

Abstract
Comprehensive functional annotation of vertebrate genomes is fundamental to biological discovery. Reverse genetic screening has been highly useful for determination of gene function, but is untenable as a systematic approach in vertebrate model organisms given the number of surveyable genes and observable phenotypes. Unbiased prediction of gene-phenotype relationships offers a strategy to direct finite experimental resources towards likely phenotypes, thus maximizing de novo discovery of gene functions. Here we prioritized genes for phenotypic assay in zebrafish through machine learning, predicting the effect of loss of function of each of 15,106 zebrafish genes on 338 distinct embryonic anatomical processes. Focusing on cardiovascular phenotypes, the learning procedure predicted known knockdown and mutant phenotypes with high precision. In proof-of-concept studies we validated 16 high-confidence cardiac predictions using targeted morpholino knockdown and initial blinded phenotyping in embryonic zebrafish, confirming a significant enrichment for cardiac phenotypes as compared with morpholino controls. Subsequent detailed analyses of cardiac function confirmed these results, identifying novel physiological defects for 11 tested genes. Among these we identified tmem88a, a recently described attenuator of Wnt signaling, as a discrete regulator of the patterning of intercellular coupling in the zebrafish cardiac epithelium. Thus, we show that systematic prioritization in zebrafish can accelerate the pace of developmental gene function discovery.
Keywords: Systems biology, Zebrafish, Cardiovascular, tmem88a
INTRODUCTION
De novo gene function discovery has been greatly facilitated by systematic gene deletion and observation of resulting phenotypes in scalable model organisms. Indeed, systematic gene disruptions in S. cerevisiae (Costanzo et al., 2010; Giaever et al., 2002), C. elegans (Kamath et al., 2003) and Drosophila (Boutros et al., 2004) have each revealed molecular functions for thousands of genes. However, given the breadth and complexity of observable phenotypes in vertebrates, comprehensive assessment of gene function through serial observation of all possible phenotypes following gene disruption remains infeasible. A more efficient alternative would be to use gene function prediction to prioritize gene candidates for more detailed phenotypic testing based on a variety of known gene and protein properties and relationships.
Computational prediction of molecular function has been effective in assigning physiological roles to genes across eukaryotic model organisms (Deng et al., 2004; Guan et al., 2008; Huttenhower et al., 2009; Karaoz et al., 2004; Lee et al., 2004; Mostafavi et al., 2008; Tasan et al., 2012; Tasan et al., 2008; Troyanskaya et al., 2003). Similar prediction frameworks have been applied to predict associated phenotypes in yeast (King et al., 2003; Saha and Heber, 2006) and worm (Lee et al., 2008) and to identify putative human disease gene candidates (Linghu et al., 2009; Woods et al., 2013) but have not been systematically coupled with in vivo validation in a vertebrate model organism.
Predictions of gene function or phenotype have generally used at least one of two basic strategies. The first strategy, termed guilt-by-profiling (GBP), begins by identifying gene properties (features, e.g. ‘gene is expressed in the brain’) that are common to the genes currently associated with a particular function of interest (training set, e.g. ‘genes known to be important for cognition’). Additional, untested genes (test set) exhibiting the hallmarks of the function of interest are then predicted to have that function. A second general strategy, termed guilt-by-association (GBA), identifies gene-gene relationships (e.g. a physical interaction between the corresponding proteins) that tend to connect genes that share a function. Untested genes that are ‘well-connected’ to the set of genes known to hold a particular function are then prioritized as top candidates.
Although there are many effective prediction algorithms, a recent benchmarking comparison (Peña-Castillo et al., 2008) ranked the Funckenstein approach (Tian et al., 2008) highly, based on estimates of precision of top-ranked function predictions. Funckenstein uses both GBA and GBP, combining results to output a single confidence score for each potential gene-function or gene-phenotype association. These predicted relationships can then be directly validated experimentally.
Zebrafish embryos are transparent, small, fast growing and are developmentally similar to higher vertebrates (Howe et al., 2013), making them ideal for large-scale study of vertebrate developmental gene functions. Zebrafish are amenable to forward genetics via chemical (Driever et al., 1996; Haffter et al., 1996) or retroviral-based (Amsterdam et al., 1999; Golling et al., 2002) mutagenesis, and to reverse genetics via knockdown (Nasevicius and Ekker, 2000), small molecules (Burns et al., 2005; Peterson et al., 2000) or, more recently, targeted gene deletion (Huang et al., 2011; Hwang et al., 2013; Meng et al., 2008). Morpholinos, which are oligomers that block translation through steric transcript inhibition, are particularly efficient (Nasevicius and Ekker, 2000), offering scalable, specific gene product inhibition lasting for up to 72 hours.
Here we predicted the effects of gene knockdown on 338 zebrafish embryonic anatomical processes (Bradford et al., 2011), with the results of these predictions being made publicly available as a community resource through two searchable online gene browsers: FuncBase and GeneMANIA. We deliberately focused on the prediction of morphant rather than mutant phenotypes so as to allow direct confirmation of prediction results at a reasonable scale. Because extant data indicated high performance for predictions of cardiac phenotypes, we aimed to experimentally validate this framework for cardiovascular phenotype prediction. Expecting a broad range of potential cardiac phenotypes, we used a five-parameter manual evaluation system to assess cardiac function, and applied it in a blinded fashion to predictions and to positive and negative controls. Subsequent quantitative assessments of cardiac physiology largely confirmed the predictions, identifying novel myocardial effects for 11 surveyed genes. These 11 include tmem88a, a known attenuator of Wnt signaling (Lee et al., 2010), shown here to influence the physiological coupling of embryonic cardiomyocytes.
RESULTS
A combined guilt-by-association and guilt-by-profiling learning procedure predicts a broad range of gene-phenotype associations
We implemented a machine learning procedure based on a diverse collection of large-scale datasets to predict phenotypes resulting from loss of gene function across the zebrafish genome (Fig. 1). We then used each of the generated computational models (one for each of the 338 anatomical process terms) to score the 15,103 zebrafish genes with available gene feature information. The over 5 million assigned scores each reflect the likelihood of observing a given phenotype (disruption of a given anatomical process) following loss of function of a specific gene.
Fig. 1.

Combined guilt-by-association and guilt-by-profiling prediction technique. Overview of the computational strategy that we followed to prioritize genes for phenotypic testing. For over 5 million phenotype-gene combinations, we estimated the likelihood that the given gene will affect a given anatomical process upon knockdown.
Gene features most important for the prediction process differed for guilt-by-association (GBA) and guilt-by-profiling (GBP) predictors. For GBP, feature importance varied based on the phenotype being predicted, but tissue expression and phylogenetic relationships appeared to consistently be the most relied upon features (see supplementary material Table S1 for gene feature rankings). In GBA, where only a single random forest classifier was used to create a gene-gene functional linkage network, phylogenetic profile similarity was the most important feature, followed by similarity in GO terms (especially those describing cellular compartments), and then similarity in protein family IDs. These feature importance results are consistent with the observed usefulness of phylogenetic profiles in predicting gene function and phenotype in other species (Levesque et al., 2003; Pellegrini et al., 1999).
Examining the training data for the 338 phenotype predictors, the broadest phenotypic terms assigned to the most genes (e.g. anatomical system, whole organism) had poor performance, either because of the limited number of negative training examples or because the predictive features we used simply held little value in predicting function at this level of abstraction. Predictions also appeared highly variable when only a small number of positive training examples were present (see supplementary material Table S2 for a full list of prediction scores by phenotype). Filtering out anatomical process terms on both extremes of the specificity spectrum (supplementary material Table S2) resulted in 242 retained terms.
Examining remaining predictions by anatomical process term, the prediction process appeared to be most effective for neuronal, sensory and cardiac phenotypes (Fig. 2A), and performed most poorly for hematopoietic phenotypes (supplementary material Table S2). Additionally, within each phenotypic category there was substantial variation in the precision of predictors. For example, among cardiovascular phenotypes (Fig. 2B), predictors of cardiac structures (e.g. heart tube, cardiac muscle cell, myocardium) showed greater performance than terms abstractly describing vascular structure and the endocardium. This did not appear to be a ‘rich-getting-richer’ scenario of terms with many training examples showing greater performance, as the top five scoring cardiovascular phenotype terms had 15 or fewer training examples. We hypothesize instead that performance stems from the fidelity of observation of anatomical structures in the transparent embryo, which might have allowed these defects to be more rigorously and consistently characterized. For example, in the case of hematopoietic phenotypes, abnormal blood flow or blood accumulation is often reported as a secondary consequence of another phenotype, which might have led to inconsistency among associated genes and poor performance for the term ‘blood’. Conversely, the term ‘nucleate erythrocyte’, which is more stringently defined, showed better performance (supplementary material Table S2).
Fig. 2.

Cross-validation suggests high prediction accuracy over a range of phenotypes. Plotting relative precision [precision at 20% recall divided by the fraction of positive training examples (the prior probability); see Materials and methods] for phenotype predictors (A) reveals a diversity in high-scoring terms, with terms related to neural, sensory or cardiovascular phenotypes seeming to appear most frequently. Relative precision also shows substantial diversity among terms in the same general phenotypic category, as illustrated using cardiovascular phenotype terms (B). A precision recall plot of the term describing general cardiovascular phenotypes (‘cardiovascular system’) (C) shows a precision of 0.72 at 20% recall (blue line), with precision approaching the prior as recall approaches 100% (green line).
Examining individual gene-phenotype predictions in the test set, 8742 gene/term predictions involving 2459 genes and 93 terms were above an 80% estimated precision cut-off. These high-confidence predictions (see supplementary material Table S3) are searchable online in the context of the FuncBase Genome Browser [http://zfunc.mshri.on.ca (Beaver et al., 2010)] and the GeneMANIA functional annotation browser [http://www.genemania.org (Mostafavi et al., 2008)]. The majority (87%) of these 2459 genes were predicted to affect five or fewer anatomical processes at this precision level, with genes encoding ribosomal constituents frequently being most pleiotropic. Indeed, using gene set enrichment analysis (Subramanian et al., 2005) we found that genes annotated as being ribosomal were significantly enriched for participation in a greater number of phenotypes (q<0.001). This is consistent with the observed phenotypic importance of ribosomal proteins in other organisms (Giaever et al., 2002), as well as in prior genetic screens in zebrafish (Amsterdam et al., 2004).
External evidence confirms high-confidence cardiovascular phenotype predictions
As substantial literature support exists for cardiovascular phenotypes, we examined the literature surrounding our cardiovascular predictions in more detail. The phenotype term broadly describing the cardiovascular system exhibited a precision at 20% recall value of 0.72 (Fig. 2C). Examination of independent lines of evidence for top hits also suggested prediction results for ‘cardiovascular system’ to be of high quality. Indeed, six of the ten top-scoring predictions were already known to be associated with cardiovascular defects. These genes had not been used as positive training examples either because the phenotype was observed through genetic mutation rather than morpholino, because a morpholino had been injected into a zebrafish line not used for training purposes, or because the publications were curated in the Zebrafish Information Network (ZFIN) database (Bradford et al., 2011) subsequent to the download of phenotypic data to establish a training set. Examples of literature-validated predictions include vmhc, in which a premature stop codon causes ventricular failure (Auman et al., 2007), and apobec2a, which causes decreased heart rate upon knockdown (Etard et al., 2010).
To more systematically determine whether the predictive value of our framework extends beyond morpholino-mediated gene knockdown, we obtained all mutant-phenotype association data from ZFIN (using an identical approach to that used above for morpholino-phenotype data). In total, 818 genes in our test set had associated mutants with phenotype information. We used our ‘cardiovascular system’ phenotype predictor, trained exclusively using morpholino experimental data, to rank these genes based on likelihood to elicit a cardiovascular phenotype. Of the 50 highest ranked genes, 41 had associated mutants with a cardiovascular phenotype (P<1×10-9, Fisher’s exact test).
Phenotyping confirms predictions of cardiovascular function
To more directly test the results of our cardiovascular phenotype predictions, we selected all genes exceeding an estimated 95% precision cut-off for targeted morpholino knockdown, excluding genes for which morpholinos targeting the transcription start site could not be designed (as a result of high GC content or self-complementarity). To increase the potential for novel discovery, we also excluded genes that had existing deletion mutants or morpholino experimentation confirming cardiac function, or which, despite not having experimental evidence, had an apparent cardiac function (e.g. vmhcl). This resulted in a list of 16 target genes (Table 1). To determine how well our procedure could discriminate against genes unlikely to cause a phenotype, the five genes with the lowest prediction score were screened as well. Finally, a morpholino targeting a gene with a known severe cardiac developmental phenotype [slc8a1a, which encodes a sodium calcium exchanger (Langenbacher et al., 2005; Stainier et al., 1996)] was screened as a positive control (see supplementary material Table S4 for a list of all morpholinos used).
Table 1.
Quantitative scoring system differentiates high-ranking genes from low
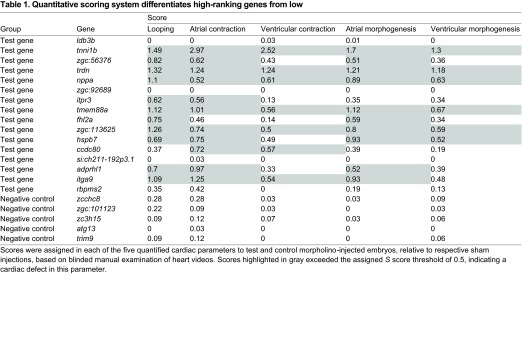
We used a simple five-parameter categorical system to score cardiac effects (‘S score’ hereafter; see Materials and methods and supplementary material Fig. S1), comparing S scores for all five cardiac parameters between test genes and negative controls. We reasoned that a manual, blinded evaluation of cardiac function would be more sensitive in identifying a range of potential cardiac defects than other comparable quantitative approaches. Using a cut-off value of S>0.5 to identify cardiovascular defects, 12 of 16 test genes were found to have a defect in at least one quantified parameter of heart function following knockdown, as compared with zero of five negative control genes (P<0.05, Fisher’s exact test; Table 1). Our results were not sensitive to the S score threshold used, in that test genes scored significantly higher than negative controls at a range of cut-offs spanning 0.3 to 0.8 (supplementary material Fig. S2A). Additionally, all five parameters of heart function scored significantly higher in test genes than negative controls (P<0.05; supplementary material Fig. S2B). Together, these data confirm that a machine learning procedure can be successfully used to differentiate genes for which loss of function elicits a cardiovascular phenotype.
One area of experimental concern given our deliberate focus on morphants was whether the cardiac effects observed could be due to morpholino toxicity or non-specific binding effects. Morpholino toxicity occurs randomly for reasons thought to be related to p53-mediated activation of apoptosis (Robu et al., 2007) and can induce a phenotype with a cardiovascular component. Additionally, although morpholino sequences were vetted for binding fidelity, off-target effects remain a possibility. The phenotypes observed appeared diverse with respect to the cardiac parameters affected, suggesting that these responses were not due to a shared general morpholino effect (supplementary material Fig. S2C). Based on the lack of phenotype observed in any of our five negative control morpholinos (each targeting a characterized gene and thus potentially subject to both morpholino toxicity and non-specific binding effects), we estimated the prevalence of cardiac phenotypes due to non-specific effects to be below 20%, which is in keeping with prior work (Ekker and Larson, 2001; Heasman, 2002; Nasevicius and Ekker, 2000). However, even if three of 12 hits (25%) were due to non-specific effects, highly ranked test genes would remain significantly differentiated from low-ranked controls (P<0.05, Fisher’s exact test).
Quantifiable decreases in cardiac function underlie screen results
We next used more detailed physiological measures to further characterize observed cardiac defects. We first examined alterations in cardiac output (CO) following injection of all test morpholinos. Here we followed a previously described approach to further confirm that observed defects were not due to non-specific toxicity: all CO experimentation was performed following co-injection of the test morpholino with a morpholino targeting p53 (also known as tp53) (Robu et al., 2007).
Of the 12 test morpholinos identified through the S score (S>0.5) as inducing a cardiac phenotype, eight caused a significant reduction in CO (supplementary material Table S5). Among these eight was tnni1b, which has known cardiac expression (Fu et al., 2009) and high similarity to human troponin I type 1 (skeletal, slow), but no known phenotype. Knockdown of tnni1b caused virtually complete asystole (supplementary material Movies 1, 2) with no obvious effects on zebrafish motility, a phenotype similar to that observed upon knockdown of cardiac troponin T (tnnt2a), the gene underlying the contractile defect in the silent heart mutant (Sehnert et al., 2002). A second morpholino targeting tnni1b splicing also demonstrated near asystole (supplementary material Movie 3) with no apparent motility defects, supporting this as a gene-specific effect.
Also among the set of genes causing reduced CO was hspb7, the human ortholog of which has been associated with heart failure through large-scale genome-wide association screening (Stark et al., 2010; Villard et al., 2011). A reduction in CO was confirmed using an additional morpholino targeting an mRNA splice site in the hspb7 transcript. However, unlike tnni1b, the phenotype following hspb7 knockdown did not appear exclusively cardiac. Most obviously, at the highest dose nearly half of all splice morpholino-injected embryos had a shortened tail, abridged at the trunk region (a phenotype not seen with any other morpholino injection; supplementary material Fig. S3). Supporting the generality of the systematic gene prioritization strategy, hspb7, in addition to its predicted cardiac effects, was associated with the phenotypic term ‘trunk’ at an estimated precision of 0.89.
Although they were confirmed as having a cardiac phenotype according to the S score, knockdown of nppa, itpr3, tmem88a and adprhl1 did not cause a significant reduction in CO. However, valvular regurgitation was observed in 55% of nppa morphant embryos examined at the highest injected dose (supplementary material Movie 4), as compared with 3% of wild-type embryos (as assessed through blinded manual evaluation; P<1×10-5, Fisher’s exact test). A regurgitation phenotype was confirmed using a second morpholino targeting the nppa transcription start site through an alternative sequence (31% of injected embryos versus 4% in controls, P<0.05).
Next, itpr3 (inositol 1,4,5-triphosphate receptor, type 3) knockdown caused a notable alteration in atrial contraction, without a corresponding significant reduction in CO (CO is largely a metric of ventricular function). Because itpr3 is known to mediate the release of intracellular calcium in epithelial cells (Maranto, 1994) we went on to examine calcium dynamics in isolated hearts following itpr3 knockdown. We found a significant decrease in diastolic calcium concentration in the atrium of morphants following injection of both the initial itpr3 morpholino and a second morpholino targeting an itpr3 mRNA splice site (Fig. 3).
Fig. 3.
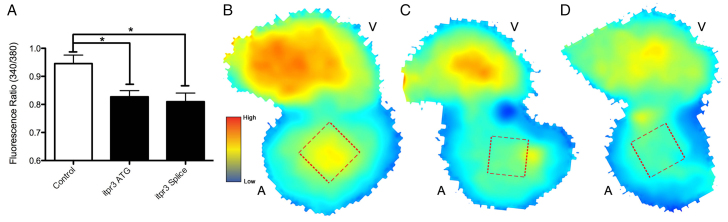
itpr3 knockdown reduces cardiac calcium availability. Diastolic calcium concentration was determined in isolated zebrafish hearts at 72 hpf following knockdown of itpr3 through either ATG-targeting or splice-blocking morpholinos. Calcium availability, measured as the 340/380 fluorescence ratio following staining with the ratiometric dye Fura-2 (see Materials and methods) was determined at regions of fixed size at the center of the atrium and ventricle (red squares), and was significantly lowered in atria (but not the ventricles) of morpholino-injected embryos (A). A general decrease in diastolic calcium was obvious throughout the atrium as shown by heatmaps indicating calcium levels in control hearts (B) or hearts isolated from ATG (C) or splice (D) morpholino-injected embryos. *P<0.05 (Student’s t-test, n>5 for each condition). Error bars indicate standard error. V, ventricle; A, atrium.
Finally, unlike nppa, itpr3 and tmem88a (tmem88a knockdown caused quantifiable alterations in cardiac conduction; more detail below), the adprhl1 knockdown phenotype was largely abrogated by co-injection with p53 morpholino. This suggests that the initially observed adprhl1 phenotype is either p53 dependent or is due to morpholino toxicity.
Thus, of 12 test genes that were both computationally predicted and subsequently identified through the S score as having a cardiac phenotype, 11 were further validated to have underlying alterations in atrial function, valvular function, CO or cardiac conduction.
Tmem88a regulates coupling in the developing ventricular myocardium
The transmembrane protein Tmem88 is a recently reported inhibitor of the Wnt signaling pathway in multiple species, acting through interaction with Dishevelled (Dvl) via a C-terminal tri-peptide VWV interaction motif (Lee et al., 2010). Knockdown of the zebrafish tmem88 ortholog tmem88a caused observable alterations in heart function at the injected dose (Table 1), albeit with no significant corresponding change in CO. Similarly, mosaic overexpression of a tmem88a transcript lacking the VWV motif (see Materials and methods) resulted in severe, systematic growth defects for 17% of embryos and observable cardiac defects for an additional 20% of embryos. By contrast, mosaic expression of the full tmem88a transcript resulted in no observable phenotypes.
Given the known effect of the Tmem88 VWV motif on Wnt signaling (Lee et al., 2010), the described role of wnt11 in establishing physiological myocardial electrical polarities in the developing zebrafish ventricle (Panáková et al., 2010), and the apparent cardiac defects following tmem88a knockdown, we next sought to identify any functional synergy between wnt11 and tmem88a. Co-injection of either the tmem88a ATG morpholino tested above or a second morpholino blocking tmem88a splicing [both independently validated as targeting tmem88a (Cannon et al., 2013)] with a previously validated wnt11 morpholino (Panáková et al., 2010) showed sensitization of the wnt11 cyclopia phenotype (Fig. 4). This suggests a synergistic genetic interaction between tmem88a and wnt11. Quantifying cell coupling, we found an increase in impulse propagation velocity in the body of the ventricle following tmem88a knockdown by both the ATG and splice-blocking morpholinos (Fig. 5A-D). This finding is consistent with an effect that enhances the intercellular coupling gradient induced by physiological levels of Wnt11 (Panáková et al., 2010). We found no appreciable difference in epithelial morphogenesis or ventricular connexin 43 expression following tmem88a inhibition that could otherwise account for this alteration in impulse propagation (Fig. 5E-L).
Fig. 4.
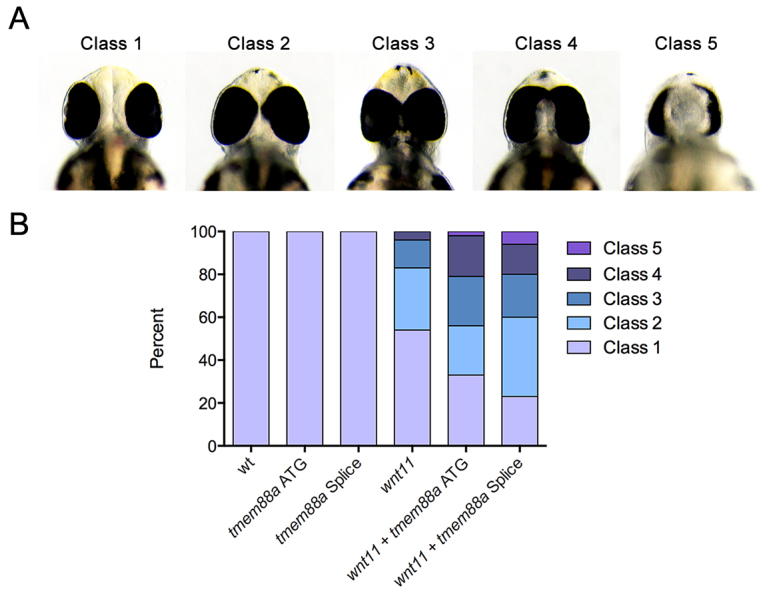
tmem88a sensitizes the wnt11 cyclopia phenotype. Embryonic zebrafish were injected with low doses of morpholinos targeting either wnt11 or tmem88a (both ATG targeting and splice blocking) and morphants scored for cyclopia at 48 hpf. Cyclopia classes (A) are as previously defined (Marlow et al., 1998), such that in class 1 eye spacing is comparable to that of wild type, class 2 the spacing is decreased, class 3 eyes are marginally fused, class 4 eyes are completely fused, and class 5 have one eye. The proportion by class (B) is significantly different (P<0.05, Chi-squared test) for both wnt11 + tmem88a ATG and wnt11 + tmem88a splice morpholino co-injections as compared with wnt11 morpholino alone.
Fig. 5.

tmem88a inhibition alters cardiac ventricular polarity. Following knockdown of tmem88a by ATG-targeting or splice-blocking morpholinos, conduction velocity as measured in isolated hearts following staining with Di-8-ANEPPS was significantly increased along the outer curvature of the ventricle (A). This difference is also illustrated through isochronal maps showing voltage propagation along a control heart (B) versus hearts from embryos injected with the ATG (C) or splice-blocking (D) morpholinos (isochrons are 5 mseconds apart, conduction goes from blue to red). There was no apparent change in connexin 43 expression or cell number accompanying this alteration in cell coupling. Expression of connexin 43 (E) was strong in the atria, but weak in the ventricles in control hearts (F), and did not appear to change with inhibition of tmem88a (ATG-blocking morpholino in G, splice-blocking morpholino in H). Cell number as measured by manual counting of ventricular nuclei (I) revealed no obvious differences between control hearts (J) and hearts from embryos injected with the ATG (K) or splice (L) morpholinos (β-catenin in green, DAPI in blue). *P<0.05 (Student’s t-test, n>4 for each condition). Error bars indicate standard error.
We next examined the expression of several key developmental markers following tmem88a knockdown to confirm that the observed effect of tmem88a on cardiac cell coupling is not a secondary consequence of a generalized effect on development (Fig. 6). fli1 served as an in situ hybridization marker for the integrity of endothelial development, myoD (also known as myod1) enabled monitoring of muscle formation, and beta-globin E3 (hbbe3) provided a readout for functional circulation and timing of hematopoiesis, as its expression is reduced by 48 hours postfertilization (hpf). Expression of fli1 and myoD in tmem88a morphant embryos was comparable to that in the wild-type reference. beta-globin E3 showed an apparent upregulation at 48 hpf in morphants and a slightly longer persistence of expression at 72 hpf when voltage measurements were taken (Fig. 6). These results suggest that tmem88a morpholinos might induce alterations in hematopoiesis at the injected dose [consistent with recent findings (Cannon et al., 2013)], but do not cause any obvious systematic developmental abnormalities that might otherwise account for the cardiac effects observed at the time point surveyed.
Fig. 6.

Expression analysis confirms the lack of a systematic developmental delay following tmem88a knockdown. Markers specific to the endothelium (fli1), developing muscle (myoD) and hematopoiesis (beta-globin E3) were visualized using in situ hybridization at 24 (A-I), 48 (J-R) and 72 (S-AA) hpf in wild-type embryos (A-C,J-L,S-U) and following injection of either the ATG (D-F,M-O,V-X) or splice-blocking (G-I,P-R,Y-AA) tmem88a morpholinos. Whereas fli1 and myoD expression appears unaltered, beta-globin E3 shows a slight elevation in expression following tmem88a knockdown at 48 hpf, which is largely alleviated by 72 hpf. High-resolution images of any panel are available upon request.
DISCUSSION
In this study we used high-confidence predictions of gene-phenotype association across over 15,000 genes to prioritize the screening of novel gene candidates for developmental functions. Through rigorous experimental assay we identified genes affecting a range of observable cardiac phenotypes. This approach to large-scale phenotype prediction and in vivo validation in a vertebrate establishes a framework for de novo discovery of gene functions across a broad spectrum of vertebrate developmental phenotypes.
Using machine learning, we identified patterns in gene expression, conservation and interaction to generate over 5 million predictions of gene-phenotype association in embryonic zebrafish. We have made the resulting high-confidence predictions of gene-phenotype association available through two public browsers: FuncBase and GeneMANIA. GeneMANIA is capable of integrating existing predictions with new gene feature data as they become available, thereby acting as a dynamic resource for future experimentation. The precision of phenotype predictions appeared highest for consistently defined anatomical phenotypes, suggesting that effective gene prioritization can be achieved for any standardized phenotype with sufficient rigorous training information.
We used morpholino knockdown to both train and test our computational approach. Morpholinos enable gene knockdown at a reasonable scale, but can have appreciable phenotypic noise due to non-specific toxicity. Despite these confounders, a clear phenotypic enrichment was seen using our in silico prediction strategy.
To directly test the fidelity of our gene-phenotype association predictions, we chose to focus on the observation of cardiovascular phenotypes. From modeling basic processes such as cellular migration (Lazic and Scott, 2011; Zhou et al., 2011) and intercellular coupling (Panáková et al., 2010), to complex arrhythmogenic (Arnaout et al., 2007) and structural (Vogel et al., 2009) cardiac disorders, zebrafish are a powerful model organism for the study of cardiac development. Zebrafish have over 1000 genes with detectable cardiac expression [as annotated by ZFIN (Bradford et al., 2011)] and thus systematic screening of each gene for associated phenotypes would require tremendous experimental resources. Alternatively, our method allowed focus on a set of genes enriched for potential phenotypes.
Through testing and validation, we were able to identify 11 genes with cardiac phenotypes. Notably, despite the fact that cardiac specificity of predicted phenotypes was not included as a criterion for initial survey, the majority of these 11 genes had an apparently cardiac-specific phenotype upon knockdown (supplementary material Table S4). Eight of these genes were demonstrated to reduce CO and the remaining three (nppa, itpr3 and tmem88a) were shown to alter other quantifiable aspects of cardiac function.
nppa, which was known to have variant expression associated with alteration in valve tissue formation in zebrafish (Camarata et al., 2010), was shown here to be associated with regurgitant defects in vivo. Additionally, itpr3, recently shown to be dysregulated in human arrhythmia (Hasdemir et al., 2010), was observed to have a direct role in atrial calcium handling. Last, tmem88a, a proposed attenuator of Wnt signaling (Lee et al., 2010), was demonstrated here to regulate ventricular cell coupling.
We observed the influence of tmem88a on wnt11 in two separate, quantifiable phenotypes (cyclopia and myocardial cell coupling), suggesting a functional synergy between these two genes. Both wnt11 overexpression and knockdown are associated with a disruption of the myocardial electrical gradient (Panáková et al., 2010). tmem88a knockdown enhanced this gradient, suggesting that the relationship between tmem88a and wnt11, potentially mediated by the known interaction between tmem88a and dvl (Lee et al., 2010), appears more complex than a simple activation or inactivation. Notably, two additional studies have been published during the review of our manuscript (Novikov and Evans, 2013; Palpant et al., 2013), both confirming a Wnt-dependent role for tmem88a in cardiomyocyte differentiation.
The number of vertebrate phenotypes potentially observable even within the finite timeframe of embryonic development is vast. When combined with the enormous number of potential genotypes in a genome, it is clear that comprehensive experimental mapping from genotype to phenotype is unlikely without informed direction of experimental effort. Here we demonstrate that systematic data integration and objective gene prioritization can successfully direct limited experimental resources for high-resolution phenotyping to ranked subsets of genes for each phenotype, accelerating the potential for gene function discovery in vertebrates. With large-scale projects underway to generate and phenotype zebrafish carrying null alleles (Kettleborough et al., 2013) and other allelic classes, future iterations of our approach will be crucial in deciphering the complex relationship between genotype and phenotype.
MATERIALS AND METHODS
Collection of feature and training data
We obtained gene-phenotype associations to train our learning procedure from the Zebrafish Information Network (ZFIN) database (Bradford et al., 2011) and included only experimentally verified gene-phenotype associations that satisfied the following two criteria: (1) gene inhibition was via a morpholino that had only one identifiable target annotated by ZFIN; and (2) the morpholino causing the observed phenotype was injected into a wild-type (i.e. non-transgenic) fish line, as we anticipated carrying out experimental verification using wild-type lines. Phenotypes used in the learning procedure were based on a hierarchical list of anatomical terms developed by a consortium of researchers together with ZFIN (Bradford et al., 2011). Through literature curation, ZFIN has captured the anatomical structures in embryonic development that are affected by injection of a given morpholino. Existing gene-phenotype associations were propagated to ancestral terms within the ontology prior to training.
Gene feature data used for training and prediction included: expression data [both tissue localization (in situ data collected by ZFIN) and microarray experimental data (more below)], phylogenetic profiles, annotated protein domains, Gene Ontology (GO) (Ashburner et al., 2000) and KEGG (Kanehisa et al., 2008) annotations, and genetic and protein interactions mapped through orthology from human, mouse and yeast. Only systematically obtained interaction data were mapped from additional model organisms so as to reduce the impact of ascertainment bias. As functional annotations can be derived directly from phenotypic evidence and thus introduce circularity into our prediction procedure, annotations derived from GO underwent stringent filtering before use. Expression data included both cell type-specific expression (annotated by ZFIN) and an experimental expression compendium generated for this purpose. To generate this compendium, we combined over 60 experimental datasets that had been published using the Affymetrix zebrafish platform from the Gene Expression Omnibus (Barrett and Edgar, 2006), after renormalizing each set using genechip robust multiarray averaging (Wu et al., 2004) via Bioconductor (Gentleman et al., 2004).
Learning procedure
The phenotype prediction procedure used here was based on the Funckenstein approach previously used to make predictions of gene function in yeast (Tian et al., 2008), mouse (Tasan et al., 2008) and human (Tasan et al., 2012). Briefly, separate classifiers were used to infer putative gene-phenotype associations based on correlations between gene features and phenotypes (Guilt-By-Profiling; GBP), and to transfer known phenotype associations between genes (Guilt-By-Association; GBA) (Fig. 1). Both GBA and GBP used random forest classifiers for prediction. A random forest classifier is an ensemble-based learner in which collections of decisions trees are constructed, each using a randomly selected subset of training information and features.
For GBP, separate random forest classifiers were constructed for each ZFIN anatomical term, resulting in a gene by anatomical term matrix of confidence values. Positive training examples for each anatomical term were genes for which inhibition through morpholino affected that term (‘phenotype’ hereafter). Negative training genes were all other genes annotated with at least one phenotype term. We used out of bag (OOB) scores to evaluate prediction accuracy for each phenotype, such that each gene is assigned a score using only models that had not been trained using information about that gene.
For GBA, gene feature data were used to construct a functional linkage network (FLN), a graph in which gene-gene linkage weight corresponds to the strength of association between two genes, as estimated via a random forest classifier. Positive training examples were all genes with any shared phenotype, while negative examples were all other gene pairs drawn from the same set that failed to exhibit any shared phenotypes. OOB scores were used to generate performance estimates for gene-gene associations. Gene-phenotype associations were then derived from the FLN probabilistically. Specifically, edge weights within an FLN clique containing all genes annotated as causing a particular phenotype (herein referred to as the ‘core’ set) were used to derive a probability density function (PDF), an estimate of the distribution of edge weights expected of gene pairs sharing the phenotype of interest. A separate PDF was created using edge weights between the core set and all other genes not having the phenotype of interest. Edge weights between each candidate gene and the core set were then obtained, and a gene-phenotype association score was assigned to be the log of the likelihood of obtaining the observed edge-weight distribution under the core PDF model relative to the likelihood under the non-core PDF, as in previous approaches (Tasan et al., 2012). GBA and GBP scores were then combined for each term using a logistic regression model optimized to maximize the cross-validated estimates of area under the cumulative precision recall curve.
Gene feature data were used differently for GBA and GBP. For example, while phylogenetic data were used as a feature for both GBA and GBP, GBP contained all species used in phylogenetic profiling as individual features, while GBA used only the similarity in phylogenetic profile between any two genes. Similarly, correlation in expression over the 60 combined experimental datasets was used as an individual feature in GBA. However, in GBP, genes were clustered based on correlation in expression over these 60 datasets, and the presence in any of these expression clusters was used as an individual feature. In total, there were 65 features used to train the GBA predictor, while 3034 were used for GBP. Feature importance for both GBA and GBP was calculated as the root mean square error introduced by excluding the given feature.
Efficacy of each of the phenotype predictors was assessed according to the precision at 20% recall using cross-validation. This measure quantifies precision (the estimated fraction of predicted gene-phenotype associations that are true, i.e. the positive predictive value) at a given recall (the fraction of known gene-phenotype associations that were correctly predicted, i.e. the true positive rate or sensitivity). Precision-recall curves for each phenotype were generated using ROCR (Sing et al., 2005), and interpolated precision at 20% recall was calculated as previously described (Manning et al., 2008). Where the precision at 20% recall values were compared for multiple predictors, a relative precision score was calculated as the precision at 20% recall divided by the prior expectation (fraction positive training examples). This was done when comparing across multiple phenotypes to reduce the influence of differences in prevalence (i.e. the fraction of genes known to be associated with the phenotype). To generate the high confidence gene-phenotype association list, a likelihood score corresponding to a precision of 0.8 was determined for each phenotype, and all genes not in the original training data scoring above this threshold were included.
Cross-validation
For GBA, feature data were used to construct a functional linkage network (FLN), a graph in which edge weight corresponds to the strength of association between two genes as estimated via a random forest classifier. This classifier was trained to predict edge weights based on known examples of shared phenotype (positive), versus all other gene pair combinations in the training set (negative), with all possible gene-gene pairings acting as a test set. To avoid the inflated performance estimates that can arise from overfitting, we used out of bag (OOB) scores (such that each gene is assigned a score using only models that had not been trained using information about that gene) to generate performance estimates for gene-gene associations. Gene-phenotype associations were then derived from the FLN probabilistically. Specifically, edge weights within an FLN clique containing all genes annotated as causing a particular phenotype (herein referred to as the ‘core’ set) were used to derive a probability density function (PDF), an estimate of the distribution of edge weights expected of gene pairs sharing the phenotype of interest. A separate PDF was created using edge weights between the core set and all other genes not having the phenotype of interest. Edge weights between each candidate gene and the core set were then obtained, and a gene-phenotype association score was assigned to be the log of the likelihood of obtaining the observed edge-weight distribution under the core PDF model relative to the likelihood under the non-core PDF, as in previous approaches (Tasan et al., 2012). Performance estimates for the gene-phenotype association predictions were evaluated using leave-one-out cross-validation.
For GBP, separate random forest classifiers were constructed for each phenotype, resulting in a gene by phenotype matrix of confidence values. OOB scores were used to evaluate prediction accuracy for each phenotype. GBA and GBP scores were then combined for each term using a logistic regression model optimized to maximize the cross-validated estimates of area under the cumulative precision versus recall curve.
Empiric testing of predictions using morpholinos
ATG-blocking morpholinos were designed using Gene Tools oligo design service for uniformity. To ensure a lack of off-target binding, the resultant morpholino sequences were aligned against the zebrafish genome using BLAST (Altschul et al., 1990) with settings optimized for small sequences.
Male and female wild-type (AB) fish were housed and embryos bred for microinjection using standard protocols. Embryos were collected, pooled and immediately used for injection. Techniques used for morpholino injection follow those previously outlined (Westerfield, 2000). Briefly, morpholinos (Gene Tools) were resuspended in sterile water at 1 mM and diluted to working concentration with Danieau’s solution [58 mM NaCl, 0.7 mM KCl, 0.4 mM MgSO4, 0.6 mM Ca(NO3)2, 5 mM HEPES]. Morpholinos were introduced into the zebrafish yolk via microinjection no later than the two-cell developmental stage. Injected embryos were then kept at 28.5°C in E3 solution (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4).
Evaluation of cardiac defects
Each morpholino was injected into zebrafish embryos at a range of doses high enough to elicit an observable phenotype (each initially at three doses: 0.5, 0.25 and 0.125 pmol). If any morpholino caused frequent, systematic growth deformities, or resulted in greater than 30% mortality at the highest dose, a lower dose was used for further experimentation. Once a suitable morpholino dose was found, injections were performed in triplicate (at least 40 embryos per injection, three sets of embryos resulting from separate mate pairings).
Images of live zebrafish hearts were acquired at 48 hpf on an Axioplan (Zeiss) upright microscope with a 5× objective lens using integrated incandescent illumination and a FastCam-PCI high-speed digital camera (Photron) with a 512×480 pixel grayscale image sensor. Images were obtained at 250 frames per second, with 1088 frames (′8 cardiac cycles) being acquired per condition. Our cardiovascular function classification system evaluated five parameters from beating hearts using sequential image files: atrial morphogenesis, ventricular morphogenesis, looping, atrial contraction, and ventricular contraction (see supplementary material Fig. S1 for a full score description). Raw scores between 1 and 4 (with 1 being normal and 4 being a severe defect) were assigned to each parameter manually with the evaluator (G.M.) being blinded as to the identity of the heart video file (ten videos per condition, in triplicate). The final score S for each of these parameters was calculated as:
 |
where n is the number of embryos analyzed per experiment, T the score for a morpholino-injected embryo, and C the score for a sham-injected control embryo from the same clutch. In order to be considered substantially affected, a cardiac parameter would need to have an average score of greater than 0.5. This would correspond, for example, to an excess of phenotypic observations in treated embryos relative to controls of 16.67% for severe defects, 25% for moderate defects, or 50% for mild defects.
Heart rate and cardiac output
Custom software (implemented in MATLAB; freely available upon request) was used to determine heart rate from sequential image files (obtained as above), while measurements of ventricular long and short axis in both diastole and systole were obtained manually for each video using ImageJ (http://rsbweb.nih.gov/ij/) and used to estimate chamber volume using standard geometric assumptions. Cardiac output was then calculated as diastolic minus systolic ventricular volume multiplied by heart rate, in a method analogous to previous approaches (Shin et al., 2010), for at least ten embryos per morpholino dose.
In situ hybridization
In situ hybridization was performed as previously described (Thisse and Thisse, 2008).
Immunohistochemistry
For β-catenin and Connexin 43 analysis, isolated hearts from 72 hpf embryos were fixed in Prefer fixative (Anatech) and incubated with primary antibodies mouse β-catenin (1:200; BD Biosciences, 610154) or rabbit Connexin 43 (1:50; Sigma-Aldrich, C6219) followed by goat anti-mouse Alexa Fluor 488 (1:1000; Invitrogen, A11029) or donkey anti-rabbit Alexa Fluor 555 (1:1000; Invitrogen, A31572), respectively. Stained hearts were mounted in ProLong Gold antifade reagent with DAPI (Invitrogen) and imaged using a Leica SP5X laser-scanning confocal microscope at 63× magnification. Images were analyzed using ImageJ.
Voltage and calcium transient mapping
Hearts were isolated from embryos at 72 hpf and stained with either the transmembrane potential-sensitive dye di-8-ANEPPS (Invitrogen) or the calcium-sensitive ratiometric dye Fura-2AM (Invitrogen) for measurement of voltage or calcium transients, respectively. Resulting fluorescence intensities were recorded with a high-speed charge-coupled device camera (RedShirtImaging) and images analyzed using software implemented in MATLAB (Panáková et al., 2010). Pacing at a constant rate of 80 beats per minute was used for calcium mapping to eliminate heart rate effects. For all comparisons, regions of interest were determined and compared between morpholino-injected embryos and controls from at least two separate mate pairings using a two-sided Student’s t-test.
Overexpression of tmem88a
RNA was isolated from 48 hpf wild-type embryos and used to synthesize cDNA as previously described (Peterson and Freeman, 2009). tmem88a-specific primers containing the MultiSite Gateway attB1 and attB2 recognition sites were then used to amplify tmem88a from cDNA by PCR. To generate the abridged tmem88a transcript (tmem88aΔVWV), the reverse primer carried a stop codon immediately before the C-terminal VWV motif. The verified PCR product was combined with pDONR221 in a BP reaction (BP Clonase II; Invitrogen) to generate MultiSite Gateway entry vectors pENTR_tmem88a and pENTR_tmem88aΔVWV. These entry vectors were combined with p3E-polyA [Tol2kit #302 (Kwan et al., 2007)], a Tol2 destination vector containing an alpha-crystallin:YFP selection marker, and pENTR5′_ubi [containing the ubiquitin promoter (Mosimann et al., 2011)], in an LR reaction (LR Clonase Plus II; Invitrogen) to generate the ubi:tmem88a and ubi:tmem88aΔVWV expression vectors. Zebrafish embryos were injected with 20 pg of either of these vectors and 10 pg transposase at the one-cell developmental stage, and screened for fluorescence and developmental phenotypes at 48 hpf.
Evaluation of Wnt11 synergy
Test morpholinos were injected either singly or in combination with a previously verified morpholino targeting wnt11 (Panáková et al., 2010) at low doses to identify potential synergistic effects. Resulting morphants were scored for cyclopia at 48 hpf based on the five classes previously described (Marlow et al., 1998). The evaluator (G.M.) was blinded as to the identity of the morphants at the time of survey. Percentages reflect the total embryos in each cyclopia class summed over three separate mate pairings. Results were compared using a Chi-squared test.
Supplementary Material
Acknowledgments
We thank all members of the C.A.M. and F.P.R. labs for helpful discussion, especially Dr Daniela Panakova and Dr Andreas Werdich, as well as Jason Montojo from the Q.M. lab. We also thank Dr Rahul Deo (UCSF), Dr Martha Marvin (Williams College), Dr Andrew Emili (University of Toronto) and Dr Natalie Morson (University of Toronto) for helpful discussions and critical reading of the manuscript.
Footnotes
Competing interests
The authors declare no competing financial interests.
Author contributions
G.M., C.A.M. and F.P.R. designed this study and wrote the manuscript. G.M., M.T. and J.E.B. adapted and implemented the phenotype prediction method under the supervision of F.P.R. All zebrafish experiments were performed by G.M., E.P., C.M. and L.A.C. under the supervision of C.A.M. and L.Z. Prediction data were integrated with public databases by H.N.C., J.D., K.Z. and H.R. under the supervision of Q.M. and F.P.R.
Funding
F.P.R. was supported by National Institutes of Health (NIH) grants [HG003224, HG004233 and HL107440]; by an Ontario Research Fund - Research Excellence Award; by the Canada Excellence Research Chairs Program; and by a Canadian Institute for Advanced Research Fellowship. M.T. was supported by an NIH grant [HG004098]. C.M. received support through a Human Frontier Science Program (HFSP) long-term fellowship and a Swiss National Science Foundation (SNSF) advanced researcher fellowship. C.A.M. was supported in this work by an NIH grant [HL098938]; the Leducq Foundation; and the Harvard Stem Cell Institute. L.Z. is a HHMI Investigator. Deposited in PMC for release after 6 months.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.099796/-/DC1
References
- Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- Amsterdam A., Burgess S., Golling G., Chen W., Sun Z., Townsend K., Farrington S., Haldi M., Hopkins N. (1999). A large-scale insertional mutagenesis screen in zebrafish. Genes Dev. 13, 2713–2724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amsterdam A., Nissen R. M., Sun Z., Swindell E. C., Farrington S., Hopkins N. (2004). Identification of 315 genes essential for early zebrafish development. Proc. Natl. Acad. Sci. USA 101, 12792–12797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaout R., Ferrer T., Huisken J., Spitzer K., Stainier D. Y., Tristani-Firouzi M., Chi N. C. (2007). Zebrafish model for human long QT syndrome. Proc. Natl. Acad. Sci. USA 104, 11316–11321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., et al. ; The Gene Ontology Consortium (2000). Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auman H. J., Coleman H., Riley H. E., Olale F., Tsai H. J., Yelon D. (2007). Functional modulation of cardiac form through regionally confined cell shape changes. PLoS Biol. 5, e53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T., Edgar R. (2006). Gene expression omnibus: microarray data storage, submission, retrieval, and analysis. Methods Enzymol. 411, 352–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaver J. E., Tasan M., Gibbons F. D., Tian W., Hughes T. R., Roth F. P. (2010). FuncBase: a resource for quantitative gene function annotation. Bioinformatics 26, 1806–1807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros M., Kiger A. A., Armknecht S., Kerr K., Hild M., Koch B., Haas S. A., Paro R., Perrimon N.; Heidelberg Fly Array Consortium (2004). Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science 303, 832–835 [DOI] [PubMed] [Google Scholar]
- Bradford Y., Conlin T., Dunn N., Fashena D., Frazer K., Howe D. G., Knight J., Mani P., Martin R., Moxon S. A., et al. (2011). ZFIN: enhancements and updates to the Zebrafish Model Organism Database. Nucleic Acids Res. 39, D822–D829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns C. G., Milan D. J., Grande E. J., Rottbauer W., MacRae C. A., Fishman M. C. (2005). High-throughput assay for small molecules that modulate zebrafish embryonic heart rate. Nat. Chem. Biol. 1, 263–264 [DOI] [PubMed] [Google Scholar]
- Camarata T., Krcmery J., Snyder D., Park S., Topczewski J., Simon H. G. (2010). Pdlim7 (LMP4) regulation of Tbx5 specifies zebrafish heart atrio-ventricular boundary and valve formation. Dev. Biol. 337, 233–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon J. E., Place E. S., Eve A. M., Bradshaw C. R., Sesay A., Morrell N. W., Smith J. C. (2013). Global analysis of the haematopoietic and endothelial transcriptome during zebrafish development. Mech. Dev. 130, 122–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M., Baryshnikova A., Bellay J., Kim Y., Spear E. D., Sevier C. S., Ding H., Koh J. L., Toufighi K., Mostafavi S., et al. (2010). The genetic landscape of a cell. Science 327, 425–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng M., Chen T., Sun F. (2004). An integrated probabilistic model for functional prediction of proteins. J. Comput. Biol. 11, 463–475 [DOI] [PubMed] [Google Scholar]
- Driever W., Solnica-Krezel L., Schier A. F., Neuhauss S. C., Malicki J., Stemple D. L., Stainier D. Y., Zwartkruis F., Abdelilah S., Rangini Z., et al. (1996). A genetic screen for mutations affecting embryogenesis in zebrafish. Development 123, 37–46 [DOI] [PubMed] [Google Scholar]
- Ekker S. C., Larson J. D. (2001). Morphant technology in model developmental systems. Genesis 30, 89–93 [DOI] [PubMed] [Google Scholar]
- Etard C., Roostalu U., Strähle U. (2010). Lack of Apobec2-related proteins causes a dystrophic muscle phenotype in zebrafish embryos. J. Cell Biol. 189, 527–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu C. Y., Lee H. C., Tsai H. J. (2009). The molecular structures and expression patterns of zebrafish troponin I genes. Gene Expr. Patterns 9, 348–356 [DOI] [PubMed] [Google Scholar]
- Gentleman R. C., Carey V. J., Bates D. M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J., et al. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G., Chu A. M., Ni L., Connelly C., Riles L., Véronneau S., Dow S., Lucau-Danila A., Anderson K., André B., et al. (2002). Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391 [DOI] [PubMed] [Google Scholar]
- Golling G., Amsterdam A., Sun Z., Antonelli M., Maldonado E., Chen W., Burgess S., Haldi M., Artzt K., Farrington S., et al. (2002). Insertional mutagenesis in zebrafish rapidly identifies genes essential for early vertebrate development. Nat. Genet. 31, 135–140 [DOI] [PubMed] [Google Scholar]
- Guan Y., Myers C. L., Lu R., Lemischka I. R., Bult C. J., Troyanskaya O. G. (2008). A genomewide functional network for the laboratory mouse. PLoS Comput. Biol. 4, e1000165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffter P., Granato M., Brand M., Mullins M. C., Hammerschmidt M., Kane D. A., Odenthal J., van Eeden F. J., Jiang Y. J., Heisenberg C. P., et al. (1996). The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Development 123, 1–36 [DOI] [PubMed] [Google Scholar]
- Hasdemir C., Aydin H. H., Celik H. A., Simsek E., Payzin S., Kayikcioglu M., Aydin M., Kultursay H., Can L. H. (2010). Transcriptional profiling of septal wall of the right ventricular outflow tract in patients with idiopathic ventricular arrhythmias. Pacing Clin. Electrophysiol. 33, 159–167 [DOI] [PubMed] [Google Scholar]
- Heasman J. (2002). Morpholino oligos: making sense of antisense? Dev. Biol. 243, 209–214 [DOI] [PubMed] [Google Scholar]
- Howe K., Clark M. D., Torroja C. F., Torrance J., Berthelot C., Muffato M., Collins J. E., Humphray S., McLaren K., Matthews L., et al. (2013). The zebrafish reference genome sequence and its relationship to the human genome. Nature 496, 498–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P., Xiao A., Zhou M., Zhu Z., Lin S., Zhang B. (2011). Heritable gene targeting in zebrafish using customized TALENs. Nat. Biotechnol. 29, 699–700 [DOI] [PubMed] [Google Scholar]
- Huttenhower C., Haley E. M., Hibbs M. A., Dumeaux V., Barrett D. R., Coller H. A., Troyanskaya O. G. (2009). Exploring the human genome with functional maps. Genome Res. 19, 1093–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang W. Y., Fu Y., Reyon D., Maeder M. L., Tsai S. Q., Sander J. D., Peterson R. T., Yeh J. R., Joung J. K. (2013). Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 31, 227–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath R. S., Fraser A. G., Dong Y., Poulin G., Durbin R., Gotta M., Kanapin A., Le Bot N., Moreno S., Sohrmann M., et al. (2003). Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421, 231–237 [DOI] [PubMed] [Google Scholar]
- Kanehisa M., Araki M., Goto S., Hattori M., Hirakawa M., Itoh M., Katayama T., Kawashima S., Okuda S., Tokimatsu T., et al. (2008). KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, D480–D484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaoz U., Murali T. M., Letovsky S., Zheng Y., Ding C., Cantor C. R., Kasif S. (2004). Whole-genome annotation by using evidence integration in functional-linkage networks. Proc. Natl. Acad. Sci. USA 101, 2888–2893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettleborough R. N., Busch-Nentwich E. M., Harvey S. A., Dooley C. M., de Bruijn E., van Eeden F., Sealy I., White R. J., Herd C., Nijman I. J., et al. (2013). A systematic genome-wide analysis of zebrafish protein-coding gene function. Nature 496, 494–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King O. D., Lee J. C., Dudley A. M., Janse D. M., Church G. M., Roth F. P. (2003). Predicting phenotype from patterns of annotation. Bioinformatics 19 Suppl. 1, i183–i189 [DOI] [PubMed] [Google Scholar]
- Kwan K. M., Fujimoto E., Grabher C., Mangum B. D., Hardy M. E., Campbell D. S., Parant J. M., Yost H. J., Kanki J. P., Chien C. B. (2007). The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev. Dyn. 236, 3088–3099 [DOI] [PubMed] [Google Scholar]
- Langenbacher A. D., Dong Y., Shu X., Choi J., Nicoll D. A., Goldhaber J. I., Philipson K. D., Chen J. N. (2005). Mutation in sodium-calcium exchanger 1 (NCX1) causes cardiac fibrillation in zebrafish. Proc. Natl. Acad. Sci. USA 102, 17699–17704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazic S., Scott I. C. (2011). Mef2cb regulates late myocardial cell addition from a second heart field-like population of progenitors in zebrafish. Dev. Biol. 354, 123–133 [DOI] [PubMed] [Google Scholar]
- Lee I., Date S. V., Adai A. T., Marcotte E. M. (2004). A probabilistic functional network of yeast genes. Science 306, 1555–1558 [DOI] [PubMed] [Google Scholar]
- Lee I., Lehner B., Crombie C., Wong W., Fraser A. G., Marcotte E. M. (2008). A single gene network accurately predicts phenotypic effects of gene perturbation in Caenorhabditis elegans. Nat. Genet. 40, 181–188 [DOI] [PubMed] [Google Scholar]
- Lee H. J., Finkelstein D., Li X., Wu D., Shi D. L., Zheng J. J. (2010). Identification of transmembrane protein 88 (TMEM88) as a dishevelled-binding protein. J. Biol. Chem. 285, 41549–41556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque M., Shasha D., Kim W., Surette M. G., Benfey P. N. (2003). Trait-to-gene: a computational method for predicting the function of uncharacterized genes. Curr. Biol. 13, 129–133 [DOI] [PubMed] [Google Scholar]
- Linghu B., Snitkin E. S., Hu Z., Xia Y., Delisi C. (2009). Genome-wide prioritization of disease genes and identification of disease-disease associations from an integrated human functional linkage network. Genome Biol. 10, R91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning C. D., Raghavan P., Schutze H. (2008). Introduction to Information Retrieval. Cambridge: Cambridge University Press; [Google Scholar]
- Maranto A. R. (1994). Primary structure, ligand binding, and localization of the human type 3 inositol 1,4,5-trisphosphate receptor expressed in intestinal epithelium. J. Biol. Chem. 269, 1222–1230 [PubMed] [Google Scholar]
- Marlow F., Zwartkruis F., Malicki J., Neuhauss S. C., Abbas L., Weaver M., Driever W., Solnica-Krezel L. (1998). Functional interactions of genes mediating convergent extension, knypek and trilobite, during the partitioning of the eye primordium in zebrafish. Dev. Biol. 203, 382–399 [DOI] [PubMed] [Google Scholar]
- Meng X., Noyes M. B., Zhu L. J., Lawson N. D., Wolfe S. A. (2008). Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nat. Biotechnol. 26, 695–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosimann C., Kaufman C. K., Li P., Pugach E. K., Tamplin O. J., Zon L. I. (2011). Ubiquitous transgene expression and Cre-based recombination driven by the ubiquitin promoter in zebrafish. Development 138, 169–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafavi S., Ray D., Warde-Farley D., Grouios C., Morris Q. (2008). GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 9 Suppl. 1, S4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasevicius A., Ekker S. C. (2000). Effective targeted gene ‘knockdown’ in zebrafish. Nat. Genet. 26, 216–220 [DOI] [PubMed] [Google Scholar]
- Novikov N., Evans T. (2013). Tmem88a mediates GATA-dependent specification of cardiomyocyte progenitors by restricting WNT signaling. Development 140, 3787–3798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palpant N. J., Pabon L., Rabinowitz J. S., Hadland B. K., Stoick-Cooper C. L., Paige S. L., Bernstein I. D., Moon R. T., Murry C. E. (2013). Transmembrane protein 88: a Wnt regulatory protein that specifies cardiomyocyte development. Development 140, 3799–3808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panáková D., Werdich A. A., Macrae C. A. (2010). Wnt11 patterns a myocardial electrical gradient through regulation of the L-type Ca(2+) channel. Nature 466, 874–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini M., Marcotte E. M., Thompson M. J., Eisenberg D., Yeates T. O. (1999). Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc. Natl. Acad. Sci. USA 96, 4285–4288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña-Castillo L., Tasan M., Myers C. L., Lee H., Joshi T., Zhang C., Guan Y., Leone M., Pagnani A., Kim W. K., et al. (2008). A critical assessment of Mus musculus gene function prediction using integrated genomic evidence. Genome Biol. 9 Suppl. 1, S2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson S. M., Freeman J. L. (2009). RNA isolation from embryonic zebrafish and cDNA synthesis for gene expression analysis. J. Vis. Exp. 30, 1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson R. T., Link B. A., Dowling J. E., Schreiber S. L. (2000). Small molecule developmental screens reveal the logic and timing of vertebrate development. Proc. Natl. Acad. Sci. USA 97, 12965–12969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robu M. E., Larson J. D., Nasevicius A., Beiraghi S., Brenner C., Farber S. A., Ekker S. C. (2007). p53 activation by knockdown technologies. PLoS Genet. 3, e78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S., Heber S. (2006). In silico prediction of yeast deletion phenotypes. Genet. Mol. Res. 5, 224–232 [PubMed] [Google Scholar]
- Sehnert A. J., Huq A., Weinstein B. M., Walker C., Fishman M., Stainier D. Y. (2002). Cardiac troponin T is essential in sarcomere assembly and cardiac contractility. Nat. Genet. 31, 106–110 [DOI] [PubMed] [Google Scholar]
- Shin J. T., Pomerantsev E. V., Mably J. D., MacRae C. A. (2010). High-resolution cardiovascular function confirms functional orthology of myocardial contractility pathways in zebrafish. Physiol. Genomics 42, 300–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sing T., Sander O., Beerenwinkel N., Lengauer T. (2005). ROCR: visualizing classifier performance in R. Bioinformatics 21, 3940–3941 [DOI] [PubMed] [Google Scholar]
- Stainier D. Y., Fouquet B., Chen J. N., Warren K. S., Weinstein B. M., Meiler S. E., Mohideen M. A., Neuhauss S. C., Solnica-Krezel L., Schier A. F., et al. (1996). Mutations affecting the formation and function of the cardiovascular system in the zebrafish embryo. Development 123, 285–292 [DOI] [PubMed] [Google Scholar]
- Stark K., Esslinger U. B., Reinhard W., Petrov G., Winkler T., Komajda M., Isnard R., Charron P., Villard E., Cambien F., et al. (2010). Genetic association study identifies HSPB7 as a risk gene for idiopathic dilated cardiomyopathy. PLoS Genet. 6, e1001167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taşan M., Tian W., Hill D. P., Gibbons F. D., Blake J. A., Roth F. P. (2008). An en masse phenotype and function prediction system for Mus musculus. Genome Biol. 9 Suppl. 1, S8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasan M., Drabkin H. J., Beaver J. E., Chua H. N., Dunham J., Tian W., Blake J. A., Roth F. P. (2012). A resource of quantitative functional annotation for homo sapiens genes. G3 (Bethesda) 2, 223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thisse C., Thisse B. (2008). High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 3, 59–69 [DOI] [PubMed] [Google Scholar]
- Tian W., Zhang L. V., Taşan M., Gibbons F. D., King O. D., Park J., Wunderlich Z., Cherry J. M., Roth F. P. (2008). Combining guilt-by-association and guilt-by-profiling to predict Saccharomyces cerevisiae gene function. Genome Biol. 9 Suppl. 1, S7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troyanskaya O. G., Dolinski K., Owen A. B., Altman R. B., Botstein D. (2003). A Bayesian framework for combining heterogeneous data sources for gene function prediction (in Saccharomyces cerevisiae). Proc. Natl. Acad. Sci. USA 100, 8348–8353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villard E., Perret C., Gary F., Proust C., Dilanian G., Hengstenberg C., Ruppert V., Arbustini E., Wichter T., Germain M., et al. ; Cardiogenics Consortium (2011). A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur. Heart J. 32, 1065–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel B., Meder B., Just S., Laufer C., Berger I., Weber S., Katus H. A., Rottbauer W. (2009). In-vivo characterization of human dilated cardiomyopathy genes in zebrafish. Biochem. Biophys. Res. Commun. 390, 516–522 [DOI] [PubMed] [Google Scholar]
- Westerfield M. (2000). The Zebrafish Book: A Guide for the Laboratory use of Zebrafish (Danio rerio), 4th edn Eugene, OR: University of Oregon Press; [Google Scholar]
- Woods J. O., Singh-Blom U. M., Laurent J. M., McGary K. L., Marcotte E. M. (2013). Prediction of gene-phenotype associations in humans, mice, and plants using phenologs. BMC Bioinformatics 14, 203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z., Irizarry R. A., Gentleman R., Martinez-Murillo F., Spencer F. (2004). A model-based background adjustment for oligonucleotide expression arrays. J. Am. Stat. Assoc. 99, 909–917 [Google Scholar]
- Zhou Y., Cashman T. J., Nevis K. R., Obregon P., Carney S. A., Liu Y., Gu A., Mosimann C., Sondalle S., Peterson R. E., et al. (2011). Latent TGF-β binding protein 3 identifies a second heart field in zebrafish. Nature 474, 645–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.