

Abstract
During asymmetric stem cell division, both the daughter stem cell and the presumptive intermediate progenitor cell inherit cytoplasm from their parental stem cell. Thus, proper specification of intermediate progenitor cell identity requires an efficient mechanism to rapidly extinguish the activity of self-renewal factors, but the mechanisms remain unknown in most stem cell lineages. During asymmetric division of a type II neural stem cell (neuroblast) in the Drosophila larval brain, the Brain tumor (Brat) protein segregates unequally into the immature intermediate neural progenitor (INP), where it specifies INP identity by attenuating the function of the self-renewal factor Klumpfuss (Klu), but the mechanisms are not understood. Here, we report that Brat specifies INP identity through its N-terminal B-boxes via a novel mechanism that is independent of asymmetric protein segregation. Brat-mediated specification of INP identity is critically dependent on the function of the Wnt destruction complex, which attenuates the activity of β-catenin/Armadillo (Arm) in immature INPs. Aberrantly increasing Arm activity in immature INPs further exacerbates the defects in the specification of INP identity and enhances the supernumerary neuroblast mutant phenotype in brat mutant brains. By contrast, reducing Arm activity in immature INPs suppresses supernumerary neuroblast formation in brat mutant brains. Finally, reducing Arm activity also strongly suppresses supernumerary neuroblasts induced by overexpression of klu. Thus, the Brat-dependent mechanism extinguishes the function of the self-renewal factor Klu in the presumptive intermediate progenitor cell by attenuating Arm activity, balancing stem cell maintenance and progenitor cell specification.
Keywords: Apc2, Brain tumor, Drosophila, Wnt signaling, Intermediate progenitor cell, Neuroblast
INTRODUCTION
Tissue-specific stem cells often undergo asymmetric cell division to self-renew and to generate an intermediate progenitor cell, which possesses limited developmental potential and functions to generate differentiated cell types (Ming and Song, 2011; Pierfelice et al., 2011; Weng and Lee, 2011; Homem and Knoblich, 2012). Thus, an efficient mechanism to abrogate the activity of self-renewal factors in the presumptive intermediate progenitor cell is pivotal for the maintenance of stem cell homeostasis and the generation of the requisite number of differentiated progeny. Failure to attenuate the function of self-renewal factors may contribute to the creation of tumor-initiating cells (Krivtsov et al., 2006; Wei et al., 2008; Liu et al., 2011; Haenfler et al., 2012; Xiao et al., 2012; Schwitalla et al., 2013). Thus, understanding the mechanisms that extinguish the activity of self-renewal factors in the presumptive intermediate progenitor cell is likely to provide novel insight into both normal development and tumor initiation.
The type II neuroblast lineage in the fly larval brain provides an excellent model for investigating the mechanisms that regulate the proper specification of intermediate progenitor cell identity in vivo (Bayraktar et al., 2010; Weng et al., 2010; Song and Lu, 2011; Haenfler et al., 2012; Xiao et al., 2012). A type II neuroblast can be unambiguously identified by the presence of Deadpan (Dpn) and the absence of Asense (Ase) expression (Dpn+ Ase-) (Bowman et al., 2008). Type II neuroblasts undergo repeated asymmetric cell division to self-renew and to generate early stage immature intermediate neural progenitors (INPs) (Dpn- Ase-) that mature into late stage immature INPs (Dpn- Ase+) (Bello et al., 2008; Boone and Doe, 2008; Bowman et al., 2008; Bayraktar et al., 2010; Xiao et al., 2012). These late stage immature INPs acquire the functional identity of an INP (Dpn+ Ase+) and subsequently undergo limited rounds of asymmetric division to generate differentiated progeny. Thus, understanding the mechanisms that specify INP identity in immature INPs will provide insight into the proper specification of intermediate progenitor cell identity.
TRIM32 and TRIM3, which are vertebrate orthologs of Drosophila Brain tumor (Brat), have been shown to play important roles in regulating neural stem cells during normal brain development and brain tumor formation (Boulay et al., 2009; Schwamborn et al., 2009). Brat contains two B-boxes and a coiled-coil domain in the N-terminus and an NHL domain in the C-terminus (Arama et al., 2000). The NHL domain is essential for Brat function in various developmental processes, including repression of mRNA translation and regulation of microRNA (Sonoda and Wharton, 2001; Neumüller et al., 2008; Harris et al., 2011). In mitotic neuroblasts, the NHL domain mediates the binding of Brat to the scaffolding protein Miranda (Mira), which partitions Brat exclusively into the progenitor cell (Betschinger et al., 2006; Lee et al., 2006a). Most of the previously isolated brat mutant alleles carry mutations in the NHL domain and exhibit defects in the maturation of immature INPs, leading to the formation of supernumerary type II neuroblasts (Arama et al., 2000; Bowman et al., 2008; Xiao et al., 2012). However, it is unclear whether the NHL domain contributes to the specification of INP identity directly or indirectly by promoting Brat protein accumulation in immature INPs.
The activation of Wnt signaling plays crucial roles in both the regulation of stem cell self-renewal and the generation of differentiated cells (Merrill, 2012; Habib et al., 2013). In the absence of Wnt ligand, Wnt signaling is negatively regulated by the destruction complex, which includes the kinases GSK3β and CKI, the scaffolding protein Axin, and the tumor suppressor Adenomatous polyposis coli (Apc) (Aoki and Taketo, 2007; MacDonald et al., 2009; Niehrs, 2012). This destruction complex phosphorylates β-catenin/Armadillo (Arm) and targets it for proteosomal degradation. Binding of the Wnt ligand to the Frizzled receptor inactivates the destruction complex, leading to the accumulation and nuclear translocation of β-catenin/Arm, where it complexes with Tcf/LEF family transcription factors and activates Wnt target gene expression. Despite its essential roles in regulating stem cell self-renewal and, ultimately, the generation of differentiated cells, little is known about the role of Wnt signaling in the specification of progenitor cell identity. Previous studies revealed that Drosophila Apc2 is cortically enriched asymmetrically in larval brain neuroblasts, such that a portion of the Apc2 is partitioned into the progenitor cell (McCartney et al., 1999). Furthermore, Apc2 becomes enriched in the cortex of the progenitor cell following neuroblast asymmetric division, suggesting that Wnt signaling activity might be negatively regulated in the progenitor cell (Akong et al., 2002). However, the functional significance of Wnt signaling in the progenitor cell has never been established.
Here, we report that Brat specifies INP identity in immature INPs by downregulating Arm via a novel mechanism that is separable from the mechanism that regulates the asymmetric segregation of Brat. We identified that the B-boxes are dispensable for asymmetric partitioning of Brat into the presumptive immature INP but are necessary for Brat-dependent specification of INP identity. We further demonstrated that proper specification of INP identity by the Brat-mediated mechanism is critically dependent on Apc2, a key destruction complex component. Consistent with Apc2 negatively regulating Arm, reducing the function of the destruction complex or overexpressing constitutively active Arm enhances the supernumerary neuroblast phenotype in brat hypomorphic mutant brains. Furthermore, reducing arm function in immature INPs suppresses the supernumerary neuroblast phenotype in brat hypomorphic mutant brains, whereas increasing arm function in immature INPs strongly enhances the supernumerary neuroblast phenotype. These data strongly suggest that Brat specifies INP identity in immature INPs by attenuating the function of Arm through the destruction complex, thereby preventing the activation of Wnt target. Finally, removing arm function also strongly suppresses the supernumerary neuroblast phenotype induced by overexpression of the self-renewal factor Klumpfuss (Klu). Thus, Brat specifies INP identity in immature INPs by attenuating the function of the self-renewal factor Klu in part through extinguishing the transcriptional activity of Arm.
RESULTS
The B-boxes of Brat are dispensable for asymmetric protein segregation
Since Brat unequally partitions into the immature INP following the asymmetric division of neuroblasts, the domains required for the asymmetric segregation of Brat are also likely to function to promote the specification of INP identity. We identified the domains required for asymmetric segregation of Brat in mitotic neuroblasts in brat null mutant brains by overexpressing a series of UAS-brat transgenes inserted into an identical docking site in the fly genome (Fig. 1A). Brat always colocalized with Mira in the basal cortex of telophase neuroblasts and co-segregated with Mira exclusively into the future progenitor cell (Fig. 1B; 100%, N=12). BratΔB-boxes also colocalized with Mira in telophase neuroblasts and co-segregated with Mira asymmetrically into the future progenitor cell (Fig. 1C; 60%, N=12). Thus, the B-boxes are dispensable for unequal partitioning of Brat during the asymmetric division of neuroblasts. By contrast, BratΔNHL and BratΔC-coil never colocalized with Mira in telophase neuroblasts and always segregated symmetrically into the cytoplasm of both daughter cells (Fig. 1D,E; 100%, N=12 per genotype). These results indicate that the NHL domain and the coiled-coil domain are essential for the asymmetric segregation of Brat in mitotic neuroblasts.
Fig. 1.

The coiled-coil domain and the NHL domain are both required to promote the asymmetric segregation of Brat in mitotic neuroblasts. (A) Schematics of Brat and the Brat deletion mutants used to identity the domain required to asymmetrically segregate Brat in mitotic neuroblasts in transgenic Drosophila and to map the Mira-binding domain in yeast two-hybrid analysis. Dashed lines indicate deletions. (B-C′) Brat and BratΔB-boxes are cortically localized and segregate asymmetrically into the future immature INP in mitotic type II neuroblasts in brat null brains. (D-E′) Deletion of either the coiled-coil or the NHL domain (BratΔC-coil or BratΔNHL) results in cytoplasmic localization and symmetric segregation into both daughter progeny. All Brat transgenic proteins are Myc tagged. The segregation pattern of the Brat transgenic proteins was determined based on the colocalization of the Myc epitope and Mira in telophase neuroblasts. Wor> is a neuroblast-specific driver. Phh3, phosphohistone H3. (F) Yeast two-hybrid analysis showing that Brat and BratΔB-boxes interact with both Mira369-506 and Mira527-638, but BratΔC-coil only interacts with Mira527-638. (G) Summary of the domains required for segregating Brat uniquely into the future immature INP during the asymmetric division of neuroblasts. Scale bar: 5 μm.
The binding of Brat to the cargo-binding domain of Mira (amino acids 405-830) is essential for its segregation into the future progenitor cell following the asymmetric division of neuroblasts (Betschinger et al., 2006; Lee et al., 2006a). Independently of our domain analyses, we identified that Mira369-506 and Mira527-638 (Fig. 1A) bind to both full-length Brat and BratΔB-boxes in a yeast two-hybrid screen using a brat mutant brain cDNA library (H.K. and C.-Y.L., unpublished). We confirmed the interaction between full-length Brat or BratΔB-boxes with both the Mira369-506 and Mira527-638 fragments in a one-to-one yeast two-hybrid interaction assay (Fig. 1F). These results indicate that the B-boxes are dispensable for the binding of Brat to Mira, consistent with BratΔB-boxes colocalizing and co-segregating with Mira into the immature INP during the asymmetric division of neuroblasts (Fig. 1C). We next tested whether the coiled-coil domain of Brat mediates direct interaction with Mira369-506 or Mira527-638. Interestingly, BratΔC-coil only interacts with Mira527-638 but not with the Mira369-506 fragment, indicating that the coiled-coil domain of Brat mediates the binding to amino acids 369-506 of Mira (Fig. 1F). These data also suggest that the NHL domain most likely interacts with amino acids 527-638 of Mira and are consistent with our previous study showing that glycine 774 and tyrosine 829 in the NHL domain of Brat mediate direct interaction with the cargo-binding domain of Mira (Lee et al., 2006a). We were unable to directly test the interaction between BratΔNHL and Mira527-638 due to excessive auto-activation. Taken together, we propose that the coiled-coil domain and the NHL domain function cooperatively to target Brat into the future progenitor cell during the asymmetric division of type II neuroblasts.
The B-boxes of Brat are uniquely required for the specification of INP identity
We next tested whether the coiled-coil domain or the NHL domain is required for Brat-dependent specification of INP identity by overexpressing the UAS-brat transgene driven by a neuroblast-specific Wor-Gal4 driver in brat null brains (Fig. 1A, Fig. 2A). We previously showed that Ase- immature INPs rapidly revert into supernumerary type II neuroblasts at the expense of INP formation in brat null brains (Fig. 2B,C,I; supplementary material Fig. S1A) (Xiao et al., 2012). Overexpression of brat suppressed the supernumerary neuroblast phenotype and restored INP formation in brat null brains (Fig. 2D,I; supplementary material Fig. S1B,C; N=10). Thus, restoring wild-type brat function in type II neuroblasts is sufficient to rescue defects in the specification of INP identity in brat null brains. Surprisingly, overexpression of bratΔC-coil or bratΔNHL also suppressed the supernumerary neuroblast phenotype in brat null brains (Fig. 2E,F,I; supplementary material Fig. S1D,E; N=10 per genotype). These results indicate that neither the coiled-coil domain nor the NHL domain is essential for Brat-dependent specification of INP identity. By contrast, overexpression of bratΔB-boxes failed to suppress the supernumerary neuroblast phenotype in brat null brains (Fig. 2G,I; supplementary material Fig. S1F,G; N=10 per genotype). Thus, the B-boxes are indispensable for Brat-dependent specification of INP identity. Because the Brat protein has two B-boxes, we tested whether they might function redundantly to mediate Brat-dependent specification of INP identity. Overexpression of either bratΔB-box 1 or bratΔB-box 2 efficiently suppressed the supernumerary neuroblast phenotype and restored INP formation in brat null brains (Fig. 2H,I; N=10; data not shown). These data indicate that the B-boxes indeed function redundantly to mediate Brat-dependent specification of INP identity (Fig. 2J).
Fig. 2.

The B-boxes of Brat function uniquely in the specification of INP identity. (A) Summary of the UAS-brat transgenes used to test for B-box redundancy in the rescue of supernumerary neuroblasts in brat null brains. (B) Summary of the cell types in the type II neuroblast lineage in the Drosophila larval brain. Dpn, Deadpan; Ase, Asense; Pros, Prospero; GMC, ganglion mother cell; INP, intermediate neural progenitor; imm INP, immature INP; neurob, neuroblast. (C-H′) Overexpression of full-length brat, bratΔC-coil, bratΔNHL or bratΔB-box 2 rescues the supernumerary neuroblast phenotype in brat null brains, but overexpression of bratΔB-boxes does not. The effects on the supernumerary neuroblast phenotype in brat mutant brains were determined based on total type II neuroblasts per brain lobe. The dotted line separates the brain from the optic lobe where both are visible in the optical section. White arrows, type II neuroblasts (Dpn+ Ase-); yellow arrows, Ase-immature INPs (Dpn- Ase-) and Ase+ immature INPs (Dpn- Ase+); yellow arrowheads, INPs (Dpn+ Ase+). (I) Quantification of total type II neuroblasts per brain lobe in brat mutant brains overexpressing various Brat transgenic proteins. Error bars indicate standard deviation of the mean (s.d.). *P<0.05 versus control (Student’s t-test). (J) Summary of the domain required for Brat-dependent specification of INP identity. Scale bar: 20 μm.
Although all TRIM family proteins contain at least one B-box, the role of this domain in the function of these proteins is unknown. We tested whether the B-boxes might mediate the function of Brat in repressing the translation of hunchback mRNA in the posterior of the preblastoderm embryo (Sonoda and Wharton, 2001). We overexpressed UAS-brat transgenes driven by nanos-Gal4 in bratfs1/Df mutant embryos. Consistent with previously published observations, overexpression of brat, but not bratΔNHL, rescued defects in abdominal segmentation in bratfs1/Df embryos (supplementary material Fig. S1H-J). Importantly, overexpression of bratΔB-boxes or bratΔC-coil also rescued defects in abdominal segmentation in bratfs1/Df embryos (supplementary material Fig. S1K-M). Thus, the B-boxes and the coiled-coil domain are dispensable for the function of Brat in repressing mRNA translation. The B-boxes, therefore, uniquely elicit the function of Brat in the specification of INP identity (Fig. 2J).
The mechanism by which Brat specifies INP identity is sensitive to reduced Apc2 function
Our data thus far indicate that Brat specifies INP identity via a novel mechanism that is independent of the domains required for asymmetric protein segregation (Figs 1, 2). To gain mechanistic insight into how Brat specifies INP identity, we screened for haploinsufficient loci that modify the supernumerary neuroblast phenotype in a sensitized brat mutant background (Xiao et al., 2012). Briefly, a wild-type larval brain lobe contained 8±0 type II neuroblasts (data not shown). Whereas a hypomorphic bratDG19310 homozygous mutant brain lobe possessed 12±3 type II neuroblasts (supplementary material Fig. S2A,B; N=10), a genetically null brat11/K06028 mutant brain lobe contained hundreds of type II neuroblasts (supplementary material Fig. S2A,C; N=10). A bratDG19310/11 mutant brain lobe possessed 24.5±4.7 type II neuroblasts and provides a sensitized genetic background for identifying the genetic modifiers of brat (Fig. 3A; supplementary material Fig. S2A; N=10). Through this screen, we identified Apc2 as a genetic enhancer of the supernumerary neuroblast phenotype in bratDG19310/11 mutant brains. We confirmed that reducing Apc2 function consistently enhanced the supernumerary neuroblast phenotype in bratDG19310/11 brains using multiple independently generated Apc2 mutant alleles, including a null allele [Apc2g10 (McCartney et al., 2006)] and two hypomorphic alleles [Apc2N175K and Apc2d40 (McCartney et al., 2001; Hamada and Bienz, 2002)] (Fig. 3B; supplementary material Fig. S2A,D,E; N=10 per genotype). Furthermore, overexpression of Apc2 rescued the enhancement of the supernumerary neuroblast phenotype in bratDG19310/11 mutant brains induced by the heterozygosity of Apc2 (Fig. 3C; N=10). Thus, reduced Apc2 function enhances the supernumerary neuroblast phenotype in bratDG19310/11 brains. Despite sharing extensive sequence homology and functional redundancy with Apc2 (Ahmed et al., 2002; Akong et al., 2002; Hamada and Bienz, 2002), reducing Apc1 (Apc - FlyBase) did not enhance the supernumerary neuroblast phenotype in bratDG19310/11 mutant brains (supplementary material Fig. S2A,F; N=10). In addition, reducing Apc1 did not significantly worsen the supernumerary neuroblast phenotype in bratDG19310/11 brains induced by the heterozygosity of Apc2 (supplementary material Fig. S2A,G,H; N=10 per genotype). Lack of effect of reducing Apc1 function on the supernumerary neuroblast phenotype in bratDG19310/11 mutant brains might be due to the differential expression patterns of Apc1 and Apc2 in the developing larval brain (Akong et al., 2002). Together, these data strongly suggest that Brat specifies INP identity via an Apc2-dependent mechanism.
Fig. 3.

Brat specifies INP identity by maintaining Apc2 cortical localization. (A,B) Reduced Apc2 function enhances the supernumerary neuroblast phenotype in brat mutant brains. (C) Increased Apc2 expression rescues the enhancement of the supernumerary neuroblast phenotype in brat mutant brains induced by Apc2 heterozygosity. (D-O′) Overexpression of full-length brat, bratΔC-coil and bratΔNHL, but not bratΔB-boxes, restores Apc2 localization in interphase type II neuroblasts in brat mutant brains. (F,H,J,L,N) The expression pattern of Apc2 in type II neuroblasts in brat mutant brains. (G,I,K,M,O) The localization pattern of Apc2 in telophase type II neuroblasts (Mira+ Ase-) in brat mutant brains. Boxed regions in D-N′ are magnified in D′-N′′. See Fig. 2 for description of labels. (P,Q) Quantitative analyses of the level of Apc2 mRNA (P) and Apc2 protein (Q) in brat mutant brains. α-tubulin (Tub) provides a loading control. Scale bars: 20 μm in A-C; 10 μm in D-N′; 5 μm in D′-O′.
Interestingly, neither the overexpression of Apc2 nor the removal of Apc2 function perturbed the identity of progeny generated by type II neuroblasts in an otherwise wild-type background (data not shown). Thus, we hypothesized that Apc2 regulates the specification of INP identity in a Brat-dependent context. Consistent with previous reports (McCartney et al., 1999; Akong et al., 2002), Apc2 was detected in both the cortex and cytoplasm of interphase type II neuroblasts and became enriched in the basal cortex of mitotic neuroblasts in wild-type brains (Fig. 3D,E; N=10 per experiment), such that Apc2 is partitioned into both daughters. However, the expression and the cortical localization of Apc2 were dramatically reduced in type II neuroblasts in brat null mutant brains (Fig. 3F,G; N=10 per experiment). Importantly, overexpression of full-length brat restored Apc2 protein expression and cortical localization in brat null type II neuroblasts (Fig. 3H,I; N=10 per experiment). These data strongly suggest that Brat specifies INP identity by maintaining Apc2 protein expression and/or cortical localization. Because the B-boxes function uniquely to elicit Brat-dependent specification of INP identity, we tested whether overexpression of bratΔB-boxes can restore Apc2 protein expression and/or cortical localization in brat null neuroblasts. Consistent with its inability to suppress the supernumerary neuroblast phenotype in brat null brains, overexpressing bratΔB-boxes failed to restore Apc2 protein expression and cortical localization in brat mutant type II neuroblasts (Fig. 3J,K; N=10 per experiment). By contrast, overexpression of bratΔC-coil or bratΔNHL efficiently restored Apc2 protein expression and cortical localization in brat null mutant type II neuroblasts (Fig. 3L-O; N=10 per experiment). Thus, Brat specifies INP identity by maintaining Apc2 protein expression and/or cortical localization through the B-boxes.
We tested whether Brat directly regulates the expression of the Apc2 gene. Although we detected a ∼30% reduction in the level of Apc2 transcript in extracts from dissected brat null brains compared with wild-type larval brains, there was no appreciable difference in the level of Apc2 protein (Fig. 3P,Q). Hence, it is most likely that Brat specifies INP identity by maintaining Apc2 cortical localization rather than by regulating Apc2 protein expression.
Brat specifies INP identity via a novel Arm-sensitive mechanism
Apc2 together with Axin, Gsk3β [Shaggy (Sgg)] and Casein kinase Iα (CKI) form the destruction complex that targets β-catenin/Arm for degradation in the absence of Wnt pathway activation (Aoki and Taketo, 2007; MacDonald et al., 2009; Niehrs, 2012). Loss of Apc2 reduces destruction complex activity, leading to elevation of the Arm levels and activation of Wnt target genes (McCartney et al., 1999). We tested if reduced function of the destruction complex can indeed enhance the supernumerary neuroblast phenotype in bratDG19310/11 mutant brains. Similar to the effect of reduced Apc2 function, heterozygosity of Axin, sgg or CKI enhanced the supernumerary neuroblast phenotype in bratDG19310/11 brains (Fig. 4A-E; N=10 per genotype). Hence, reduced function of the destruction complex enhances the supernumerary neuroblast phenotype in brat mutant brains.
Fig. 4.

Increased Arm function enhances supernumerary neuroblast formation in brat mutant brains. (A-D) Like Apc2, increasing Arm activity by reducing the function of other components of the destruction complex enhances the supernumerary neuroblast phenotype in brat mutant brains. See Fig. 2 for description of labels. (E) Quantification of total type II neuroblasts per brain lobe in brat mutant brains. (F-I) The supernumerary neuroblast phenotype in brat mutant brains is sensitive to changes in arm function. (F,G) Reduced arm function suppresses the enhancement of the supernumerary neuroblast phenotype in brat mutant brains induced by the heterozygosity of Apc2. (H,I) Increased arm function enhances the supernumerary neuroblast phenotype in brat mutant brains. (J) Quantification of total type II neuroblasts per brain lobe in brat mutant brains. *P<0.05 versus control (Student’s t-test). Error bars indicate s.d. Scale bars: 20 μm.
We next tested whether increased Arm activity contributes to the enhancement of the supernumerary neuroblast phenotype in bratDG19310/11 mutant brains induced by heterozygosity of Apc2. Indeed, reducing the function of arm suppressed the enhancement of the supernumerary neuroblast phenotype in bratDG19310/11 brains heterozygous for Apc2 (Fig. 4F,G,J; N=10 per genotype). Consistent with this finding, overexpression of armS10, which encodes a stabilized form of Arm that is refractory to destruction complex activity (Pai et al., 1997), strongly enhanced the supernumerary neuroblast phenotype in bratDG19310/11 mutant brains (Fig. 4H,J; N=10 per genotype). Unexpectedly, overexpression of armS10 alone was not sufficient to induce supernumerary neuroblasts in wild-type brains (Fig. 4J; N=10 per genotype). Thus, Brat specifies INP identity by attenuating the activity of Arm via a destruction complex-dependent mechanism. These results also indicate that increased Arm activity induces supernumerary neuroblasts in a manner that is dependent upon a parallel mechanism that acts downstream of Brat.
Brat specifies INP identity in immature INPs by attenuating the transcriptional activity of Arm
Our previous study strongly suggests that Brat specifies INP identity in Ase- immature INPs (Xiao et al., 2012), prompting us to test whether Brat attenuates Arm activity specifically in Ase- immature INPs. Consistent with this model, overexpression of Apc2 in either neuroblasts or Ase- immature INPs [using Erm-Gal4(II)] suppressed the enhancement of the supernumerary neuroblast phenotype in bratDG19310/11 mutant brains induced by heterozygosity of Apc2 (Fig. 5A-D,F; N=10 per genotype). By contrast, overexpression of Apc2 in Ase+ immature INPs [using Erm-Gal4(III)] did not have any effect on the enhancement of the supernumerary neuroblast phenotype in bratDG19310/11 brains induced by Apc2 heterozygosity (Fig. 5E,F; N=10). Furthermore, overexpression of armS10 in neuroblasts or Ase- immature INPs also enhanced the supernumerary neuroblast phenotype in bratDG19310/11 mutant brains, whereas overexpression of armS10 in Ase+ immature INPs did not (Fig. 5G-K; N=10 per genotype). Together, these results strongly suggest that Brat specifies INP identity in Ase- immature INPs by attenuating Arm activity through the destruction complex.
Fig. 5.

Brat specifies INP identity by antagonizing Arm-dependent gene transcription in immature INPs. (A) Summary of the expression patterns of the Gal4 drivers within the type II neuroblast lineage (Xiao et al., 2012). (B-E) Increased function of Apc2 early in the lineage suppresses the enhancement of the supernumerary neuroblast phenotype in brat mutant brains induced by Apc2 heterozygosity. (F) Quantification of total type II neuroblasts per brain lobe in brat mutant brains. (G-J) Increased function of arm early in the lineage enhances the supernumerary neuroblast phenotype in brat mutant brains. (K) Quantification of total type II neuroblasts per brain lobe in brat mutant brains. (L-P) Decreased arm function via arm gene heterozygosity or overexpression of TcfDN in type II neuroblasts or Ase- immature INPs suppresses the supernumerary neuroblast phenotype in brat mutant brains. By contrast, overexpression of TcfDN in Ase+ immature INPs does not suppress the supernumerary neuroblast phenotype in brat mutant brains. (Q) Quantification of total type II neuroblasts per brain lobe in brat mutant brains. Error bars indicate s.d. *P<0.05 versus control (Student’s t-test). See Fig. 2 for description of labels. Scale bars: 20 μm.
During canonical Wnt signaling, β-catenin/Arm forms a transcriptional activator complex by binding to Tcf and activates Wnt target gene expression (Aoki and Taketo, 2007; MacDonald et al., 2009; Niehrs, 2012). We tested whether Brat specifies INP identity in Ase- immature INPs by attenuating the transcriptional activator function of Arm. Reducing the function of arm suppressed the supernumerary neuroblast phenotype in bratDG19310/11 mutant brains (Fig. 5L,M,Q; N=10 per genotype). Similarly, overexpression of TcfDN, which encodes a dominant-negative form of Tcf (Pangolin - FlyBase), in either neuroblasts or Ase- immature INPs also suppressed the supernumerary neuroblast phenotype in bratDG19310/11 brains (Fig. 5N,O,Q; N=10 per genotype). By contrast, overexpression of TcfDN in Ase+ immature INPs failed to suppress the supernumerary neuroblast phenotype in bratDG19310/11 brains (Fig. 5P,Q; N=10). Thus, Brat specifies INP identity in Ase- immature INPs by attenuating Arm-activated gene transcription. Because increased Arm activity alone is not sufficient to induce supernumerary neuroblast formation (Fig. 4L), Wnt signaling promotes neuroblast identity in a context that is dependent on parallel mechanisms acting downstream of Brat.
Klu induces supernumerary neuroblast formation via an Arm-dependent mechanism
We previously showed that Brat specifies INP identity in Ase- immature INPs by antagonizing the neuroblast self-renewal factor Klu (Xiao et al., 2012). Thus, we tested whether Wnt signaling promotes neuroblast identity in a context dependent on Klu. Control clones derived from single type II neuroblasts overexpressing klu contained mostly supernumerary neuroblasts, whereas all arm mutant clones overexpressing klu consistently contained fewer supernumerary neuroblasts (Fig. 6A-D; N=10 clones). More specifically, ∼50% of arm mutant clones overexpressing klu contained greater than one neuroblast per clone, while the remaining arm mutant clones reproducibly possessed only one neuroblast per clone (Fig. 6B-D). Thus, these data strongly suggest that reducing the function of arm dampens the capacity of klu to induce supernumerary neuroblasts. Consistently, neuroblast clones co-expressing Apc2 and klu also possessed far fewer supernumerary neuroblasts and more INPs than the control clones overexpressing klu alone (Fig. 6E-G). Because overexpression of Apc2 alone did not affect the specification of cell identity in the type II neuroblast lineage (data not shown), these results indicate that increased Apc2 function also dampens the capacity of klu to induce supernumerary neuroblasts. Taken together, we conclude that Arm functions in a Klu-dependent context to promote neuroblast identity.
Fig. 6.

arm is required for the formation of supernumerary neuroblasts induced by overexpression of klu. (A-C′) The removal of arm function significantly reduces the formation of supernumerary neuroblasts induced by overexpression of klu. Larvae carrying GFP-positive type II neuroblast mosaic clones (outlined by the yellow dotted line) were aged for 72 hours after clone induction, and brains were stained for the markers indicated. (D) The frequency of clones containing one or more Dpn+ type II neuroblasts in larvae of the genotype indicated. (E-F′) Co-expression of Apc2 suppresses supernumerary type II neuroblasts induced by overexpression of klu. (G) The total number of neuroblasts per clone in larvae of the genotype indicated. Error bars indicate s.d. *P<0.05 versus control (Student’s t-test). See Fig. 2 for description of labels. Scale bars: 10 μm.
DISCUSSION
Asymmetric stem cell division provides an efficient mechanism to simultaneously self-renew a stem cell and to generate a progenitor cell that produces differentiated progeny. Because self-renewal proteins segregate into both daughter progeny of the dividing parental stem cell through the inheritance of its cytoplasmic content, rapidly downregulating the activity of these proteins is essential for the specification of progenitor cell identity. Brat plays a central role in specifying INP identity in the Ase- immature INP by antagonizing the function of the self-renewal transcription factor Klu (Xiao et al., 2012). We have extended our previous findings to show that Brat specifies INP identity in the Ase- immature INP through two separable, but convergent, mechanisms. We identified a novel Brat-dependent mode of Wnt pathway regulation that prevents Ase- immature INPs from reverting into supernumerary neuroblasts. We showed that Brat specifies INP identity by attenuating the transcriptional activity of Arm through its N-terminal B-boxes (Fig. 2). This negative regulation of Arm is achieved through the activity of Apc2 and the destruction complex (Fig. 7). Because increased arm function alone was insufficient to induce supernumerary neuroblasts (Fig. 4H-J), the ability of Wnt signaling to promote neuroblast identity is dependent on other signaling mechanisms that act downstream of Brat. Indeed, Arm function is essential for Klu to induce supernumerary neuroblasts. These two Brat-regulated mechanisms function to safeguard against the accidental reversion of an uncommitted progenitor cell into a supernumerary stem cell and to ensure that an uncommitted progenitor cell can only adopt progenitor cell identity.
Fig. 7.
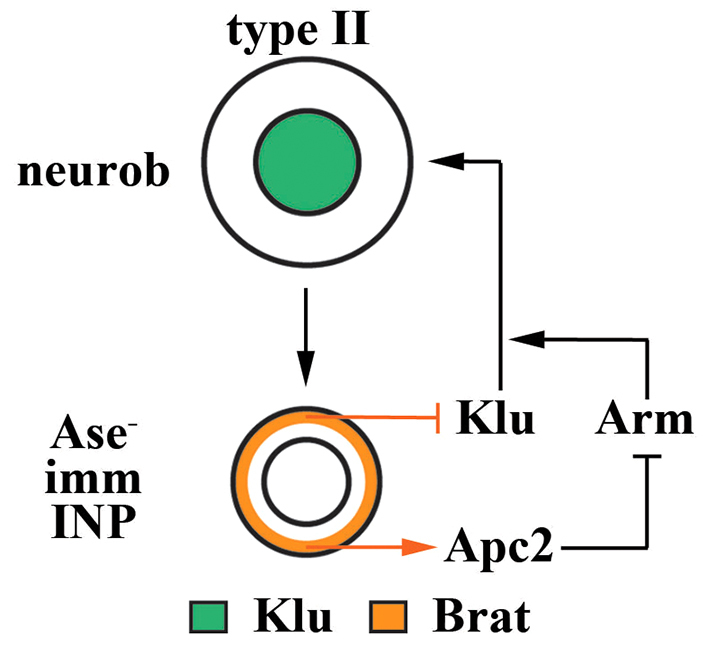
Model summarizing the mechanism by which Brat specifies INP identity by antagonizing both Klu and Arm function in immature INPs. Brat specifies INP identity in Ase- immature INPs by antagonizing Arm function via an Apc2-dependent mechanism and repressing the activity of the self-renewal factor Klu. In the absence of brat function, the increased activity of klu and arm cooperatively induce the reversion of Ase- immature INPs into supernumerary type II neuroblasts.
Asymmetric protein segregation and asymmetric cell fate specification are mechanistically separable
Physical interaction with the cargo-binding domain of Mira is essential for the unequal segregation of Brat into the immature INP following the asymmetric division of neuroblasts (Betschinger et al., 2006; Lee et al., 2006a). Previous studies concluded that the NHL domain of Brat directly interacts with the cargo-binding domain of Mira, but the roles of the B-boxes and the coiled-coil domain in the asymmetric segregation of Brat were unknown due to a lack of specific mutant alleles. By combining a yeast two-hybrid interaction assay and in vivo functional validation, we conclude that both the coiled-coil domain and the NHL domain are indeed required for the asymmetric segregation of Brat into the Ase- immature INP following the asymmetric division of neuroblasts (Fig. 1). We speculate that the coiled-coil domain and the NHL domain of Brat function cooperatively to provide a more stable binding platform for Mira to ensure efficient protein segregation.
The severity of the supernumerary neuroblast phenotype in various brat mutant allelic combinations correlates with the level of endogenous brat inherited by the Ase- immature INP. The bratDG19310 mutation carries a transposable P-element inserted in the 5′ regulatory region of the brat gene (http://flybase.org/reports/FBal0215708.html). The brat11 mutation, however, results in a premature stop codon at amino acid 779, leading to a truncated form of the protein that lacks most of the NHL domain and is predicted to be unable to interact with Mira (Arama et al., 2000; Napolitano and Meroni, 2012). The bratDG19310 or bratDG19310/11 allelic combination most likely reduces Brat expression without affecting its binding to Mira. Thus, the minimal threshold of Brat necessary for the proper specification of INP identity in Ase- immature INPs is met most of the time, leading to a mild supernumerary neuroblast phenotype in bratDG19310 or bratDG19310/11 brains (supplementary material Fig. S2A,B). By contrast, the brat11 homozygous or brat11/Df mutant allelic combination impairs the binding of Brat to Mira, rendering the Mira-based asymmetric protein-sorting mechanism unable to segregate Brat into the Ase- immature INP. As such, the threshold of Brat necessary for proper specification of INP identity in Ase- immature INPs is rarely met, leading to a severe supernumerary neuroblast phenotype in brat11 or brat11/Df brains (Fig. 2C,L). Overexpression of the bratΔC-coil or bratΔNHL transgene using the UAS/Gal4 system almost certainly results in an abnormally high level of the transgenic protein in the cytoplasm of neuroblasts. Thus, inheriting a portion of the neuroblast cytoplasm containing an overwhelming abundance of the mutant transgenic protein is likely to be sufficient to reach the threshold of Brat necessary for proper specification of INP identity in Ase- immature INPs. We conclude that the mechanism that causes Brat to asymmetrically segregate into the Ase- immature INP is functionally separable from the mechanism that specifies INP identity.
Could the asymmetric protein segregation mechanism promote the specification of INP identity by depleting Brat from the neuroblast? Type II neuroblasts overexpressing brat, bratΔC-coil or bratΔNHL maintained their identity and generated similar numbers of progeny as wild-type control neuroblasts (supplementary material Fig. S3). Thus, it is unlikely that Brat-dependent specification of INP identity occurs through asymmetric depletion of Brat from the neuroblast. We also tested whether Brat acts redundantly with other asymmetrically segregating determinants to specify INP identity in Ase- immature INPs. Numb also exclusively segregates into the immature INP during asymmetric divisions of type II neuroblasts (Haenfler et al., 2012). However, asymmetric segregation of Numb is not dependent on Brat, and Numb-dependent specification of INP identity also occurs independently of Brat (supplementary material Fig. S4). Thus, it is unlikely that Brat acts redundantly with other asymmetric segregating determinants to specify INP identity in Ase- immature INPs.
The B-boxes function uniquely to promote Brat-dependent specification of INP identity
A surprising finding revealed by the current study is that the B-boxes are uniquely required for the specification of INP identity. This raises a series of interesting questions. What are the roles of B-boxes in the function of Brat in embryonic neuroblasts? Embryos lacking both maternal and zygotic function of brat often lack RP2 neurons but never possess supernumerary neuroblasts (Betschinger et al., 2006). Since brat mutant alleles that specifically affect the function of B-boxes are unavailable, the roles of B-boxes in the function of Brat during the asymmetric division of embryonic neuroblasts remain unknown. Brat regulates embryonic pattern formation by repressing mRNA translation through the ternary complex that also contains Nanos and Pumilio (Sonoda and Wharton, 2001). However, it is unlikely that Brat specifies INP identity through the Nanos-Pumilio-Brat translational repression complex for the following reasons. First, the NHL domain of Brat is required for binding to Pumilio and Nanos and for the assembly of the translational repressor complex (Sonoda and Wharton, 2001). However, the NHL domain is dispensable for Brat-dependent specification of INP identity (Fig. 2). Second, Nanos expression is undetectable in larval brains, and pumilio mutant larval brains do not possess supernumerary type II neuroblasts (data not shown). Together, these results are consistent with our conclusion that Brat specifies INP identity via a novel Arm-mediated mechanism.
The amino acid sequence of the B-boxes is highly conserved among all TRIM family proteins, including Brat, and is predicted to adopt a ‘RING-like’ fold tertiary structure (Massiah et al., 2006; Massiah et al., 2007; Tao et al., 2008). The RING-like fold might facilitate protein-protein interactions (Massiah et al., 2006; Massiah et al., 2007; Napolitano and Meroni, 2012). This is a particularly intriguing hypothesis in light of the fact that Apc2 and Brat both localize to the basal cortex in type II neuroblasts, and overexpression of brat, but not bratΔB-boxes, can restore Apc2 protein localization in neuroblasts (Fig. 3). However, we were unable to co-immunoprecipitate epitope-tagged Brat and endogenous Apc2 from the brain lysate extracted from brat null mutant larvae overexpressing a Myc-tagged Brat transgenic protein (data not shown). Thus, Brat might maintain Apc2 protein localization indirectly through other factors. Future biochemical analyses of the Brat protein and identification of the proteins that directly interact with the B-boxes will provide insight into how Brat controls Apc2 localization.
Brat specifies INP identity by downregulating Arm activity via Apc2 and the destruction complex
The destruction complex targets β-catenin/Arm for degradation during canonical Wnt signaling, so reduced function of the destruction complex will lead to an increase in β-catenin/Arm, which forms a complex with Tcf/LEF family transcription factors to activate Wnt target gene expression (Aoki and Taketo, 2007; MacDonald et al., 2009; Niehrs, 2012). Our study led us to conclude that the Brat-Apc2 mechanism specifies INP identity by preventing aberrant activation of Wnt target gene expression in Ase- immature INPs (Figs 4, 5). We tested the role of the Wnt ligand in the Brat-dependent specification of INP identity by removing the function of the Wnt ligand using a temperature-sensitive mutant allele or by overexpressing a dominant-negative form of Frizzled (FzDN or GPI-dFz2) in bratDG19310/11 mutant brains. Interestingly, neither of these manipulations modified the supernumerary neuroblast phenotype in the sensitized brat genetic background (data not shown). These results suggest that the Wnt ligand and its receptor Fz are irrelevant in the Brat-dependent specification of INP identity and that the Brat-Apc2 mechanism prevents Wnt target gene expression in Ase- immature INPs by negatively regulating the activity of Arm. However, our data do not exclude the possibility that a novel activating mechanism of Wnt signaling might be present in type II neuroblasts in Drosophila larval brains.
We also sought to directly demonstrate that loss of brat function indeed leads to derepression of Wnt target gene expression in supernumerary neuroblasts. We examined the expression of two distinct Wnt reporter transgenes, WRE-lacZ and Notum-lacZ (Chang et al., 2008) in brat mutant brains. However, we were unable to detect the expression of these transgenes in supernumerary neuroblasts in brat null mutant brains (data not shown). Because genetic manipulations altering the activity of Arm efficiently modify the supernumerary neuroblast phenotype in brat mutant brains, these two transgenes are unlikely to have the necessary regulatory elements to reflect Wnt target gene activity in this tissue. Thus, we propose that the Brat-Apc2 mechanism specifies INP identity by antagonizing the transcriptional activity of Arm in Ase- immature INPs via a receptor-independent mechanism.
Wnt signaling plays a permissive role in regulating the functional output of the self-renewal factor Klu in uncommitted intermediate progenitor cells
Wnt signaling regulation plays key roles in both stem cell renewal and the differentiation of progenitor cell types (Merrill, 2012; Habib et al., 2013). In the mammalian intestinal epithelium, for example, loss of Apc and activation of Wnt signaling results in the maintenance of stem cell properties in the progenitor cells, a failure to differentiate, and the production of intestinal polyps that progress to malignant tumors (Schwitalla et al., 2013). In the intestine, the inappropriate activation of Wnt signaling is sufficient to elicit stem cell properties. In the progenitor cells of larval type II neuroblasts, the activation of Wnt signaling alone, through either the expression of stabilized Arm (Fig. 4) or the loss of Apc2 (data not shown), does not drive stem cell renewal in otherwise wild-type immature INPs. In this system, Brat is the key regulator attenuating self-renewal through two independent, but convergent, mechanisms in its regulation of both Klu and Wnt signaling. Although Arm activity is required for Klu-dependent self-renewal in immature INPs (Fig. 6), its inability to promote self-renewal alone suggests that Wnt signaling is likely to be playing a permissive role rather than an instructive role in eliciting the neuroblast identity. We propose that Brat downregulates the function of Klu through both Arm-dependent and -independent mechanisms (Fig. 7). Previous studies have demonstrated that TRIM32 and TRIM3, which are vertebrate orthologs of Brat, are essential regulators of neural stem cells during brain development and brain tumor formation (Boulay et al., 2009; Schwamborn et al., 2009). It will be interesting to test whether TRIM32 and TRIM3 regulate neural stem cells via a β-catenin-dependent mechanism.
MATERIALS AND METHODS
Fly genetics and transgenes
We used Oregon R as the wild-type control and the following mutant alleles: bratk06028, bratfs1 and brat11 (Arama et al., 2000), bratDG19310 (Xiao et al., 2012), Apc2N175K (Hamada and Bienz, 2002), Apc2d40 (McCartney et al., 2001), Apc2g10 (McCartney et al., 2006), Apc1Q8 (Ahmed et al., 1998), arm8 (Peifer and Wieschaus, 1990), AxinS044230 (Tolwinski and Wieschaus, 2001), CKIα8B12 (Legent et al., 2012), sgg8E22 (Legent et al., 2012), wgCX4 (Baker, 1987), wgts (Bejsovec and Martinez Arias, 1991), Nanos-GAL4 (Van Doren et al., 1998), UAS-Apc2-GFP (Akong et al., 2002), UAS-armS10 (Pai et al., 1997), UAS-GPI-dfz2 (Rulifson et al., 2000), UAS-dTcfΔN (van de Wetering et al., 1997), Wor-GAL4 (Lee et al., 2006b), Elav-GAL4 (C155) (Brand and Perrimon, 1993), Act-FRT-CD2-FRT-GAL4 (Pignoni and Zipursky, 1997), pumMsc (Lehmann and Nusslein-Volhard, 1987), pum13 (Lehmann and Nusslein-Volhard, 1987), WRE-lacZ and Notum-lacZ (Chang et al., 2008). The following stocks were obtained from the Bloomington Drosophila Stock Center: hs-flp, UAS-mCD8-GFP, FRT40A, tub-gal80, FRT82B, FRT19A, Df(2L)Exel8040 and UAS-GFP.
The following transgenic lines were generated in this study: UAS-brat-myc, UAS-bratΔB-boxes-myc, UAS-bratΔB-box 1-myc, UAS-bratΔB-box 2-myc, UAS-bratΔC-coil-myc and UAS-bratΔNHL-myc. The cDNA was cloned into p{UAST}attB vector and the transgenic fly lines were generated via ϕC31 integrase-mediated transgenesis (Bischof et al., 2007).
Immunofluorescent staining and antibodies
Larval brains were dissected in Schneider’s medium (Sigma) and fixed in 100 mM PIPES (pH 6.9), 1 mM EGTA, 0.3% Triton X-100, 1 mM MgSO4 containing 4% formaldehyde for 23 minutes. We have empirically determined that fixation for 23 minutes allows us to obtain the most robust signal-to-noise ratio to visualize larval brain neuroblasts stained with the Dpn and Ase antibodies. Larval brains were then washed for 20 minutes in 1× PBS containing 0.3% Triton X-100 (PBST) and incubated in primary antibodies diluted in PBST for 3 hours at room temperature. Antibodies used include guinea pig anti-Ase (1:100), rat anti-Dpn (1:2), rabbit anti-Ase (1:400), rat anti-Mira (1:100), mouse anti-Prospero (MR1A; 1:100), mouse anti-Elav (1:50; DSHB, 9F8A9), mouse anti-β-gal (1:100; Sigma, G4644), mouse anti-phosphohistone H3 (1:2000; Upstate Biotechnology, 06-570), chicken anti-GFP (1:2000; Aves Labs, GF-1020), rabbit anti-Scrib (1:2500), rabbit anti-Apc2 (1:100; Y. Yamashita, University of Michigan, Ann Arbor, MI, USA), rabbit anti-cMyc (1:200; Santa Cruz, sc-789), guinea pig anti-Numb (1:2500; J. Skeath, Washington University, St Louis, MO, USA) and rabbit anti-Nos (1:2000; Y. Tao and D. Chen, Chinese Academy of Sciences, China). Species-specific fluorophere-conjugated secondary antibodies (Jackson ImmunoResearch, 703-545-155, 112-605-167; Life Technologies, A-11034, A-11035, A-11074, A31553, A-31556) were used at 1:500. We used Rhodamine phalloidin (Invitrogen) to visualize cortical actin. The confocal images were acquired on a Leica SP5 scanning confocal microscope.
Yeast two-hybrid interaction assay
The bait comprising full-length Brat, BratΔB-boxes or BratΔC-coil was PCR amplified and cloned into the LexA DNA-binding domain cloning vector. BratΔNHL was excluded from the screen due to excessive auto-activation. The prey comprising Mira369-506 or Mira527-638 was cloned into the Gal4 activation domain cloning vector. All constructs were checked by sequencing. The bait and prey constructs were transformed, respectively, into L40ΔGal4 (mata) and Y187 (matα) haploid yeast cells. Interaction pairs were tested in duplicate, as two independent clones from each mating were picked for the growth assay. The clones were tested for their growth as calibrated drops to provide a qualitative measurement of the affinity and a comparison between the various assays. For each interaction, several dilutions (10-1, 10-2, 10-3 and 10-4) of the diploid yeast cells (culture normalized at 5×104 cells) and cells expressing both bait and prey constructs were spotted on several selective media. The DO-2 selective medium lacking tryptophan and leucine was used as a growth control and to verify the presence of both the bait and prey plasmids. The different dilutions were also spotted on a selective medium without tryptophan, leucine and histidine (DO-3). Four different concentrations of 3A-T (1, 5, 10 and 50 mM; Sigma), an inhibitor of the HIS3 gene product, were added to the DO-3 plates to increase stringency and reduce possible auto-activation by Brat or its variants.
Identification of type II neuroblasts
Type II neuroblasts were identified by a combination of molecular marker expression (Dpn+ Ase-) and their location on the dorsal surface of the larval brain lobe (Bello et al., 2008; Boone and Doe, 2008; Bowman et al., 2008).
Clonal analyses
Clones were induced following published methods (Lee and Luo, 2001; Weng et al., 2010).
Real-time PCR
Late third instar larval brains were dissected free of surrounding tissues. Total RNA was extracted following the standard Trizol RNA isolation protocol and cleaned using the Qiagen RNeasy Kit. cDNA was reverse transcribed using the First Strand cDNA Synthesis Kit for RT-PCR (AMV) (Roche). Quantitative PCR was performed using ABsolute QPCR SYBR Green ROX Mix (Thermo Scientific). Resulting data were analyzed by the comparative CT method, and relative mRNA expression is presented.
Western blot
Proteins were extracted from late third instar larval brains. Cell lysates separated by SDS-PAGE were blotted onto an Immobilon transfer membrane (Millipore) and then incubated with antibodies to Apc2 (1:1000) or α-tubulin (1:5000; Sigma, T6199). Blots were incubated with HRP-conjugated secondary antibodies (Millipore) and proteins were detected by Pierce ECL Western Blotting Substrate according to the manufacturer’s protocol.
Supplementary Material
Acknowledgments
We thank Drs Y. Ahmed, K. Cadigan, D. Chen, C. Doe, J. Skeath, Y. Tao, J. Treisman, R. Xi and Y. Yamashita for fly stocks and antibody reagents; the Bloomington Drosophila Stock Center and the Vienna Drosophila RNAi Center for fly stocks; the Drosophila Genomics Resource Center for the brat cDNA clone; BestGene for generating the transgenic fly lines; Hybrigenics for performing the yeast two-hybrid interaction assays; and the members of the C.-Y.L. laboratory for reading the manuscript and providing critical comments.
Footnotes
Competing interests
The authors declare no competing financial interests.
Author contributions
H.K., Q.X. and C.-Y.L. designed experiments. H.K. and Q.X. performed experiments. H.K., Q.X. and C.-Y.L. analyzed and interpreted data. H.K., Q.X., B.M.M. and C.-Y.L. wrote the manuscript.
Funding
H.K. was supported by a fellowship from the Japan Society for the Promotion of Science. C.-Y.L. is supported by a National Institutes of Health grant [R01NS077914]. Deposited in PMC for release after 12 months.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.099382/-/DC1
References
- Ahmed Y., Hayashi S., Levine A., Wieschaus E. (1998). Regulation of armadillo by a Drosophila APC inhibits neuronal apoptosis during retinal development. Cell 93, 1171–1182 [DOI] [PubMed] [Google Scholar]
- Ahmed Y., Nouri A., Wieschaus E. (2002). Drosophila Apc1 and Apc2 regulate Wingless transduction throughout development. Development 129, 1751–1762 [DOI] [PubMed] [Google Scholar]
- Akong K., McCartney B. M., Peifer M. (2002). Drosophila APC2 and APC1 have overlapping roles in the larval brain despite their distinct intracellular localizations. Dev. Biol. 250, 71–90 [DOI] [PubMed] [Google Scholar]
- Aoki K., Taketo M. M. (2007). Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene. J. Cell Sci. 120, 3327–3335 [DOI] [PubMed] [Google Scholar]
- Arama E., Dickman D., Kimchie Z., Shearn A., Lev Z. (2000). Mutations in the beta-propeller domain of the Drosophila brain tumor (brat) protein induce neoplasm in the larval brain. Oncogene 19, 3706–3716 [DOI] [PubMed] [Google Scholar]
- Baker N. (1987). Molecular cloning of sequences from wingless a segment polarity gene in Drosophila the spatial distribution of a transcript in embryos. EMBO J. 6, 1765–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayraktar O. A., Boone J. Q., Drummond M. L., Doe C. Q. (2010). Drosophila type II neuroblast lineages keep Prospero levels low to generate large clones that contribute to the adult brain central complex. Neural Dev. 5, 26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejsovec A., Martinez Arias A. (1991). Roles of wingless in patterning the larval epidermis of Drosophila. Development 113, 471–485 [DOI] [PubMed] [Google Scholar]
- Bello B. C., Izergina N., Caussinus E., Reichert H. (2008). Amplification of neural stem cell proliferation by intermediate progenitor cells in Drosophila brain development. Neural Dev. 3, 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betschinger J., Mechtler K., Knoblich J. A. (2006). Asymmetric segregation of the tumor suppressor brat regulates self-renewal in Drosophila neural stem cells. Cell 124, 1241–1253 [DOI] [PubMed] [Google Scholar]
- Bischof J., Maeda R. K., Hediger M., Karch F., Basler K. (2007). An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc. Natl. Acad. Sci. USA 104, 3312–3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone J. Q., Doe C. Q. (2008). Identification of Drosophila type II neuroblast lineages containing transit amplifying ganglion mother cells. Dev. Neurobiol. 68, 1185–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulay J. L., Stiefel U., Taylor E., Dolder B., Merlo A., Hirth F. (2009). Loss of heterozygosity of TRIM3 in malignant gliomas. BMC Cancer 9, 71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman S. K., Rolland V., Betschinger J., Kinsey K. A., Emery G., Knoblich J. A. (2008). The tumor suppressors Brat and Numb regulate transit-amplifying neuroblast lineages in Drosophila. Dev. Cell 14, 535–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand A. H., Perrimon N. (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–415 [DOI] [PubMed] [Google Scholar]
- Chang J. L., Chang M. V., Barolo S., Cadigan K. M. (2008). Regulation of the feedback antagonist naked cuticle by Wingless signaling. Dev. Biol. 321, 446–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib S. J., Chen B. C., Tsai F. C., Anastassiadis K., Meyer T., Betzig E., Nusse R. (2013). A localized Wnt signal orients asymmetric stem cell division in vitro. Science 339, 1445–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haenfler J. M., Kuang C., Lee C. Y. (2012). Cortical aPKC kinase activity distinguishes neural stem cells from progenitor cells by ensuring asymmetric segregation of Numb. Dev. Biol. 365, 219–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada F., Bienz M. (2002). A Drosophila APC tumour suppressor homologue functions in cellular adhesion. Nat. Cell Biol. 4, 208–213 [DOI] [PubMed] [Google Scholar]
- Harris R. E., Pargett M., Sutcliffe C., Umulis D., Ashe H. L. (2011). Brat promotes stem cell differentiation via control of a bistable switch that restricts BMP signaling. Dev. Cell 20, 72–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homem C. C., Knoblich J. A. (2012). Drosophila neuroblasts: a model for stem cell biology. Development 139, 4297–4310 [DOI] [PubMed] [Google Scholar]
- Krivtsov A. V., Twomey D., Feng Z., Stubbs M. C., Wang Y., Faber J., Levine J. E., Wang J., Hahn W. C., Gilliland D. G., et al. (2006). Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 442, 818–822 [DOI] [PubMed] [Google Scholar]
- Lee C. Y., Wilkinson B. D., Siegrist S. E., Wharton R. P., Doe C. Q. (2006a). Brat is a Miranda cargo protein that promotes neuronal differentiation and inhibits neuroblast self-renewal. Dev. Cell 10, 441–449 [DOI] [PubMed] [Google Scholar]
- Lee C. Y., Robinson K. J., Doe C. Q. (2006b). Lgl, Pins and aPKC regulate neuroblast self-renewal versus differentiation. Nature 439, 594–598 [DOI] [PubMed] [Google Scholar]
- Lee T., Luo L. (2001). Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci. 24, 251–254 [DOI] [PubMed] [Google Scholar]
- Legent K., Steinhauer J., Richard M., Treisman J. E. (2012). A screen for X-linked mutations affecting Drosophila photoreceptor differentiation identifies Casein kinase 1α as an essential negative regulator of wingless signaling. Genetics 190, 601–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann R., Nusslein-Volhard C. (1987). Involvement of the pumilio gene in the transport of an abdominal signal in the Drosophila embryo. Nature 329, 167–170 [Google Scholar]
- Liu C., Sage J. C., Miller M. R., Verhaak R. G., Hippenmeyer S., Vogel H., Foreman O., Bronson R. T., Nishiyama A., Luo L., et al. (2011). Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 146, 209–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald B. T., Tamai K., He X. (2009). Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massiah M. A., Simmons B. N., Short K. M., Cox T. C. (2006). Solution structure of the RBCC/TRIM B-box1 domain of human MID1: B-box with a RING. J. Mol. Biol. 358, 532–545 [DOI] [PubMed] [Google Scholar]
- Massiah M. A., Matts J. A., Short K. M., Simmons B. N., Singireddy S., Yi Z., Cox T. C. (2007). Solution structure of the MID1 B-box2 CHC(D/C)C(2)H(2) zinc-binding domain: insights into an evolutionarily conserved RING fold. J. Mol. Biol. 369, 1–10 [DOI] [PubMed] [Google Scholar]
- McCartney B. M., Dierick H. A., Kirkpatrick C., Moline M. M., Baas A., Peifer M., Bejsovec A. (1999). Drosophila APC2 is a cytoskeletally-associated protein that regulates wingless signaling in the embryonic epidermis. J. Cell Biol. 146, 1303–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney B. M., McEwen D. G., Grevengoed E., Maddox P., Bejsovec A., Peifer M. (2001). Drosophila APC2 and Armadillo participate in tethering mitotic spindles to cortical actin. Nat. Cell Biol. 3, 933–938 [DOI] [PubMed] [Google Scholar]
- McCartney B. M., Price M. H., Webb R. L., Hayden M. A., Holot L. M., Zhou M., Bejsovec A., Peifer M. (2006). Testing hypotheses for the functions of APC family proteins using null and truncation alleles in Drosophila. Development 133, 2407–2418 [DOI] [PubMed] [Google Scholar]
- Merrill B. J. (2012). Wnt pathway regulation of embryonic stem cell self-renewal. Cold Spring Harb. Perspect. Biol. 4, a007971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming G. L., Song H. (2011). Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 70, 687–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano L. M., Meroni G. (2012). TRIM family: Pleiotropy and diversification through homomultimer and heteromultimer formation. IUBMB Life 64, 64–71 [DOI] [PubMed] [Google Scholar]
- Neumüller R. A., Betschinger J., Fischer A., Bushati N., Poernbacher I., Mechtler K., Cohen S. M., Knoblich J. A. (2008). Mei-P26 regulates microRNAs and cell growth in the Drosophila ovarian stem cell lineage. Nature 454, 241–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehrs C. (2012). The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 13, 767–779 [DOI] [PubMed] [Google Scholar]
- Pai L. M., Orsulic S., Bejsovec A., Peifer M. (1997). Negative regulation of Armadillo, a Wingless effector in Drosophila. Development 124, 2255–2266 [DOI] [PubMed] [Google Scholar]
- Peifer M., Wieschaus E. (1990). The segment polarity gene armadillo encodes a functionally modular protein that is the Drosophila homolog of human plakoglobin. Cell 63, 1167–1176 [DOI] [PubMed] [Google Scholar]
- Pierfelice T., Alberi L., Gaiano N. (2011). Notch in the vertebrate nervous system: an old dog with new tricks. Neuron 69, 840–855 [DOI] [PubMed] [Google Scholar]
- Pignoni F., Zipursky S. L. (1997). Induction of Drosophila eye development by decapentaplegic. Development 124, 271–278 [DOI] [PubMed] [Google Scholar]
- Rulifson E. J., Wu C. H., Nusse R. (2000). Pathway specificity by the bifunctional receptor frizzled is determined by affinity for wingless. Mol. Cell 6, 117–126 [PubMed] [Google Scholar]
- Schwamborn J. C., Berezikov E., Knoblich J. A. (2009). The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell 136, 913–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwitalla S., Fingerle A. A., Cammareri P., Nebelsiek T., Göktuna S. I., Ziegler P. K., Canli O., Heijmans J., Huels D. J., Moreaux G., et al. (2013). Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 152, 25–38 [DOI] [PubMed] [Google Scholar]
- Song Y., Lu B. (2011). Regulation of cell growth by Notch signaling and its differential requirement in normal vs. tumor-forming stem cells in Drosophila. Genes Dev. 25, 2644–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda J., Wharton R. P. (2001). Drosophila Brain Tumor is a translational repressor. Genes Dev. 15, 762–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao H., Simmons B. N., Singireddy S., Jakkidi M., Short K. M., Cox T. C., Massiah M. A. (2008). Structure of the MID1 tandem B-boxes reveals an interaction reminiscent of intermolecular ring heterodimers. Biochemistry 47, 2450–2457 [DOI] [PubMed] [Google Scholar]
- Tolwinski N. S., Wieschaus E. (2001). Armadillo nuclear import is regulated by cytoplasmic anchor Axin and nuclear anchor dTCF/Pan. Development 128, 2107–2117 [DOI] [PubMed] [Google Scholar]
- van de Wetering M., Cavallo R., Dooijes D., van Beest M., van Es J., Loureiro J., Ypma A., Hursh D., Jones T., Bejsovec A., et al. (1997). Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF. Cell 88, 789–799 [DOI] [PubMed] [Google Scholar]
- Van Doren M., Williamson A. L., Lehmann R. (1998). Regulation of zygotic gene expression in Drosophila primordial germ cells. Curr. Biol. 8, 243–246 [DOI] [PubMed] [Google Scholar]
- Wei J., Wunderlich M., Fox C., Alvarez S., Cigudosa J. C., Wilhelm J. S., Zheng Y., Cancelas J. A., Gu Y., Jansen M., et al. (2008). Microenvironment determines lineage fate in a human model of MLL-AF9 leukemia. Cancer Cell 13, 483–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng M., Lee C. Y. (2011). Keeping neural progenitor cells on a short leash during Drosophila neurogenesis. Curr. Opin. Neurobiol. 21, 36–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng M., Golden K. L., Lee C. Y. (2010). dFezf/Earmuff maintains the restricted developmental potential of intermediate neural progenitors in Drosophila. Dev. Cell 18, 126–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q., Komori H., Lee C. Y. (2012). klumpfuss distinguishes stem cells from progenitor cells during asymmetric neuroblast division. Development 139, 2670–2680 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.