

Abstract
Background
Synthetic triterpenoids are potent anticancer agents, but their therapeutic efficacy or mechanism of action for prostate cancer has not been investigated. The goal of this study was to determine the antitumor activity and the mechanism of action of methyl-2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oate (CDDO-Me), a oleanane-derived synthetic triterpenoid for human prostate cancer cells.
Methods
The antitumor activity of CDDO-Me for hormone-refractory PC-3 (AR−) and C4-2 (AR+) prostate cancer cell lines was determined by effects on cell growth and induction of apoptosis, identification of molecular targets, and therapeutic efficacy in vivo in PC-3 xenograft model.
Results
CDDO-Me inhibited the growth and induced apoptosis in PC-3 and C4-2 cells at extremely low concentrations. The antitumor activity of CDDO-Me was associated with the inhibition of p-Akt, mammalian target of rapamycin (mTOR), and nuclear factor kappa B (NF-κB) signaling proteins and their downstream targets such as p-Bad and p-Foxo3a (Akt); p-S6K1, p-eIF-4E and p-4E-BP1 (mTOR); and COX-2, VEGF and cyclin D1(NF-κB). Silencing of Akt sensitized the PC-3 cells to CDDO-Me, whereas overexpression of Akt induced resistance to CDDO-Me. Targeted silencing of Akt showed that Akt does not regulate mTOR activation in PC-3 cells, but targeted silencing of mTOR sensitized PC-3 cells to CDDO-Me mediated growth inhibition. Further, treatment with CDDO-Me inhibited the growth of PC-3 xenografts in nude mice.
Conclusions
This study demonstrated potent antitumor activity of CDDO-Me against prostate cancer cells both in vitro and in vivo. Data also identified Akt and mTOR as molecular targets of CDDO-Me in prostate cancer cells.
Keywords: CDDO-Me, prostate cancer, apoptosis, Akt, mTOR, xenograft
Introduction
Carcinoma of the prostate (CaP) is the most commonly diagnosed cancer and the second leading cause of cancer related mortality in American males. Current therapies, such as radical prostatectomy, local radiotherapy or brachytherapy while successful for treating localized prostate cancer are of limited efficacy against the metastatic disease [1,2]. Androgen deprivation therapy produces objective responses; however, responses are usually temporary and eventually proceed to the hormone-refractory disease [3]. Because the incidence of CaP increases with advancing age, prostate cancer is expected to become an increasingly greater medical problem as life expectancy improves. Thus, clinical development of safe and effective chemopreventive agents for prostate cancer could significantly reduce disease-related morbidity and mortality and improve prognosis.
Triterpenes or triterpenoids are members of a larger family of structurally related compounds known as cyclosqualenoids that are widely distributed in the plant kingdom [4]. Oleanolic acid and ursolic acid are naturally occurring triterpenoids that have been used in traditional medicine for centuries as antibacterial, antifungal, anti-cancer, and anti-inflammatory agents [5–7]. Recently, synthetic derivatives of oleanolic acid have been tested for anti-inflammatory and anticancer activity. 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acid (CDDO), a synthetic oleanane triterpenoid and its C-28 methyl ester derivative methyl-2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oate (CDDO-Me) and C-28 imidazole derivative 1-(2-cyano-3,12-dioxooleana-1,9-dien-28-oyl) imidazole (CDDO-Im) exhibit stronger anti-inflammatory activity than natural oleanolic acid [8–10].
Synthetic CDDOs have been shown to inhibit the proliferation of diverse types of tumor cell lines in vitro [11–13]. Although the mechanisms of the anticancer effects of CDDOs are not fully understood, cancer cell differentiation and activation of caspase-dependent and independent apoptosis contribute to the antitumor activity of CDDOs [14–16]. In addition, CDDOs inhibit MAPK (Erk1/2) and NF-κB signaling, and modulate TGF-β/Smad and PPARγ signaling [17–19]. CDDOs have also exhibited the antitumor activity in vivo in several mouse model systems, including leukemia, melanoma, breast cancer, and lung cancer [20–22]. The in vivo efficacy of CDDOs for prostate cancer has not been investigated.
We have previously shown that CDDO-Me inhibits the growth of hormone-sensitive (LNCaP) and hormone-refractory (PC-3 and DU145) human prostate cancer cell lines in vitro by inducing apoptosis through activation of caspases 3, 8, and 9, and causing mitochondrial depolarization and the release of cytochrome c [23]. Furthermore, induction of apoptosis was associated with the inhibition of prosurvival Akt, NF-κB and mammalian target of rapamycin (mTOR) signaling proteins. However, the functional relevance of these molecular targets in mediating the antitumor effects of CDDOs has remained undetermined. In the present study, we investigated the role of Akt/mTOR signaling axis in response of prostate cancer cells to CDDO-Me. Our data demonstrate that CDDO-Me independently targets Akt and mTOR in inducing apoptosis and inhibiting the growth of prostate cancer cells in vitro. Furthermore, CDDO-Me potently inhibits the growth of prostate cancer xenografts in vivo.
Materials and Methods
Materials
CDDO-Me was obtained from the National Cancer Institute, Bethesda, MD through the Rapid Access to Intervention Development Program. A 100 mM stock solution of CDDO-Me was prepared in DMSO, which was subsequently diluted in tissue culture medium to obtain the working concentrations. Antibodies against p-Akt (ser473), NF-κB (p65), mTOR, p-mTOR (Ser2448), S6K1, p-S6K1 (thr421/ser424), 4E-BP1, p-4E-BP1 (thr37/46), p-eIF4E (ser209), p-Bad (ser136), p-Foxo3a (ser2531), VEGF, and cyclin D1 were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
Cell Lines
PC-3 human prostate cancer cell line was obtained from American Type Culture Collection (ATCC, Rock-ville, MD). PC-3 cells were grown in F-12K nutrient mixture (Gibco BRL, Rockville, MD) supplemented with 10% fetal calf serum, 1% penicillin/streptomycin, and 25 mM HEPES buffer. C4-2 cells, a subline of LNCaP that is androgen independent but expresses a functional androgen receptor (AR+), were obtained from Dr. Svend Freytag, Henry Ford Health System, and were grown in RPMI-1640 with 10% FBS. Both cell lines were cultured at 37°C in a humidified atmosphere consisting of 5% CO2 and 95% air, and maintained by subculturing cells twice a week.
Measurement of Cell Viability (MTS Assay)
2 × 104 cells in 100 μl of cell culture medium were seeded into each well of a 96-well plate. After incubation for 24 h, cells were treated with CDDO-Me for additional 72 h. Cell viability was then determined by the colorimetric MTS assay using CellTiter 96 AQueous One Solution Proliferation Assay System from Promega (Madison, WI). After incubation for 2 hr at 37°C, the absorbance, which is directly proportional to the number of viable cells in cultures, was measured at 490 nm using a microplate reader.
Annexin V-FITC Binding
Induction of apoptosis was assessed by the binding of annexin V to phosphotidylserine, which is externalized to the outer leaflet of the plasma membrane early during induction of apoptosis. Briefly, PC-3 and C4-2 cells treated or not with CDDO-Me were resuspended in the binding buffer provided in the annexin V-FITC apoptosis detection kit II (BD Biosciences, Pharmingen). Five microliters of annexin V-FITC reagent and 5 μl of propidium iodide solution (PI) was added to cell suspension and then incubated for 30 min at room temperature in the dark. Stained cells were analyzed by flow cytometry using a FACScan flow cytometer (Becton Dickinson).
Isolation of Nuclear Proteins
Following treatment with CCDO-Me for 24 hr, cells were washed with PBS and incubated on ice for 15 min in hypotonic buffer A (10 mM HEPES, pH 7.9, 10 mM KCl,0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 0.5 mM PMSF, and 0.6% NP40). Cells were vortexed gently for lysis and nuclei were separated from the cytosol by centrifugation at 12,000g for 1 min. Nuclei were resuspended in buffer C (20 mM HEPES, pH 7.9, 25% glycerol, 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 0.5 mM PMSF) and shaken for 30 min at 4°C. Nuclear extracts were obtained by centrifugation at 12,000g and protein concentration measured by Bradford assay (Bio-Rad, Richmond, CA). NF-κB in nuclear extracts was detected by Western blotting as described below.
Western Blotting
Cellular lysates were prepared by detergent lysis (1% Triton-X 100 (v/v), 10 mM Tris-HCl (pH 7.5), 5 mM EDTA, 150 mM NaCl, 10% glycerol, 2 mM sodium vanadate, 5 μg/ml leupeptin, 1 μg/ml aprotinin, 1 μg/ml pepstatinin, and 10 μg/ml 4-2-aminoethyl-benzenesulfinyl fluoride). Lysates were clarified by centrifugation at 14,000g for 10 min at 4°C, and protein concentrations were determined by Bradford assay. Samples (50 μg) were boiled in an equal volume of sample buffer (20% glycerol, 4% SDS, 0.2% Bromo-phenol Blue, 125 mM Tris-HCl (pH 7.5), and 640 mM 2-mercaptoethanol) and separated on SDS-polyacry-lamide gels. Proteins resolved on the gels were transferred to nitrocellulose membranes. Membranes were blocked with 5% milk in 10 mM Tris-HCl (pH 8.0), 150 mM NaCl with 0.05% Tween 20 (TPBS) and probed using protein specific antibodies to p-Akt (ser473), p-mTOR (ser2448), NF-κB (p65), p-Bad (ser136), p-Foxo3a (ser2531), p-S6K1 (thr421/ser424), p-eIF-4E (ser209) and p-4E-BP1 (thr37/46), COX-2, VEGF, cyclin D1 or β-actin (loading control) and HRP-conjugated secondary antibody. Immune complexes were visualized with enhanced chemiluminescence (ECL) detection system.
DNA Transfection
For overexpression of Akt, semi-confluent cultures of PC-3 cells in 60 mm2 cell culture dishes were transfected with 10 μg of empty or Akt expression vector (pUSEamp) DNA containing Myc-His tagged mouse Akt1 (activated) under the control of CMV promoter (Upstate Cell Signaling, Lake Placid, NY) using LipofectAMINE Plus reagent. After incubation for 24 hr, cells were analyzed for the expression of exogenous Akt1/PKBα by immunoblotting using anti-Myc Tag antibody.
For silencing of Akt or mTOR, PC-3 cells were transfected with double stranded siRNA of Akt or mTOR using SignalSilence siRNA kits from Cell Signaling Technology (Beverly, MA). Briefly, 106 cancer cells were plated in 60 mm Petri dish for 24 hr and treated with 3 ml of transfection medium containing 20 μg LipofectAMINE and 100 nM siRNA for 24 hr. Gene silencing in transfected cells was confirmed by Western blotting.
In Vivo Antitumor Activity of CDDO-Me
The in vivo antitumor activity of CDDO-Me was tested in PC-3 xenograft model. 2 × 106 PC-3 cells in 50 μl of medium mixed with 50 μl of matrigel (BD Biosciences) were injected s.c. in the flank of Balb/c nude mice. On day 8, tumor dimensions (length, width and height) were measured with a caliper and CDDO-Me treatment started. In treatment group, mice (n = 5) were gavaged daily with 10 μmol/kg CDDO-Me in 0.1 ml of vehicle consisting of cremaphor EL:DMSO:PBS (1:1:8), 5 days a week for 5 weeks. In control group, mice (n = 5) were gavaged with vehicle without CDDO-Me. Tumor dimensions were measured at 4-day intervals and tumor volume calculated by the formula: length × width × height × 0.5236 mm3.
Statistical Analysis
Data are presented as means ± SD. The differences between control and treatment groups were analyzed using Dunnett multiple comparison test and differences with P<0.05 were considered statistically significant.
Results
Effect of CDDO-Me on the Growth of PC-3 and C4-2 Prostate Cancer Cells
To determine the effect of CDDO-Me on the growth of prostate cancer cells, hormone-refractory PC-3 (AR−) or C4-2 (AR+) cells were treated with CDDO-Me at concentrations ranging from 0.625 to 10 μM for 72 hr and cell viability determined by MTS assay. As shown in Figure 1A, insignificant reduction in viability was observed at 0.62 μM CDDO-Me. However, significant reduction in viability was caused at 1.25 μM (PC-3,68%; C4-2, 73%), which further increased to 77% and 88% reduction in viability at 10 μM CDDO-Me. In contrast to prostate cancer cell lines, non-transformed prostate epithelial cell line BPH1, a fibroblast cell line and normal splenic lymphocytes were significantly more resistant to CDDO-Me. For instance, there was no reduction in the growth of BPH1 cells at 1.25 mM CDDO-Me and only about 27% reduction at 2.5 mM compared to more than 75% inhibition of tumor cell growth. Even at higher concentrations of CDDO-Me (e.g.,5–10mM) growth inhibition of BPH1 cells (44–49%) was significantly less than that of tumor cells (80–88%). Fibroblasts were resistant to CDDO-Me up to 10 mM. Similarly, splenic lymphocytes were also resistant to CDDO-Me with only 24% reduction in viable cells at 10 mM CDDO-Me.
Fig. 1.
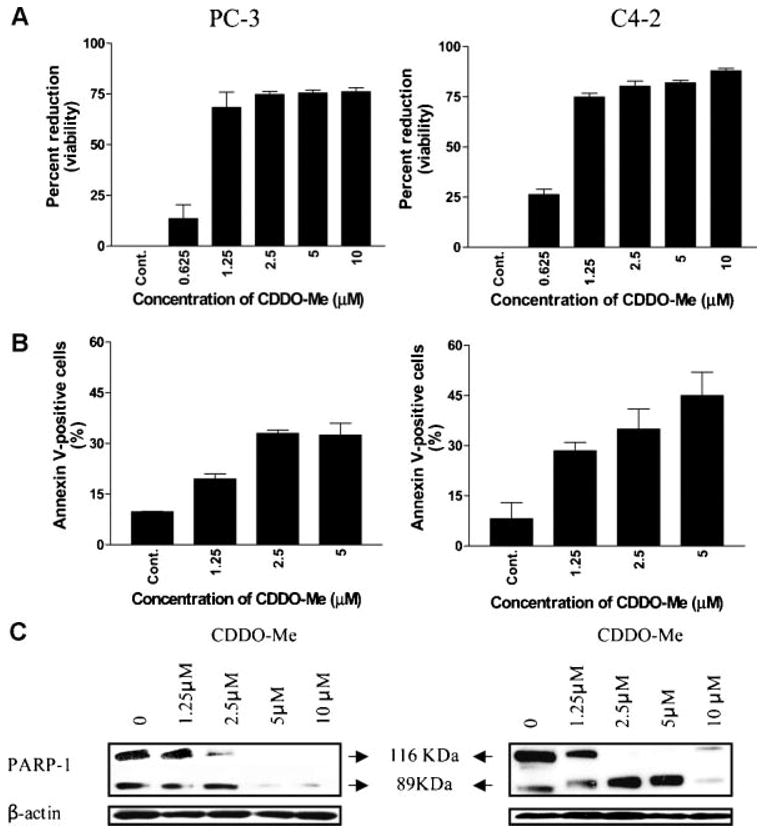
A:CDDO-Meinhibits the growth of prostate cancer cells. 2 × 104 PC-3orC4-2 cells were seeded in each well of amicrotiter plate in 0.1 ml of culture medium. Cells were allowed to adhere for 24 hr before treating with CDDO-Me at concentrations ranging from 0 to 10 μM for72hr. Cell viability was measured by MTS assay using CellTiter AQueous assay system from Promega. Data are presented as Mean±SD percent reduction in viability obtained in three independent experiments. B: Treatment with CDDO-Me increases binding of annexin V-FITC. PC-3 or C4-2 cells were treated with CDDO-Me at concentrations of 1.25-5 μM for 24 hr. Cells were then incubated with 5 μl of annexin V-FITC reagent and 5 μl of PI for 30 min at room temperature. Annexin V-FITC-binding was assessed by flow cytometry. Results are presented as percentage of annexin V-FITC positive cells. Bar graph represents mean±SD of three separate experiments. C: CDDO-Me cleaves PARP-1 in prostate cancer cells. PC-3 or C4-2 cells were treated with CDDO-Me at concentrations of 1.25-10 μM for24hr. Cellular lysate prepared from untreated and treated cells was analyzed for the cleavage of native PARP-1by Western blotting as described in Materials and Methods Section. Immunoblots showing native (116 kDa) and cleaved (89 kDa) PARP-1 protein bands are presented. Similar results were obtained in three independent experiments.
CDDO-Me Induces Apoptosis in Prostate Cancer Cells
Whether growth inhibition of prostate cancer cells by CDDO-Me was due to the induction of apoptosis was investigated next. Induction of apoptosis was determined by the binding of annexin V-FITC to PC-3 and C4-2 cells treated with CDDO-Me. As shown in Figure 1B, a small percentage of untreated PC-3 and C2-4 cells bound annexin V-FITC (7% and 5%, respectively). In contrast, the percentage of annexin V-FITC binding PC-3 and C4-2 cells markedly increased after treatment with CDDO-Me at concentrations of 1.25, 2.5, and 5 μM for 24 hr (PC-3, 39%, 33% and 34%; C4-2, 29%, 35% and 45%, respectively).
In another measure to confirm induction of apoptosis, we evaluated the cleavage of PARP-1 in cells treated with CDDO-Me at 1–25 to 10 μM for 24 hr by Western blotting. Figure 1C clearly demonstrates the cleavage of PARP-1 by CDDO-Me in both cell lines as identified by the disappearance of the 116 kDa native protein and the presence of an 89 kDa cleaved PARP-1 fragment at concentrations of 1.25–10 mM. Thus, increase in annexin V-FITC-binding and cleavage of PARP-1 indicate that reduction in the viability of PC-3 and C4-2 cells by CDDO-Me is caused by induction of apoptosis.
CDDO-Me Inhibits Akt, mTOR, and NF-κB Signaling Proteins
Akt/mTOR and Akt/NF-κB are major antiapoptotic pathways that confer survival advantage and resistance of cancer cells to various forms of anticancer therapies. We investigated whether induction of apoptosis in prostate cancer cells by CDDO-Me involved the inhibition of Akt, mTOR and NF-κB and downstream effectors regulated by these signaling molecules. For this, cellular lysate or nuclear extract of PC-3 and C4-2 cells treated with CDDO-Me (1.25–10 μM) for 24 hr was analyzed for p-Akt and p-mTOR (cellular lysate) and NF-κB (nuclear extract), respectively. CDDO-Me significantly reduced the levels of p-Akt, p-mTOR and NF-κBat1.25 μM and almost completely inhibited these signaling proteins at 2.5 μM and above in both cell lines (Fig. 2).
Fig. 2.

CDDO-Me inhibits Akt, mTOR and NF-κB and the expression of downstream intermediaries. PC-3 and C4-2 cells were treated with CDDO-Me at concentrations of 1.25–10 μM for 24 hr. After treatment, cell lysates and nuclear extracts were prepared and analyzed for p-Akt, p-mTOR, and NF-κB (p65) by Western blotting. Cell lysates were also analyzed for downstream targets of p-Akt (p-Bad, p-Foxo3a), p-mTOR (p-S6K1, p-eIF-4E, p-4E-BP1) and NF-κB (COX-2, VEGF, cyclin D1). The uniformity of sample loading was determined by anti-β-actin antibody. Similar results were obtained in three independent experiments.
Further analysis of cells treated with CDDO-Me showed that it also inhibits the major downstream intermediary targets of Akt, mTOR and NF-κB. For instance, CDDO-Me inhibited p-Bad and p-Foxo-3a, the downstream targets of activated Akt involved in apoptosis, at concentrations of 1.25–2.5 μM in both cell lines (Fig. 2). Similarly, the levels of mTOR pathway mediators such as p-S6K1, p-eIF-4E, and p-4E-BP-1 were either reduced or completely inhibited by CDDO-Me in a concentration dependent manner. The levels of COX-2, VEGF and cyclin D1 which are transcriptionally regulated by NF-κB were markedly reduced or completely inhibited by CDDO-Me at concentrations of 1.25–5 μM. Together, these data suggest that the inhibition of p-Akt, p-mTOR and NF-μB might be necessary for induction of apoptosis by CDDO-Mein prostate cancer cells.
Akt Is a Target of CDDO-Me in PC-3 Cells
Since CDDO-Me inhibited both p-Akt and p-mTOR in PC-3 cells we determined whether Akt/mTOR signaling axis regulates the response of PC-3 cells to CDDO-Me. First, we evaluated whether modulation of Akt would alter the response of cancer cells to CDDO-Me. For this, PC-3 cells were transfected with either siRNA-Akt or an Akt expression plasmid (myr-Akt1-pUSE) to decrease or increase the levels of Akt. The response of PC-3 cells transfected with siRNA-Akt was then tested at CDDO-Me concentrations that were in active in MTS assay (e.g.,0.312 or 0.625 μM).As shown in Figure 3A, control PC-3 cells were unresponsive to CDDO-Me at 0.312 μM with a small reduction in viability occurring at 0.625 μM. On the other hand, PC-3 cells in which Akt has been silenced by transfection with siRNA-Akt (inset) showed increased susceptibility to CDDO-Me at 0.312 or 0.625 μM (28% and 49% reduction in viability, respectively). On the other hand, transfection with a non-targeting siRNA did not sensitize PC-3 cells to CDDO-Me (not shown). In contrast, overexpression of Akt in cells transfected with Akt1 expression plasmid (Fig. 3B) markedly reduced the susceptibility to CDDO-Me at concentrations of 1.25, 2.5, and 5 μM, (52%, 59%, and 63% reduction, respectively).
Fig. 3.

Akt regulates the response of prostate cancer cells to CDDO-Me. To determine the functional relevance of Akt in response to CDDO-Me, the Akt expression was either inhibited or increased by transfecting PC-3 cells with siRNA-Akt (A) or Aktl expression plasmid (myr-Aktl-pUSE,B) using Lipofect AMINE Plus reagent. After confirming the change in Akt expression, the susceptibility of transfected cells to CDDO-Me was measured in MTS assay. PC-3 cells transfected with siRNA-Akt were treated with CDDO-Me at concentrations of 0.312-0.625 μM, which are ordinarily inactive. Cells transfected with Aktl expression plasmid were treated with CDDO-Me at concentrations of 0.625-5 μM. Bargraphs (A,B) represent mean ± SD of two separate experiments.
Akt Does Not Regulate mTOR Activationin PC-3 Cells
In many tumor model systems, Akt has been shown to regulate the activation of mTOR [24,25]. In order to determine whether Akt regulation of response to CDDO-Me is mediated through mTOR, we first examined whether Akt regulates mTOR activation in PC-3 cells. For this, PC-3 cells were treated with SH-6, an inhibitor of p-Akt or transfected with siRNA-Akt and levels of p-Akt and p-mTOR were analyzed by Western blotting. As shown in Figure 4A, treatment with SH-6 not only inhibited p-Akt but also inhibited p-mTOR, suggesting that Akt regulates the expression of mTOR in PC-3 cells. However, targeted inhibition of Akt through genetic ablation with siRNA-Akt resulted in no decrease in the levels of p-mTOR (Fig. 4B), arguing that mTOR expression in prostate cancer cells is not regulated by Akt. The inhibition of p-mTOR by Akt inhibitor SH-6 probably resulted from non-Akt associated effects of SH-6 on PC-3 cells.
Fig. 4.

Effect of Akt inhibition on p-mTOR expression in PC-3 cells. A:PC-3 cells were treated with Akt inhibitor SH-6 at concentrations of 5,10, or 20 mM for 24 hr. Cell lysates were analyzed for levels of p-Aktandp-mTOR. B:PC-3 cells were transfected with siRNA-Akt for 48 hr using Lipofect AMINE Plus reagent. Cell lysates were analyzed for the expression of p-Akt, p-mTOR and β-actin (loading control).
Silencing of mTOR With siRNA-mTOR Sensitizes PC-3 Cells to CDDO-Me
The functional relevance of inhibition of p-mTOR in the antitumor activity of CDDO-Me in PC-3 cells was investigated next. PC-3 cells were transfected with siRNA-mTOR and response to inactive concentrations of CDDO-Me was measured. As shown in Figure 5A, transfection with siRNA-mTOR inhibited p-mTOR expression in PC-3 cells without a significant change in p-Akt level. Further, the inhibition of p-mTOR increased the susceptibility of PC-3 cells to CDDO-Me. Control PC-3 cells were unresponsive to CDDO-Me at concentrations of 0.312 and 0.625 μM, but silencing of mTOR by siRNA-mTOR sensitized them to CDDO-Me (27% and 48% growth inhibition). Transfection with a non-targeting siRNA had no effect on the response of PC-3 cells to CDDO-Me. This result indicated that mTOR is a target of CDDO-Me in killing of prostate cancer cells.
Fig. 5.
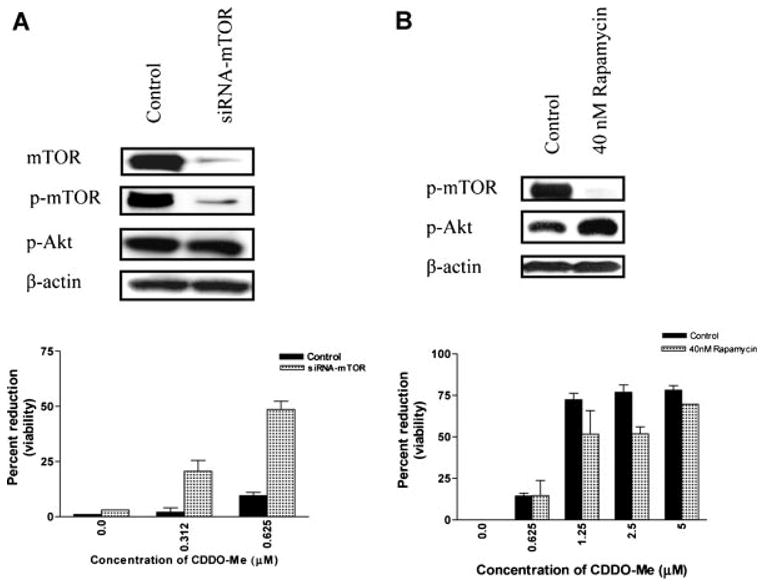
Transfection with siRNA-mTOR but not the treatment with rapamycin sensitizes PC-3 cells to CDDO-Me. A: PC-3 cells were trans-fected with siRNA-mTOR (100 nM) using Lipofect AMINE Plus reagent. After 24 hr, inhibition of p-mTOR expression was confirmed by Western blotting. Transfected or control cells were treated with inactive concentrations of CDDO-Me (0.312 and 0.625 μM) for 48 hr and viability measured by MTS assay. Bar graph represents mean ± SD of two separate experiments. B: PC-3 cells were treated with rapamycin (40 nM) for 24 hr and the inhibition of p-mTOR was confirmed by Western blotting. PC-3 cells treated with rapamycin were treated with CDDO-Me (0.625-5 μM) for 48 hr and viability determined by MTS assay.
To further analyze the role of mTOR in response to CDDO-Me, PC-3 cells were pre-treated with rapamycin for 24 hr before applying CDDO-Me (0.625–5μM) for 48 hr. As shown in Figure 5B, treatment with rapamycin inhibited p-mTOR but it increased the level of p-Akt. Further, in contrast to the sensitization of PC-3 cells following silencing of mTOR with siRNA-mTOR, rapamycin failed to sensitize PC-3 cells to CDDO-Me. In fact, PC-3 cells pretreated with rapamycin were measurably less sensitive to CDDO-Me compared to control cells.
CDDO-Me Inhibits the Growth of PC-3 Xenografts
Whether CDDO-Me inhibits tumor growth in vivo was examined in PC-3 xenograft model system. Figure 6 compares tumor growth progression in vehicle control and CDDO-Me treated animals. Tumors in vehicle control group progressively increased in size over a period of 6 weeks and one mouse was terminated on day 39 because of large tumor size. In the CDDO-Me treatment group, after an initial increase in tumor size for about 2 weeks, tumors began to shrink and decreased in size. After 5 weeks of treatment with CDDO-Me, the average tumor size was significantly less than the size on the day treatment was started (P < 0.01). On day 50, the experiment was terminated and tumors were harvested and weighed. Tumors of mice treated with CDDO-Me weighed significantly less than those of the untreated mice (untreated = 524.5 ± 35 mg; treated = 85.8 ± 28 mg; P < 0.001). These data demonstrated significant antitumor activity of CDDO-Me in vivo. Furthermore, dosing of animals at 10 μmol/kg CDDO-Me, 5 days a week for 5 weeks was tolerated well without significant change in body weight or peripheral blood cell count.
Fig. 6.

CDDO-Me inhibits the growth of PC-3 xenografts. Balb/c nude mice were injected with 2 × 106 PC-3 cells s.c. in matrix gel (50%) in the right flank. On day 8, mice with measurable tumors were randomly assigned to two groups: vehicle control (n = 5) and CDDO-Metreatment (n = 5). Mice were treated with CDDO-Meat a dose of 10μmol/kg, 5 days a week for 5 weeks. Tumor dimensions were measured at 4-day intervals and tumor volumes estimated. Data are presented as mean tumor volume±SD CDDO-Me significantly inhibited the growth of PC-3 xenograft (P < 0.01).
Discussion
Malfunction of apoptosis has been implicated in tumor development and resistance to cancer therapies [26]. Thus, promotion of apoptosis in prostate cancer cells could potentially lead to the regression and improved prognosis of the refractory disease. Our present study demonstrated that CDDO-Me, a synthetic triter-penoid derived from oleanolic acid is a potent inducer of apoptosis in hormone-refractory prostate cancer cells. Killing of prostate cancer cells by CDDO-Me was associated with significant increase in annexin-V binding and cleavage of PARP-1, indicating that apoptosis is part of the mechanism by which CDDO-Me inhibits the growth of prostate cancer cells. Furthermore, the inhibition of prosurvival/progrowth signaling proteins such as p-Akt, p-mTOR and NF-κB suggested that Akt, mTOR and NF-κB are potential targets of CDDO-Me.
In a previous report we demonstrated that among the three synthetic CDDOs (e.g., CDDO, CDDO-Im, and CDDO-Me), CDDO-Me was most active against prostate cancer cells regardless of their hormonal status [23]. Data in this report demonstrate that the response of prostate cancer cells to CDDO-Me may also be independent of androgen receptor (AR) expression. Hormone-refractory PC-3 and C4-2 differ in their expression of AR. PC-3 cells are AR− and C4-2 cells are AR+; however, both were equally sensitive to CDDO-Me. Thus, CDDO-me is active against both hormone-refractory AR− and AR+ prostate cancer cells.
PI3K/Akt/mTOR and Akt/NF-κB are major antiapoptotic/prosurvival pathways that control cell proliferation, survival and malignant transformation [27,28]. Akt, mTOR and NF-κB are frequently hyper-activated in most cancers [29-31]. Activated p-Akt promotes cell growth and survival by inactivating downstream substrates such as Bad, procaspase-9, and Forkhead transcription factors [32,33]. NF-κB family of transcription factors controls the expression of numerous genes involved in immune and inflammatory responses, cell proliferation, oncogenesis, angiogenesis, and apoptosis [28,30]. mTOR is a 290 kDa serine-threonine kinase, which controls cell growth, survival, division and motility [34,35]. Our results demonstrate that PC-3 and C4-2 cells express p-Akt, p-mTOR and NF-κB, and their expression is inhibited by CDDO-Me. CDDO-Me also reduced/inhibited the downstream targets of Akt (p-Bad, Foxo3a), mTOR (p-S6K1, p-eIF-4E, p-4E-BP1) and NF-κB (COX-2, VEGF, cyclin D1) in both cell lines. Thus, each of the three major growth promoting/survival pathways and their downstream intermediates were inhibited by CDDO-Me in prostate cancer cells, suggesting that the inhibition of these antiapoptotic signaling proteins is critical for induction of apoptosis by CDDO-Me.
The role of Akt and NF-kB tumor cell survival and resistance to anticancer therapies through inhibition of apoptosis has been well established in a number of tumor model systems [36,37], but it is less well understood in case of mTOR. In the presentstudy, wefocused on the role of Akt/mTOR signaling axis in mediating the antitumor effects of CDDO-Me. Initially we evaluated the functional significance of inhibition of Akt in the growth inhibitory activity of CCDO-Me. Results of the transfection experiments to regulate the expression of Akt showed that inhibition of Akt with siRNA-Akt increased the sensitivity of PC-3 cells to CDDO-Me at concentrations which were otherwise inactive. In contrast, overexpression of Akt by transfection with an expression vector rendered PC-3 cells resistant to the active concentrations of CDDO-Me. These findings identified Akt as a target of CDDO-Me in inducing apoptosis in prostate cancer cells.
In multicellular organisms mTOR plays a major role in mediating the downstream effects of Akt, regulating protein synthesis, cell proliferation and apoptosis [24,25]. Akt activates mTOR either directly by phosphorylating it at Ser2448 or indirectly by inhibiting the tumor suppressor activity of tuberous sclerosis complex (TSC1/TSC2) through phosphorylation of TSC2 [38,39]. Since CDDO-Me inhibited p-Akt in prostate cancer cells, we determined whether inhibition of p-mTOR resulted because of the inhibition of Akt or independent of Akt. Pharmacological inhibition of p-Akt with SH-6 inhibited not only p-Akt but also p-mTOR. The inhibition of p-Akt was not due to the inhibition of basal Akt since SH-6 does not affect basal Akt expression as shown in a previous study [40]. The inhibition of p-mTOR by SH-6 indicated that Akt regulates mTOR activation in PC-3 cells. In contrast, however, specific silencing of Akt with siRNA-Akt blocked p-Akt without affecting p-mTOR, arguing that mTOR activation in PC-3 cells was not dependent on Akt. Perhaps, additional mechanisms such as energy or nutritional stress account for the activation of mTOR in PC-3 cells.
Since CDDO-Me inhibited p-mTOR, we investigated the relevance of mTOR inhibition in mediating the antitumor activity of CDDO-Me. Selective inhibition of mTOR with siRNA-mTOR increased the sensitivity of PC-3 cells to CDDO-Me. These data demonstrated that mTOR is a functionally relevant molecular target of CDDO-Me that mediates the apoptosis-inducing effects of CDDO-Me independent of Akt. In contrast to the sensitizing effect of siRNA-mTOR, treatment with rapamycin, a pharmacological inhibitor mTOR, failed to sensitize PC-3 cells. The discrepancy in the effect of mTOR inhibition by siRNA and rapamycin on the sensitization of PC-3 cells to CDDO-Me was attributed to the levels of p-Akt. The inhibition of mTOR by siRNA-mTOR did not significantly alter p-Akt expression, whereas the inhibition of mTOR with rapamycin increased p-Akt level three-fold. The latter finding is consistent with the results of other studies showing that in some tumor model systems, the inhibition of mTOR with rapamycin activates PI3K/Akt pathway through a feedback loop resulting in increased p-Akt expression [41]. Furthermore, since knockdown of Akt or mTOR only partially sensitized PC-3 cells to CDDO-Me, indicated that Akt and mTOR are independent targets of CDDO-Me.
The antitumor activity of CDDO-Me was not just an in vitro effect on prostate cancer cells but was also observed in vivo. Treatment with CDDO-Me inhibited the growth of PC-3 xenografts. In fact, in a small fraction of mice, tumors were completely eradicated after treatment with CDDO-Me for 5 weeks. Thus, inhibition of growth of prostate cancer cells in vitro and in vivo demonstrates the therapeutic potential of CDDO-Me for prostate cancer. Our studies have shown that Akt and mTOR are major targets of CDDO-Me for suppression of growth and induction of apoptosis in vitro. We have also established that PC-3 xenografts maintain expression of p-Akt, p-mTOR and NF-κB in tumor tissue. However, whether Akt and mTOR are also the molecular targets of CDDO-Me for tumor growth inhibition in vivo remains to be established.
Although rapamycin derivatives (RAD001, CCI-779, and AP23573) are in clinical trials for anticancer effects, the existence of the feedback loop in which mTOR inhibitors enhance the activation of PI3K/Akt survival pathway compromises the therapeutic benefit of mTOR specific inhibitors [41]. Since CDDO-Me independently targets Akt and mTOR, because of this dual anti-kinase activity CDDO-Me promises to be a more potent antineoplastic agent for prostate cancer and perhaps other cancers than mTOR specific inhibitors.
Supplementary Material
Acknowledgments
Authors gratefully acknowledge receiving C4-2 cells from Dr. Svend Freytag.
Grant sponsor: NIH; Grant number: 1R01 CA130948-01.
Footnotes
Additional supporting information may be found in the online version of this article.
References
- 1.Garnick MB. Hormonal therapy in the management of prostate cancer: From Higgins to the present. Urology. 1997;49:5–15. doi: 10.1016/s0090-4295(97)00163-5. [DOI] [PubMed] [Google Scholar]
- 2.Hanks GE. Long-term control of prostate cancer with radiation. Urol Clin North Am. 1996;23:605–616. doi: 10.1016/s0094-0143(05)70339-6. [DOI] [PubMed] [Google Scholar]
- 3.Crawford ED, Rosenblum M, Ziada AM, Lange PH. Overview: Hormone refractory prostate cancer. Urology. 1999;54:1–7. doi: 10.1016/s0090-4295(99)00447-1. [DOI] [PubMed] [Google Scholar]
- 4.Dzubak P, Hajduch M, Vydra D, Hustova A, Kvasnica M, Biedermann D, Markova L, Urban M, Sarek J. Pharmacological activities of natural triterpenoids and therapeutic implications. Nat Prod Rep. 2006;23:394–411. doi: 10.1039/b515312n. [DOI] [PubMed] [Google Scholar]
- 5.Huang MT, Ho CT, Wang ZY, Ferraro T, Lou YR, Stauber K, Ma W, Georgiadis C, Laskin JD, Conney AH. Inhibition of skin tumorigenesis by rosemary and its constituents carnosol and ursolic acid. Cancer Res. 1994;54:701–707. [PubMed] [Google Scholar]
- 6.Nishino H, Nishino A, Takayasu J, Hasegawa T, Iwashima A, Hirabayashi K, Iwata S, Shibata S. Inhibition of the tumor-promoting action of 12-O-tetradecanoylphorbol-13-accetate by some oleanane-type triterpenoid compounds. Cancer Res. 1988;48:5210–5215. [PubMed] [Google Scholar]
- 7.Ryu SY, Oak MH, Yoon SK, Cho DI, Yoo GS, Kim TS, Kim KM. Anti-allergic and anti-inflammatory triterpenes from the herb of Prunella vulgaris. Planta Med. 2000;66:358–360. doi: 10.1055/s-2000-8531. [DOI] [PubMed] [Google Scholar]
- 8.Honda T, Rounds BV, Gribble GW, Suh N, Wang Y, Sporn MB. Design and synthesis of 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, anovel and highly active inhibitor of nitric oxide production in mouse macrophages. Biorg Med Chem Lett. 1998;8:2711–2714. doi: 10.1016/s0960-894x(98)00479-x. [DOI] [PubMed] [Google Scholar]
- 9.Suh N, Honda T, Finlay HJ, Barchowsky A, Williams C, Benoit NE, Xie QW, Nathan C, Gribble GW, Sporn MB. Triterpenoids suppress inducible nitric oxide synthase (iNOS) and inducible cyclooxigenase (COX-2) in mouse macrophages. Cancer Res. 1998;58:717–723. [PubMed] [Google Scholar]
- 10.Honda T, Rounds BV, Bore L, Favaloro FG, Gibble GW, Suh N, Wang Y, Sporn MB. Novel synthetic oleanane triterpenoids: A series of highly active inhibitors of nitric oxide production in mouse macrophages. Bioorg Chem Lett. 1999;9:3429–3434. doi: 10.1016/s0960-894x(99)00623-x. [DOI] [PubMed] [Google Scholar]
- 11.Gao X, Deeb D, Jiang H, Liu Y, Dulchavsky S, Gautam S. Synthetic triterpenoids inhibit growth and induce apoptosis in human glioblastoma and neuroblastoma cells through inhibition of prosurvival Akt, NF-κB and Notch1 signaling. J Neurooncol. 2007;84:147–157. doi: 10.1007/s11060-007-9364-9. [DOI] [PubMed] [Google Scholar]
- 12.Konopleva M, Tsao T, Ruvolo P, Stiouf I, Estrov Z, Leysath CE, Zhao S, Harris D, Chang S, Jackson CE, Munsell M, Suh N, Gribble G, Honda T, May WS, Sporn MB, Andreeff M. Novel triterpenoid CDDO-Me is a potent inducer of apoptosis and differentiation in acute myelogenous leukemia. Blood. 2002;99:326–335. doi: 10.1182/blood.v99.1.326. [DOI] [PubMed] [Google Scholar]
- 13.Shishodia S, Sethi G, Konopleva M, Andreeff M, Aggarwal BB. A synthetic triterpenoid, CDDO-Me, inhibits IkappaBalpha kinase and enhances apoptosis induced by TNF and chemother-apeutic agents through down-regulation of expression of nuclear factor kappaB-regulated gene products in human leukemic cells. Clin Cancer Res. 2006;12:1828–1838. doi: 10.1158/1078-0432.CCR-05-2044. [DOI] [PubMed] [Google Scholar]
- 14.Ikeda T, Sporn M, Honda T, Gribble GW, Kufe D. The novel triterpenoid CDDO and its derivatives induce apoptosis by disruption of intracellular redox balance. Cancer Res. 2003;63:5551–5558. [PubMed] [Google Scholar]
- 15.Konopleva M, Tsao T, Estrov Z, Lee RM, Wang RY, Jackson CE, McQueen T, Monaco G, Munsell M, Belmont J, Kantarjian H, Sporn MB, Andreeff M. The synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid induces caspase-dependent and -independent apoptosis in acute myelogenous leukemia. Cancer Res. 2004;64:7927–7935. doi: 10.1158/0008-5472.CAN-03-2402. [DOI] [PubMed] [Google Scholar]
- 16.Ito Y, Pandey P, Sporn MB, Datta R, Kharbanda S, Kufe D. The novel triterpenoid CDDO induces apoptosis and differentiation of human osteosarcoma cells by a caspase-8 dependent mechanism. Mol Pharmacol. 2001;59:1094–1099. doi: 10.1124/mol.59.5.1094. [DOI] [PubMed] [Google Scholar]
- 17.Konopleva M, Contractor R, Kurinna SM, Chen W, Andreeff M, Ruvolo PP. The novel triterpenoid CDDO-Me suppresses MAPK pathways and promotes p38 activation in acute myeloid leukemia cells. Leukemia. 2005;19:1350–1354. doi: 10.1038/sj.leu.2403828. [DOI] [PubMed] [Google Scholar]
- 18.Suh N, Roberts AB, Birkey Reffey S, Miyazono K, Itoh S, ten Dijke P, Heiss EH, Place AE, Risingsong R, Williams CR, Honda T, Gribble GW, Sporn MB. Synthetic triterpenoids enhance transforming growth factor beta/Smad signaling. Cancer Res. 2003;63:1371–1376. [PubMed] [Google Scholar]
- 19.Chintharlapalli S, Papineni S, Konopleva M, Andreef M, Samudio I, Safe S. 2-Cyano-3,12-dioxoolean-1,9-dien-28-oic acid and related compounds inhibit growth of colon cancer cells through peroxisome proliferator-activated receptor gamma-dependent and -independent pathways. Mol Pharmacol. 2005;68:119–128. doi: 10.1124/mol.105.011437. [DOI] [PubMed] [Google Scholar]
- 20.Place AE, Suh N, Williams CR, Risingsong R, Honda T, Honda Y, Gribble GW, Leesnitzer LM, Stimmel JB, Willson TM, Rosen E, Sporn MB. The novel synthetic triterpenoid, CDDO-imidazolide, inhibits inflammatory response and tumor growth in vivo. Clin Cancer Res. 2003;9:2798–2806. [PubMed] [Google Scholar]
- 21.Xiaoyang L, Konopleva M, Zeng Z, Ruvolo V, Stephens LC, Schober W, McQueen T, Dietrich M, Madden TL, Andreeff M. The novel triterpenoid C-28 methyl ester of 2-cyano-3, 12-dioxoolen-1, 9-dien-28-oic acid inhibits metastatic murine breast tumor growth through inactivation of STAT3 signaling. Cancer Res. 2007;67:4210–4218. doi: 10.1158/0008-5472.CAN-06-3629. [DOI] [PubMed] [Google Scholar]
- 22.Liby K, Royce DB, Williams CR, Risingsong R, Yore MM, Honda T, Gribble GW, Dmitrovsky E, Sporn TA, Sporan MB. The synthetic triterpenoid CDDO-methyl ester and CDDO-amide prevent lung cancer induced by vinyl carbomate in A/J mice. Cancer Res. 2007;67:2414–2419. doi: 10.1158/0008-5472.CAN-06-4534. [DOI] [PubMed] [Google Scholar]
- 23.Deeb D, Gao X, Dulchavsky SA, Gautam SC. CDDO-Me induces apoptosis and inhibits Akt, mTOR and NF-κB in prostate cancer cells. Anticancer Res. 2007;27:3035–3044. [PubMed] [Google Scholar]
- 24.Sekulic A, Hudson CC, Homme JL, Yin P, Otterness DM, Karnitz LM, Abraham RT. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res. 2000;60:3504–3513. [PubMed] [Google Scholar]
- 25.Hay N, Sonenberg N. Upstream and downstream of mTOR. Gene Dev. 2006;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 26.Kerr JF, Winterford CM, Harmon BV. Apoptosis. Its significance in cancer and cancer therapy. Cancer. 1994;73:2013–2026. doi: 10.1002/1097-0142(19940415)73:8<2013::aid-cncr2820730802>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 27.Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 28.Mayo MW, Baldwin AS. The transcription factor NF-κB: Control of oncogenesis and cancer therapy resistance. Biochim Biophys Acta. 2000;1470:M55–M62. doi: 10.1016/s0304-419x(00)00002-0. [DOI] [PubMed] [Google Scholar]
- 29.Altomare DA, Testa JR. Perturbations of the Akt signaling pathway in human cancer. Oncogene. 2005;24:7455–7464. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- 30.Karin M, Cao Y, Greten FR, Li ZW. NF-κB in cancer. From innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 31.Bjornsti MA, Houghton PJ. The TOR pathway; a target for cancer therapy. Nat Rev Cancer. 2004;4:335–348. doi: 10.1038/nrc1362. [DOI] [PubMed] [Google Scholar]
- 32.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of Bad couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 33.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 34.Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C, Blenis J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotictranslation initiation factor 4E. Mol Cell Biol. 2004;24:200–216. doi: 10.1128/MCB.24.1.200-216.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu L, Li F, Cardelli JA, Martin KA, Blenis J, Huang S. Rapamycin inhibits cell motility by suppression of mTOR-mediated S6K1 and 4E-BP1 pathway. Oncogene. 2006;25:7029–7040. doi: 10.1038/sj.onc.1209691. [DOI] [PubMed] [Google Scholar]
- 36.Weaver KD, Yeyeodu S, Cusack JC, Jr, Baldwin AS, Jr, Ewend MG. Potentiation of chemotherapeutic agents following antagonism of nuclear factor kappa B in human gliomas. J Neurooncol. 2003;61:187–196. doi: 10.1023/a:1022554824129. [DOI] [PubMed] [Google Scholar]
- 37.Cusack JC., Jr Overcoming antiapoptotic responses to promote chemosensitivity in metastatic colorectal cancer to the liver. Ann Surg Oncol. 2003;10:852–862. doi: 10.1245/aso.2003.07.518. [DOI] [PubMed] [Google Scholar]
- 38.Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005;15:702–713. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 39.Huang J, Manning BD. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem J. 2008;412:179–190. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deeb D, Jiang H, Gao X, Al-Holou S, Danyluk AL, Dulchavsky SA, Gautam SC. Curcumin [1,7-bis(4-hydroxy-3-methoxyphenyl)-1–6-heptadine-3,5-dione; C21H20O6] sensitizes human prostate cancer cells to tumor necrosis factor-related apoptosis-inducing ligand/Apo2L-induced apoptosis by suppressing nuclear factor-kappaB via inhibition of the prosurvival Akt signaling pathway. J Pharmacol Exp Ther. 2007;321:616–625. doi: 10.1124/jpet.106.117721. [DOI] [PubMed] [Google Scholar]
- 41.OReilly KE, Rojo F, She Q, Solit D, Mills D, Smith D, Lane H, Hofman F, Hicklin DJ, Lugwig DL, Baselga J, Rosen N. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.