

Abstract
The simple copolymerization of N-carboxyanhydride (NCA) monomers is utilized to generate copolypeptides having a combination of α-helix and β-sheet sub-structures that, when grown from a solvophilic synthetic polymer block segment, are capable of driving mechano-responsive supramolecular sol-to-gel-to-sol and sol-to-gel-to-gel transitions reversibly, which allow also for injection-based processing and self-healing behaviors. A new type of polypeptide-based organogelator, methoxy poly(ethylene glycol)-block-poly(γ-benzyl-l-glutamate-co-glycine) (mPEG-b-P(BLG-co-Gly)), is facilely synthesized by statistical ring-opening copolymerizations (ROPs) of γ-benzyl-l-glutamate (BLG) and glycine (Gly) NCAs initiated by mPEG-amine. These systems exhibit tunable secondary structures and result in sonication stimulus responsiveness of the organogels with the polypeptide segment variation, controlled by varying the ratio of BLG NCA to Gly NCA during the copolymerizations. Attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR) studies indicate the α-helical component decreases while the β-sheet content increases systematically with a higher mole fraction of Gly in the polypeptide segment. The supramolecular assembly of β-sheet nanofibrils, having a tunable width over the range of 10.4 – 14.5 nm with varied BLG to Gly ratio, are characterized by transmission electron microscopy (TEM). The further self-assembly of these nanostructures into 3-D gel networks within N,N-dimethylformamide (DMF) occurs at low critical gelation concentrations (CGC) (lowest ca. 0.6 wt %). Increased BLG to Gly ratios lead to an increase of the α-helical component in the secondary structures of the polypeptide segments, resulting in wider and more flexible nanofibrils. The presence of α-helical component in the polymers enhances the stability of the organogels against sonication, and instantaneous gel-to-gel transitions are observed as in situ reconstruction of networks occurs within the gelled materials after sonication. In marked contrast, the β-sheet-rich gel, prepared from mPEG-b-PGly, exhibits an instant gel-to-sol transition after sonication is applied. The CGC concentration and stiffness of this mPEG-b-P(BLG-co-Gly) organogel system can be tuned by simply varying the percentages of α-helix and β-sheet in the secondary structures through control of the BLG to Gly ratio during synthesis. The mechanical properties of these organogels are studied by dynamic mechanical analyses (DMA), having storage moduli of ca. 12.1 kPa at room temperature. The injectability and self-healing capabilities are demonstrated by direct observation of the macroscopic self-healing behavior experiment.
Introduction
Stimuli-responsive materials have attracted significant attention in the last decades for their potential applications in the construction of adaptive materials, art conservation, sensor design, and controlled release systems.1 Light,2 temperature,3 pH,4 magnetic flux,5 and oxidation-reduction6 are commonly used stimuli to trigger the reorganization of self-assembled nano- or microstructures. These triggers often focus on controlling supramolecular noncovalent interactions, such as hydrogen-bonding, π-π stacking and van der Waals interactions. Recently, it has been found that sonication can be a novel stimulus to tune the self-assembly behavior and aggregate morphology7 within organogel systems, by disrupting hydrogen-bonding and π-π stacking interactions, resulting in sol-to-gel,8 gel-to-sol,9 and gel-to-gel10 transitions. However, elaborate designs having precise control on the molecular level and, consequently, tedious multi-step syntheses are required to obtain these subtle transitions. Thus, sonication-responsive organogels remain limited to primarily low-molecular-weight gelators (LMWGs),7 few studies of systems developed from macromolecular hydrogelators and organogelators have been performed.11
Polypeptide-based materials have been widely used in supramolecularly-assembled hydrogels12 and organogels.13 Their broad applicability is due to the precisely-defined nano- and microstructures derived from supramolecular hierarchical assemblies of polypeptides and the variety of stimuli responses available to natural and synthetic polypeptides, which are often unobtainable from non-polypeptide-based materials. Secondary structures from the self-assembly of polypeptides, especially α-helix and β-sheet conformations, have been proposed as driving forces for the construction of organogels. For example, Kim et al. reported poly(ferrocenysilane)-block-poly(γ-benzyl-l-glutamate) (PFS-b-PBLG) as an organogelator in toluene with thermoreversible gel-to-sol behavior, originating from the monolayer 1-D stacking of the α-helical polypeptide segments (PBLG) into nanoribbons.13a In 2008, Cameron and co-worker reported thermoreversible organogelators, poly(O-benzyl-l-threonine)-block-poly(γ-benzyl-l-glutamate) (PBnT-b-BnE) and poly(O-benzyl-l-threonine)-block-poly(ɛ-N-Boc-l-lysine) (PBnT-b-BocK); both of which contained one α-helix- and one β-sheet-producing polypeptide segment.13c The gelation was found to be driven by β-sheet formation and stacking into nanotapes by the PBnT segments. At the same time the α-helical structure from the PBnE or PBocK block, in an energetically-unfavorable parallel configuration, triggered twisting of the nanotapes in order to minimize these interactions. At present, tunable sonication-responsive organogels from polypeptides have not been explored, and we were interested in investigating such systems, including the self-assembly and gelation behavior of hybrid statistical copolypeptides.
Recently, our group reported a chemically-reactive polypeptide-based organogelator, poly(ethylene glycol)-block-poly(dl-allylglycine) (PEG-b-PDLAG), containing a racemic PDLAG homopolypeptide block.14 Gel formation was driven by supramolecular assembly of β-sheet secondary structures into nano-fibrils in the nanodomain, and which underwent gel-to-sol transition when subjected to sonication stimulus. To explore the effect of constituency control of the secondary structures formed, particularly α-helix and β-sheet configurations, on the physical and mechanical properties, this current work involves a new type of organogelator, methoxy poly(ethylene glycol)-block-poly(γ-benzyl-l-glutamate-co-glycine) (mPEG-b-P(BLG-co-Gly)) These organogel systems exhibited tunable sonication-responsive properties by variation of the secondary structures from the hybrid copolypeptide without the need of elaborate design and multi-step syntheses. Secondary structures were modified by systematically altering the ratios of BLG to Gly, which, respectively, have preferences for α-helix and β-sheet configurations.15 The organogels from α-helix-rich diblock copolymers showed higher stability against sonication and instantaneous gel-to-gel transitions were observed. However, the β-sheet-rich organogel formulations exhibited immediate gel-to-sol transition when sonication was applied. Further, the injectability and self-healing abilities of these organogel systems were directly observed through assessment of the bulk macroscopic self-healing responses.
Experimental section
Materials
Ethyl acetate, n-hexane, tetrahydrofuran (THF), diethyl ether, N,N-dimethylformamide (DMF, anhydrous, ≥ 99.8%), γ-benzyl-l-glutamate (≥ 99%), bis(trichloromethyl) carbonate (≥ 98%), glycine (≥ 99%), trichloromethyl chloroformate (≥ 97%) and Alizarin Red S were purchased from Sigma-Aldrich company (USA). Monomethoxy-monoamino-terminated poly(ethylene glycol) (mPEG45-NH2, Mn = 2000 g/mol) was purchased from Rapp Polymere (Germany). All chemicals were used without further purification, unless otherwise noted.
Instrumentation
1H and 13C NMR spectra were recorded on Varian Inova 300 MHz spectrometers interfaced to a UNIX computer using VnmrJ software. Chemical shifts were referenced to the solvent resonance signals. Attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR) spectra were recorded on an IR Prestige 21 system (Shimadzu Corp.) and analyzed using IRsolution v. 1.40 software.
Thermogravimetric analysis (TGA) was performed under Argon atmosphere using a Mettler-Toledo model TGA/SDTA851e (Mettler-Toledo, Inc., Columbus, OH), with a heating rate of 10 °C/min. Measurements were analyzed using Mettler-Toledo STARe v. 7.01 software. Glass transition temperatures (Tg) were measured by differential scanning calorimetry (DSC) on a Mettler-Toredo DSC822®, with a heating rate of 10 °C/min. Measurements were analyzed using Mettler-Toledo STARe v. 7.01 software. The Tg was taken as the midpoint of the inflection tangent, upon the second heating scan.
Wide-angle X-ray scattering (WAXS) was performed on a Bruker D8 Bragg-Brentano X-ray powder diffractometer. The sample was placed in the sample holder of a two circle goniometer, enclosed in a radiation safety enclosure. The X-ray source was a 2.2 kW Cu X-ray tube, maintained at an operating current of 40 kV and 40 mA. The X-ray optics was the standard Bragg-Brentano para-focusing mode with the X-ray diverging from a DS slit (1 mm) at the tube to strike the sample and then converging at a position sensitive X-ray Detector (Lynx-Eye, Bruker-AXS). The two-circle 250 mm diameter goniometer was computer controlled with independent stepper motors and optical encoders for the θ and 2θ circles with the smallest angular step size of 0.0001° 2θ. The software suit for data collection and evaluation was window based. Data collection was automated COMMANDER program by employing a DQL file and analysed by the program EVA.
Transmission electron microscopy (TEM) images were collected on a JEOL 1200 EX operating at 100 kV and micrographs were recorded at calibrated magnifications using a SLA-15C CCD camera. The final pixel size was 0.42 nm/pixel. Samples for TEM measurements were prepared as follows: 10 µL of the dilute solution (with a polymer concentration of 1 mg/mL) was deposited onto a carbon-coated copper grid, and after 2 min, the excess of the solution was quickly wicked away by a piece of filter paper. The samples were then negatively stained with 1 wt % phosphotungstic acid (PTA) aqueous solution. After 30 s, the excess staining solution was quickly wicked away by a piece of filter paper and the samples were left to dry under vacuum overnight.
Sonication responsive experiments were performed in an ultrasonic homogenizer (maximum power, 150W, 20KHz, Model 150 V/T, Biologics, Inc.) equipped with a micro tip with a diameter of 3.81 mm, employing the power outlet of 45 W in the frequency of 20 KHz at room temperature.
Dynamic mechanical analysis (DMA) was performed on a Mettler Toledo TT-DMA system. DMA of 5 wt % samples in DMF were performed over 3 h in compression on a 3.2 mm thick, 10 mm diameter cylinder. Dynamic measurements were recorded over a range of 0.1 to 10 Hz at room temperature with static stress modulated to 2% compression and a dynamic force applied to provide ±1% deformation. Kinetic data presented were obtained as a single exponential decay using Origin Pro 8.1 software. The gel stiffness was quantified by the evaluation of the compression storage modulus (E′).16
Confocal images were taken using a FV 1000 system (Olympus) with a 10x objective (Olympus) and a 543 nm He-Ne laser excitation source. Emission was collected using a monochrometer set to a 100 nm band pass (560 – 660 nm) and a photomultiplier tube. Image analysis was performed in Fluoviow software (Olympus) and a 1.1 gamma correction was used to enhance feature contrast after scaling image contrast intensity to minimum and maximum threshold set points of 1% above dark count and 1% below maximum counts for both fluorescence and DIC images. Slides were generated by lightly compressing the gels between a cleaned glass microscope slide and a cleaned glass cover slip after dye addition.
Synthesis of γ-benzyl-l-glutamate N-carboxyanhydride (BLG NCA) monomer 1
The BLG NCA monomer was prepared following the literature method.17 In a 500 mL three-necked round bottom flask equipped with a magnetic stir bar, condenser and nitrogen inlet, γ-benzyl-l-glutamate (10.0 g, 42 mmol) and bis(trichloromethyl) carbonate (4.2 g, 14 mmol) were added and suspended in 300 mL ethyl acetate at 65 °C under nitrogen bubbling for 4 h. After cooling to 5 °C, the crude product was extracted with nanopure water (100 mL, 5 °C) and 0.5 wt % sodium bicarbonate solution (100 mL, 5 °C), respectively. The organic layer was dried over MgSO4, filtrated and concentrated. The resulting solid was recrystallized three times from ethyl acetate/n-hexane 1:1 (v/v), and dried in vacuum to obtain a white crystal (2.56 g, yield: 70%). 1H NMR (300 MHz, DMSO, ppm): δ 1.99 (m, 2H, CH2CH2COOCH2), 2.52 (t, J = 6.8 Hz, 2H, CH2CH2COOCH2), 4.47 (m, 1H, COCHNH), 5.10 (s, 2H, COOCH2Ar), 7.37 (m, 5H, ArH), 9.10 (br, 1H, COCHNH). 13C NMR (75 MHz, DMSO, ppm): δ 26.4, 29.1, 56.2, 65.7, 128.0, 128.1, 128.4, 136.0, 151.9, 171.3, 171.7. FTIR (cm−1): 3331, 3251, 2931, 1859, 1773, 1703, 1250, 1185, 1112, 930. HRMS: calculated [M−-H] for C13H13NO5: 262.0715, found: 262.0710.
Synthesis of glycine NCA (Gly NCA) monomer 2
In a 250 mL three-necked round bottom flask equipped with a magnetic stir bar, condenser and nitrogen inlet, glycine (5.0 g, 66.6 mmol) was suspended in 100 mL THF and heated to 50 °C. Trichloromethyl chloroformate (4.39 g, 22.2 mmol) was added into the reaction solution by syringe and the reaction mixture was refluxed for another 4 h under nitrogen. The reaction mixture was filtered before cooling to room temperature. The filtrate was concentrated, recrystallized three times from THF/n-hexane 2:1 (v/v) and dried in vacuum to obtain a white crystal (1.50 g, yield: 67%). The product was stored in −20 °C freezer under nitrogen atmosphere. 1H NMR (300 MHz, DMSO, ppm): δ 4.18 (s, 2H, COCH2NH), 8.83 (br, 1H, COCH2NH). 13C NMR (75 MHz, DMSO, ppm): δ 46.3, 153.0, 169.4. FTIR (cm−1): 3500-3100, 2955, 1876, 1740, 1275, 1113, 1061, 925. HRMS: calculated [M−-H] for C3H3NO3: 100.0035, found: 100.0039.
Synthesis of mPEG-b-P(BLG-co-Gly) diblock copolymer
As a representative example, the synthesis of mPEG45-b-P(BLG16-co-Gly16) was described. In a 10 ml flame dried Schlenk flask equipped with a magnetic stir bar, mPEG45-NH2 (53.3 mg, 0.027 mmol) was added into a solution of BLG NCA (105.3 mg, 0.4 mmol) and Gly NCA (40.4 mg, 0.4 mmol) in 5.8 ml anhydrous DMF solution. The reaction mixture was stirred with a stir rate of 400 rpm under continuous nitrogen flow (100 mL/min) at room temperature for 72 h. The quantitative consumption of monomers was confirmed by measuring the intensity of the NCA anhydride peak at 1788 cm−1 using ATR-FTIR. The Schlenk flask was capped with a rubber stopper with a needle outlet connected with a drying tube. The polymerization gelled generally after 24 h polymerization. The gel was precipitated into diethyl ether under vigorous stirring and a white powder was obtained after centrifuged and dried in vacuum at room temperature (yield: 75%). 1H (300 MHz, TFA-D, ppm): δ 2.04 (br, 32H, CH2CH2COOCH2), 2.48 (br, 32H, CH2CH2COOCH2), 3.50 (s, 3H, CH3O), 3.83 (s, 180H, CH2CH2O), 4.15 (br, 30H, NHCH2CO), 4.65 (br, 16H, NHCHCO), 5.07 (br, 32H, COOCH2), 7.22 (m, 80H, ArH). 13C NMR (75 MHz, TFA-D, ppm): δ 26.7, 30.0, 42.5, 53.2, 68.4, 69.5, 127.9, 128.3, 128.5, 134.1, 173.3, 175.7, 176.1. FTIR (cm−1): 3289, 2870, 1732, 1701, 1649, 1626, 1545, 1510, 1452, 1437, 1387, 1248, 1159, 1098, 849, 737, 696. DSC: Tg = 18 °C, Tg = 103 °C. TGA in N2: 20–325 °C, 32% mass loss; 325–422 °C, 30% mass loss; 422–500 °C, 6% mass loss; 32% mass remaining above 500 °C.
Results and discussion
With our general interest in the creation of polymer-based stimuli-responsive materials with simple molecular design and synthetic feasibility, for fundamental studies and future applications, we began investigation into the preparation of hybrid diblock copolymers having statistical copolypeptides as one of the block segments, to incorporate the potential for stimuli-triggered supramolecular assembly of secondary structures within the polypeptide segments. In order to understand the relationship between the polypeptide composition and the secondary structure in the resulting self-assembled organogels, the mPEG-b-P(BLG-co-Gly) diblock copolymers were synthesized via facile statistical ring-opening copolymerizations (ROPs) of BLG (1) and Gly (2) NCAs by using mPEG45-NH2 as the macroinitiator (Scheme 1). ROPs of amino acid NCAs have been demonstrated to be a convenient and efficient technique for the preparation of high molecular weight synthetic polypeptides in large quantity, while maintaining the capability of self-assembly into organogels.18 In fact, NCA ROPs can be conducted under normal Schlenk techniques, with the rate of polymerization being controlled by a straightforward nitrogen flow method.19 A series of mPEG-b-P(BLG-co-Gly) diblock copolymers with altered compositions in the polypeptide segments was synthesized by systematically varying the feed ratio of BLG and Gly NCA monomers during the copolymerizations (Table 1). For each copolymerization, the required amounts of monomers and macroinitiator were dissolved in anhydrous DMF and the ROP was allowed to proceed for three days at room temperature, under a continuous nitrogen flow (flow rate = 100 mL/min). When Gly NCA occupied a mol % higher than 15% in the monomer mixture, spontaneous gelation occurred during the polymerization (6 – 12 in Table 1) within 24 h. With the mol % of Gly NCA less than 15%, the reaction mixtures remained in the solution state throughout the polymerizations (4 and 5 in Table 1). Each reaction mixture was precipitated into diethyl ether and dried under vacuum to yield the targeted diblock copolymer as a white powder. This processing capability of the mPEG-b-P(BLG-co-Gly) diblock copolymer materials, in the form of either gels or powders, allowed further exploration of the compositions, structures and properties.
Scheme 1.
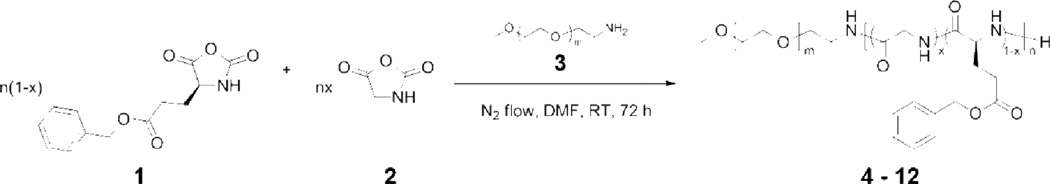
Synthesis of mPEG-b-P(BLG-co-Gly) diblock copolymer.
Table 1.
Average degree of polymerization (DPn), number of repeat unit, mole fraction of Gly (χGly) in the polypeptide segment, number-average molecular weight (Mn) and critical gelation concentration (CGC) for the mPEG-b-P(BLG-co-Gly) diblock copolymers synthesized with different monomer feed ratios.
| Polymer | 1 : 2a) | DPnb) | Number of repeat unitb) | χGly (%)b) |
Mn (kDa)b) |
CGC (wt %)c) |
|
|---|---|---|---|---|---|---|---|
| 1, BLG | 2, Gly | ||||||
| 4 | 1 : 0 | 33 | 33 | 0 | 0 | 9.2 | -d) |
| 5 | 6 : 1 | 33 | 29 | 4 | 12 | 8.6 | -d) |
| 6 | 3 : 1 | 31 | 25 | 6 | 19 | 7.8 | 2.5 |
| 7 | 2 : 1 | 31 | 22 | 9 | 29 | 7.3 | 1.5 |
| 8 | 1.5 : 1 | 30 | 19 | 11 | 37 | 6.8 | 1.2 |
| 9 | 1.2 : 1 | 30 | 17 | 13 | 43 | 6.5 | 0.9 |
| 10 | 1 : 1 | 32 | 16 | 16 | 50 | 6.4 | 0.6 |
| 11 | 1 : 4 | 32 | 6 | 26 | 81 | 4.8 | 0.8 |
| 12 | 0 : 1 | 33 | 0 | 33 | 100 | 3.9 | 2.0 |
The mPEG45-NH2 (3) were used as macroinitiators in all polymerizations with monomer : initiator = 30 : 1;
Calculated by 1H NMR spectroscopy;
CGCs of organogels in DMF were determined by the test tube inversion method at room temperature;
No gelation was observed with the concentration of polymer up to 10 wt %
To investigate the incorporated ratios of the two monomers in the copolymers under different feed ratios, 1H NMR spectroscopy characterization was employed using deuterated trifluoroacetic acid (TFA-D) as the solvent, which was capable of breaking the strong hydrogen-bonding in these systems and maintaining the polymers in the solution state (Fig. 1a). The average degrees of polymerization, numbers of repeat units for BLG and Gly, mole fractions of Gly in the polypeptide segments, and number-average molecular weights of the block copolymers were determined by comparison of the methylene proton intensities of the mPEG chain resonating at ca. 3.83 ppm (b in Fig. 1a) with the intensities of the PBLG benzyl methylene protons resonating at ca. 5.07 ppm (j in Fig. 1a), the PBLG methine proton at ca. 4.65 ppm (f in Fig. 1a), and the PBLG phenyl protons at ca. 7.22 ppm (k in Fig. 1a), and the integration of the Gly methylene protons at ca. 4.15 ppm (d in Fig. 1a), and were found to be in agreement with the stoichiometry used in the polymer syntheses (Table 1). For both BLG (Fig. 1b) and Gly (Fig. 1c), the calculated numbers of repeat units from 1H NMR spectroscopy showed linear correlations with the theoretical numbers of repeat units, which were obtained according to the feed ratios of the two monomers and the ratios of monomers to macroinitiator, indicating that BLG and Gly NCA monomers had similar reactivities and were fully consumed during the polymerizations.20
Fig. 1.

(a) 1H NMR spectrum of polymer 10 dissolved in TFA-D. Calculated numbers of (b) BLG and (c) Gly repeat units as a function of theoretical numbers of repeat units in the polymers 4 – 12.
The ability of mPEG-b-P(BLG-co-Gly) diblock copolymer to form organogels in different solvents was investigated and the properties of DMF gels were studied comprehensively. To prepare the organogel systems, the denoted weight fractions of polymer and DMF were mixed and stable organogels were generated after standing at room temperature for up to 3 days. To investigate the solvent effect in the gel formation, several organic solvents were screened. The polymers could not be dissolved into non-polar or low-polar organic solvents, such as hexane, dichloromethane (DCM), chloroform and tetrahydrofuran (THF), while gelation was observed in relatively high-polarity organic solvents, such as dioxane and dimethyl sulfoxide (DMSO). The critical gel concentrations (CGCs, Table 1) were obtained by preparing different polymer concentrations in DMF and measured by the test tube inversion method at room temperature.21 By comparing polymer 11 (0.8 wt %) with polymer 10 (0.6 wt %), it could be concluded that the CGC decreased with increased Gly content in the polypeptide segment when the Gly content was less than 50 mol %, while this trend reversed with Gly at greater than 50 mol %. The turbidities of the organogels at 2.5 wt % also changed from being transparent to opaque with increased Gly content in the polypeptide segments (Fig. S1 in ESI†).
In order to further understand the properties of these organogel systems, the supramolecular structures of the dried gels from polymers 4 – 12 were investigated by ATR-FTIR (Fig. 2a). The characteristic absorbances of secondary structures were clearly observed in the FTIR spectra, especially in the region of amide I band (1700 – 1600 cm−1). The absorbances at 1650 cm−1 (amide I) and at 1547 cm−1 (amide II) were attributed to α-helical conformations.22 The β-sheet secondary structure was observed at 1675 cm−1 and 1626 cm−1 (both in amide I), in combination with a peak at 1520 cm−1 (amide II).23 Other secondary structure populations, such as random coils or turns, showed absorbances at 1600 – 1620 cm−1, 1640 – 1650 cm−1, 1660 – 1670 cm−1 and 1680 – 1700 cm−1.24 The peak at 1728 cm−1 corresponded to the C=O stretch of the benzyl ester group in the side chain of BLG.16 Qualitatively, the absorption intensity assigned to the peaks of β-sheet secondary structures increased with higher mole fraction of Gly in the polypeptide segment, while the intensities of peaks of α-helices decreased correspondingly.
Fig. 2.

(a) ATR-FTIR spectra and (b) secondary structure populations of polymers 4 – 12 in the solid state.
Further quantitative analyses of secondary structures were conducted by employing second derivative and deconvolution strategies to extract relative peak intensities of individual bands in the ATR-FTIR spectra. These analyses provided extraction of secondary structural components in the heavily populated amide I regions, followed by curve fitting to determine signal amplitude for each band (Fig. S2 in ESI†), as summarized in Fig. 2b.16, 25. For each polymer, the supramolecular structures were composed of α-helix, β-sheet and other secondary structures, regardless of the difference in the chemical compositions. As the mole fraction of Gly in the polypeptide segment increased from 0% to 100%, the α-helical content decreased systematically from 65% to 6% of total band intensity and the β-sheet component raised gradually from 14% to 66%. The β-sheet contribution to secondary structure became dominant over the α-helix contribution with Gly mole fraction greater than 19% in the polypeptide segment. Other secondary structure band intensities remained relatively constant for all formulations.
The higher-order self-assembled structures of the dried gels from mPEG-b-P(BLG-co-Gly) diblock copolymers were also investigated by WAXS. The WAXS pattern for gel from polymer 12 (Fig. S3 in ESI†) gave a d spacing of 4.4 Å with stronger signals for PEG crystallization reflection, which was also demonstrated by the melting and crystallizing peaks in the DSC trace of polymer 12 (Fig. S4b in ESI†). This d spacing of 4.4 Å has been observed and assigned to antiparallel β-sheet formation in poly(L-alanylglycine)26 and the self-assembled PEG-b-β-strand-peptides.27 The WAXS patterns for gels from polymers 7, 10 and 11 had similar d spacings at 4.6 Å, 4.6 Å and 4.5 Å, respectively. These slightly increased d spacings suggested higher α-helix content in the secondary structures, which could enlarge the distance between adjacent polypeptide chains. The presence of α-helical content also appeared to have disrupted PEG chain order, observed as a lack of PEG crystallization peaks in the WAXS patterns of the gels from polymers 7, 10 and 11 and a lack of melting transition in the DSC trace of polymer 11 (Fig. S4a in ESI†).
TEM was employed to study the nanostructures of the organogels as related to the copolypeptide composition. The TEM samples were prepared with a polymer concentration of 1.0 mg/mL in DMF and stained by PTA aqueous solution. Bright-field TEM images of organogels from polymers 7, 10, 11 and 12 (Fig. 3a, 3c, 3e and 3g, respectively) showed fibrillar nanostructures with average widths of 14.5, 12.1, 11.5 and 10.4 nm. The narrow width distributions of nanofibrils from polymers 7, 10, 11 and 12 (Fig. 3b, 3d, 3f and 3h, respectively), obtained by counting 100 nanofibrils in the TEM images, indicated that all of the organogels had well-defined nanofibrillar morphologies. The average diameters of nanofibrils increased with a larger α-helical content in the secondary structures, which might be attributed to the formation of twisted fibrils (Inset images at the upper right corners of Fig. 3a, 3c, 3e and 3g, respectively), originating from a combination of increased steric hindrance and chirality change with the introduced BLG component.13c, 28 Furthermore, the β-sheet-rich polymer 12 showed more rigid nanofibrils when compared with the other fibril nanostructures from polymers with increased α-helical components.
Fig. 3.

TEM images and fiber width distributions of gels from polymers 7, 10, 11 and 12, obtained by counting 100 fibers. (a) TEM image and (b) fiber width distribution of 7. (c) TEM image and (d) fiber width distribution of 10. (e) TEM image and (f) fiber width distribution of 11. (g) TEM image and (h) fiber width distribution of 12. The inserted images at the upright corners of (a), (c), (e) and (g) are the proposed twisted fibrils.
Additional studies of the thermal properties and external stimuli-responsive features of the organogel systems from polymers 7, 10, 11 and 12 in DMF were also performed. Gels from all four polymers, with a concentration of 5 wt % in DMF, could withstand temperatures of 90 °C over 1 h, however, the four gels behaved quite differently after 30 s sonication at room temperature. The gels from polymers 7, 10 and 11 remained in the gel state, even with longer sonication time (ca. 30 min), while the gel from polymer 12 was transformed to a solution state after 30 s sonication (Fig. 4). This transition was reversible, with the gel reforming after being placed at room temperature for 3 h. TEM was also used to characterize the changes of nanostructural morphologies within the gels before and after sonication was applied. The gels from polymers 11 and 12, with an initial concentration of 5 wt % in DMF, were diluted to polymer solutions of 1.0 mg/mL after 30 s sonication, drop deposited, and stained with PTA aqueous solution to prepare the TEM samples. Alterations in the nanostructures for gels from polymers 11 and 12 were observed by comparison of TEM images before (Fig. 3e and 3g) and after (Fig. 5a and 5b) sonication. In the gel from polymer 11, longer nanofibrils disappeared after sonication and, instead, short nano-rods were observed. The ability of the gel states to reform and for the gel assemblies to undergo toughening was also demonstrated by dynamic mechanical analysis (DMA), in which a 20% increase in storage modulus was detected for the gel after sonication (Fig. S5 in ESI†). This sonication-promoted gel-to-gel transition with increased modulus implied that a temporary disruption of nanostructures occurred for polymer 11 during sonication and resulted in ordering that was even more stable, akin to an annealing process. Immediate reassembly of short nano-rods, in situ after sonication, facilitated the rebuilding of 3-D networks and resulted in a sonication-triggered gel-to-gel transition. However, in the gel produced from polymer 12, the longer nanofibrils were expressly converted into short nano-rods, which were well-aligned in nanodomians, but lacked longer range connectivity between clusters, ultimately resulting in a gel-to-sol transition. When the solution was maintained at room temperature for 3 h, the one-dimensional growth of nano-rods to longer nanofibrils resulted in reconstruction of the gel.
Fig. 4.

Images of organogels from polymers 7, 10, 11 and 12 in DMF (5.0 wt %) before (left) and after (right) sonication.
Fig. 5.

TEM images of organogels from polymers (a) 11 and (b) 12 after 30 s sonication.
While the nano- and supramolecular-scale properties could be correlated to the macroscale gel-to-gel and gel-to-sol transitions, it is also important to study the micro- and meso- scale structures of the gels in order to fully understand the gel behavior through all scales of self-interaction. To this effect, dye incubation (1 wt % 0.1 mM Alizarin Red S in DMF) in 5 wt % gels from polymers 11 and 12 were studied by laser scanning confocal microscopy (Fig. 6). It was observed that the gel homogeneity on the micro- and meso-scales was much higher in the organogel from polymer 12, both before and after sonication (Fig. 6.3 and 6.4) when compared to the gel of polymer 11 (Fig. 6.1 and 6.2). Observing the organogel from polymer 11 in the bulk (Fig. 6.1a), as well as in isolated regions (Fig. 6.1b–e), it was evident that both larger mesoscale domains (ca. 50 µm) and microscale (ca. 1–10 µm) structures could be observed. On sonication of the organogel from polymer 11 (Fig. 6.2), both types of structures remained on these scales and longer range interactions did not appear to be overtly changed in the gel, correlating well to the tube inversion and DMA findings. Pre-sonication, organogel from polymer 12 possessed a homogeneous background emission with a collection of proximate microdomains (ca. <1 to 5 µm). Also of note, the fluorescent image showed diffused (Fig. 6.3b) structure edges coincident with the differential imaging contrast (DIC) micrograph, but significantly broader than those in the gels from polymer 11, indicating extradomain interactions beyond that observed in DIC. Considering organogel from polymer 12 post-sonication (Fig. 6.4), it was observed that the higher emission regions were smaller and more numerous in the fluorescence channel, while in DIC the majority of microstructures had been eliminated. These findings indicated the generation of isolated, sub-diffraction scale domains, which lacked intrinsic long-range interactions.
Fig. 6.

Confocal imaging of dyed organogel from polymers 11 in DMF (5 wt %) before (1a–e) and after (2a–e) sonication. Confocal imaging of dyed organogel from polymer 12 in DMF (5 wt %) before (3a–b) and after (4a–b) sonication. Fluorescence images were greenscale and DIC images were greyscale. Scale bars: 100 µm for 1a–c, 2a–c, 3a and 4a; 50 µm for 1d, 1e, 2d, 2e; 20 µm for 3b, 4b
The mechanical properties of the organogel systems were characterized by DMA on polymers 7, 10, 11 and 12 in the gel state at a concentration of 5 wt %, which was sufficiently high to produce a robust gel that could withstand high frequency operation. Frequency modulation showed systematic changes in the storage (E′) and loss (E′′) moduli of the organogels for polymers 7, 10, 11 and 12 (Fig. 7a). For each polymer, the loss modulus remained smaller than the storage modulus within the frequencies observed (0.1 to 10 Hz), demonstrating no gel-to-sol transition in this frequency range, which was also observed in the tan(δ) data (Fig. S6 in ESI†). The storage moduli at 1 Hz of the four gels were 3.0, 8.4, 11.6 and 3.9 kPa for the samples 7, 10, 11 and 12. With the mole fraction of Gly changing from 19% to 81%, the gel moduli increased by a factor of 4, demonstrating an enhancement of the strength of the gel systems. However, the storage modulus dropped significantly for the 100% Gly (polymer 12) system (Fig. 7b), in comparison to those copolypeptide block copolymer materials containing proportions of BLG.
Fig. 7.

(a) Moduli of organogels from polymers 7, 10, 11 and 12 in DMF (5 wt %) as a function of frequency conducted by DMA. E′and E′′ indicate storage and loss modulus, respectively. (b) Storage modulus of organogels from polymers 7, 10, 11 and 12 in DMF (5 wt %) as a function of mole fraction of Gly (χGly) in polypeptide segment under the static frequency of 1 Hz in DMA tests.
Taking together the results of NMR, FTIR, WAXS, DSC, TEM, confocal and DMA analyses, a mechanism for organogel formation from the mPEG-b-P(BLG-co-Gly) diblock copolymers and the relative stabilities of the matrices to sonication is proposed (Scheme 2). The supramolecular self-assembly of the polypeptide segments into well-defined nanofibrils was driven by the formation of β-sheet secondary structure, as seen spectrally and microscopically, resulting in the construction of organogels. The presence of α-helical content in the secondary structures broadened the distance between the polypeptide chains and the width of nanofibrils. In the gel state, after sonication, the nanofibrillar structures were deconstructed into short nano-rods by cleaving the supramolecular interactions between polypeptide segments. In the α-helix-rich organogels, the reassembly of relatively less locally ordered and more flexible nano-rods, in combination with the maintenance of long-range interactions, resulted in the immediate reconstruction of organogels in situ as a sonication- triggered gel-to-gel transition. However, the failure to reconstruct 3-D network from more locally aligned and rigid nano-rods, and the disappearance of micro- and mesoscale interactions for the β-sheet-rich polymer, led to a sonication-triggered gel-to-sol transition. Ultimately, the effect on the external sonication-responsive property due to secondary structure control of statistical copolypeptide-based organogels provides a novel and facile method to modify the properties of stimuli-responsive materials by tuning the self-assembled nano- or microstructures without the need of precise control on the molecular level.
Scheme 2.

A schematic illustration of the nanofibrils formed in the network structure of organogels form polymers 11 (upper) and 12 (lower) and also the gel-to-gel and gel-to-sol transformations after sonication.
With the construction of organogels from mPEG-b-P(BLG-co-Gly) diblock copolymers being driven by β-sheet formation in the polypeptide segments, a self-healing property of the gel system was expected and demonstrated by a macroscopic self-healing experiment conducted on polymer 11 in DMF at a concentration of 20 wt %._ENREF_5129 Half of the organogel was blended with 0.01 mL of Alizarin Red S DMF solution for visibility (Fig. 8a). Both of the gel fractions were then loaded into syringes (Fig. 8b), extruded into a glass petri dish and cut into pieces (Fig. 8c). Self-healing behavior began immediately after two pieces of organogel were placed into contact on the cut faces (Fig. 8d). After 3 min, the combined gel was sufficiently strong to be picked up by tweezers (Fig. 8e). Multiple pieces of cut gel could be reconnected together by attaching them one-by-one (Fig. 8f). Thizarin Red S was observed in the self-healed gel at room temperature, and afforded a single gel matrix with a homogeneous color after 3 days (Fig. 8g).
Fig. 8.

Macroscopic self-healing behavior of organogels from polymer 11 in DMF (20 wt %). (a) Gel was mixted with Alizarin Red S DMF solution, (b) loaded into syringe, (c) cut into pieces, (d) attached together, (e) picked up by tweezer, (f) then connected one by one and (g) diffusion of dye complete after 3 days.
Conclusions
Sonication-responsive macromolecular statistical copolypeptide-based organogels exhibit unique properties that are tuned by secondary structural control that is offered by a series of robust statistical copolymerizations with variation in the copolypeptide feedstock selection. This strategy circumvents the elaborate designs and multi-step syntheses. The effect of the self-assembled secondary structures, especially α-helix and β-sheet configurations, on the properties of organogels are explored by using a serious of mPEG-b-P(BLG-co-Gly) diblock copolymers with varied chemical compositions in the polypeptide segments. Detailed characterization studies reveal that gelation in DMF is driven by supramolecular assembly of β-sheets into well-defined fiber-like nanostructures, further generating a 3-D network through longer-range interactions. The sonication-triggered gel-to-gel transition observed in the case of organogels from α-helix-rich systems is shown to originate from an immediate reconstruction into a 3-D network of nano-rods. However, for the β-sheet-rich polymer, an gel-to-sol transition results from the failure of network rebuilding, instead showing reorganization of nano-rods into locally aligned nanodomains with a small amount of interactions between them. The CGC and stiffness of these organogels can be tuned by simply varying the ratios of α-helix to β-sheet in the secondary structures, which is in-turn modulated by the feed ratios of BLG and Gly NCA monomers during the polymerization. These results illustrate the influence of secondary structures on supramolecular assembly of hybrid diblock synthetic polypeptides in the gel state and demonstrate a novel, facile approach to fabricate nanostructures with stimuli-responsive properties. Moreover, the sonication-responsive properties, the injectability and self-healing capabilities of the organogels enable further study and potential applications in sensor design, construction of adaptive materials and controlled release systems.
Supplementary Material
Acknowledgements
This work was supported in part from the National Heart Lung and Blood Institute of the National Institutes of Health as a Program of Excellence in Nanotechnology (HHSN268201000046C) and the National Science Foundation under grant number DMR-1105304. The Welch Foundation is gratefully acknowledged for support through the W. T. Doherty-Welch Chair in Chemistry, Grant No. A-0001. The microscopy & imaging center (MIC) at Texas A&M University is also gratefully acknowledged.
Footnotes
Electronic Supplementary Information (ESI) available: Fig. S1–S5. See DOI:10.1039/b000000x/
References
- 1.a Stuart MAC, Huck WTS, Genzer J, Muller M, Ober C, Stamm M, Sukhorukov GB, Szleifer I, Tsukruk VV, Urban M, Winnik F, Zauscher S, Luzinov I, Minko S. Nature Mater. 2010;9:101. doi: 10.1038/nmat2614. [DOI] [PubMed] [Google Scholar]; b Capadona JR, Shanmuganathan K, Tyler DJ, Rowan SJ, Weder C. Science. 2008;319:1370. doi: 10.1126/science.1153307. [DOI] [PubMed] [Google Scholar]; c Capito RM, Azevedo HS, Velichko YS, Mata A, Stupp SI. Science. 2008;319:1812. doi: 10.1126/science.1154586. [DOI] [PubMed] [Google Scholar]; d Du JZ, O'Reilly RK. Soft Matter. 2009;5:3544. [Google Scholar]; e Carretti E, Bonini M, Dei L, Berrie BH, Angelova LV, Baglioni P, Weiss RG. Acc. Chem. Res. 2010;43:751. doi: 10.1021/ar900282h. [DOI] [PubMed] [Google Scholar]; f Randolph LM, Chien MP, Gianneschi NC. Chem. Sci. 2012;3:1363. doi: 10.1039/C2SC00857B. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Grigoryev A, Sa V, Gopishetty V, Tokarev I, Kornev KG, Minko S. Adv. Funct. Mater. 2013 [Google Scholar]
- 2.a Goodwin AP, Mynar JL, Ma YZ, Fleming GR, Frechet JMJ. J. Am. Chem. Soc. 2005;127:9952. doi: 10.1021/ja0523035. [DOI] [PubMed] [Google Scholar]; b Haines LA, Rajagopal K, Ozbas B, Salick DA, Pochan DJ, Schneider JP. J. Am. Chem. Soc. 2006;127:17025. doi: 10.1021/ja054719o. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Liu XK, Jiang M. Angew. Chem. Int. Ed. 2006;45:3846. doi: 10.1002/anie.200504364. [DOI] [PubMed] [Google Scholar]
- 3.a Park MH, Joo MK, Choi BG, Jeong B. Acc. Chem. Res. 2012;45:424. doi: 10.1021/ar200162j. [DOI] [PubMed] [Google Scholar]; b Jiang Y, Zeng F, Gong R, Guo Z, Chen C-F, Wan X. Soft Matter. 2013;9:7538. [Google Scholar]; c Pester CW, Konradi A, Varnholt B, van Rijn P, Boker A. Adv. Funct. Mater. 2012;22:1724. [Google Scholar]
- 4.a Zou J, Zhang SY, Shrestha R, Seetho K, Donley CL, Wooley KL. Polym. Chem. 2012;3:3146. doi: 10.1039/C2PY20324C. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Du JZ, Armes SP. J. Am. Chem. Soc. 2005;127:12800. doi: 10.1021/ja054755n. [DOI] [PubMed] [Google Scholar]; c Gensel J, Dewald I, Erath J, Betthausen E, Muller AHE, Fery A. Chem. Sci. 2013;4:325. [Google Scholar]
- 5.Borase T, Ninjbadgar T, Kapetanakis A, Roche S, O'Connor R, Kerskens C, Heise A, Brougham DF. Angew. Chem. Int. Ed. 2013;52:3164. doi: 10.1002/anie.201208099. [DOI] [PubMed] [Google Scholar]
- 6.Kramer JR, Deming TJ. J. Am. Chem. Soc. 2012;134:4112. doi: 10.1021/ja3007484. [DOI] [PubMed] [Google Scholar]
- 7.Cravotto G, Cintas P. Chem. Soc. Rev. 2009;38:2684. doi: 10.1039/b901840a. [DOI] [PubMed] [Google Scholar]
- 8.a Naota T, Koori H. J. Am. Chem. Soc. 2005;127:9324. doi: 10.1021/ja050809h. [DOI] [PubMed] [Google Scholar]; b Isozaki K, Takaya H, Naota T. Angew. Chem. Int. Ed. 2007;46:2855. doi: 10.1002/anie.200605067. [DOI] [PubMed] [Google Scholar]; c Wu JC, Yi T, Shu TM, Yu MX, Zhou ZG, Xu M, Zhou YF, Zhang HJ, Han JT, Li FY, Huang CH. Angew. Chem. Int. Ed. 2013;47:1063. doi: 10.1002/anie.200703946. [DOI] [PubMed] [Google Scholar]; d Afrasiabi R, Kraatz HB. Chem. Eur. J. 2013;19:1769. doi: 10.1002/chem.201202268. [DOI] [PubMed] [Google Scholar]
- 9.Paulusse JMJ, van Beek DJM, Sijbesma RP. J. Am. Chem. Soc. 2007;129:2392. doi: 10.1021/ja067523c. [DOI] [PubMed] [Google Scholar]
- 10.Yu XD, Liu QA, Wu JC, Zhang MM, Cao XH, Zhang S, Wang Q, Chen LM, Yi T. Chem. Eur. J. 2010;16:9099. doi: 10.1002/chem.201000187. [DOI] [PubMed] [Google Scholar]
- 11.b Wang XQ, Kluge JA, Leisk GG, Kaplan DL. Biomaterials. 2008;29:1054. doi: 10.1016/j.biomaterials.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liu ZX, Feng Y, Yan ZC, He YM, Liu CY, Fan QH. Chem. Mater. 2012;24:3751. [Google Scholar]
- 12.a Nowak AP, Breedveld V, Pakstis L, Ozbas B, Pine DJ, Pochan D, Deming TJ. Nature. 2002;417:424. doi: 10.1038/417424a. [DOI] [PubMed] [Google Scholar]; b Huang J, Hastings CL, Duffy GP, Kelly HM, Raeburn J, Adams DJ, Heise A. Biomacromolecules. 2013;14:200. doi: 10.1021/bm301629f. [DOI] [PubMed] [Google Scholar]; c Cheng YL, He CL, Xiao CS, Ding JX, Cui HT, Zhuang XL, Chen XS. Biomacromolecules. 2010;14:468. doi: 10.1021/bm3017059. [DOI] [PubMed] [Google Scholar]; d Chen Y, Pang XH, Dong CM. Adv. Funct. Mater. 2010;20:579. [Google Scholar]
- 13.a Kim KT, Park C, Vandermeulen GWM, Rider DA, Kim C, Winnik MA, Manners I. Angew. Chem. Int. Ed. 2005;44:7964. doi: 10.1002/anie.200502809. [DOI] [PubMed] [Google Scholar]; b Jeong Y, Joo MK, Sohn YS, Jeong B. Adv. Mater. 2007;19:3947. [Google Scholar]; c Gibson MI, Cameron NR. Angew. Chem. Int. Ed. 2011;47:5160. doi: 10.1002/anie.200801056. [DOI] [PubMed] [Google Scholar]; d Tang HY, Lee CU, Zhang DH. J. Polym. Sci., Part A: Polym. Chem. 2011;49:3228. [Google Scholar]
- 14.Zou J, Zhang F, Chen Y, Raymond JE, Zhang S, Fan J, Zhu J, Li A, Seetho K, He X, Pochan DJ, Wooley KL. Soft Matter. 2013;9:5951. doi: 10.1039/C3SM50582K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kricheldorf HR. Angew. Chem. Int. Ed. 2006;45:5752. doi: 10.1002/anie.200600693. [DOI] [PubMed] [Google Scholar]
- 16.Kotharangannagari VK, Sanchez-Ferrer A, Ruokolainen J, Mezzenga R. Macromolecules. 2012;45:1982. [Google Scholar]
- 17.Poche DS, Moore MJ, Bowles JL. Synth. Commun. 1999;29:843. [Google Scholar]
- 18.a Hadjichristidis N, Iatrou H, Pitsikalis M, Sakellariou G. Chem. Rev. 2009;109:5528. doi: 10.1021/cr900049t. [DOI] [PubMed] [Google Scholar]; b Huang J, Heise A. Chem. Soc. Rev. 2013;42:7373. doi: 10.1039/c3cs60063g. [DOI] [PubMed] [Google Scholar]
- 19.Zou J, Fan J, He X, Zhang S, Wang H, Wooley KL. Macromolecules. 2013;46:4223. doi: 10.1021/ma4007939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a Wamsley A, Jasti B, Phiasivongsa P, Li XL. J. Polym. Sci., Part A: Polym. Chem. 2004;42:317. [Google Scholar]; b Hayashi S, Ohkawa K, Yamamoto H. Macromol. Biosci. 2006;6:228. doi: 10.1002/mabi.200500211. [DOI] [PubMed] [Google Scholar]
- 21.Yu L, Zhang H, Ding JD. Angew. Chem. Int. Ed. 2006;45:2232. doi: 10.1002/anie.200503575. [DOI] [PubMed] [Google Scholar]
- 22.Kuo SW, Lee HF, Huang WJ, Jeong KU, Chang FC. Macromolecules. 2009;42:1619. [Google Scholar]
- 23.Aggeli A, Bell M, Boden N, Keen JN, Knowles PF, McLeish TCB, Pitkeathly M, Radford SE. Nature. 1997;386:259. doi: 10.1038/386259a0. [DOI] [PubMed] [Google Scholar]
- 24.Miyazawa T, Blout ER. J. Am. Chem. Soc. 1961;83:712. [Google Scholar]
- 25.a Kauppinen JK, Moffatt DJ, Mantsch HH, Cameron DG. Anal. Chem. 1981;53:1454. [Google Scholar]; b Kauppinen JK, Moffatt DJ, Mantsch HH, Cameron DG. Appl. Spectrosc. 1981;35:271. doi: 10.1364/AO.20.001866. [DOI] [PubMed] [Google Scholar]
- 26.Panitch A, Matsuki K, Cantor EJ, Cooper SJ, Atkins EDT, Fournier MJ, Mason TL, Tirrell DA. Macromolecules. 1997;30:42. [Google Scholar]
- 27.Rosler A, Klok HA, Hamley IW, Castelletto V, Mykhaylyk OO. Biomacromolecules. 2003;4:859. doi: 10.1021/bm034058s. [DOI] [PubMed] [Google Scholar]
- 28.a Blake C, Serpell L. Structure. 1996;4:989. doi: 10.1016/s0969-2126(96)00104-9. [DOI] [PubMed] [Google Scholar]; b Adamcik J, Mezzenga R. Macromolecules. 2012;45:1137. [Google Scholar]
- 29.a Liu D, Wang D, Wang M, Zheng Y, Koynov K, Auernhammer GK, Butt H-J, Ikeda T. Macromolecules. 2013;46:4617. [Google Scholar]; b Phadke A, Zhang C, Arman B, Hsu CC, Mashelkar RA, Lele AK, Tauber MJ, Arya G, Varghese S. Proc. Natl. Acad. Sci. USA. 2012;109:4383. doi: 10.1073/pnas.1201122109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.