

Abstract
Specialized microenvironments, or niches, provide signaling cues that regulate stem cell behavior. In the Drosophila testis, the JAK-STAT signaling pathway regulates germline stem cell (GSC) attachment to the apical hub and somatic cyst stem cell (CySC) identity. Here, we demonstrate that chickadee, the Drosophila gene that encodes profilin, is required cell autonomously to maintain GSCs, possibly facilitating localization or maintenance of E-cadherin to the GSC-hub cell interface. Germline specific overexpression of Adenomatous Polyposis Coli 2 (APC2) rescued GSC loss in chic hypomorphs, suggesting an additive role of APC2 and F-actin in maintaining the adherens junctions that anchor GSCs to the niche. In addition, loss of chic function in the soma resulted in failure of somatic cyst cells to maintain germ cell enclosure and overproliferation of transit-amplifying spermatogonia.
Keywords: Profilin, Stem cells, APC2
INTRODUCTION
Adult stem cells are responsible for tissue homeostasis, generating short-lived but highly differentiated cell types and replenishing differentiated cells lost to injury. The molecular mechanisms that control stem cell function in vivo are crucial for regenerative medicine, and defects in these mechanisms may underlie degenerative diseases, tumor formation and aging. Short-range signals from the local microenvironment, the stem cell niche, maintain populations of adult stem cells over time through a balance between self-renewal and differentiation. The mechanisms by which stem cells recognize, attach to and orient towards their niche are essential for maintenance of regenerative capacity throughout the life of an individual.
The Drosophila testis stem cell niche supports germline stem cells (GSCs) and somatic cyst stem cells (CySCs), both of which are attached to a group of non-dividing somatic cells: the hub. Hub cells express a secreted ligand, Unpaired, which activates the Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway in both GSCs and CySCs (Kiger et al., 2001; Tulina and Matunis, 2001; Leatherman and Dinardo, 2008). Although a key role of activated STAT in male germ cells may be to maintain GSC-hub attachment (Leatherman and Dinardo, 2010), the STAT targets that mediate attachment are not yet known.
The chickadee gene, which encodes the only Drosophila homolog of profilin, an actin-binding protein that regulates microfilament polymerization (Cooley et al., 1992; Theriot and Mitchison, 1993; Verheyen and Cooley, 1994), was identified in genetic screens as being required for maintenance of early germ cell populations in testes (Castrillon et al., 1993; Gönczy and DiNardo, 1996). Here, we show that the chic locus is bound by activated STAT in Drosophila testes and required cell autonomously in germ cells to maintain GSCs at the hub, probably through effects on cell adhesion. In addition, chic function is required in somatic cyst cells for neighboring germ cells to differentiate.
RESULTS
chic is required cell autonomously for maintenance of germline stem cells in their niche
Loss of function of the single Drosophila profilin homolog, chic, caused failure to maintain male germline stem cells (GSCs) over developmental time, consistent with the effects of chic mutations on adult testes reported by Gönczy and DiNardo (1996). In third instar larvae, GSC number was markedly decreased in chic mutants compared with wild type (Fig. 1). Although null mutant combinations of chic alleles were embryonic lethal (Verheyen and Cooley, 1994; Baum and Perrimon, 2001), chic animals transheterozygous for either the hypomorphic chic11 and the strong loss-of-function chic1320, or for chic1320 and the null chic221 survived to adulthood, so testes from these animals could be scored at larval stages. In wild-type late larval testes, a rosette of 12.0±2.6 GSCs (n=26 testes) surrounded the hub (Fig. 1A). However, only 3.6±2.8 GSCs (n=36 testes) were detected next to the hub in age-matched larval testes from chic11/chic1320 hypomorphs (Fig. 1B), and only 0.7±1.0 GSCs (n=12 testes) were observed in larval testes from chic221/chic1320 strong loss-of-function mutants (Fig. 1C). In most testes from third instar chic221/chic1320 larvae, the earliest germ cells observed were spermatocytes, suggesting that GSCs had been present at earlier stages in development but that GSCs were lost from the testis tip during larval development (Fig. 1C). Consistent with progressive loss of GSCs over time, the number of GSCs touching the hub in chic11/chic1320 hypomorphs dropped from 3.6±2.8 GSCs per testis (n=36 testes) in late third instar larvae to 0.7±1.9 GSCs per testis (n=104 testes) in newly eclosed adults. As noted in other cases of germ cell loss, somatic cells expressing the cyst cell nuclear marker Tj moved in to fill the area adjacent to the hub when GSCs were lost in chic mutants (Fig. 1A′-C′).
Fig. 1.
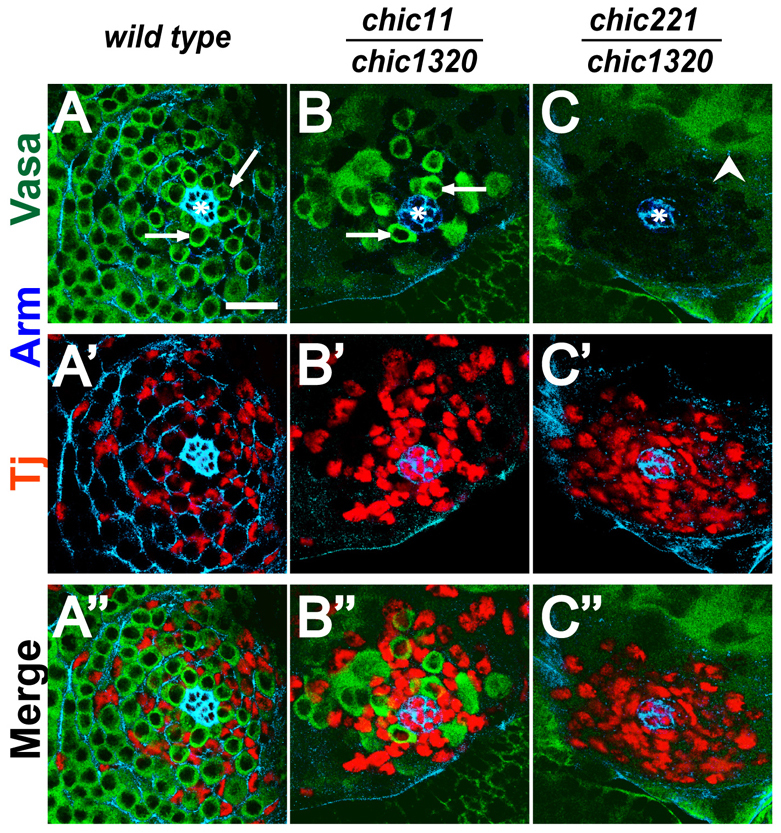
Loss of germline stem cells in chic mutants. (A-C′) Larval testis tips from (A-A′) wild-type, (B-B′) chic11/chic1320 hypomorph, (C-C′) chic221/chic1320 strong loss-of-function animals with anti-Arm/β-catenin (blue) to mark hub cells (asterisk), anti-Vasa (green) to mark germ cells and anti-Tj (red) to mark early cyst cell nuclei. Arrows indicate Vasa-positive cells touching the hub that were scored as GSCs. Arrowhead indicates differentiating spermatocytes. Scale bar: 20 μm.
Analysis of germline clones indicated that chic is required cell autonomously for GSC maintenance. GSCs were made homozygous mutant for chic and simultaneously marked by loss of green fluorescent protein (GFP) by FLP-mediated recombination induced by heat shock. For two different null alleles of chic, GFP-negative chic mutant GSC clones were detected next to the hub at 3 days post-clone induction (dpci) in 80% (chic221) and 96% (chicP5205) of testes. However, the percentage of testes maintaining GFP-negative chic mutant GSC clones next to the hub decreased over time. By 11 dpci, none of the testes contained chic mutant GSC clones (Fig. 2A). By contrast, control GSC clones induced in a genetic background wild type for chic were maintained over the 11-day period of observation (Fig. 2A). The chic mutant germ cells initiated differentiation and progressed to spermatocytes. However, no chic-null spermatid clones were observed, suggesting that mutant spermatocytes failed to complete terminal differentiation (supplementary material Fig. S1). Likewise, when function of chic was knocked down specifically in germ cells throughout development by RNAi under control of nanos-GAL4 at 18°C, testes from newly eclosed adults completely lacked GSCs, similar to the chic strong loss-of-function phenotype (Fig. 2B-G′). Wild-type testes displayed a gradient of differentiating germ cells beginning with GSCs at the apical tip and progressing through spermatogonia and spermatocytes (Fig. 2B,E-E′) to mature spermatid bundles at the distal end of the testis. By contrast, in testes in which chic function had been knocked down in early germ cells by RNAi, elongated spermatid bundles extended into the apical tip (Fig. 2C) in newly eclosed males, and spermatogonia and even spermatocytes were rare (Fig. 2F-F′) or not detected, similar to the phenotype observed in adult testes from chic1320/chic221 strong loss-of-function mutants (Fig. 2D,G-G′).
Fig. 2.
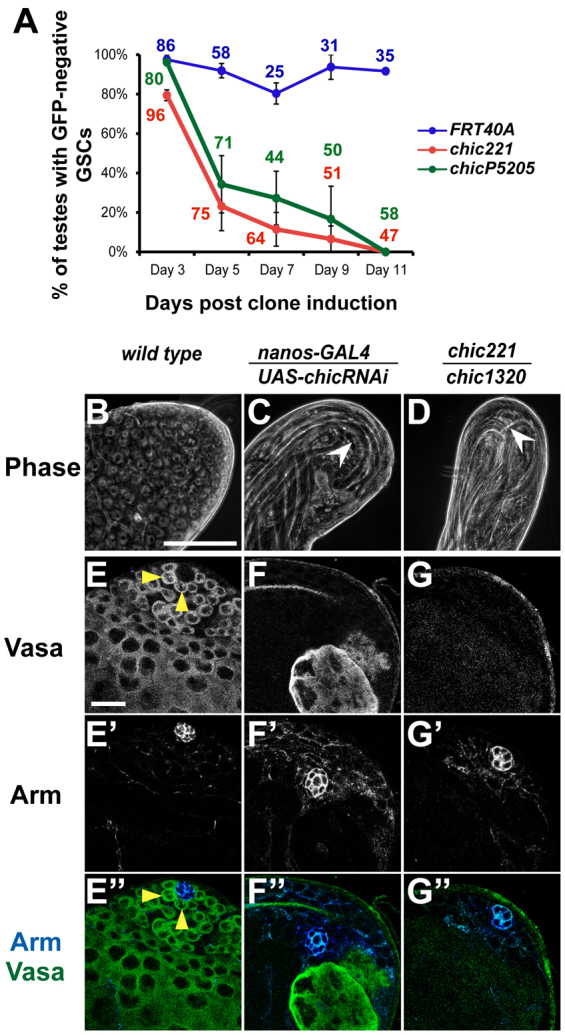
chic is cell autonomously required for maintenance of germline stem cells. (A) Percentage of testes with at least one GFP-negative, homozygous control FRT40A (blue), chic221 (red) or chicP5205 (green) GSC over time after clone induction. Numbers next to data points reflect the number of testes scored for each genotype at each time point. Error bars indicate s.e.m. See also supplementary material Fig. S1. (B-G′) Adult testis tips from (B,E-E′) wild type, (C,F-F′) from nanos-GAL4/UAS-chicRNAi resulting in knockdown of chic in the germ line and from (D,G-G′) chic221/chic1320 strong loss-of-function mutant. (B-D) Phase-contrast images. White arrowheads indicate elongated spermatid bundles. Scale bar: 10 μm. (E-G′) Anti-Vasa to mark germ cells. Anti-Arm to mark hub cells. Yellow arrowheads indicate GSCs abutting the hub. Scale bar: 20 μm.
Overexpression of APC2 in early germ cells rescued loss of GSCs in chic hypomorphs
The requirement of profilin for GSC maintenance raised the possibility that microfilament assembly and/or function might help maintain GSCs in their niche at the testis apical tip. Consistent with this, analysis of GFP-tagged F-actin expressed in GSCs under control of nanos-GAL4 revealed actin-GFP localized to the cell cortex under the GSC-hub interface (Fig. 3A′, arrowhead). Analysis of F-actin by phalloidin staining of wild-type testes also showed F-actin localized to the interface between GSCs and hub cells, juxtaposed with β-catenin/Armadillo (Fig. 3B′). In age-matched larval testes from chic11/chic1320 hypomorphs, F-actin localized to regions where GSCs touched hub cells but F-actin localization was decreased or lost where GSCs were absent (Fig. 3C,C′,C′). In testes completely lacking GSCs from strong loss-of-function chic221/chic1320 larvae, F-actin localization around the perimeter of the hub was completely absent (Fig. 3D,D′,D′). Forced expression of actin-GFP subunits in germ cells of chic11/chic1320 or chic221/chic1320 animals was not sufficient to rescue GSC loss (data not shown), consistent with the known role of profilin in F-actin assembly from G-actin subunits.
Fig. 3.
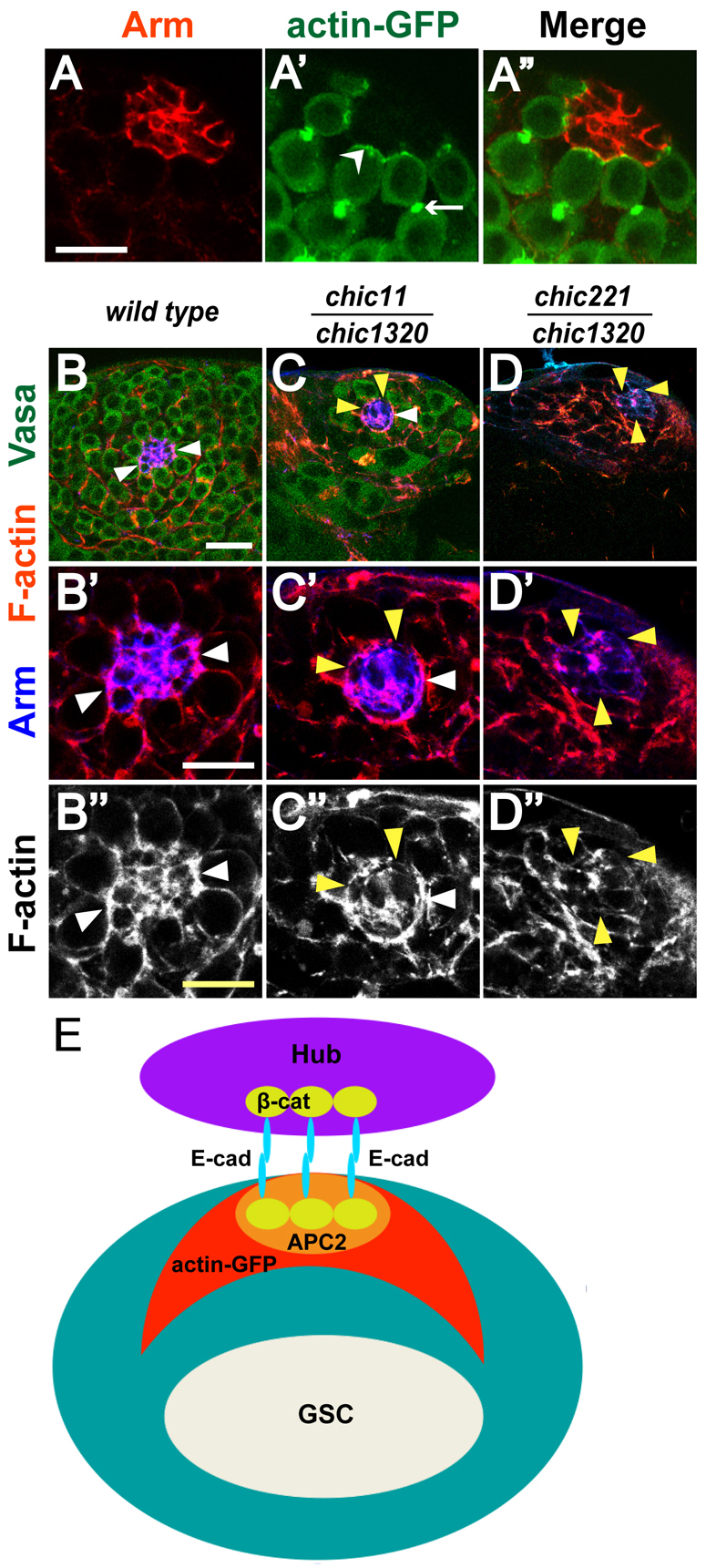
F-actin is enriched at the hub-GSC interface. (A-A′) Localization of actin-GFP in GSCs. Adult testis tip from a nanos-GAL4; UAS-actin-GFP fly stained with (A) anti-Arm (red), (A′) anti-GFP to reveal actin-GFP expression in germ cells. (A′) Merge. Arrowhead indicates GSC-hub interface. Arrow indicates fusome. Scale bar: 10 μm. (B-D′) Larval testis tips from (B-B′) wild type, (C-C′) chic11/chic1320 hypomorph and (D-D′) chic221/chic1320 strong loss-of-function animals with anti-Vasa (green) to mark germ cells, phalloidin to highlight F-actin (red) and anti-Arm (blue) to mark hub cells. White arrowheads indicate F-actin localized to hub-GSC interface. Yellow arrowheads indicate loss of F-actin at the hub perimeter. Scale bars: 20 μm in B-D; 10 μm in B′-D′. (E) Localization patterns of adherens junction components: E-cadherin and β-catenin, APC2 and actin-GFP.
Notably, in control larval testes, the border of the hub in contact with GSCs appeared concave, with V-shaped protrusions between GSCs (Fig. 1A-A′, Fig. 3A′,B-B′, Fig. 4A). In larval testes from chic11/chic1320 hypomorphs, however, the hub border appeared more rounded (Fig. 1B-B′, Fig. 3C-C′, Fig. 4B), suggesting defects in adherence between GSCs and hub cells.
Fig. 4.

APC2 protein is distributed around the cell cortex of chic mutant GSCs. (A-B′′) Larval testis tips from (A-A′′) chic/+ controls and (B-B′′) chic11/chic1320 hypomorphs stained with anti-APC2 (red), anti-Vasa (green) to mark germ cells and anti-Arm (blue) to outline hub cells. (C-D′′) Adult testis tips from males 4 days post-clone induction. (C-C′′) chicP5205 and (D-D′′) chic221 clonal testes stained with anti-APC2 (red), anti-GFP (green), anti-Arm (blue) and anti-Tj (blue) to highlight early cyst cell nuclei. Magenta arrowheads indicate homozygous chicP5205 (C′,C′′) and chic221 (D′,D′′) mutant GSCs compared with heterozygous control GSCs (blue arrowheads). Yellow lines indicate cortical localization of APC2 in GSCs. Asterisk indicates the hub. Scale bars: 10 μm. (E,F) Percentage of GSCs with APC2 localized to hub-GSC interface (yellow) or around GSC cell cortex (blue) in mutant animals (E) or clonal testes (F). Two-tailed P values of Fisher’s exact test are shown.
Loss of profilin function also affected distribution of APC2, one of the fly homologs of the mammalian tumor suppressor protein Adenomatous Polyposis Coli, suggesting that functional chic in GSCs may promote enrichment of APC2 at the hub-GSC interface. GSCs are attached to hub cells by E-cadherin-based adherens junctions, which provide an anchoring platform for concentration of APC2 (Fig. 3E) (Yamashita et al., 2003). Immunofluorescence analysis of testes from chic/+ control third instar larvae revealed APC2 protein localized mainly to the hub-GSC interface in GSCs but with localization observed around the GSC cell cortex in some cells (Fig. 4A′,E). In 66.7% of GSCs in chic/+ control larval testes, APC2 localized to the hub-GSC interface (Fig. 4E). By contrast, in larval testes from chic11/chic1320 hypomorphs, only 9.5% of GSCs scored displayed APC2 localized to the hub-GSC interface (Fig. 4B′,E). Similarly, immunofluorescence analysis of germline clones from adult males 4 dpci demonstrated that 87.5% of homozygous chicP5205 and 84.2% of homozygous chic221 mutant GSC clones showed APC2 distributed around the cell cortex rather than localized to the hub-GSC interface, compared with 46.2% and 38.7%, respectively, of adjacent heterozygous control GSC clones from the same testes (Fig. 4C′,D′,F).
Strikingly, forced expression of APC2 in germ cells rescued male GSC loss in chic hypomorphs. When APC2 was expressed in the germ line of chic11/chic1320 hypomorphs under control of nanos-GAL4, testes from newly eclosed adults had the normal spatial gradient of germline differentiation from early germ cells to mature sperm restored (Fig. 5B), similar to chic/+ controls (Fig. 5C) and in contrast to the loss of early germ cells observed in chic11/chic1320 mutant siblings grown under the same conditions (Fig. 5A). Immunofluorescence revealed Vasa-positive GSCs (4.9±1.6 GSCs per testis; n=114 testes) attached to the hub in the rescued testes (Fig. 5F), compared with 0.7±1.9 GSCs per testis (n=104 testes) in chic11/chic1320 siblings (Fig. 5E). There was no significant difference in the number of Vasa-positive GSCs in chic/+ sibling controls without added APC2 (7.9±1.6 GSCs per testis; n=114 testes) (Fig. 5G) compared with chic/+ sibling controls with forced expression of APC2 (7.9±1.8 GSCs per testis; n=111 testes) (Fig. 5H), indicating that whereas forced expression of APC2 increased GSC number in chic11/chic1320 mutants, ectopic APC2 did not increase GSC number in control testes.
Fig. 5.
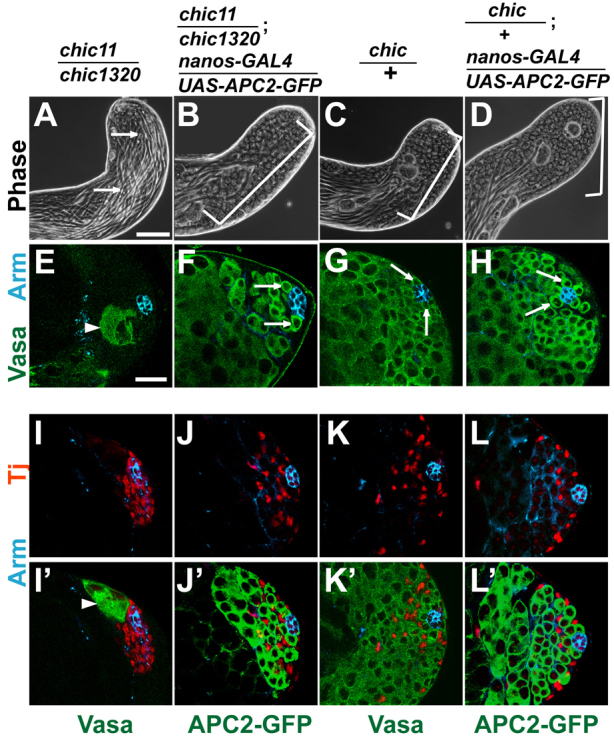
Forced expression of APC2 in the germ line rescued GSC loss in chic hypomorphs. (A-D) Phase images of testes from newly eclosed adults of (A,E,I,I′) chic11/chic1320 hypomorph (arrows indicate elongated spermatid bundles occupy the testis tip), (B,F,J,J′) chic11/chic1320; nanos-GAL4/UAS-APC2-GFP, (C,G,K,K′) chic/+ control and (D,H,L,L′) chic/+; nanos-GAL4/UAS-APC2-GFP testes. Brackets in B-D indicate gradient of germ cells from GSCs at the apical tip differentiating to spermatogonia, spermatocytes and elongated spermatids. (E-H) Immunofluorescence images showing germ cells (green, anti-Vasa) and hub cells (blue, anti-Arm). Arrows indicate GSCs surrounding the hub. (I-L) Testis tips stained with anti-Arm (blue) and anti-Tj (red) to mark early cyst cell nuclei. (I′-L′) Same fields showing either anti-Vasa or anti-GFP (green) to mark germ cells. Arrowheads in E,I′ indicate spermatocytes near apical tip. Scale bars: 20 μm.
Expression of APC2 in the germ line also restored normal distribution of somatic cyst cells. Rather than many Tj-positive cyst cells clustered next to the hub in the absence of germ cells observed in testes from chic11/chic1320 mutants (Fig. 5I,I′), testis tips from chic11/chic1320; nanos-GAL4/UAS-APC2-GFP males had Tj-positive cells dispersed throughout the apical region (Fig. 5J,J′) in a pattern similar to chic/+ controls (Fig. 5K,K′) and chic/+ controls with added APC2 (Fig. 5L,L′). Forced expression of APC2 in germ cells of strong loss-of-function chic221/chic1320 animals failed to rescue GSC loss (data not shown).
Function of profilin/chic was required cell intrinsically in germ cells for proper localization and/or stabilization of the adherens junction component E-cadherin to the hub-GSC interface. Expression of an E-cadherin-GFP transgene specifically in early germ cells using nanos-GAL4 allowed E-cadherin in the plasma membrane of GSCs to be distinguished from E-cadherin in the membrane of hub cells at the hub-GSC interface (Fig. 6). In early germ cells in late third instar larval testes from animals wild-type for chic, the E-cadherin-GFP localized to the GSC-hub interface (Fig. 6A-Aa′′) as observed previously for adult testes (Srinivasan et al., 2012). Forced expression of E-cadherin-GFP in early germ cells of chic11/chic1320 (Fig. 6B-Ba′′) or chic221/chic1320 animals (data not shown) failed to rescue GSC loss. Adult testes dissected from newly eclosed chic11/chic1320 or chic221/chic1320 animals with nanos-GAL4; UAS-E-cad-GFP stained with anti-Vasa lacked early germ cells (data not shown). Likewise, testes from late third instar chic11/chic1320; nanos-GAL4/UAS-Ecad-GFP larvae contained similar GSC numbers (4.75±2.0 GSCs; n=20 testes) to age-matched testes from chic11/chic1320 larvae without expression of E-cadherin-GFP (3.6±2.8 GSCs; n=36 testes, see Fig. 1), further demonstrating that forced expression of E-cadherin was not sufficient to rescue GSC loss in chic mutants.
Fig. 6.

chic is required in germ cells for E-cadherin localization to the hub-GSC interface. (A-Ba′′) Larval testes expressing E-cad-GFP in early germ cells. (A-Aa′′) +/+; nanos-GAL4/UAS-Ecad-GFP controls, (B-Ba′′) chic11/chic1320; nanos-GAL4/UAS-Ecad-GFP animals. Anti-GFP (green) to highlight expression of the E-cad-GFP transgene, anti-Vasa (red) and anti-Arm (blue) to outline hub cells, and anti-Tj (blue) to mark somatic cyst cells. Yellow arrowheads indicate E-cadherin-GFP localized to hub-GSC interface. Scale bars: 20 μm in A-A′′,B-B′′; 10 μm in Aa-Aa′′,Ba-Ba′′.
Notably, E-cadherin-GFP failed to localize to the GSC-hub interface in chic mutant GSCs. Analysis of 78 control GSCs in nine late larval testes showed E-cadherin-GFP localized to the hub-GSC interface (Fig. 6A,Aa). By contrast, 96.8% of chic11/chic1320; nanos-GAL4/UAS-Ecad-GFP mutant GSCs analyzed (92/95) in 20 late larval testes displayed no detectable E-cadherin-GFP signal (Fig. 6B,Ba).
Taken together, these observations suggest that F-actin underlying adherens junctions may help maintain GSC-hub attachment by localizing and anchoring junctional components such as E-cadherin and APC2, and that lowered activity of the actin regulator profilin in GSCs may weaken adherens junctions and allow somatic cells to outcompete germ cells for association with the hub. Increased levels of APC2 in germ cells may strengthen adherens junctions between GSCs and the hub, making up for partial impairment of F-actin assembly in chic hypomorphs.
The transcription factor STAT binds near the chic promoter region
The chic gene has three predicted alternate transcription start sites (TSS) (Fig. 7A), two of which were shown by 5′ RACE to be active in upd-overexpressing testes, which contain an abundance of GSCs and CySCs. A reporter construct containing a genomic DNA fragment extending from the ATG of chic to 2256 bp upstream of the ATG and including all three TSSs (Fig. 7A) was sufficient to drive expression of GFP in both germline and somatic lineages in the testis (Fig. 7C-C′′), consistent with the ubiquitous role of profilin in regulating the actin cytoskeleton.
Fig. 7.

The STAT transcription factor binds near the chic promoter region. (A) Screen shot of chic locus from UCSC Genome Browser. Black boxes indicate exons. Hatched lines indicate introns. Purple circles indicate transcription start sites active in upd-overexpressing testes. Red bar indicates consensus STAT site. Black square indicates quantitative PCR (qt-PCR) amplicon. chic reporter containing a genomic DNA fragment extending from the ATG of chic to 2256 bp upstream, EGFP-coding sequence and SV40 3′UTR/terminator sequence. (B) Average enrichment for P-STAT (red) or normal rabbit serum (green) binding in the region flanking the STAT site relative to thioredoxin for three biological replicates. Error bars indicate s.e.m. P values calculated using the two-tailed Student’s t-test. (C-D′′) Adult testes from chic-EGFP transgenic flies containing (C-C′′) wild-type or (D-D′′) mutated consensus STAT-binding motifs stained with anti-GFP to highlight reporter expression, with anti-Vasa to mark germ cells, with anti-Tj to mark early cyst cell nuclei and with anti-FasIII to mark hub cells (blue asterisk). Scale bar: 50 μm.
Chromatin immunoprecipitation (ChIP) analysis suggested that chic may be a direct target of the transcription factor STAT in GSCs and/or CySCs. A consensus STAT-binding motif was present in the chic promoter region, just downstream of the first TSS (Fig. 7A). ChIP from extracts of upd-overexpressing testes with antibodies against STAT (P-STAT) followed by quantitative PCR showed 13.7±3.8-fold enrichment of the genomic region spanning the consensus STAT motif relative to a control gene thioredoxin, whereas ChIP with normal rabbit serum showed no enrichment (0.8±0.2) relative to thioredoxin (Fig. 7B). However, mutation of the STAT motif in the reporter construct did not eliminate E-GFP expression in GSCs and CySCs in vivo (Fig. 7D-D′′), suggesting either that STAT is not the only transcriptional regulator of chic expression in stem cells, or STAT may direct an increase in the levels of chic undetected by immunostaining of the GFP reporter.
Furthermore, forced expression of profilin in stat-depleted GSCs failed to rescue loss of attachment of GSCs to the apical hub, suggesting that chic is not the only important target of STAT for GSC attachment. To evaluate the role of STAT in chic-mediated GSC maintenance, germline-specific RNAi was performed to knock down function of stat in GSCs while profilin was ectopically expressed at the same time (supplementary material Fig. S2). Flies were grown to adulthood at 18°C then shifted to 30°C to induce expression of the RNAi hairpin. Testes were dissected at 14 days post-shift. As previously reported (Leatherman and Dinardo, 2010), stat-depleted GSCs detached from the hub and were displaced by somatic cells that surrounded the hub (supplementary material Fig. S2A-A′). stat-depleted GSCs in which profilin was overexpressed also failed to maintain attachment to the hub and were found displaced away from the hub by somatic cells (supplementary material Fig. S2B-B′). Similarly, forced expression of APC2 in GSCs failed to rescue loss of GSC attachment to hub cells (supplementary material Fig. S2C-C′), suggesting that neither profilin nor APC2 is sufficient to maintain stat-depleted GSCs at the hub.
Loss of chic in somatic cyst cells resulted in germ cell over-proliferation
Tissue-specific knockdown by RNAi revealed that loss of chic function in somatic cyst cells of the testis caused germ cells to over-proliferate at the expense of differentiation. When function of chic was knocked down in somatic cells by driving expression of a hairpin RNA directed against chic under control of the somatic-specific driver c587-GAL4, testes became filled with small cells. Flies were grown at 18°C until eclosion then adult males were shifted to 30°C to upregulate expression of the RNA hairpin. At day 1 post-shift, all stages of spermatogenesis were observed by phase-contrast microscopy. However, by days 4/5 post-shift, spermatocytes and mature spermatids were markedly reduced in number and testes were filled with clusters of small, DAPI-bright cells (Fig. 8B,D). Remaining spermatocyte cysts were occasionally found interspersed between the clusters of small cells. Testes expressing the chicRNAi hairpin sometimes appeared wider than sibling controls, likely due to the massive numbers of small cells.
Fig. 8.
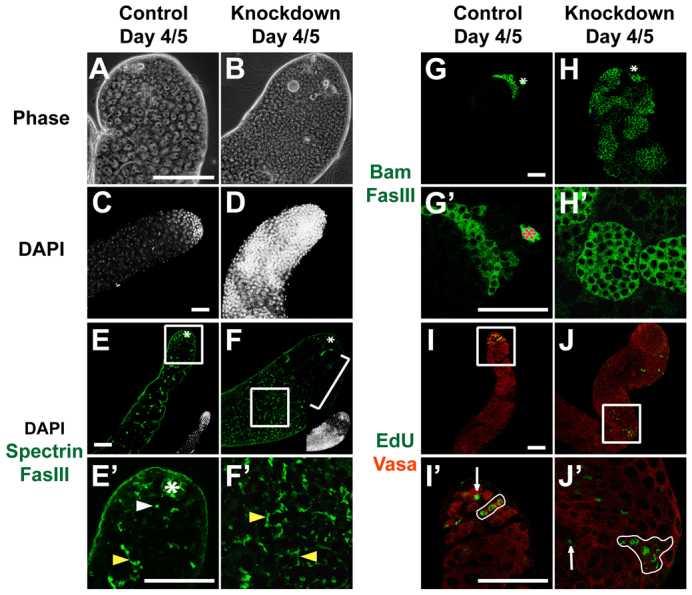
Loss of chic function in the soma caused germ cell over-proliferation. Adult testes from (A,C,E,E′,G,G′,I,I′) c587-GAL4;+ controls and (B,D,F,F′,H,H′,J,J′) c587-GAL4; UAS-chicRNAi animals resulting in knockdown of chic in somatic cyst cells. (A,B) Phase-contrast images of testis tips. Scale bar: 10 μm. (C-J′) Immunofluorescence images. Scale bars: 50 μm. (C,D) DAPI highlights cell nuclei. (E,F) Anti-FasIII to outline hub cells (asterisk) and anti-Spectrin to show fusomes (green). Insets show DAPI versions of images. Rectangle indicates region magnified in E′,F′. Bracket indicates spermatocytes. (E′,F′) High magnification view of different confocal plane of testes in E and F. White arrowhead indicates dot fusome. Yellow arrowheads indicate branched fusomes. (G,H) Anti-Bam to highlight spermatogonial cysts (green). (G′,H′) Bam-positive germ cells (green) at higher magnification. (I-J′) Anti-Vasa (red) to mark germ cells and EdU (green) to mark cells in S phase. Rectangle indicates region magnified in I′,J′. Arrows indicate single germ cells in S phase. Loops indicate clusters of germ cells undergoing S phase in synchrony.
Fusome morphology, expression of cytoplasmic Bam and synchronous cell cycle progression all indicated that transit-amplifying germ cells failed to stop proliferation and differentiate into spermatocytes when profilin function was knocked down in somatic cyst cells. Although immunostaining of control testes with anti-Spectrin showed GSCs and gonialblasts with dot fusomes, and spermatogonial and spermatocyte cysts with branched fusomes (Fig. 8E,E′), the small cells that accumulated in c587-GAL4; UAS-chicRNAi animals mostly contained branched fusomes (Fig. 8F,F′), indicating their identity as transit-amplifying spermatogonia rather than GSCs or gonialblast-like cells.
Likewise, the spermatogonial protein, bag of marbles (Bam), normally expressed in 4-, 8- and 16-cell spermatogonial cysts but not detected in GSCs, gonialblasts or spermatocytes in controls (Fig. 8G,G′), was expressed in the small cells that accumulated throughout the testis in c587-GAL4; UAS-chicRNAi animals (Fig. 8H,H′). Consistent with failure of spermatogonia to cease mitotic division after loss of chic function in somatic cyst cells, analysis of cells in S phase after a short pulse of 5-ethynyl-2′-deoxyuridine (EdU), showed EdU incorporation in germ cells far away from the apical tip in testes from c587-GAL4; UAS-chicRNAi animals, in contrast to EdU incorporation only near the testis apical tip in GSCs, 2-, 4- and 8-cell spermatogonial cysts in controls (Fig. 8I,I′). EdU-positive germ cells away from the tip in c587-GAL4; UAS-chicRNAi animals included singletons and doublets, suggesting some cells resembled GSCs or gonialblasts. However, the majority of EdU-positive germ cells divided in 4-, 8-, 16- and >16-cell clusters (Fig. 8J,J′), indicating spermatogonial cell identity and failure to exit the transit-amplifying program.
Function of chic is required for maintenance of the somatic cyst cell lineage
Testes from c587-GAL4; UAS-chicRNAi adults shifted to restrictive temperature showed loss of cyst cells over time. By day 4/5 post-shift, zinc-finger homeodomain-1 (Zfh-1)-positive somatic cyst stem cells (CySCs) in testes from c587-GAL4; UAS-chicRNAi adults were mostly absent (Fig. 9B′) compared with sibling controls (Fig. 9A′). Tj-positive cyst cells also decreased in number and sometimes exhibited abnormal morphology: nuclei appeared larger than normal and had de-condensed chromatin (Fig. 9B′). The number of Eya-positive cyst cells also declined (Fig. 9B′′) compared with sibling controls (Fig. 9A′′).
Fig. 9.
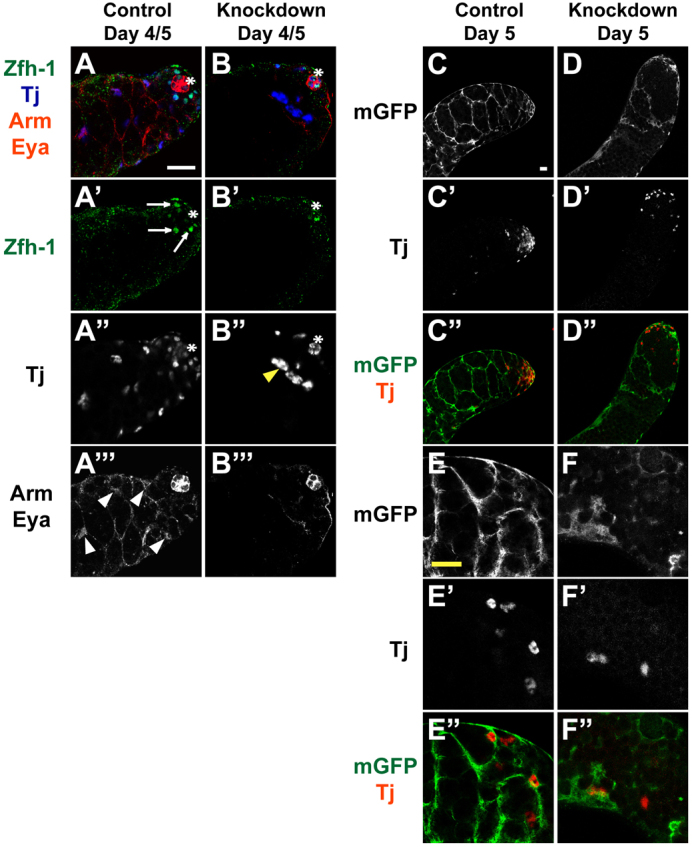
chic is required for maintenance of the somatic cell lineage. (A-B′′) Testes from (A) c587-GAL4;+ controls and (B) c587-GAL4; UAS-chicRNAi animals. (A,B) Merged images showing somatic cell markers. anti-Zfh-1 (green) to mark somatic cyst stem cell (CySC) nuclei (arrows in A′). anti-Tj (blue) to label early cyst cell nuclei. Yellow arrowhead indicates aberrant cyst cells. anti-Arm and anti-Eya (red) mark hub cells (asterisk) and late cyst cells (white arrowheads), respectively. (C-F′) Testes from (C-E′) c587-GAL4; UAS-mGFP/+ controls and (D-F′) c587-GAL4; UAS-mGFP/UAS-chicRNAi animals. Anti-GFP (green) highlights membrane-bound GFP in the cell membranes of cyst cells and anti-Tj (red) labels early cyst cell nuclei. Scale bars: 20 μm. See supplementary material Figs S3 and S4.
Somatic cells lacking chic function were unable to maintain germ cell enclosure, visualized by labeling cyst cell membranes using a membrane-bound UAS-Green Fluorescent Protein (UAS-mGFP). UAS-chicRNAi and UAS-mGFP were both expressed under control of somatic-specific c587-GAL4, enabling evaluation of cyst cell enclosure of germ cells. In control testes, GFP was detected on intact, continuous membranes of somatic cells surrounding germ cells (Fig. 9C,C′,E,E′). By contrast, in testes from c587-GAL4/UAS-mGFP; UAS-chicRNAi/+ animals held at restrictive temperature for 5 days, the membrane-targeted GFP revealed either truncated, diffuse strands or complete failure to surround the clusters of small germ cells (Fig. 9D,F) whether a Tj-positive somatic cell was present or not (Fig. 9D′,F′), suggesting cyst cell cytoplasmic extensions were fragmented, not established or had disintegrated. Similar results were obtained by staining c587-GAL4; UAS-chicRNAi testes for Arm, which labels the cell membranes of cyst cells (Fig. 9A′′,B′′; supplementary material Fig. S3A′,B′).
DISCUSSION
Profilin/Chickadee in the germ line
Chickadee, which encodes the only Drosophila profilin homolog, is required cell intrinsically for GSC maintenance in the testis. As profilin is a regulator of actin filament polymerization (Cooley et al., 1992; Theriot and Mitchison, 1993; Verheyen and Cooley, 1994) and filamentous actin (F-actin) plays a crucial role in the development and stabilization of cadherin-catenin-mediated cell-cell adhesion (Desai et al., 2013; Nola et al., 2011; Jamora and Fuchs, 2002), profilin likely maintains attachment of Drosophila male GSCs to the hub through its effect on F-actin, which concentrates at the hub-GSC interface where localized adherens junctions anchor GSCs to hub cells. We propose that profilin-dependent stabilization of F-actin at the GSC cortex next to the hub may help localize E-cadherin and APC2 to the junctional region. E-cadherin and APC2 in turn may recruit β-catenin/Armadillo, stabilizing the adherens junctions that attach GSCs to the hub. Chickadee may thus facilitate maintenance of GSCs through a cascade of interactions leading to localization and/or retention of both E-cadherin and β-catenin at the hub-GSC interface.
E-cadherin plays a crucial role in maintaining hub-GSC attachment. GSC clones mutant for E-cadherin are not maintained (Voog et al., 2008; Inaba et al., 2010). In addition, germline overexpression of E-cadherin delayed GSC loss in stat-depleted GSCs (Leatherman and Dinardo, 2010). Our results indicate that profilin function is required in GSCs for proper localization of E-cadherin to the hub-GSC interface. Several studies have shown that the actin cytoskeleton plays a crucial role in assembly and stability of adherens junctions (Carramusa et al., 2007; Chu et al., 2004; Imamura et al., 1999). A favored model in the field is that actin filaments indirectly anchor and reinforce E-cadherin-mediated cell junctions by forming an intracellular scaffold for E-cadherin molecules (Lambert et al., 2007; Mège et al., 2006). Indeed, binding to F-actin stabilized E-cadherin and promoted its clustering (Hong et al., 2013). Furthermore, the actin cytoskeleton participates in proper localization of E-cadherin molecules to cell-cell contacts (Collinet and Lecuit, 2013). In chic/profilin mutant GSCs, disruption of actin polymerization at the cell cortex leading to local F-actin disorganization may destabilize E-cadherin and reduce its ability to localize to the GSC-hub junction, form clusters and build adequate adherens junctions.
Destabilization of E-cadherin may contribute to the mislocalization of APC2 seen in chic mutant GSCs, as E-cadherin recruits APC2 to cortical sites in GSCs (Inaba et al., 2010). Raising possibilities of a more direct link, actin filaments have been shown to be required for association of APC2 with adherens junctions in the Drosophila embryo and ovary (Yu et al., 1999; Townsley and Bienz, 2000). Treatment of embryos with actin-depolymerizing drugs resulted in complete delocalization of APC2 from adhesive zones and diffuse APC2 staining throughout the cell (Townsley and Bienz, 2000). Moreover, in ovaries of chic1320/chic221 females, APC2 was substantially delocalized from the plasma membranes of nurse cells and their ring canals, and increased levels of cytoplasmic APC2 staining were observed (Townsley and Bienz, 2000). Similarly, we found that APC2 was delocalized from the hub-GSC interface in larval testes of chic11/chic1320 hypomorphs.
In several studies, delocalization of APC2 from junctional membranes correlated with detachment of β-catenin/Armadillo from adherens junctions. APC2 co-localizes with Armadillo and E-cadherin at adherens junctions of Drosophila epithelial cells (Yu et al., 1999), nurse cells in Drosophila ovaries (Townsley and Bienz, 2000) and at the hub-GSC interface in Drosophila testes (Yamashita et al., 2003). Disruption of APC2 function resulting in significant reduction of junctional APC2 was accompanied by delocalization of junctional Armadillo and increased levels of free cytoplasmic Armadillo in embryonic epithelial cells (Yu et al., 1999) and ovaries (Townsley and Bienz, 2000). In the Townsley and Bienz study, which used chic1320/chic221 strong loss-of-function mutants, the delocalizing effect on junctional Armadillo was variable, presumably due to incomplete penetrance of chic mutant effects. Although we did not observe significant disruption in Armadillo staining along the hub-GSC interface of testes from chic hypomorphs, this may be due to incomplete penetrance. In addition, the Armadillo protein detected could be localized to the cortex of hub cells rather than GSCs.
Our finding that germline specific overexpression of APC2 in chic11/chic1320 hypomorphs partially rescued GSC loss is consistent with a previously proposed model that actin filaments shuttle APC2 to adherens junctions and APC2 in turn recruits cytoplasmic Armadillo to junctional membranes, reinforcing the adherens junctions (Townsley and Bienz, 2000). It is possible that in chic11/chic1320 hypomorphs, residual actin filaments associated with adherens junctions between the hub and GSCs are sufficient to shuttle the increased amounts of cytoplasmic APC2 to adherens junctions. This APC2 may in turn recruit free cytoplasmic Armadillo to the hub-GSC interface, locally stabilizing the adherens junctions and anchoring GSCs to their niche. Notably, however, germline specific overexpression of APC2 in testes of strong loss-of-function chic1320/chic221 mutants failed to rescue GSC loss. Thus either, adequate levels of actin filament polymerization may be required for the proposed translocation of junctional proteins to the plasma membrane, or APC2 function/localization may not be the only or even the major cell-autonomous target of profilin function important for maintaining GSCs. Indeed, loss of APC2 function did not lead to GSC loss (Yamashita et al., 2003). We suggest that the localized cortical F-actin underlying adherens junctions at the GSC-hub interface, best candidate for the most direct target of chic function, strongly stabilizes adherens junctions between GSCs and the hub, with high levels of cortical APC2 able to in part make up for weak chic function by also stabilizing adherens junctions.
Maintenance of hub-GSC attachment appears to be a key role of STAT in GSCs (Leatherman and Dinardo, 2010). Our finding that STAT binds to a site near the upstream promoter of the chic gene raises the possibility that STAT might foster GSC attachment to the hub in part by ensuring high levels of transcription of profilin in GSCs. However, activation of STAT is clearly not the only regulatory influence on profilin expression as profilin is an essential gene expressed in many cell types, including those in which STAT is not active or detected. It is likely that transcription factors other than STAT turn on profilin expression in many cell types and that STAT acts along with other regulators to reinforce profilin expression in GSCs and CySCs. Conversely, overexpression of profilin was not sufficient to re-establish attachment of stat-depleted GSCs, suggesting that STAT probably regulates a number of genes to ensure that GSCs remain within the stem cell niche.
Loss of chic in the soma affects germ cells
Loss of chic function in somatic cyst cells impaired the ability of cyst cells to build and/or maintain the cytoplasmic extensions through which they embrace and enclose spermatogonial cysts. Two somatic cyst cells normally surround each gonialblast and enclose its mitotic and meiotic progeny throughout Drosophila spermatogenesis. The cyst cells co-differentiate with the germ cells they enclose (Gönczy and DiNardo, 1996). Several lines of evidence support the model that either the ability of somatic cyst cells to enclose germ cells or their ability to send signals to adjacent germ cells is important to restrict proliferation and promote differentiation of germ cells (Kiger et al., 2000; Tran et al., 2000; Lim and Fuller, 2012). In either case, activation of EGFR in cyst cells is required for cyst cells to enclose germ cells and/or send the signals for germ cells to differentiate (Kiger et al., 2000; Tran et al., 2000; Sarkar et al., 2007). The similarities in phenotype between loss of chic function and loss of EGFR activation in somatic cyst cells raise the possibility that chic/profilin may act downstream of activated EGFR to modulate the actin cytoskeleton for the remodeling of cyst cells to form or maintain the cytoplasmic extensions that enclose germ cells. Indeed, activated EGFR is known in other systems to tyrosine phosphorylate phospholipase C-γ1 (PLC-γ1), a soluble enzyme in quiescent cells like daughter cyst cells, activating it to catalyze hydrolysis of the membrane lipid phosphatidylinositol 4,5-bisphosphate (PIP2) (Goldschmidt-Clermont et al., 1991; Chen et al., 1994; Chou et al., 2002), which binds profilin protein with high affinity, which inhibits the interaction between profilin and actin (Goldschmidt-Clermont et al., 1991). The hydrolysis of PIP2 by activated PLC-γ1 results in localized release of profilin and other actin-binding proteins, enabling them to interact with actin and participate in cytoskeletal rearrangement and membrane protrusion (Chen et al., 1996; Chou et al., 2002). Thus, based on biochemical analysis in other systems, a link between EGFR activation and profilin leading to local remodeling of the actin cytoskeleton is plausible in somatic cyst cells, although it remains to be directly tested.
MATERIALS AND METHODS
Fly stocks and husbandry
Flies were grown on standard cornmeal molasses agar medium with crosses performed at 25°C unless indicated otherwise. Strains are described in FlyBase (http://flybase.org) or otherwise specified. y1w1 flies or sibling controls were used as wild type. P{A92}chic11 [Bloomington Drosophila Stock Center (BDSC)] is a hypomorphic allele generated by P-element insertion (Castrillon et al., 1993). P{PZ}chic01320 (BDSC), a strong loss-of-function allele generated by P-element insertion (Cooley et al., 1992), did not behave as strongly as a null in heteroallelic combinations in our studies. The null allele chic221 (BDSC) is a deletion of 5′ non-coding and some chic-coding sequences (Verheyen and Cooley, 1994). chic05205a is a null allele (Baum and Perrimon, 2001). The chic UAS-dsRNA strain (#102759) was obtained from the Vienna Drosophila RNAi Center (VDRC). To drive expression of UAS transgenes in early germ cells Sco/CyO; nanos-Gal4VP16 (Van Doren et al., 1998) or chic1320/CyO; nanos-Gal4VP16 virgin females were crossed to chic11/CyO; UAS-transgene or chic221/CyO; UAS-transgene males at 25°C. Transgenes included UAS-APC2-GFP (BDSC), UAS-Actin-GFP (BDSC), UAS-EGFP-chic from A. Prokop (Gonçalves-Pimentel et al., 2011); and UAS-DEFL#6-1, i.e. UAS-Ecadherin-GFP (Oda and Tsukita, 1999), from the Drosophila Genetic Resource Center, Kyoto.
Immunofluorescence
Immunostaining was performed as described previously (Srinivasan et al., 2012). Primary antibodies used were: rabbit anti-GFP (1:500-1:3000, Invitrogen); sheep anti-GFP (1:1000, AbD Serotec); rabbit anti-APC2 (1:5000, from M. Bienz, Cambridge, UK); rabbit anti-zinc-finger homeodomain-1 (Zfh-1) (1:5000, from R. Lehmann, New York, USA); guinea pig anti-Traffic Jam (Tj) (1:5000, from D. Godt, Toronto, Canada); goat anti-Vasa (1:100-1:200, DC-13 from Santa Cruz Biotech); mouse anti-Chickadee chi 1J [1:5, Developmental Studies Hybridoma Bank (DSHB)]; mouse anti-Armadillo (Arm) (1:10, DSHB); mouse anti-3A9 Alpha Spectrin (1:10, DSHB); mouse anti-Fas III 7G10 (1:10, DSHB); mouse anti-Eya 10H6 (1:10, DSHB); mouse anti-Bam (1:5, DSHB); and rat E-cadherin (1:40, DSHB). Secondary antibodies (Jackson Immunolabs) were used at 1:200-1:500. All images were taken using a Leica SP2 AOBS confocal system and processed with Adobe Photoshop.
Clonal analysis
For clonal analyses using FLP/FRT mitotic recombination, y,w, hs-FLP122; FRT40A, Ubi-GFP/Cyo virgin females were crossed to w; FRT40A/CyO or chic221, FRT40A/CyO or chicP5205, FRT40A/CyO males at 25°C. Clones were induced by heat shocking pupae for 2 hours on days eight and nine post-lay in a 37°C circulating water bath. Male flies were collected 2 days post-clone induction (dpci). The following genotypes were scored: y,w, hs-FLP122; FRT40A, Ubi-GFP/FRT40A (control clones were wild type for chic function); y,w, hs-FLP122; FRT40A, Ubi-GFP/chic221, FRT40A (chic-null clones) and y,w, hs-FLP122; FRT40A, Ubi-GFP/chicP5205, FRT40A (chic-null clones). GSCs divide approximately every 12-16 hours (Yadlapalli et al., 2011; Tran et al., 2012); therefore, germline clones were assessed up to 11 dpci, allowing for multiple rounds of GSC self-renewal to have occurred. GSC clones were defined as GFP-negative, Vasa-positive cells touching the hub. APC2 localization in GSC clones was evaluated in testes dissected from 2-day-old males (4 dpci). GSCs with APC2 detected at the hub-GSC interface but not along other regions of the cell cortex were scored as GSCs with APC2 localized to hub-GSC interface. GSCs with APC2 detected along regions of the cortex whether or not also at the hub-GSC interface were scored as GSCs with APC2 localized around the cell cortex. All clonal analyses were repeated three independent times with data shown an average of the independent experiments.
Inducible RNAi knockdowns
For germline RNAi knockdown, w, UAS-dicer2;nanos-Gal4VP16/TM3Sb virgin females were crossed to w; UAS-chicRNAi (VDRC 102759) males at 18°C, allowing the RNAi hairpin to be expressed throughout progeny development. Testes from newly eclosed males were analyzed in live squashes by phase contrast and in immunostained preparations by confocal microscopy. Knockdown of chic expression in early germ cells by RNAi appeared effective because it caused loss of GSCs, similar to the results seen in homozygous mutant clones. We could not assess Chic protein or RNA levels because early germ cells were lost in the knockdown testes. For somatic RNAi knockdown, c587-Gal4; Sco/CyO; tubGAL80ts/TM6B virgin females were crossed to w; UAS-chicRNAi males at 18°C. Progeny were shifted to 30°C upon eclosion. Testes from 4- to 5-day-old adults were analyzed. To test RNAi efficiency in somatic cells, immunostaining with anti-Profilin was compared between knockdown and control testes in which cell membranes of somatic cyst cells were marked by expression of a membrane-targeted GFP. No anti-Profilin staining was detected in cell membranes of cyst cells in knockdown testes even where membrane-GFP indicated that somatic cell processes were present. By contrast, anti-Profilin staining was observed in cell membranes of cyst cells in control testes (supplementary material Fig. S4).
5′ RACE
Total RNA was isolated from upd overexpressing testes using TRIzol Reagent (Invitrogen). 5′ RACE was performed using the FirstChoice RLM-RACE Kit (Ambion). Nested PCR reactions were performed using gene-specific primer sequences: 5′-cacgaaattggcagggagac-3′ and 5′-tcttgccagctcatggtgct-3′. Gel-purified PCR products were ligated into sequencing vectors using TOPO-TA (Invitrogen), and positive clones sequenced.
Chromatin immunoprecipitation (ChIP) from upd overexpressing testes
upd overexpressing testes (w; UAS-upd/+; nos-Gal4VP16/+) were dissected into Schneider’s Media on ice, cross-linked in 1% formaldehyde in Schneider’s Media for 15 minutes at 37°C, washed twice in 1×PBS and pellets were flash-frozen in liquid nitrogen then stored at -80°C.
ChIP was performed using a modified version of the ChIP protocol described previously (Chen et al., 2005) with 100 testis pairs per biological replicate. Lysates were sonicated 4×10 seconds on ice with an output setting of 1 and constant duty cycle, with ≥50 seconds rest between pulses, using a Branson Sonifier 250 fitted with a 1/8 inch stepped tip. Sheared chromatin ranged from 250 bp to 1 kb. Following dilution in ChIP Dilution Buffer, 1% of whole-cell extract by volume was reserved for input DNA extraction. Pooled lysates were precleared by incubation with 150 μl 1:1 Protein A agarose beads blocked with salmon sperm DNA for 1 hour at 4°C. Following overnight incubation with anti-P-STAT or non-specific rabbit IgG (for mock pull-downs), lysates were incubated with 30 μl 1:1 Protein A agarose beads for 1 hour at 4°C, the beads were washed, the bound DNA was recovered, pooled eluates were reverse cross-linked (Chen et al., 2005) and DNA was extracted twice with 200 μl Ultrapure Phenol:Chloroform:Isoamyl Alcohol (25:24:1) and recovered by ethanol precipitation. Samples were resuspended in 30 μl 10 μM Tris-HCl (pH 7.5) and yield was measured using a Nanodrop ND-1000 spectrophotometer.
Quantitative PCR
ChIP DNA was analyzed by q-PCR to assess relative enrichment at predicted STAT-binding sites versus an unoccupied site in the thioredoxin gene. Primers that amplify 80-100 bp genomic DNA centered over the predicted target or non-target site were designed. Primer sequences listed 5′ to 3′ included: chic target (S), accaccccacgttcagtaag; chic target (A), cagcgtgtgaacagtgtgag; thioredoxin (S), caaattcgcatgctgtcagt; and thioredoxin (A), ggctgctggctgttctttac. Production of real-time PCR products was monitored using SYBR Green. Reactions containing either 10 ng input or ChIP DNA were run in triplicate using the default 40-cycle PCR protocol with dissociation curves on an Applied Biosystems 7300 thermocycler, and data analyzed using Microsoft Excel. Enrichment, defined as (IP/Input)target site/(IP/Input)non-target site, where the non-target site is thioredoxin, was calculated independently for P-STAT and IgG reactions.
Transgenic reporters
pattB-chic-EGFP consisted of genomic region (chr2L: 5979503-5981759) extending from the ATG of chic to 2,256 bp upstream PCR-amplified from y1w1 genomic DNA, subcloned upstream of E-GFP coding sequence and SV-40 3′UTR/terminator. pattB-Δchic-EGFP was derived from pattB-chic-EGFP by amplifying the chic sequence in two segments using primers containing engineered mutations that mutate the predicted STAT-binding site from the consensus TTCCAGGAA to GGACAGTCG. The two resulting PCR products were ligated, subcloned into pBS-KS+, sequenced to ensure the necessary mutation was introduced, then moved into pattB-EGFP. Clones were sequenced following each subcloning step to ensure that sequences, orientation and number of inserts were correct. Transgenic fly lines were generated by BestGene (Chino Hills, CA) using the PhiC31 integrase system (http://flyc31.frontiers-in-genetics.org/). Independent transgenic lines containing insertions on chromosomes X, 2 and 3, respectively, were analyzed. Immunostaining was performed on testes carrying wild-type and mutated versions of the transgenic reporter constructs, and samples were imaged sequentially using the same settings on a Leica SP2 AOBS confocal microscope.
Supplementary Material
Acknowledgments
We thank the Bloomington Drosophila Stock Center, Drosophila Genetic Resource Center and Vienna Drosophila RNAi Center for fly stocks; and the Developmental Studies Hybridoma Bank for antibodies. We thank Anthony Mahowald, Sangbin Park and members of the Fuller lab for helpful discussions and advice.
Footnotes
Competing interests
The authors declare no competing financial interests.
Author contributions
A.R.S., A.S.C., Y.M.Y. and E.L.D. performed experiments. A.S.C. and E.L.D. designed and interpreted ChIP experiments. A.R.S. and M.T.F. designed experiments, interpreted data and prepared the manuscript.
Funding
This work was supported by the National Institutes of Health (NIH) Interdisciplinary Training Grant in Regenerative Medicine Predoctoral Fellowship [R90 DK071508-05] and by the Stanford University Diversifying Academia, Recruiting Excellence Doctoral Fellowship to A.R.S.; by the Robert Wood Johnson Foundation Harold Amos Medical Faculty Development Program to A.C.S.; by the Searle Scholar Program and NIH [R01 GM086481 to Y.M.Y.]; by an American Heart Association Western States Predoctoral Fellowship, Stanford Graduate Fellowship and National Science Foundation Predoctoral Fellowship to E.L.D.; and by the NIH [R01 GM080501 to M.T.F.]. M.T.F. acknowledges the Reed-Hodgson Professorship in Human Biology at Stanford University, which partly supported this work. Deposited in PMC for release after 12 months.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.101931/-/DC1
References
- Baum B., Perrimon N. (2001). Spatial control of the actin cytoskeleton in Drosophila epithelial cells. Nat. Cell Biol. 3, 883–890 [DOI] [PubMed] [Google Scholar]
- Carramusa L., Ballestrem C., Zilberman Y., Bershadsky A. D. (2007). Mammalian diaphanous-related formin Dia1 controls the organization of E-cadherin-mediated cell-cell junctions. J. Cell Sci. 120, 3870–3882 [DOI] [PubMed] [Google Scholar]
- Castrillon D. H., Gönczy P., Alexander S., Rawson R., Eberhart C. G., Viswanathan S., DiNardo S., Wasserman S. A. (1993). Toward a molecular genetic analysis of spermatogenesis in Drosophila melanogaster: characterization of male-sterile mutants generated by single P element mutagenesis. Genetics 135, 489–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P., Xie H., Sekar M. C., Gupta K., Wells A. (1994). Epidermal growth factor receptor-mediated cell motility: phospholipase C activity is required, but mitogen-activated protein kinase activity is not sufficient for induced cell movement. J. Cell Biol. 127, 847–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P., Murphy-Ullrich J. E., Wells A. (1996). A role for gelsolin in actuating epidermal growth factor receptor-mediated cell motility. J. Cell Biol. 134, 689–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Hiller M., Sancak Y., Fuller M. T. (2005). Tissue-specific TAFs counteract Polycomb to turn on terminal differentiation. Science 310, 869–872 [DOI] [PubMed] [Google Scholar]
- Chou J., Stolz D. B., Burke N. A., Watkins S. C., Wells A. (2002). Distribution of gelsolin and phosphoinositol 4,5-bisphosphate in lamellipodia during EGF-induced motility. Int. J. Biochem. Cell Biol. 34, 776–790 [DOI] [PubMed] [Google Scholar]
- Chu Y. S., Thomas W. A., Eder O., Pincet F., Perez E., Thiery J. P., Dufour S. (2004). Force measurements in E-cadherin-mediated cell doublets reveal rapid adhesion strengthened by actin cytoskeleton remodeling through Rac and Cdc42. J. Cell Biol. 167, 1183–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinet C., Lecuit T. (2013). Stability and dynamics of cell-cell junctions. Prog. Mol. Biol. Transl. Sci. 116, 25–47 [DOI] [PubMed] [Google Scholar]
- Cooley L., Verheyen E., Ayers K. (1992). chickadee encodes a profilin required for intercellular cytoplasm transport during Drosophila oogenesis. Cell 69, 173–184 [DOI] [PubMed] [Google Scholar]
- Desai R., Sarpal R., Ishiyama N., Pellikka M., Ikura M., Tepass U. (2013). Monomeric α-catenin links cadherin to the actin cytoskeleton. Nat. Cell Biol. 15, 261–273 [DOI] [PubMed] [Google Scholar]
- Goldschmidt-Clermont P. J., Kim J. W., Machesky L. M., Rhee S. G., Pollard T. D. (1991). Regulation of phospholipase C-γ 1 by profilin and tyrosine phosphorylation. Science 251, 1231–1233 [DOI] [PubMed] [Google Scholar]
- Gonçalves-Pimentel C., Gombos R., Mihály J., Sánchez-Soriano N., Prokop A. (2011). Dissecting regulatory networks of filopodia formation in a Drosophila growth cone model. PLoS ONE 6, e18340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gönczy P., DiNardo S. (1996). The germ line regulates somatic cyst cell proliferation and fate during Drosophila spermatogenesis. Development 122, 2437–2447 [DOI] [PubMed] [Google Scholar]
- Hong S., Troyanovsky R. B., Troyanovsky S. M. (2013). Binding to F-actin guides cadherin cluster assembly, stability, and movement. J. Cell Biol. 201, 131–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura Y., Itoh M., Maeno Y., Tsukita S., Nagafuchi A. (1999). Functional domains of alpha-catenin required for the strong state of cadherin-based cell adhesion. J. Cell Biol. 144, 1311–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba M., Yuan H., Salzmann V., Fuller M. T., Yamashita Y. M. (2010). E-cadherin is required for centrosome and spindle orientation in Drosophila male germline stem cells. PLoS ONE 5, e12473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamora C., Fuchs E. (2002). Intercellular adhesion, signalling and the cytoskeleton. Nat. Cell Biol. 4, E101–E108 [DOI] [PubMed] [Google Scholar]
- Kiger A. A., White-Cooper H., Fuller M. T. (2000). Somatic support cells restrict germline stem cell self-renewal and promote differentiation. Nature 407, 750–754 [DOI] [PubMed] [Google Scholar]
- Kiger A. A., Jones D. L., Schulz C., Rogers M. B., Fuller M. T. (2001). Stem cell self-renewal specified by JAK-STAT activation in response to a support cell cue. Science 294, 2542–2545 [DOI] [PubMed] [Google Scholar]
- Lambert M., Thoumine O., Brevier J., Choquet D., Riveline D., Mège R. M. (2007). Nucleation and growth of cadherin adhesions. Exp. Cell Res. 313, 4025–4040 [DOI] [PubMed] [Google Scholar]
- Leatherman J. L., Dinardo S. (2008). Zfh-1 controls somatic stem cell self-renewal in the Drosophila testis and nonautonomously influences germline stem cell self-renewal. Cell Stem Cell 3, 44–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leatherman J. L., Dinardo S. (2010). Germline self-renewal requires cyst stem cells and stat regulates niche adhesion in Drosophila testes. Nat. Cell Biol. 12, 806–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J. G., Fuller M. T. (2012). Somatic cell lineage is required for differentiation and not maintenance of germline stem cells in Drosophila testes. Proc. Natl. Acad. Sci. USA 109, 18477–18481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mège R. M., Gavard J., Lambert M. (2006). Regulation of cell-cell junctions by the cytoskeleton. Curr. Opin. Cell Biol. 18, 541–548 [DOI] [PubMed] [Google Scholar]
- Nola S., Daigaku R., Smolarczyk K., Carstens M., Martin-Martin B., Longmore G., Bailly M., Braga V. M. (2011). Ajuba is required for Rac activation and maintenance of E-cadherin adhesion. J. Cell Biol. 195, 855–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda H., Tsukita S. (1999). Nonchordate classic cadherins have a structurally and functionally unique domain that is absent from chordate classic cadherins. Dev. Biol. 216, 406–422 [DOI] [PubMed] [Google Scholar]
- Sarkar A., Parikh N., Hearn S. A., Fuller M. T., Tazuke S. I., Schulz C. (2007). Antagonistic roles of Rac and Rho in organizing the germ cell microenvironment. Curr. Biol. 17, 1253–1258 [DOI] [PubMed] [Google Scholar]
- Srinivasan S., Mahowald A. P., Fuller M. T. (2012). The receptor tyrosine phosphatase Lar regulates adhesion between Drosophila male germline stem cells and the niche. Development 139, 1381–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot J. A., Mitchison T. J. (1993). The three faces of profilin. Cell 75, 835–838 [DOI] [PubMed] [Google Scholar]
- Townsley F. M., Bienz M. (2000). Actin-dependent membrane association of a Drosophila epithelial APC protein and its effect on junctional Armadillo. Curr. Biol. 10, 1339–1348 [DOI] [PubMed] [Google Scholar]
- Tran J., Brenner T. J., DiNardo S. (2000). Somatic control over the germline stem cell lineage during Drosophila spermatogenesis. Nature 407, 754–757 [DOI] [PubMed] [Google Scholar]
- Tran V., Lim C., Xie J., Chen X. (2012). Asymmetric division of Drosophila male germline stem cell shows asymmetric histone distribution. Science 338, 679–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulina N., Matunis E. (2001). Control of stem cell self-renewal in Drosophila spermatogenesis by JAK-STAT signaling. Science 294, 2546–2549 [DOI] [PubMed] [Google Scholar]
- Van Doren M., Williamson A. L., Lehmann R. (1998). Regulation of zygotic gene expression in Drosophila primordial germ cells. Curr. Biol. 8, 243–246 [DOI] [PubMed] [Google Scholar]
- Verheyen E. M., Cooley L. (1994). Profilin mutations disrupt multiple actin-dependent processes during Drosophila development. Development 120, 717–728 [DOI] [PubMed] [Google Scholar]
- Voog J., D’Alterio C., Jones D. L. (2008). Multipotent somatic stem cells contribute to the stem cell niche in the Drosophila testis. Nature 454, 1132–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadlapalli S., Cheng J., Yamashita Y. M. (2011). Drosophila male germline stem cells do not asymmetrically segregate chromosome strands. J. Cell Sci. 124, 933–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita Y. M., Jones D. L., Fuller M. T. (2003). Orientation of asymmetric stem cell division by the APC tumor suppressor and centrosome. Science 301, 1547–1550 [DOI] [PubMed] [Google Scholar]
- Yu X., Waltzer L., Bienz M. (1999). A new Drosophila APC homologue associated with adhesive zones of epithelial cells. Nat. Cell Biol. 1, 144–151 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.