

Abstract
Previously, we reported that RTP801, a stress regulated protein, is induced in multiple cellular models of Parkinson's disease (PD), in an animal model of PD and in dopaminergic neurons of PD patients. In cellular PD models, RTP801 is both sufficient and necessary for death. We further showed that RTP801 and PD mimetics such as 6-OHDA trigger neuron death by suppressing activation of the key kinase mammalian target of rapamycin (mTOR). Here, we report that as a consequence of mTOR signaling blockade, 6-OHDA suppresses the phosphorylation and activation of Akt, a major supporter of neuron survival. This effect is mediated by RTP801 and appears to underlie neuron death induced by 6-OHDA. Examination of postmortem dopaminergic neurons reveals a consistent depletion of phospho-Akt, but not of total Akt in PD patients. These observations support a sequential mechanism in which PD-associated stresses induce RTP801, suppress mTOR signaling, deplete phosphorylated/activated Akt and permit neuron degeneration and death.
Keywords: Parkinson's disease, Akt, RTP801, neurodegeneration, 6-hydroxydopamine, mTOR
Introduction
Parkinson's disease (PD) is characterized by degeneration of specific populations of central and peripheral neurons, including substantia nigral dopaminergic neurons (Fahn, 1998; Marras and Lang, 2008). The mechanisms underlying neuron death in PD are incompletely understood and current therapies largely address symptoms rather than neuron loss.
Because neuron death, regardless of initiating causes, generally requires proapoptotic gene activation, we used serial analysis of gene expression to identify transcripts induced in a cellular model of PD (Ryu et al., 2002, 2005). The most induced transcript (100-fold) was that encoding the protein RTP801/REDD1. RTP801 is induced by stresses including DNA damage, oxidative stress, hypoxia, ER stress and energy depletion (Ellisen et al., 2002; Shoshani et al., 2002; Wang et al., 2003; Sofer et al., 2005) that have been raised as causes of neuron degeneration in PD (Marras and Lang, 2008). RTP801 can be pro or antiapoptotic and in neuronal cells promotes death (Shoshani et al., 2002; Malagelada et al., 2006). We found that multiple PD mimetics (6-OHDA, MPP+ and rotenone) induced RTP801 in neuronal cells and that it mediated death in each case (Malagelada et al., 2006). Moreover, RTP801 was induced in an animal model of PD and is elevated in dopaminergic neurons of PD patients. Such findings establish RTP801 as a potential contributor to neuron degeneration in PD.
The mechanisms by which RTP801 kills neurons have been unclear. The only presently described action of RTP801 is to suppress activation of the kinase mammalian target of rapamycin (mTOR) via a mechanism dependent on TSC2 (tuberous sclerosis protein 2) (DeYoung et al., 2008). In consonance with such observations, we showed that 6-OHDA inhibits mTOR activity and that this was mediated by RTP801 (Malagelada et al., 2006). Moreover, TSC2 shRNA suppressed killing of neuronal cells by RTP801 and 6-OHDA. Such findings implicate blockade of mTOR signaling in the mechanism by which RTP801 causes neuron death.
We considered how mTOR activity might be relevant to neuron survival. mTOR forms two complexes with distinct substrate specificities. The mTORC1 complex controls protein translation by phosphorylating substrates such as S6 kinase and 4EBP1 (Hara et al., 2002; Kim et al., 2002; Thedieck et al., 2007). mTORC2, in contrast, phosphorylates the protein kinase Akt at Ser473 (Sarbassov et al., 2005; Jacinto et al., 2006). mTORC2, by incompletely understood mechanisms, also affects Akt phosphorylation at Thr308 (Sarbassov et al., 2005). While Akt phosphorylation at either site is activating, full activation requires phosphorylation at both Ser473 and Thr308 (Alessi et al., 1996). Akt is a major promoter of neuron survival (Dudek et al., 1997) and does so by phosphorylating substrates including BAD, GSK3, NFkB and forkhead proteins (Cross et al., 1995; del Peso et al., 1997; Kulik et al., 1997; Brunet et al., 2001).
In light of (1) RTP801 induction in PD models and PD, (2) the proapoptotic action of RTP801 in neurons, (3) the relationships between RTP801, mTOR and Akt activity and (4) the central action of Akt in neuron survival, we examine here the role of RTP801 in suppressing Akt activity and promoting neuron death in PD.
Materials and Methods
Antibodies, plasmids and materials.
Anti-RTP801 antiserum was purchased from Millipore or from Proteintech Group. Anti-Erk1 antibody was obtained from Santa Cruz Biotechnology. Anti-horseradish peroxidase-, Alexa fluor 568 or 483 conjugated secondary antibodies were obtained from Pierce and Invitrogen, respectively.
Antibodies against phospho-(Ser473) Akt, phospho-(Thr308) Akt, phospho-(Ser253) Foxo3a, phospho-(Ser2448)-mTOR and phospho-(Ser136) BAD were obtained from Cell Signaling Technology. Monoclonal mouse anti GFP antibody was purchased from Novus Biologicals.
RTP801 constructs were generated by PCR cloning (RTP801 forward: 5′-GAATTCGAACCATGCCTAGCCTTTGGGATCG-3′; RTP801 reverse: 5′-GTCGACTCAACACTCTTCAATGAGCA-3′) from a cDNA library derived from neuronal PC12 cells and ligated to the pCMS-EGFP vector as described previously (Malagelada et al., 2006). All newly made constructs were verified by DNA sequencing. 6-OHDA was purchased from Sigma or Tocris. pIRES-CA-Akt (E17K) vector and pIRES-Dominant-negative Akt vector were a kind gift from Dr. Thomas Franke (New York University, New York, NY)
Cell culture.
PC12 cells were cultured and treated with nerve growth factor (NGF) as described previously (Greene and Tischler, 1976). For NGF treatment, the cells were cultured in RPMI 1640 medium (Cellgro) supplemented with 1% horse serum, penicillin/streptomycin, and 50 ng/ml recombinant human NGF (a kind gift from Genentech) for 8–10 d. Medium was changed every other day and immediately before treatments. 6-OHDA was prepared before use in 10 mm stocks and diluted in medium to the indicated final concentrations. Neonatal rat superior cervical ganglion sympathetic neurons were cultured as described previously (Ryu et al., 2002; Malagelada et al., 2006). Treatments with 6-OHDA was performed at day 7 in vitro, and cell viability was assessed 24 h later. Immunocytochemistry was performed 16 h after treatment. Values represent the mean ± SEM of at least three different experiments.
ShRNA production.
Two different RTP801 shRNAs and a scrambled shRNA (control shRNA) were prepared in DsRed expressing pSIREN vectors as previously described (Malagelada et al., 2006) by using BD knock-out RNAi systems according to the manufacturer's instructions (BD Biosciences) based on the following sequences: 1: 5′- AAG ACT CCT CAT ACC TGG ATG-3′; 4: 5′- AAG AGC TGC CAT AGT GTG GCT-3′. Sh scrambled RNA-eGFP, ShRTP801_1-eGFP and shRTP801_4-eGFP were constructed by excising shRNA sequence and upstream U6 promoter out of the pSIREN vector via bglII and EcoRI, and ligating into similarly cut pCMS-eGFP (which lacks the CMV promoter region but retains the EGFP cistron).
Transfection.
Neuronal PC12 cells and rat sympathetic neurons were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. 48 h after transfection, cells were treated with 6-OHDA. Twenty-four hours after the treatments, viable transfected (as judged by expression of fluorophore or by immunostaining against eGFP protein) neuronal PC12 or sympathetic neurons were scored by strip-counting or whole-well counting, respectively, under an epifluorescence microscope (Malagelada et al., 2006).
Western blot analysis.
Whole-cell extracts were analyzed as previously described (Malagelada et al., 2006).
Immunofluorescence.
Neuronal PC12 cells or sympathetic neurons were fixed with 4% paraformaldehyde in PBS at room temperature for 15 min. The cultures were washed three times with PBS and blocked with 5% horse serum (Vector Laboratories) in PBS containing 0.3% Triton X-100 for an hour at room temperature. Primary antibodies against GFP, phospho-Ser473 Akt or phospho-Thr308 Akt were diluted following supplier's instructions in PBS-0.3% Triton X-100 and incubated overnight at 4°C. Cultures were washed three times in PBS and incubated with Alexa Fluor 483 or 568 conjugated secondary antibodies against rabbit and mouse primary antibodies for an hour at room temperature, washed in PBS and mounted with ProLong Gold antifade from Invitrogen. Cells were observed under fluorescence microscopy or confocal microscopy.
Immunohistochemistry in human sections.
Sections of PD and non-PD substantia nigra were obtained from the New York Brain Bank at Columbia University with the aid of Drs. Jean-Paul Vonsattel, Serge Przedborski and Vernice Jackson-Lewis (Columbia University, New York, NY). Paraffin-embedded sections were heated for 1 h at 65°C and dewaxed in xylene. Rehydration was performed by incubating the slides in an ethanol series (100, 75 and 50%). Sections were incubated in 3% H2O2 at room temperature for 10 min to block nonspecific peroxidase-activity and then antigen was retrieved in Reveal solution from Biocare Medical in a rice cooker for 35 min at 100°C. Sections were then washed with TBS-0.1% Tween (TBST) and blocked first with Avidin/biotin solution (Vector Laboratories), and subsequently in 5% horse serum-TBST for 1 h at room temperature (RT). They were then incubated with phospho-Thr308 Akt or phospho-Ser473 Akt primary antibodies overnight at 4°C and with biotinylated secondary antibody for 1 h at 37°C. Sections were next incubated in ready-to-use ABC complex solution (Vector Laboratories) at RT for 30 min and then SG substrate (Vector Laboratories) was added. Sections were washed, dehydrated and mounted with Permount (Fisher Scientific) mounting media and observed under brightfield microscopy.
Statistics.
All experiments were performed at least in triplicate, and results are reported as means ± SEM. Student's t test was performed as unpaired, two-tailed sets of arrays and presented as probability (p) values. ANOVA with Bonferroni's multiple comparison test was performed to compare multiple groups to the control group.
Results
6-OHDA diminishes Akt phosphorylation and activity in neuronal PC12 cells and rat sympathetic neurons
We have reported that the protein RTP801 mediates neuronal death triggered by 6-OHDA and other PD mimetics and that it does so, at least in part, by blocking activation of mTOR signaling (Malagelada et al., 2006). We have therefore investigated the mechanism by which mTOR inactivation by PD mimetics and RTP801 leads to neuronal death. The mTOR complex mTORC2 has been described as possessing PDK2 activity that phosphorylates the kinase Akt at residue Ser473 and promotes its activation (Jacinto et al., 2004; Sarbassov et al., 2004). Akt phosphorylation by the kinase PDK1 at a second activating site, Thr308, is also suppressed by loss of mTORC2 signaling (Sarbassov et al., 2005). Because active Akt is a major regulator of neuron survival (Dudek et al., 1997; Franke et al., 1997a), we first examined its phosphorylation state after exposure to 6-OHDA. Western immunoblot analysis of lysates of neuronal (NGF treated for at least 5 d) PC12 cells exposed to 6-OHDA for various times showed a substantial drop in Akt phosphorylation at Ser473 (Fig. 1A). This was detectable by 8 h of treatment (a time when RTP801 induction occurs) (Fig. 1A) (Malagelada et al., 2006) and was maximal by ∼16 h, the time when 6-OHDA-promoted death begins to become apparent. This effect was also manifest at the cellular level as shown by immunostaining cells for phospho-Ser473-Akt with or without 16 h of exposure to 6-OHDA (Fig. 1B).
Figure 1.

6-OHDA promotes dephosphorylation of Akt in neuronal PC12 cells. A, Western immunoblot of extracts of neuronal PC12 cells exposed to 6-OHDA for various times. Membranes was probed with antibodies to phospho-Ser473-Akt, phospho-Thr308-Akt, and RTP801 and reprobed with antibodies to total Akt or Erk1 antibodies to show equal protein loading and constancy of Akt expression in each condition. B, Immunostaining of neuronal PC12 cells reveals loss of Akt phosphorylation at Ser 473 and Thr308 after 16 h of exposure to 6-OHDA compared with untreated cultures [Control (Ctrl)]. Nuclei were visualized by staining with Hoechst 33258. These are results are representative of at least 3 independent Western immunoblot and immunostaining experiments.
To extend these observations to primary neurons, we used cultured rat sympathetic neurons. Sympathetic neurons are affected in Parkinson's Disease (Orimo et al., 2008) and, like neuronal PC12 cells that highly resemble them, die in response to RTP801 over-expression (about a 50% of cell death) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), and undergo RTP801-dependent death after exposure to 6-OHDA (Malagelada et al., 2006). Western immunoblotting (Fig. 2A) as well as immunostaining (Fig. 2B) revealed an extensive loss of Akt phosphorylation at Ser473 following 16 h of exposure to 6-OHDA. Densitometric quantification of Western blots from 3 independent experiments showed a fall of 47.5 ± 0.5% (p < 0.01 vs control cultures). This was paralleled by an increase in RTP801 levels as well as by a decrease in phosphorylation of mTOR at its p70S6 kinase phosphorylation site, Ser2448 (Chiang and Abraham, 2005) (Fig. 2A). The latter observations indicate that mTOR kinase activity is impaired in 6-OHDA-treated sympathetic neurons, as previously seen in neuronal PC12 cells (Malagelada et al., 2006).
Figure 2.

6-OHDA promotes dephosphorylation of Akt and suppresses phosphorylation of the Akt substrates Bad and Foxo3a in rat sympathetic neurons. A, Cultured sympathetic neurons were exposed to 10 μm 6-OHDA for 16 h and cell extracts were subjected to SDS-PAGE and Western immunoblotting. Membranes were probed with the indicated immunoreagents and reprobed with antibodies for Akt and ERK1 to show that protein loading was equal in each condition. B, Immunostaining reveals that 6-OHDA treatment causes Akt dephosphorylation at both Akt residues, Ser473 and Thr308 (both in green), in cultures of rat sympathetic neurons. Treatment was for 16 h with 10 μm 6-OHDA. Nuclei were visualized by Hoechst 33258 (blue). These are representative results from at least 3 independent immunostaining experiments, each one in triplicate.
In addition to phosphorylation at Ser473, Akt activity is also positively regulated by phosphorylation at Thr308. This is mediated by PDK1 and requires activation of PI3K signaling (Dudek et al., 1997; Franke et al., 1997a). The latter is relevant to our system because both neuronal PC12 cells and sympathetic neurons require and are cultured in the presence of nerve growth factor, an activator of PI3K and of Akt phosphorylation (Philpott et al., 1997; Crowder and Freeman, 1998). As shown in Figures 1 and 2, in addition to diminishing Akt phosphorylation at Ser473, 6-OHDA also decreased phosphorylation of Akt at Thr308 in both neuronal PC12 cells and in sympathetic neurons as indicated by both Western immunoblotting and immunostaining.
For sympathetic neurons, densitometric quantification of Western blots from 3 independent experiments showed that levels of phospho-Thr308-Akt fell by 59.5 ± 16.5% (p < 0.05 vs control cultures) at 16 h of treatment.
Loss of Akt phosphorylation at both Ser473 and Thr308 would be expected to diminish cellular Akt activity. To assess this, we used Western immunoblotting to analyze phosphorylation of two of its substrates, BAD and Foxo3a (Datta et al., 1997; del Peso et al., 1997; Zheng et al., 2002) in neuronal cells exposed to 6-OHDA for 16 h. In sympathetic neurons (Fig. 2A) and neuronal PC12 cells (data not shown), there was a marked decrease in phosphorylation of BAD at Ser136 and of Foxo3a at Ser 253, the sites targeted by Akt.
Ectopic expression of RTP801 triggers loss of Akt phosphorylated at Ser473 and Thr308
Because RTP801 mediates the actions of 6-OHDA on neuronal survival, we next assessed whether RTP801 can also affect the levels of cellular Akt phosphorylation. Neuronal PC12 cells were transfected either with control vector (pCMSeGFP) or with vector expressing RTP801 (pCMSeGFP-RTP801). Two days later, the cultures were fixed and immunostained for phospho-Ser473-Akt or phospho-Thr308-Akt. Cells transfected with the control vector appeared no different from nontransfected cells with respect to expression of Akt phosphorylated at either site (Fig. 3A). In contrast, cells transfected with RTP801 (eGFP+) showed a greatly diminished level of immunostaining for both phospho-Ser473-Akt or phospho-Thr308-Akt. This was confirmed by scoring the control and RTP801-transfected cells for the presence or absence of detectable phospho-Akt staining (Fig. 3B).
Figure 3.
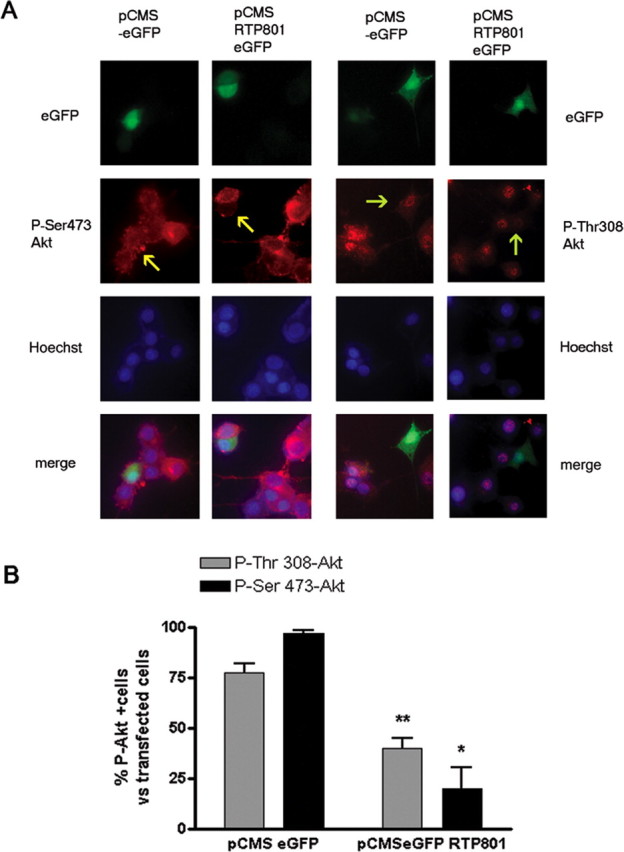
RTP801 overexpression induces dephosphorylation of Akt at Ser473 and Akt Thr308. A, Neuronal PC12 cells were transfected with pCMS-EGFP or pCMS-EGFP RTP801 and 48 h later, cultures were fixed and immunostained with antibodies to phospho-Ser473 Akt (in red, left panel), phospho-Thr308 Akt (in red, right panel) and GFP (in green, in both panels). Arrows indicate positions of transfected cells. B, Proportions of transfected (eGFP+) cells positive for expression of phospho-Ser473 or Thr308− Akt were scored under fluorescence microscopy for each condition. Values represent mean ± SEM for 3 independent experiments done in triplicate. **p < 0.05 vs eGFP+ control cells. *p < 0.01 vs eGFP+ control cells.
RTP801 mediates the effects of 6-OHDA on loss of phospho-Akt
Our past study showed that knockdown of RTP801 with multiple shRNAs protected neuronal PC12 cells and sympathetic neurons from several different PD mimetics (Malagelada et al., 2006). To assess whether the effects of 6-OHDA on Akt phosphorylation are also mediated by RTP801, rat sympathetic neurons were transfected with a construct expressing an shRNA previously shown to strongly block expression of RTP801 (pCMS eGFPshRTP801_4) or with a control construct containing a scrambled shRNA that recognizes no known rat sequence (pCMS eGFPshG) (Malagelada et al., 2006). Two days after transfection, the cultures were treated with or without 6-OHDA for 24 h and were then immunostained with phospho-Ser473- or phospho-Thr308-Akt antibodies as well as with mouse GFP antibody. Cells were also stained with Hoechst 33258 to visualize pyknotic nuclei. Transfected neurons with no sign of chromatin condensation were scored for the proportion that positively stained with the two phospho-AKT antibodies. For neurons transfected with control shRNA, 35–45% lost detectable immunostaining for phospho-Ser473 or phospho-Thr308 Akt after treatment with 6-OHDA (Fig. 4A, white bars). In contrast, for neurons transfected with RTP801 shRNA there little or no significant loss of expression of either phosphorylated form of Akt (Fig. 4A, black bars). Comparable results were achieved with neuronal PC12 cells in which transfected RTP801 shRNA blocked loss of both phospho-Ser473 and phospho-Thr308 Akt after 16 h of exposure to 6-OHDA (Fig. 4B) (data not shown). We used a second independent RTP801 shRNA (shRTP801_1) (Malagelada et al., 2006) to confirm these findings (Fig. 4A,B).
Figure 4.

RTP801 mediates 6-OHDA-induced dephosphorylation of Akt. A, Rat sympathetic neurons (SCG) were transfected with either pCMS eGFP-shG (scrambled shRNA as a control) or pCMS eGFP-shRTP801_1 or pCMS eGFP-shRTP801_4 and 2 d later were treated with or without 10 μm 6-OHDA for 24 h. Cultures were then immunostained with antibodies to phospho-Ser473 Akt or phospho-Thr308 Akt along with antiserum to GFP to visualize transfected GFP+ neurons. Proportions of transfected neurons positive for phospho-Akt (Ser473 or Thr308) were scored under fluorescence microscopy. B, Neuronal PC12 cells were transfected with either pSiren-DsRed ShLuciferase as a control or pSiren-DsRed ShRTP801_1 or pSiren-DsRed ShRTP801_4 and 2 d later were treated with or without 100 μm 6-OHDA for 24 h. Proportions of transfected cells positive for expression of phospho-Ser473 Akt were scored under fluorescence microscopy. Values from both neuronal PC12 cells and sympathetic neurons represent mean ± SEM for 3 independent experiments done in triplicate, +p < 0.01 vs untreated ShG or ShLuciferase transfected cells; *p < 0.01 vs 6-OHDA treated ShG or ShLuc transfected cells; **p < 0.05 vs 6-OHDA treated ShG transfected cells.
Constitutively active Akt protects cells from cell death induced by 6-OHDA and RTP801
Our findings indicate that 6-OHDA leads to depletion of neuronal levels of phospho-Akt. Because phospho-Akt is required for neuron survival, we next assessed whether such depletion may underlie the toxic actions of 6-OHDA. We reasoned that if this were the case, then augmentation of cellular phospho-Akt with a constitutively activatable form of this enzyme would be protective. In a past study, infection with virus expressing a constitutively activatable form of Akt (Myr-Akt) was reported to protect dopaminergic neurons in an in vivo model of 6-OHDA treatment (Ries et al., 2006). For our investigations, we used a construct of Akt1 (CA-Akt) with a glutamic acid to lysine mutation at residue 17. This mutation leads to constitutive cellular phosphorylation and activation of Akt (Carpten et al., 2007). Western immunoblotting confirmed that the mutant was phosphorylated when transfected into HEK293 cells (data not shown). Neuronal PC12 cells were transfected with either pIRES empty vector, pIRES-CA-Akt1, or pIRES-kinase-dead Akt1 (K179M) and after 48 h were exposed to 6-OHDA. One day later, the transfected cells were scored for survival. CA-Akt, but not the kinase-dead form of the enzyme protected the cells from death induced by 6-OHDA (*p < 0.01 vs pIRES-empty vector transfected cells) (Fig. 5A).
Figure 5.

Constitutively active Akt protects from death induced by both 6-OHDA and ectopic RTP801. A, Neuronal PC12 cells were transfected with pIRES, -pIRES-CA-Akt (Akt E17K) or pIRES-dominant negative Akt and after 48 h, cultures were treated with or without 100 μm 6-OHDA. 24 h later, transfected viable cells were scored under fluorescence microscopy. Values are expressed as mean ± SEM of at least three independent experiments in triplicate in each condition. *p < 0.01 compared with cells transfected with pIRES empty vector. B, Neuronal PC12 cells were cotransfected with pIRES, pIRES-CA–Akt, or pIRES-dominant negative Akt in the presence of pCMSeGFP empty vector or pCMS eGFP-RTP801. Seventy-two hours later, viable transfected cells were scored under fluorescence microscopy. Values represent means ± SEM for 3 independent experiments done in triplicate *p < 0.01 versus pCMS eGFP-RTP801+ cells.
Given that RTP801 depletes neuronal levels of phospho-Akt and mediates death induced by 6-OHDA, we next queried whether CA-Akt would also protect neuronal cells from over-expression of RTP801. To address this, neuronal PC12 cells were cotransfected with pIRES-CA–Akt, or pIRES-Akt(K179M) in the presence of pCMS-eGFP empty vector or pCMS eGFP-RTP801 and 72 h later, numbers of viable transfected cells were scored. As shown in Figure 5B, CA-Akt, but not kinase-dead Akt, provided significant protection from RTP801 over-expression. Together, these findings are consistent with the model that 6-OHDA induces RTP801 which in turn depresses intracellular levels of phospho-Akt, leading to neuronal death.
Expression levels of phospho-Ser473-Akt and phospho-Thr308-Akt are decreased in neuromelanin positive neurons in the substantia nigra of PD patients
In our previous study, we observed that RTP801 protein, in addition to its elevation in cellular and animal models of PD, was more highly expressed in neuromelanin-containing neurons in the substantia nigra (SN) of postmortem brains from PD patients than in comparable tissue from non-PD patients (Malagelada et al., 2006). We therefore wished to determine, based on our in vitro studies, whether such neurons might also show changes in expression of phospho-Akt. Neuromelanin typically accumulates in dopaminergic neurons of the SN in individuals with or without PD, and it is this class of pigmented neurons that are lost in the disease. The presence of neuromelanin, which appears brown under brightfield microscopy, thus served as a convenient marker for dopaminergic SN neurons in paraffin-embedded postmortem PD and non-PD brains in which we performed immunohistochemical staining (using a blue-gray chromogen to distinguish it from neuromelanin) of phospho-Ser473-Akt and phospho-Thr308-Akt. Sections from eight PD and seven non-PD brains were examined. Although there was variability in intensity within the population, most neuromelanin-positive neurons from the non-PD brains consistently showed positive staining for both phospho-Ser473- and phospho-Thr308-Akt (Fig. 6A; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). In contrast, although a small number of neuromelanin-positive neurons in the PD brains showed positive staining for phospho-Akt, the majority of neurons gave no detectable signal (Fig. 6A; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Immunostaining for total Akt in the same sets of brains revealed positive staining in all observed neuromelanin-positive neurons. Although there were differences in expression from neuron to neuron, there were no evident overall differences between PD and non-PD neurons (Fig. 6A; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The midbrain sections we examined also contained cells with neuron-like morphology that did not contain detectable neuromelanin and that were presumably nondopaminergic. In contrast to the majority of neuromelanin-positive neurons in PD brains, these stained positively for phospho-Ser473-Akt and phospho-Thr308-Akt in both PD and control brains (Fig. 6A).
Figure 6.
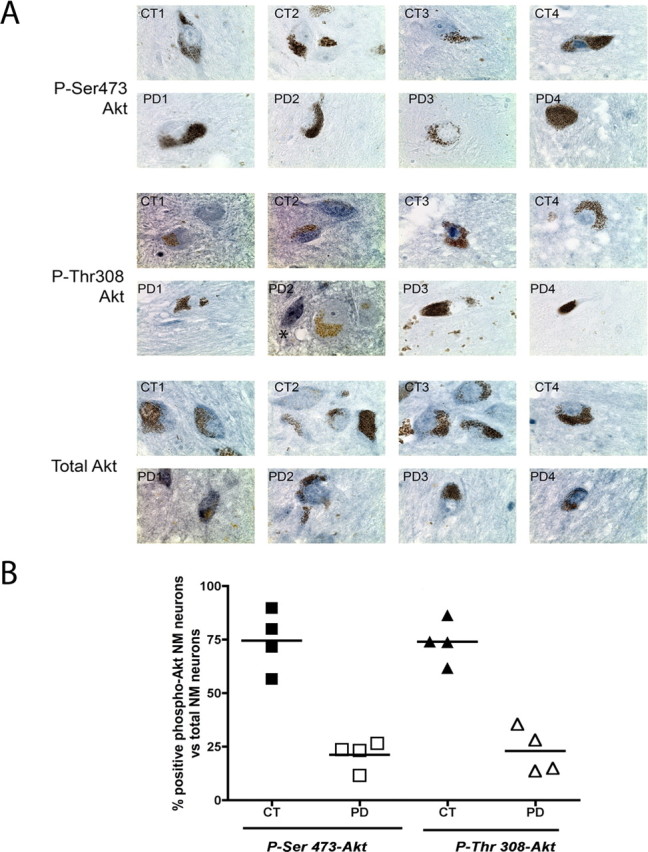
Expression of phospho-Ser473 and phospho-Thr308-Akt expression is diminished in human dopaminergic PD substantia nigral neurons. A, Sections of postmortem human SN from control (CT1, CT2,CT3 and CT4) and PD (PD1, PD2, PD3 and PD4) brains were immunostained (blue-gray) for phospho-Ser473-Akt (top) or phospho-Thr308-Akt (middle) in each set of samples. Note the presence of neuromelanin granules (brown) in the somas of dopaminergic neurons. In parallel, other sections from the same patients were immunostained with an antibody against total Akt (bottom). The asterisk in the middle panel (PD2) points to a cell with neuronal morphology that did not contain detectable neuromelanin and that positively stained for phospho-Thr308-Akt. B, Neuromelanin positive neurons in PD and control sections were scored for the presence or absence of detectable staining for phospho-Ser473 Akt and phospho-Thr308. Values show proportion of total neuromelanin-positive neurons in each brain that showed detectable immunostaining for each antigen. (Controls: black squares for Phospho-Ser473-Akt, black triangles for Phospho-Thr308 Akt. PD: white squares for Phospho-Ser473-Akt, white triangles for Phospho-Thr308 Akt). The numbers of neuromelanin positive neurons scored were between 60 and 200 per brain for controls and 10–60 for PD patients (20× magnification). Results were expressed as percentage of positive phospho-Akt neuromelanin (NM) neurons versus total NM positive neurons present per field per patient. Note that the lower neuron numbers in the PD brains reflects loss of neurons that is characteristic of this disorder.
To quantify the differences in phospho-Akt staining in PD and control SN, we scored individual neuromelanin-positive neurons from four randomly chosen PD and four randomly chosen control brains for the presence or absence of detectable phosphorylated Akt. In the sections from controls, ∼75% positively stained for Akt-Ser473 and Thr308 whereas these values were ∼25% for sections from PD brains (Fig. 6B). These findings support the conclusion that DA neurons in PD brains exhibit not only elevated expression of RTP801, but also diminished levels of the survival protein phospho-Akt.
Discussion
Our past studies have shown that the stress-responsive protein RTP801 is substantially induced in multiple cellular models of PD, in an animal model of PD and in dopaminergic neurons of patients with PD (Ryu et al., 2002; Malagelada et al., 2006). RTP801 over-expression is sufficient to promote neuron death and we have reported that this protein is required for neuron death in cellular models of PD (Malagelada et al., 2006). The focus of the present investigations was to explore the underlying mechanism by which RTP801 leads to death of neurons in the context of PD. This was performed in cellular models of PD, neuronal PC12 cells and sympathetic neurons, exposed to the PD mimetic 6-OHDA. Neuronal PC12 cells have been used in a number of PD-related studies (Malagelada and Greene, 2008) and closely resemble sympathetic neurons, a population that is adversely affected in PD (Orimo et al., 2008).
We observed that 6-OHDA and RTP801 deplete cellular levels of phosphorylated Akt, a major anti-apoptotic kinase (Dudek et al., 1997; Franke et al., 1997a,b; Philpott et al., 1997; Crowder and Freeman, 1998; Brunet et al., 2001). Significantly, this was accompanied by a fall in Akt activity as reflected in the decreased phosphorylation of the Akt substrates Foxo3a and BAD. Both Foxo3a and BAD have proapoptotic actions in neurons and such actions are suppressed when these proteins are phosphorylated by Akt (Datta et al., 1997; del Peso et al., 1997; Zheng et al., 2002). The dephosphorylated forms of these proteins, as well as of other Akt-regulated proapoptotic species may therefore contribute to the death promoted by 6-OHDA and RTP801. Although we have examined Akt phosphorylation in toxin models of PD, it is significant tht DJ-1 downregulation and Parkin loss-of-function are reported to cause impairment of the PI3K/Akt cascade and Akt dephosphorylation in Drosophila (Yang et al., 2005)
Given the key function of active Akt in maintaining neuronal survival, our findings suggest that the fall in cellular levels of phospho-Akt and of Akt activity triggered by 6-OHDA and RTP801 are also responsible for, or at least contribute to, neuron death. To test this hypothesis, we used CA-Akt, a mutant form of the enzyme that becomes constitutively phosphorylated and activated and that should therefore replace or augment endogenous activated Akt. In support of this model, CA-Akt protected cells from both 6-OHDA and RTP801. Of direct relevance, a recent study reported that virally delivered Myr-Akt, another constitutively activatable form of Akt, protects dopaminergic substantia nigra neurons from 6-OHDA in vivo (Ries et al., 2006).
Although our findings place RTP801 upstream of Akt phosphorylation, there is no evidence for the converse in our system. Basal RTP801 levels are low in cells maintained with NGF, a factor that activates Akt, and NGF deprivation, which leads to loss of PI3K activity and of Akt phosphorylation and activity, does not induce RTP801 (Malagelada et al., 2006).
An unusual feature of RTP801 is that it has both anti and proapoptotic actions, depending on the cellular context. For example, RTP801 is reported to have no effect on the viability of proliferating NGF-untreated PC12 cells and protects them as well as MCF7 cells from oxidative stress (Shoshani et al., 2002). It also protects lymphoma cells from death induced by dexamethasone (Wang et al., 2003) and desensitizes prostate tumor cells to apoptotic stimuli (Schwarzer et al., 2005). For several additional cell types, there is no indication that RTP801 over-expression alone induces death (Brugarolas et al., 2004; Corradetti et al., 2005; Ryu et al., 2005). In contrast, RTP801 over-expression promotes death of lung epithelial cells and of neuronal cells including PC12 cells exposed to NGF (Shoshani et al., 2002; Malagelada et al., 2006), sympathetic neurons and hippocampal neurons (López-Toledano et al., 2006). Downregulation of RTP801 also protected retinal ganglion cells in a mouse model of retinopathy of prematurity (Brafman et al., 2004). The selective vulnerability of some cell types such as neurons to RTP801 could be due to a differential sensitivity with respect to Akt phosphorylation. For instance, transient over-expression of RTP801 in HEK293 cells affects neither their viability nor levels of phosphorylated Akt (Corradetti et al., 2005; DeYoung et al., 2008).
The only presently described cellular response to RTP801 is blockade of mTOR activation via release of TSC2 from 14-3-3 protein (DeYoung et al., 2008). Our past work confirmed that RTP801 and 6-OHDA (in an RTP801-dependent manner) suppress mTOR signaling in neuronal cells and that death promoted by PD mimetics and RTP801 is dependent on TSC2 (Malagelada et al., 2006). It therefore follows that the depletion of phospho-Akt observed in response to 6-OHDA and RTP801 are mediated at least in part by inhibition of mTOR. In support of this, Akt is phosphorylated at Ser473 by the mTORC2 complex and phosphorylation of Akt at this site is greatly diminished in mammalian cells lacking mTOR or mTORC2 signaling components (Sarbassov et al., 2005). In addition, knockdown of mTOR or of the mTORC2 component rictor also compromises PDK1-dependent phosphorylation of Akt at Thr308 (Sarbassov et al., 2005). We therefore favor the scheme that induction of RTP801 by 6-OHDA in neurons blocks mTOR signaling and that this in turn leads to loss of Akt phosphorylation at both Ser473 and Thr308 and consequently, to cell death.
To relate out observations in cellular models of PD to the human disease, we have used immunostaining of sections from PD and non-PD postmortem brains. We have reported that RTP801 is elevated in dopaminergic neurons in the SN of PD patients (Malagelada et al., 2006). Here, we find that the same group of neurons shows diminished levels of phospho-Ser473- and phospho-Thr308-Akt. Because the majority of dopaminergic neurons examined showed elevated RTP801 and decreased phospho-Akt, we conclude that the two states largely coincide in the same neurons. These observations are consistent with a causal role (as delineated by our model studies) for elevated RTP801 and consequent loss of Akt activity in the neuronal degeneration and death that characterize PD.
Unlike the acute effects of PD mimetics in cellular and animal models, neuron degeneration and death in PD occurs over a prolonged period. Although we cannot survey the same neurons over time in PD patients, the presence of elevated RTP801 and decreased phospho-Akt in those neurons that remain at the time of death suggests that such changes do not necessarily cause rapid death and may be sustained over protracted periods at sublethal levels. Akt and mTOR signaling pathways play multiple roles in neurons aside from maintaining survival, and it is likely that even partial blockade of their function by RTP801 may impair neuron function in ways relevant to PD. For instance, mTOR and Akt regulate cell mass and Akt has trophic actions on neurons including increasing their size and degree of sprouting (Ries et al., 2006) as well as the caliber and branching of axons (Markus et al., 2002). In PD, in contrast, there is a significant atrophy of pigmented SN neurons (Rudow et al., 2008). mTOR (Swiech et al., 2008) and Akt (Jaworski et al., 2005; Kumar et al., 2005) signaling pathways also play critical roles in dendritic arborization which is markedly reduced in neurons affected by PD (Patt and Gerhard, 1993; Kumar et al., 2005). Loss of mTOR signaling also promotes autophagy (Ravikumar et al., 2004), a process that is present in PD neurons (Anglade et al., 1997) and that can be causally associated with neurodegeneration (Boland and Nixon, 2006). One may imagine, therefore, that in PD, sustained stresses (environmental or genetic in nature) will induce RTP801 and result in chronic depression of mTOR and Akt activities that, while not initially fatal, will impair neuronal function. At some point, when a critical threshold of activity loss is reached, neuron death will ensue.
Our observations raise RTP801 and the pathways up- and down-stream of it as potential targets for preventing the degeneration and death of neurons associated with PD. This includes strategies that might selectively enhance the activation of mTOR and/or Akt in affected neurons.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke–National Institutes of Health grants, the Parkinson's Disease Foundation, and the American Parkinson's Disease Foundation. We thank Dr. Thomas Franke for providing constructs and for helpful discussions, and Drs. Jean-Paul Vonsattel, Katerina Mancevska, Serge Przedborski, and Vernice Jackson-Lewis for providing human brain sections.
References
- AlessiDR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA (1996) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- AngladeP, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y (1997) Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Histopathol 12:25–31. [PubMed] [Google Scholar]
- BolandB, Nixon RA (2006) Neuronal macroautophagy: from development to degeneration. Mol Aspects Med 27:503–519. [DOI] [PubMed] [Google Scholar]
- BrafmanA, Mett I, Shafir M, Gottlieb H, Damari G, Gozlan-Kelner S, Vishnevskia-Dai V, Skaliter R, Einat P, Faerman A, Feinstein E, Shoshani T (2004) Inhibition of oxygen-induced retinopathy in RTP801-deficient mice. Invest Ophthalmol Vis Sci 45:3796–3805. [DOI] [PubMed] [Google Scholar]
- BrugarolasJ, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG Jr (2004) Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 18:2893–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BrunetA, Datta SR, Greenberg ME (2001) Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol 11:297–305. [DOI] [PubMed] [Google Scholar]
- CarptenJD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, Uhlik M, Lin A, Du J, Qian YW, Zeckner DJ, Tucker-Kellogg G, Touchman J, Patel K, Mousses S, Bittner M, et al (2007) A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 448:439–444. [DOI] [PubMed] [Google Scholar]
- ChiangGG, Abraham RT (2005) Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J Biol Chem 280:25485–25490. [DOI] [PubMed] [Google Scholar]
- CorradettiMN, Inoki K, Guan KL (2005) The stress-inducted proteins RTP801 and RTP801L are negative regulators of the mammalian target of rapamycin pathway. J Biol Chem 280:9769–9772. [DOI] [PubMed] [Google Scholar]
- CrossDA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378:785–789. [DOI] [PubMed] [Google Scholar]
- CrowderRJ, Freeman RS (1998) Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J Neurosci 18:2933–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DattaSR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91:231–241. [DOI] [PubMed] [Google Scholar]
- DeYoungMP, Horak P, Sofer A, Sgroi D, Ellisen LW (2008) Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14–3-3 shuttling. Genes Dev 22:239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del PesoL, González-García M, Page C, Herrera R, Nuñez G (1997) Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 278:687–689. [DOI] [PubMed] [Google Scholar]
- DudekH, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME (1997) Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275:661–665. [DOI] [PubMed] [Google Scholar]
- EllisenLW, Ramsayer KD, Johannessen CM, Yang A, Beppu H, Minda K, Oliner JD, McKeon F, Haber DA (2002) REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol Cell 10:995–1005. [DOI] [PubMed] [Google Scholar]
- FahnS (1998) Medical treatment of Parkinson's disease. J Neurol 245:P15–P24. [DOI] [PubMed] [Google Scholar]
- FrankeTF, Kaplan DR, Cantley LC (1997a) PI3K: downstream AKTion blocks apoptosis. Cell 88:435–437. [DOI] [PubMed] [Google Scholar]
- FrankeTF, Kaplan DR, Cantley LC, Toker A (1997b) Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science 275:665–668. [DOI] [PubMed] [Google Scholar]
- GreeneLA, Tischler AS (1976) Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci U S A 73:2424–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HaraK, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K (2002) Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110:177–189. [DOI] [PubMed] [Google Scholar]
- JacintoE, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, Hall MN (2004) Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 6:1122–1128. [DOI] [PubMed] [Google Scholar]
- JacintoE, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B (2006) SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127:125–137. [DOI] [PubMed] [Google Scholar]
- JaworskiJ, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M (2005) Control of dendritic arborization by the phosphoinositide-3′-kinase-Akt-mammalian target of rapamycin pathway. J Neurosci 25:11300–11312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KimDH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM (2002) mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110:163–175. [DOI] [PubMed] [Google Scholar]
- KulikG, Klippel A, Weber MJ (1997) Antiapoptotic signalling by the insulin-like growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Mol Cell Biol 17:1595–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KumarV, Zhang MX, Swank MW, Kunz J, Wu GY (2005) Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J Neurosci 25:11288–11299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-ToledanoMA, Malagelada C, Shelanski ML, Greene LA (2006) Downregulation of RTP801 is necessary for differentiation of neural stem cell progeny in vivo and in vitro. Soc Neurosci Abstr 32:221–27. [Google Scholar]
- MalageladaC, Greene LA (2008) PC12 cells as a model for Parkinson's disease research. Parkinson's disease: molecular and therapeutic insights from experimental models 375–389.
- MalageladaC, Ryu EJ, Biswas SC, Jackson-Lewis V, Greene LA (2006) RTP801 is elevated in Parkinson brain substantia nigral neurons and mediates death in cellular models of Parkinson's disease by a mechanism involving mammalian target of rapamycin inactivation. J Neurosci 26:9996–10005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MarkusA, Zhong J, Snider WD (2002) Raf and akt mediate distinct aspects of sensory axon growth. Neuron 35:65–76. [DOI] [PubMed] [Google Scholar]
- MarrasC, Lang A (2008) Invited article: changing concepts in Parkinson disease: moving beyond the decade of the brain. Neurology 70:1996–2003. [DOI] [PubMed] [Google Scholar]
- OrimoS, Uchihara T, Nakamura A, Mori F, Kakita A, Wakabayashi K, Takahashi H (2008) Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson's disease. Brain 131:642–650. [DOI] [PubMed] [Google Scholar]
- PattS, Gerhard L (1993) A Golgi study of human locus coeruleus in normal brains and in Parkinson's disease. Neuropathol Appl Neurobiol 19:519–523. [DOI] [PubMed] [Google Scholar]
- PhilpottKL, McCarthy MJ, Klippel A, Rubin LL (1997) Activated phosphatidylinositol 3-kinase and Akt kinase promote survival of superior cervical neurons. J Cell Biol 139:809–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RavikumarB, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC (2004) Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 36:585–595. [DOI] [PubMed] [Google Scholar]
- RiesV, Henchcliffe C, Kareva T, Rzhetskaya M, Bland R, During MJ, Kholodilov N, Burke RE (2006) Oncoprotein Akt/PKB induces trophic effects in murine models of Parkinson's disease. Proc Natl Acad Sci U S A 103:18757–18762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RudowG, O'Brien R, Savonenko AV, Resnick SM, Zonderman AB, Pletnikova O, Marsh L, Dawson TM, Crain BJ, West MJ, Troncoso JC (2008) Morphometry of the human substantia nigra in ageing and Parkinson's disease. Acta Neuropathol 115:461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RyuEJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA (2002) Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson's disease. J Neurosci 22:10690–10698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RyuEJ, Angelastro JM, Greene LA (2005) Analysis of gene expression changes in a cellular model of Parkinson disease. Neurobiol Dis 18:54–74. [DOI] [PubMed] [Google Scholar]
- SarbassovDD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM (2004) Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 14:1296–1302. [DOI] [PubMed] [Google Scholar]
- SarbassovDD, Guertin DA, Ali SM, Sabatini DM (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307:1098–1101. [DOI] [PubMed] [Google Scholar]
- SchwarzerR, Tondera D, Arnold W, Giese K, Klippel A, Kaufmann J (2005) REDD1 integrates hypoxia-mediated survival signaling downstream of phosphatidylinositol 3-kinase. Oncogene 24:1138–1149. [DOI] [PubMed] [Google Scholar]
- ShoshaniT, Faerman A, Mett I, Zelin E, Tenne T, Gorodin S, Moshel Y, Elbaz S, Budanov A, Chajut A, Kalinski H, Kamer I, Rozen A, Mor O, Keshet E, Leshkowitz D, Einat P, Skaliter R, Feinstein E (2002) Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol Cell Biol 22:2283–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SoferA, Lei K, Johannessen CM, Ellisen LW (2005) Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol Cell Biol 25:5834–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SwiechL, Perycz M, Malik A, Jaworski J (2008) Role of mTOR in physiology and pathology of the nervous system. Biochim Biophys Acta 1784:116–132. [DOI] [PubMed] [Google Scholar]
- ThedieckK, Polak P, Kim ML, Molle KD, Cohen A, Jenö P, Arrieumerlou C, Hall MN (2007) PRAS40 and PRR5-like protein are new mTOR interactors that regulate apoptosis. PLoS ONE 2:e1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WangZ, Malone MH, Thomenius MJ, Zhong F, Xu F, Distelhorst CW (2003) Dexamethasone-induced gene 2 (dig2) is a novel pro-survival stress gene induced rapidly by diverse apoptotic signals. J Biol Chem 278:27053–27058. [DOI] [PubMed] [Google Scholar]
- YangY, Gehrke S, Haque ME, Imai Y, Kosek J, Yang L, Beal MF, Nishimura I, Wakamatsu K, Ito S, Takahashi R, Lu B (2005) Inactivation of Drosophila DJ-1 leads to impairments of oxidative stress response and phosphatidylinositol 3-kinase/Akt signaling. Proc Natl Acad Sci U S A 102:13670–13675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZhengWH, Kar S, Quirion R (2002) FKHRL1 and its homologs are new targets of nerve growth factor Trk receptor signaling. J Neurochem 80:1049–1061. [DOI] [PubMed] [Google Scholar]