a preferential activation of PKCβII in the aorta and heart.6 Increasing evidence links hyperglycemia to diabetic microvascular complications via activation of PKCβ,7–9 but PKCβ-dependent mechanisms have yet to be elucidated in macrovascular complications of diabetes. We previously reported that PKCβ −/−/ApoE−/− mice displayed less atherosclerosis than ApoE−/− mice and demonstrated links between activation of PKCβ and nondiabetic atherosclerosis.10 In parallel, our previous findings revealed that activated PKCβ, especially βII isoform, is a critical upstream regulator of early growth response 1 (Egr-1) in response to vascular stress, such as hypoxia,11, 12 ischemia/reperfusion13 and nondiabetic atherosclerosis.10 Egr-1, in turn, acts as a master switch coordinating expression of proinflammatory cytokines, chemokines, procoagulant molecules, cell adhesion molecules, and matrix metalloproteinase-2 (MMP-2).10, 14, 15 However, macrovascular disease in diabetes is manifested by more accelerated and progressive atherosclerosis which is more widely distributed. As the metabolism of glucose results in production of activators of PKCβ, it was logical to investigate the role of PKCβ in modulation of atherosclerosis in diabetes.
In this study, we hypothesized that in diabetes, enhanced activation of PKCβ is critical to augmented expression of a broad range of inflammatory mediators and increased phosphorylation of signaling molecules in the diabetic vascular wall; processes that contribute to accelerated atherosclerotic lesion initiation and progression in diabetes. Here, we tested these concepts in homozygous ApoE−/− and PKCβ −/−/ApoE−/− mice made diabetic with streptozotocin (stz).16–18 In addition, we tested the effects of a PKCβ inhibitor, ruboxistaurin, on atherosclerosis in diabetic ApoE−/− mice. Furthermore, in vitro, we tested the effects of small interfering RNA (siRNA) to Egr-1 or inhibitors of PKCβ, ERK1/2, JNK and p38 mitogen-activated protein kinase (MAPK) on high glucose-induced proinflammatory gene expression in U937 macrophages.
Materials and Methods
Materials and Methods are available in the online-only Supplement.
Results
Enhanced activation of PKCβII in aorta of diabetic ApoE−/− mice
To dissect the contribution of PKCβ in the pathogenesis of diabetes-accelerated atherosclerosis, we first assessed the extent of activation of the PKCβII isoform in the aortas of diabetic ApoE−/− mice at age 10 weeks, a time point before the development of significant atherosclerosis. Compared to nondiabetic ApoE−/− mice, diabetic ApoE−/− mice displayed higher levels of phosphor-PKCβII in membranous fractions vs. nondiabetic ApoE−/− mice (Figure 1, p<0.0001), although nondiabetic ApoE−/− mice showed an increase in phosphor-PKCβII in membranous fractions vs. wild-type (WT) mice (Figure 1, p<0.0001). In contrast, no change in phosphor-PKCδ was detected in the membranous fraction from WT, nondiabetic ApoE−/− and diabetic ApoE−/− mice. Loading controls using anti-β-actin IgG demonstrated identical protein loading (Figure 1). These observations indicated enhanced activation of PKCβII in diabetic ApoE−/− mice.
Figure 1. Accelerated activation of PKCβII in aorta of diabetic ApoE−/− mice.

Membranous protein from aortas of the indicated mice at age 10 weeks was subjected to Western blot for detection of phosphor-PKCβII, phosphor-PKCδ and β-actin. Representative of N=3 mice/condition.
Effects of PKCβ deletion on accelerated atherosclerosis in diabetic ApoE−/− mice
Mice were rendered diabetic at age 6 weeks and fed normal rodent chow. At age 14 weeks, mean atherosclerotic lesion area at the aortic sinus was 34,362.17 ± 7,114.49 μm2 (n=13) in nondiabetic ApoE−/− mice. Diabetes, as expected, led to a ≈ 1.91-fold increase in lesion area in diabetic ApoE−/− mice (65,585.5 ± 4,345.13 μm2, n=14) compared to nondiabetic ApoE−/− mice; p<0.001 (Figure 2A). In contrast, atherosclerotic lesion area at the aortic sinus was ≈ 38% lower in diabetic PKCβ −/−/ApoE−/− (40,611.10 ± 4,767.72 μm2, n=15) compared to that in diabetic ApoE−/−mice; p<0.001 (Figure 2A). Diabetic PKCβ −/−/ApoE+/+ animals displayed no atherosclerotic lesions (not shown). The effects of PKCβ on diabetic atherosclerosis were sustained beyond the earliest stages of lesion formation and accelerated by aging. Specifically, at age 20 weeks, similar results were observed, however, the differences between double-null and single-null ApoE−/− mice were even more striking: mean atherosclerotic lesion area was ≈ 55% lower in diabetic PKCβ −/−/ApoE−/− mice (99,259.62 ± 11,138.17 μm2, n=13) versus diabetic ApoE−/− animals (220,952.86 ± 14,653.50 μm2, n=14); p<0.0001 (Figure 2B). Sudan IV stain for aortic en face area, at age 20 weeks, also showed greater lesion area in diabetic ApoE−/− mice compared to nondiabetic ApoE−/− mice (p<0.05, Figure 2C), and this was reduced in diabetic PKCβ −/−/ApoE−/−mice compared to diabetic ApoE−/− animals (p<0.05, Figure 2C). In parallel with decreased atherosclerotic lesion area, the lesion complexity index was also significantly lower in diabetic PKCβ −/−/ApoE−/− mice versus diabetic ApoE−/− mice at age 20 weeks (0.562 ± 0.05 versus 0.966 ± 0.006; p<0.0001, Figure 2D).
Figure 2. Impact of PKCβ deletion on diabetic atherosclerosis.

Shown are representative images of aortic root sections stained with Oil Red O at age 14 weeks (A) and 20 weeks (B) and Sudan IV stained aortic en face at age 20 weeks (C). Scale bar =200 μm. Mean atherosclerotic lesion areas (μm2) were determined in ND ApoE−/− (n=13), D ApoE−/− (n=14) and D PKCβ −/−/ApoE−/− male mice (n=15 and n= 13) at age 14 weeks (A) and 20 weeks (B). Lesion complexity index was calculated in D ApoE−/− and D PKCβ −/−/ApoE−/− male mice at age 20 weeks (n=10 and n=13) (D). D: diabetic; ND: nondiabetic.
Effects of PKCβ inhibition on atherosclerosis in diabetic ApoE−/− mice
If PKCβ activation contributed to accelerated atherosclerosis development, its inhibition should reduce lesions. We tested a selective inhibitor of this enzyme, ruboxistaurin. When ApoE−/− male mice expressing PKCβ were treated with ruboxistaurin from age 5 to 20 weeks, mean atherosclerotic lesion area at the aortic sinus was ≈ 59% lower in diabetic ApoE−/− male mice fed ruboxistaurin (106682.5 ± 13626.72 μm2, n=13) versus diabetic ApoE−/− male mice fed vehicle chow (260267.86 ± 22926.95 μm2, n=14); p<0.0001 (Figure 3A). In parallel, the complexity index was significantly lower in diabetic ApoE−/− mice fed ruboxistaurin versus vehicle (0.487 ± 0.052 versus 0.978 ± 0.009; p<0.0001, Figure 3B).
Figure 3. Impact of PKCβ inhibitor on diabetic atherosclerosis.

Shown are representative images of aortic root sections stained with Oil Red O at age 20 weeks (A). Scale bar =200 μm. Mean atherosclerotic lesion areas (μm2) were determined in D ApoE−/− male mice fed vehicle chow (VEH, n=14) or fed ruboxistaurin (RBX, n=13) at age 20 weeks (A). Lesion complexity index was calculated in D ApoE−/− male mice fed VEH chow or fed RBX at age 20 weeks (n=10 and n=14) (B). D: diabetic.
Effects of PKCβ deletion/inhibition on glycemia and lipid in diabetic ApoE−/− mice
We examined factors that might account for the beneficial effects of PKCβ deletion or inhibition. Diabetic mice displayed a significantly higher plasma glucose level, cholesterol and triglyceride than nondiabetic mice (Table I in the online-only Data Supplement). Importantly, no statistically significant differences were observed in levels of plasma glucose, cholesterol and triglyceride in diabetic PKCβ −/−/ApoE−/− animals vs. diabetic ApoE−/− mice or in diabetic ApoE−/− mice fed ruboxistaurin vs. vehicle chow at age 14 weeks (data not shown) and at age 20 weeks (Table S1). Thus, the impact of deletion or blockade of PKCβ on accelerated atherosclerosis in diabetes was independent of modulation of other key risk factors for atherosclerosis.
Effects of PKCβ deletion on vascular expression of inflammatory mediators and activation of mitogen-activated protein kinase (MAPK) in diabetic ApoE−/− mice
We sought to identify the specific mechanisms by which PKCβ contributed to early atherogenesis in diabetic ApoE−/− mice. Aortic RNA was prepared from nondiabetic and diabetic ApoE−/− and PKCβ −/−/ApoE−/− mice at age 10 weeks, a time point before atherosclerotic lesion development. The RNA samples were subjected to pathway-focused gene expression profiling using Mouse Atherosclerosis RT2 Profiler™ PCR Array. Our data revealed a progressive increase in expression of 14 out of 84 stress-responsive genes (Table II in the online-only Data Supplement), including chemokines (Ccl2, Ccr1 & Ccr2), cytokines (IL1α, IL1b, IL1r2 & Spp1), transmembrane proteins (Itgαx & Itgb2), Matrix metallopeptidase (Mmp3), signal transducer (Msr1) and adhesion molecules (Sele, Sell, Selp & Selplg) in diabetic ApoE−/− mice. In contrast, those gene transcripts were dramatically reduced in diabetic PKCβ −/−/ApoE−/− mice.
Next, to further validate a progressive increase in gene expression in diabetic ApoE−/−mice, primers were chosen for real-time PCR analysis if upregulation of the gene was ≥ 2 fold higher in aortas of diabetic ApoE−/− mice compared to that of nondiabetic ApoE−/− mice at age 10 weeks or ≤ 2 fold lower in aortas of diabetic PKCβ −/−/ApoE−/− mice compared to that of diabetic ApoE−/− mice. As indicated in Figure 4A, all of the above gene expression changes in aortas of diabetic ApoE−/− mice were verified; there was a significant increase in each of these genes in diabetic ApoE−/− aortas (#p≤0.05, *p≤0.01 and ^p≤0.001) compared to nondiabetic ApoE−/− mice at age 10 weeks; whereas these gene expression levels in diabetic aortas of PKCβ −/−/ApoE−/− mice were not significantly different compared to that of nondiabetic ApoE−/− mice at age 10 weeks. Furthermore, as indicated in Figure 4A, quantitative real-time PCR confirmed that the progressive increase in gene expression of those 14 inflammatory mediators in diabetic ApoE−/−mice was significantly reduced in diabetic PKCβ −/−/ApoE−/− mice (##p≤0.05, **p≤0.01 and ^^p≤0.001). In contrast, as indicated in Figure 4B, some representative genes, such as Ace, Itga2 and PdgFrb, were not altered in diabetic mice and not affected by PKCβ deficiency.
Figure 4. Impact of diabetes and PKCβ on the expression of inflammatory mediators and activation of MAP kinase.

Gene expression profiling was validated by real-time PCR in aortic RNA of D ApoE−/− mice vs. ND ApoE−/− mice (n≥3, #p ≤0.05, *p≤0.01 and ^p≤0.001) or D PKCβ −/−/ApoE−/− mice (n≥3, ##p≤0.05, **p≤0.01 and ^^p≤0.001) at age 10 weeks (A). Shown are some representative genes not altered in D ApoE−/− or D PKCβ −/−/ApoE−/− mice (B). Aortic RNA was subjected to real-time PCR for detection of Egr-1 RNA (C). Aortic protein was subjected to Western blot for detection of phosphor (P)-ERK1/2 and total (T)-ERK (D), P-JNK and T-JNK (E). D: diabetic; ND: nondiabetic.
In addition, we determined whether PKCβ modulated the expression of Egr-1 and activation of signaling mechanisms, such as ERK1/2 and JNK MAP kinases in the vessel wall of diabetic ApoE−/− mice at age 10 weeks. Real-time PCR revealed that transcripts for Egr-1 were significantly increased in aortas of diabetic ApoE−/− mice compared to nondiabetic ApoE−/− mice (p<0.01, Figure 4C). In contrast, transcripts for Egr-1 were significantly decreased in aortas of diabetic PKCβ −/−/ApoE−/− mice vs. diabetic ApoE−/− mice (p<0.01, Figure 4C). Immunoblots displayed that Phosphor-ERK1/2 (P-ERK1/2, p<0.01, Figure 4D) and phosphor-JNK (P-JNK, p<0.01, Figure 4E) were significantly lower in the aortas of diabetic PKCβ −/−/ApoE−/− mice vs. diabetic ApoE−/− mice. Loading controls using anti-total ERK1/2 (T-ERK) and anti-total JNK (T-JNK) IgG demonstrated identical protein loading (Figure 4D and 4E).
Effects of PKCβ deletion on inflammatory-appearing lesions in diabetic ApoE−/− mice
On the basis of the above findings that the increased expression of Itgax transcripts was dependent on PKCβ activation in aortas of diabetic ApoE−/− mice, we sought to determine whether PKCβ-dependent Itgax expression contributes to inflammatory-appearing lesions in the development of diabetic atherosclerosis. CD11c (ITGAX), encoding integrin alpha X chain protein, expressed on monocytes/macrophages and dendritic cells (DCs). 19 Immunofluorescence staining with a macrophage marker, anti-MOMA-2 IgG, showed that infiltration of macrophages in atherosclerotic lesions of aortic sinus was significantly lower in diabetic PKCβ −/−/ApoE−/− mice (Figure 5A) compared to diabetic ApoE−/− mice (Figure 5B) at age 20 weeks (p<0.01, Figure 5D). Next, colocalization of CD11c and MOMA-2 revealed that the immunoreactivity for CD11c was predominantly present in the macrophages in the atherosclerotic lesions of diabetic ApoE−/− mice at age 20 weeks (Figure 5B and 5C). However, CD11c-expressing cells were significantly lower in diabetic PKCβ −/−/ApoE−/− mice (Figure 5A) compared to diabetic ApoE−/− mice (Figure 5B) at age 20 weeks (p<0.01, Figure 5E). Isotype control immunohistochemistry with isotype-matched hamster IgG and rat IgG showed no staining (Figure 5A, 5B and 5C). These data suggest an important role of PKCβ in modulating CD11c expression in macrophages which potentially contributed to the development of diabetic atherosclerosis.
Figure 5. Impact of PKCβ on the inflammatory cells in atherosclerotic lesions in diabetes.
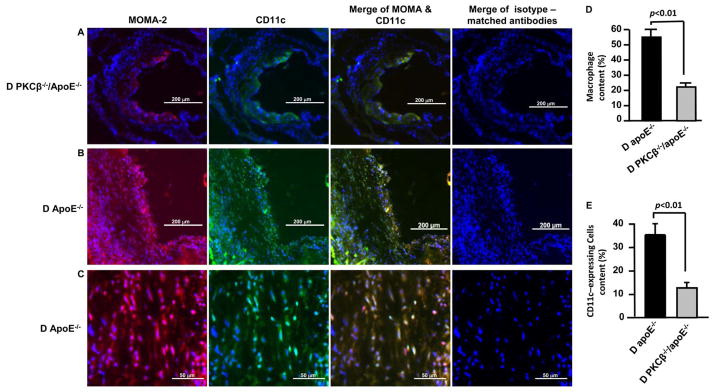
The sections of aortic root from D PKCβ −/−/ApoE−/− mice (A, 10x magnification, Scar bar=200μm) and D ApoE−/− mice (B, 10x magnification, Scar bar=200μm; & C, 40x magnification, scar bar= 50μm) at age 20 weeks were stained with anti-MOMA-2 and anti-CD11c, or isotype-matched antibodies. Nuclei were counterstained with DAPI. The merging of MOMA-2 and CD11C or merging of isotype-matched antibodies is shown. To determine the relative amounts of MOMA-2 positive macrophages (D) or CD11-expressing cells (E) in an atherosclerotic lesion, the positive area of MOMA-2 or CD11c was divided by the total lesion area of atherosclerosis. D: Diabetic.
Effects of high glucose on PKCβ-dependent signaling pathways in U937 macrophages
Stimulated by the above findings, to further dissect the molecular mechanisms by which PKCβ activation upregulated gene expression in monocytes/macrophages in settings characterized by high levels of glucose, we used human U937 macrophages as an in vitro model system. Real-time PCR analysis of total RNA from U937 cells exposed to high glucose demonstrated an approximate 3.72, 2.88, 2.95 and 3.68-fold increase in Egr-1, CD11c, CCL2 and IL-1β transcripts respectively, compared with U937 cells cultured in low glucose (p<0.0001; Figure 6A–D), in a manner significantly suppressed by the inhibition of PKCβ (LY379196, 30 nM; p<0.0001 or p<0.001, Figure 6A–D) and ERK1/2 (U0126, 5 μM; p<0.0001; Figure 6A–D). On the other hand, high glucose-induced CD11c expression in U937 cells was not suppressed by the inhibitors of JNK (SP600125, 20 μM) and p38 (SB203550, 20 μM) (Figure 6B). High-glucose-induced Egr-1 expression in U937 cells was suppressed by the inhibitor of JNK (p<0.0001; Figure 6A), not by an inhibitor of p38 (Figure 6A) and high glucose-induced increase in CCL2 and IL-1β expression was suppressed by the inhibitors of JNK and p38 (p<0.0001; Figure 6C and 6D).
Figure 6. Impact of high glucose and PKCβ-dependent signaling pathways on the expression of inflammatory mediators in U937 macrophages.
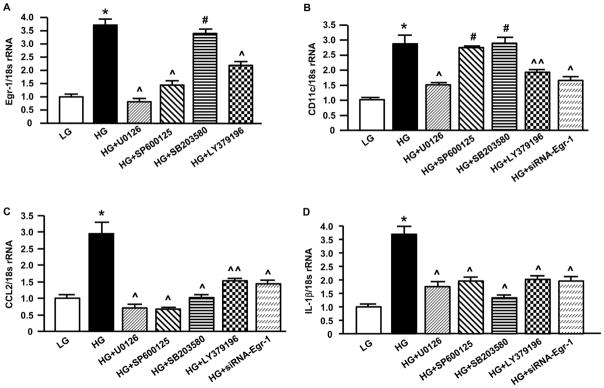
U937 cells were seeded and exposed to 5 mM (low) or 25 mM (high) glucose in the absence or presence of siRNA to Egr-1 or the inhibitors of PKCβ, ERK1/2, JNK and p38. Cells were harvested followed by RNA isolation. Real-time PCR analysis of gene expression was performed. Data are represented as the relative gene expression of Egr-1 (A), CD11c (B), CCL2 (C ) and IL-1β (D) normalized to 18s rRNA. * p<0.001, the level of gene expression in U937 macrophages exposed to high glucose vs. low glucose; ^p<0.001, ^^p<0.01 and # N.S. (Not significant), the level of gene expression in U937 macrophages exposed to high glucose with the treatment of inhibitors or siRNA vs. non-treatment.
Next, we tested whether Egr-1, a downstream target of ERK1/2 and PKCβ in vascular stress, is required for high glucose-induced expression of CD11c, CCL2 and IL-1β in U937 macrophages. Introduction of siRNA to knockdown Egr-1 expression in U937 cells blunted the high glucose-induced upregulation of CD11c, CCL2 and IL-1β transcripts (p<0.0001; Figure 6B–D) but not scramble control (data not shown). These data suggest that the PKCβ pathway modulates CD11c, CCL2 and IL-1β upregulation, at least in part via activation of ERK1/2 and Egr-1.
Discussion
We report an important role for PKCβ in acceleration of initiation and progression of atherosclerosis in diabetic ApoE−/− mice. Activation of PKCβII, indicated by translocation from the cytosol to the plasma membrane, has been shown in aortas of nondiabetic ApoE−/− mice vs. wild-type C57BL/6 animals in our previous studies.10 Our present data demonstrated a significant increase in phosphor-PKCβII in the membrane fractions from aortas of diabetic ApoE−/− mice vs. nondiabetic ApoE−/− mice at age 10 weeks, a time point before the development of significant atherosclerosis. These findings suggest that enhanced activation of PKCβII in a hyperglycemic and hyperlipidemic environment may reflect a response of the blood vessel wall to superimposed biochemical and metabolic stresses. Consistent with important roles for PKCβ in accelerated atherosclerosis in diabetes, our findings demonstrated that genetic deletion or pharmacological inhibition of PKCβ in diabetic ApoE−/− mice significantly diminished atherosclerotic lesion areas and complexity indices, but did not affect the levels of glucose, cholesterol and triglyceride. These findings suggest that modulation of diabetic atherosclerosis by PKCβ deletion or blockade was independent of changes in glucose and lipids. Because the traditional therapeutic approaches by glycemic control lacks clearly an established benefit in macrovascular disease, our findings may suggest important implications for blockade of PKCβ signaling pathway by employing the PKCβ inhibitor ruboxistaurin for the management of diabetic atherosclerosis.
Both elevated glucose and lipids contribute to increased inflammation in diabetes.20, 21 We assessed the mechanisms by which PKCβ plays a crucial role in the accelerated initiation and progression of atherosclerotic lesion areas in diabetes. Our data demonstrated that increased expression of inflammatory mediators and activation of signaling MAP kinases, including proinflammatory transcription factor Egr-1, chemokines (Ccl2, Ccr1 & Ccr2), cytokines (IL-1α, IL-1β, IL-1γ2 & Spp1), transmembrane proteins (Itgax & Itgb2), matrix metallopeptidase (Mmp3), signal transducer (Msr1), adhesion molecules (Sele, Sell, Selp, Selplg & Spp1), in early atherogenesis in the aortas of diabetic ApoE−/− mice is dependent on the state of glycemia and the state of PKCβ expression since genetic deletion of PKCβ in diabetic ApoE−/− mice dramatically attenuated expression of those inflammatory mediators and signaling molecules. Our findings reveal a critical role for PKCβ-dependent upregulation of inflammatory mediators and signaling molecules in initiating steps of atherogenesis in the diabetic vascular wall, leading to enhanced progression of atherosclerotic complications.
Given the inflammatory basis of atherosclerosis, macrophages have been considered to be important immune effector cells which contribute to and promote the development of atherosclerotic plaques.22–24 An in vitro study showed that macrophages, when treated with oxidatively modified lipids, differentiate into foam cells and in the process acquire expression of DC marker.25 One of the DC markers, CD11c which is not an exclusive marker of DCs but can also be found on macrophages,19 has been implicated as a functional integrin in human and mouse models.26, 27 Our data revealed that the increased CD11+ cells in atherosclerotic lesions in diabetic ApoE−/− mice are, at least in part, macrophages based on colocalization with MOMA-2. In concurrence with these findings, the cholesterol-rich diet-fed Ldlr−/− mice displayed foam cells expressing DC marker CD11c,28 and AopE−/− mice showed an increased number of CD11c+ cells with the progression of atherosclerosis.24, 29 Also, high fat diet (HFD)-fed ApoE−/− mice displayed increased CD11c+ cells which were colocalized with MOMA-2 in atherosclerotic lesions. 27 Interestingly, we provide the first evidence that the increase in gene expression of Itgax (CD11c) in diabetic ApoE−/− mice expressing PKCβ was suppressed in diabetic ApoE−/−mice devoid of PKCβ. In parallel, decreased % macrophages/total lesion area and % CD11c+ cells/total lesion area were evident in diabetic PKCβ −/−/ApoE−/− mice vs. diabetic ApoE−/− mice. In addition, we demonstrated that PKCβ regulated increased expression of Egr-1 and phosphorylation of ERK1/2 and JNK MAP kinases in early atherogenesis in diabetes. Consistent with these observations, high glucose-induced increase in Egr-1 transcripts in U937 macrophages was blunted by the inhibitors of PKCβ, ERK1/2 and JNK, but not by the inhibitor of p38. Since Egr-1 has been demonstrated to play an important role in nondiabetic atherosclerosis 15, 30, 31 and it is one of the key downstream target genes of PKCβ and ERK1/2 or JNK in the response to hypoxia,11, 12, 32 ischemia,13 acute vascular injury33, 34 and in nondiabetc atherosclerosis,10 our current data suggest that PKCβ mediates upregulation of Egr-1, at least in part, via increased phosphorylation of ERK1/2 and JNK in early atherogenesis in diabetes. Furthermore, as there are two putative Egr-1 binding motifs in the proximal region of the CD11c (p150,95) promoter which may participate in the regulation of CD11c expression based on our TRANSFAC database search,35 we examined the effects of high glucose-stimulated PKCβ-dependent signaling pathway on CD11 gene expression in U937 macrophages. The knockdown of Egr-1 expression by siRNA or inhibition of PKCβ and ERK1/2, but not JNK and p38, resulted in attenuation of high glucose-induced CD11c expression in U937 macrophages. These data suggest that PKCβ regulates CD11 expression in the inflammatory macrophages in atherosclerosis associated with diabetes, at least in part, via activation of Egr-1 and ERK1/2 signaling pathway. Studies are underway to further probe the CD11c promoter sites by electrophoretic mobility shift (EMSA) and supershift assays and the chromatin immunoprecipitation (ChIP) assays to establish functionality.
Consistent with our other observations, we showed ample evidence of the involvement of other inflammatory components in the inflammatory and pathological processes of atherosclerosis. One of the chemoattractants implicated in early atherogenesis is monocyte chemoattractant protein-1 (MCP-1/CCL2), which binds to C-C chemokine receptor 2 (CCR2).36 MCP-1/CCL2 is highly expressed in atherosclerotic plaques36 and mediates macrophage recruitment in the atheromatous lesion. Given the importance of MCP-1/CCR2 pathway in the process of atherosclerosis, our findings may have important implications in suppression of MCP-1/CCR2 pathway since we demonstrated that genetic deletion of PKCβ attenuated upregulation of MCP-1/CCL2 and CCR2 in vascular tissues of diabetic ApoE−/− mice at age 10 weeks, a time point linked to an early stage of atherogenesis. Since increased CD11c expression in adipose tissue and blood in both mice and humans was found as a consequence of elevated MCP-1 levels secreted by adipose tissue,37 correlations or interactions between CD11c and MCP-1 will be tested in future studies in vascular inflammation in diabetic atherosclerosis. In addition, a large number of proinflammatory cytokines has been shown to be expressed in human atherosclerotic lesions, particularly in association with infiltrating monocytes and macrophages.38 Interleukin-1 (IL-1) has been implicated as a regulatory protein in the development and clinical sequelae of atherosclerosis.39 IL-1β is produced by activated macrophages and considered a master switch of inflammation.40 IL-1β and its related parent IL-1α have both been implicated in the pathogenesis of atherosclerosis in previous studies.40 Engagement of CD11c β2 integrin by antibodies was reported to induce IL-1β production on primary huaman monocytes.41 The mechanisms instigating and perpetuating inflammatory responses have been studied extensively, however, less is known about the mechanisms governing the resolution of inflammation. We demonstrated that all of the above inflammatory mediators were modulated by vascular activation of PKCβ since they were dramatically attenuated in diabetic ApoE−/− mice devoid of PKCβ. Based on our previous findings that MCP-1/CCL2 and IL-1β have been demonstrated as downstream target genes of Egr-1 in atherosclerosis in euglycemia,15 and our current in vitro data that the knockdown of Egr-1 expression by siRNA or inhibition of PKCβ, ERK1/2, JNK and p38 led to attenuation of high glucose-induced CCL2 and IL-1β expression in U937 macrophages, we suggest that PKCβ regulates increased expression of CCL2 and IL-1β, at least in part, via activation of ERK1/2/Egr-1 and/or JNK/Egr-1 or p38 signaling pathways in atherosclerosis in diabetes. Although the exact mechanisms by which PKCβ regulates these inflammatory mediators in the development of diabetic atherosclerosis are also the subject of ongoing investigation, our present study may shed light on the effectiveness of PKCβ blockade as a strategy to protect the stressed vascular system from irreversible injury caused by inflammatory response in human atherosclerotic disease, especially in diabetes.
Although a growing body of evidence from animal and human studies indicates beneficial effects of PKCβ inhibition on microvascular parameters,9, 42, 43 our studies have examined the effects of PKCβ inhibitor, ruboxistaurin, on macrovascular disease in diabetes. Our data provide the first evidence that pharmacological blockade of PKCβ by the PKCβ inhibitor, ruboxistaurin, significantly attenuated atherosclerotic lesion areas and complexity indices in diabetic ApoE−/−mice. This work raises the possibility that blockade of the PKCβ by a pharmacologic inhibitor, ruboxistaurin, may attenuate neointimal expansion, vascular injury or damage triggered by inflammatory and pathological processes during the development of atherosclerosis in diabetes.
In summary, these data link enhanced activation of PKCβ to diabetes-accelerated atherosclerosis via a mechanism that includes modulation of vascular expression of inflammatory mediators and activation of signaling MAP kinases involved in inflammatory responses; processes that contribute to acceleration of vascular inflammation and atherosclerosis in diabetes. We have uncovered a novel role for PKCβ in modulating CD11c expression and inflammatory responses of macrophages in the development of diabetic atherosclerosis. Thus, a better understanding of the inflammatory mechanisms by which PKCβ contributes to such processes in the vessel wall could be relevant to devise preventive and therapeutic strategies for diabetic atherosclerosis. In addition, based on our demonstration that pharmacological inhibition of PKCβ reduced lesion size, we suggest that these studies may provide the basis for potential therapeutic application of PKCβ antagonism (ruboxistaurin) to diabetic atherosclerosis.
Supplementary Material
Significance.
Our findings show a critical role for PKCβ-dependent upregulaion of inflammatory mediators and signaling molecules in initiating steps of atherogenesis in the diabetic vascular wall, leading to enhanced progression of atherosclerotic complication. Interestingly, our data provide the first evidence that PKCβ modulates CD11c expression and inflammatory responses of macrophages in the development of diabetic atherosclerosis. Thus, a better understanding of PKCβ-dependent inflammatory mechanisms in the vessel wall could be relevant to devise preventive and therapeutic strategies for diabetic atherosclerosis. Furthermore, our demonstration that pharmacological inhibition of PKCβ reduced lesion size may provide the basis for potential therapeutic application of PKCβ antagonism (ruboxistaurin) to diabetic atherosclerosis.
Acknowledgments
The authors gratefully acknowledge Latoya Woods (Diabetes Research Program, Division of Endocrinology, Department of Medicine, New York University School of Medicine) for her excellent assistance in preparation of the manuscript.
Sources of Funding
This study was supported in part by grants from United States Public Health Service [National Heart, Lung, and Blood Institute Grants (RO1 HL073325 and PO1 HL60901)].
Abbreviations
- PKC
Protein kinase C
- MAP
mitogen activated protein
- Egr-1
early growth response gene-1
- MMP-2
matrix metalloproteinase-2
- stz
streptozotocin
- RT
reverse transcription
- D
diabetic
- ND
nondiabetic
- MCP-1
Chemokine (C-C motif) ligand 2 (CCL2) is also referred to as monocyte chemotactic protein-1
- CCR2
C-C chemokine receptor 2
- IL-1
Interleukin-1
- SELPLG
P-selectin glycoprotein ligand 1
- MSR
Macrophage scavenger receptors class A
- MMP3
matrix metallopeptidase
- DC
dendritric cell
Footnotes
Disclosures
None.
References
- 1.Ruderman NB, Gupta S, Sussman I. Hyperglycemia, diabetes and vascular disease: An overview. In: Ruderman N, Williamson J, Brownlee M, editors. Hyperglycemia, diabetes and vascular disease. New York, NY: Oxford University Press; 1992. pp. 3–20. [Google Scholar]
- 2.Kannel WB, McGee DL. Diabetes and cardiovascular disease. The framingham study. JAMA. 1979;241:2035–2038. doi: 10.1001/jama.241.19.2035. [DOI] [PubMed] [Google Scholar]
- 3.Ferreiro JL, Angiolillo DJ. Diabetes and antiplatelet therapy in acute coronary syndrome. Circulation. 2011;123:798–813. doi: 10.1161/CIRCULATIONAHA.109.913376. [DOI] [PubMed] [Google Scholar]
- 4.Luscher TF, Creager MA, Beckman JA, Cosentino F. Diabetes and vascular disease: Pathophysiology, clinical consequences, and medical therapy: Part ii. Circulation. 2003;108:1655–1661. doi: 10.1161/01.CIR.0000089189.70578.E2. [DOI] [PubMed] [Google Scholar]
- 5.Haffner SM, Mykkanen L, Festa A, Burke JP, Stern MP. Insulin-resistant prediabetic subjects have more atherogenic risk factors than insulin-sensitive prediabetic subjects: Implications for preventing coronary heart disease during the prediabetic state. Circulation. 2000;101:975–980. doi: 10.1161/01.cir.101.9.975. [DOI] [PubMed] [Google Scholar]
- 6.Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, King GL. Preferential elevation of protein kinase c isoform beta ii and diacylglycerol levels in the aorta and heart of diabetic rats: Differential reversibility to glycemic control by islet cell transportation. Proc Natl Acad Sci USA. 1992;89:11059–11063. doi: 10.1073/pnas.89.22.11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Danis RP, Sheetz MJ. Ruboxistaurin: Pkc-beta inhibition for complications of diabetes. Expert Opin Pharmacother. 2009;10:2913–2925. doi: 10.1517/14656560903401620. [DOI] [PubMed] [Google Scholar]
- 8.Kelly DJ, Zhang Y, Hepper C, Gow RM, Jaworski K, Kemp BE, Wilkinson-Berka JL, Gilbert RE. Protein kinase c beta inhibition attenuates the progression of experimental diabetic nephropathy in the presence of continued hypertension. Diabetes. 2003;52:512–518. doi: 10.2337/diabetes.52.2.512. [DOI] [PubMed] [Google Scholar]
- 9.Aiello LP. The potential role of pkc beta in diabetic retinopathy and macular edema. Surv Ophthalmol. 2002;47 (Suppl 2):S263–269. doi: 10.1016/s0039-6257(02)00391-0. [DOI] [PubMed] [Google Scholar]
- 10.Harja E, Chang JS, Lu Y, Leitges M, Zou YS, Schmidt AM, Yan SF. Mice deficient in pkcbeta and apolipoprotein e display decreased atherosclerosis. FASEB J. 2009;23:1081–1091. doi: 10.1096/fj.08-120345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan SF, Lu J, Zou YS, Soh-Won J, Cohen DM, Buttrick PM, Cooper DR, Steinberg SF, Mackman N, Pinsky DJ, Stern DM. Hypoxia-associated induction of early growth response-1 gene expression. J Biol Chem. 1999;274:15030–15040. doi: 10.1074/jbc.274.21.15030. [DOI] [PubMed] [Google Scholar]
- 12.Yan SF, Lu J, Zou YS, Kisiel W, Mackman N, Leitges M, Steinberg S, Pinsky D, Stern D. Protein kinase c-beta and oxygen deprivation. A novel egr-1 dependent pathway for fibrin deposition in hypoxemic vasculature. J Biol Chem. 2000;275:11921–11928. doi: 10.1074/jbc.275.16.11921. [DOI] [PubMed] [Google Scholar]
- 13.Fujita T, Asai T, Andrassy M, Stern DM, Pinsky DJ, Zou YS, Okada M, Naka Y, Schmidt AM, Yan SF. Pkcbeta regulates ischemia/reperfusion injury in the lung. J Clin Invest. 2004;113:1615–1623. doi: 10.1172/JCI19225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan SF, Fujita T, Lu J, Okada K, Shan Zou Y, Mackman N, Pinsky DJ, Stern DM. Egr-1, a master switch coordinating upregulation of divergent gene families underlying ischemic stress. Nat Med. 2000;6:1355–1361. doi: 10.1038/82168. [DOI] [PubMed] [Google Scholar]
- 15.Harja E, Bucciarelli LG, Lu Y, Stern DM, Zou YS, Schmidt AM, Yan SF. Early growth response-1 promotes atherogenosis: Mice deficient in early growth response-1 and apolipoprotein e display decreased atherosclerosis and vascular inflammation. Circ Res. 2004;94:333–339. doi: 10.1161/01.RES.0000112405.61577.95. [DOI] [PubMed] [Google Scholar]
- 16.Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ, Jr, Chow WS, Stern D, Schmidt AM. Suppression of accelarated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med. 1998;4:1025–1031. doi: 10.1038/2012. [DOI] [PubMed] [Google Scholar]
- 17.Bucciarelli LG, Wendt T, Qu W, Lu Y, Lalla E, Rong LL, Goova MT, Moser B, Kislinger T, Lee DC, Kashyap Y, Stern DM, Schmidt AM. Rage blockade stabilizes established atherosclerosis in diabetic apolipoprotein e-null mice. Circulation. 2002;106:2827–2835. doi: 10.1161/01.cir.0000039325.03698.36. [DOI] [PubMed] [Google Scholar]
- 18.Bu DX, Rai V, Shen X, Rosario R, Lu Y, D’Agati V, Yan SF, Friedman RA, Nuglozeh E, Schmidt AM. Activation of the rock1 branch of the transforming growth factor-beta pathway contributes to rage-dependent acceleration of atherosclerosis in diabetic apoe-null mice. Circ Res. 2010;106:1040–1051. doi: 10.1161/CIRCRESAHA.109.201103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tomkin GH. Atherosclerosis, diabetes and lipoproteins. Expert review of cardiovascular therapy. 2010;8:1015–1029. doi: 10.1586/erc.10.45. [DOI] [PubMed] [Google Scholar]
- 20.Kanter JE, Johansson F, LeBoeuf RC, Bornfeldt KE. Do glucose and lipids exert independent effects on atherosclerotic lesion initiation or progression to advanced plaques? Circ Res. 2007;100:769–781. doi: 10.1161/01.RES.0000259589.34348.74. [DOI] [PubMed] [Google Scholar]
- 21.Mazzone T, Chait A, Plutzky J. Cardiovascular disease risk in type 2 diabetes mellitus: Insights from mechanistic studies. Lancet. 2008;371:1800–1809. doi: 10.1016/S0140-6736(08)60768-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Butcher MJ, Galkina EV. Phenotypic and functional heterogeneity of macrophages and dendritic cell subsets in the healthy and atherosclerosis-prone aorta. Frontiers in physiology. 2012;3:44. doi: 10.3389/fphys.2012.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Randolph GJ. The fate of monocytes in atherosclerosis. Journal of thrombosis and haemostasis : JTH. 2009;7 (Suppl 1):28–30. doi: 10.1111/j.1538-7836.2009.03423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis (*) Annual review of immunology. 2009;27:165–197. doi: 10.1146/annurev.immunol.021908.132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho HJ, Shashkin P, Gleissner CA, Dunson D, Jain N, Lee JK, Miller Y, Ley K. Induction of dendritic cell-like phenotype in macrophages during foam cell formation. Physiological genomics. 2007;29:149–160. doi: 10.1152/physiolgenomics.00051.2006. [DOI] [PubMed] [Google Scholar]
- 26.Sadhu C, Ting HJ, Lipsky B, Hensley K, Garcia-Martinez LF, Simon SI, Staunton DE. Cd11c/cd18: Novel ligands and a role in delayed-type hypersensitivity. J Leukoc Biol. 2007;81:1395–1403. doi: 10.1189/jlb.1106680. [DOI] [PubMed] [Google Scholar]
- 27.Wu H, Gower RM, Wang H, Perrard XY, Ma R, Bullard DC, Burns AR, Paul A, Smith CW, Simon SI, Ballantyne CM. Functional role of cd11c+ monocytes in atherogenesis associated with hypercholesterolemia. Circulation. 2009;119:2708–2717. doi: 10.1161/CIRCULATIONAHA.108.823740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paulson KE, Zhu SN, Chen M, Nurmohamed S, Jongstra-Bilen J, Cybulsky MI. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ Res. 2010;106:383–390. doi: 10.1161/CIRCRESAHA.109.210781. [DOI] [PubMed] [Google Scholar]
- 29.Manthey HD, Zernecke A. Dendritic cells in atherosclerosis: Functions in immune regulation and beyond. Thrombosis and haemostasis. 2011;106:772–778. doi: 10.1160/TH11-05-0296. [DOI] [PubMed] [Google Scholar]
- 30.McCaffrey TA, Fu C, Du B, Eksinar S, Kent KC, Bush H, Kreiger K, Rosengart T, Cybulsky MI, Silverman ES, Collins T. High level expression of egr-1 and egr-1 inducible genes in mouse and human atherosclerosis. J Clin Invest. 2000;105:653–662. doi: 10.1172/JCI8592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rokosh G. Heme egr-1: New partners in atherosclerotic progression? Circ Res. 2008;102:6–8. doi: 10.1161/CIRCRESAHA.107.168252. [DOI] [PubMed] [Google Scholar]
- 32.Xu Y, Toure F, Qu W, Lin L, Song F, Shen X, Rosario R, Garcia J, Schmidt AM, Yan SF. Advanced glycation end product (age)-receptor for age (rage) signaling and up-regulation of egr-1 in hypoxic macrophages. J Biol Chem. 2010;285:23233–23240. doi: 10.1074/jbc.M110.117457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andrassy M, Belov D, Harja E, Zou YS, Leitges M, Katus HA, Nawroth PP, Yan SD, Schmidt AM, Yan SF. Central role of pkcbeta in neointimal expansion triggered by acute arterial injury. Circ Res. 2005;96:476–483. doi: 10.1161/01.RES.0000156903.37007.d1. [DOI] [PubMed] [Google Scholar]
- 34.Huang C, Chang JS, Xu Y, Li Q, Zou YS, Yan SF. Reduction of pkcbetaii activity in smooth muscle cells attenuates acute arterial injury. Atherosclerosis. 2010;212:123–130. doi: 10.1016/j.atherosclerosis.2010.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lopez-Cabrera M, Nueda A, Vara A, Garcia-Aguilar J, Tugores A, Corbi AL. Characterization of the p150,95 leukocyte integrin alpha subunit (cd11c) gene promoter. Identification of cis-acting elements. J Biol Chem. 1993;268:1187–1193. [PubMed] [Google Scholar]
- 36.Nelken NA, Coughlin SR, Gordon D, Wilcox JN. Monocyte chemoattractant protein-1 in human atheromatous plaques. J Clin Invest. 1991;88:1121–1127. doi: 10.1172/JCI115411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu H, Perrard XD, Wang Q, Perrard JL, Polsani VR, Jones PH, Smith CW, Ballantyne CM. Cd11c expression in adipose tissue and blood and its role in diet-induced obesity. Arterioscler Thromb Vasc Biol. 2010;30:186–192. doi: 10.1161/ATVBAHA.109.198044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ross R. The pathogenesis of atherosclerosis: A perspective for the 1990’s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 39.Moyer CF, Sajuthi D, Tulli H, Williams JK. Synthesis of il-1 alpha and il-1 beta by arterial cells in atherosclerosis. Am J Pathol. 1991;138:951–960. [PMC free article] [PubMed] [Google Scholar]
- 40.Kleemann R, Zadelaar S, Kooistra T. Cytokines and atherosclerosis: A comprehensive review of studies in mice. Cardiovasc Res. 2008;79:360–376. doi: 10.1093/cvr/cvn120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rezzonico R, Chicheportiche R, Imbert V, Dayer JM. Engagement of cd11b and cd11c beta2 integrin by antibodies or soluble cd23 induces il-1beta production on primary human monocytes through mitogen-activated protein kinase-dependent pathways. Blood. 2000;95:3868–3877. [PubMed] [Google Scholar]
- 42.Javey G, Schwartz SG, Flynn HW, Jr, Aiello LP, Sheetz M. Ruboxistaurin: Review of safety and efficacy in the treatment of diabetic retinopathy. Clinical Medical Insights: Therapeutics. 2010;2:625–631. [Google Scholar]
- 43.Clarke M, Dodson PM. Pkc inhibition and diabetic microvascular complications. Best Pract Res Clin Endocrinol Metab. 2007;21:573–586. doi: 10.1016/j.beem.2007.09.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.