

Abstract
Background
Mast cells are increased in isolated mitral regurgitation (MR) in the dog and may mediate extracellular matrix (ECM) loss and left ventricular (LV) dilatation. We tested the hypothesis that mast cell stabilization would attenuate LV remodeling and improve function in the MR dog.
Methods
MR was induced in adult dogs randomized to no treatment (MR, n = 9) or to mast cell stabilizer, ketotifen (MR+MCS, n = 6) for four months. LV hemodynamics was obtained after four months of MR and magnetic resonance imaging (MRI) was performed at sacrifice.
Results
MRI-derived serial short axis LV end-diastolic (ED) and end-systolic (ES) volumes, LVED volume/mass ratio, and LV three-dimensional radius/wall thickness were increased in MR and MR+MCS dogs compared to normal dogs (n = 6) (P < 0.05). Interstitial collagen was decreased by 30% in both MR and MR+MCS vs. normal dogs (P < 0.05). LV contractility by LV maximum time-varying elastance was significantly depressed in MR and MR + MCS dogs. Furthermore, cardiomyocyte fractional shortening was decreased in MR vs. normal dogs and further depressed in MR+MCS dogs (P < 0.05). In vitro administration of ketotifen to normal cardiomyocytes also significantly decreased fractional shortening and calcium transients.
Conclusions
Chronic mast cell stabilization did not attenuate eccentric LV remodeling or collagen loss in MR. However, MCS therapy had a detrimental effect on LV function due a direct negative inotropic effect on cardiomyocyte function.
Keywords: heart failure, mast cells, calcium, myocyte
INTRODUCTION
Isolated mitral regurgitation (MR) is characterized by left ventricular (LV) dilation and augmented stroke volume because ejection is facilitated by regurgitation into the low pressure left atrium.1 Isolated MR in the dog produces LV dilation and wall thinning, cardiomyocyte elongation, and a decrease in interstitial collagen which is also associated with an increase in mast cells.2–4 We have shown that in the volume overload of aortocaval fistula (ACF) in the rat, interstitial collagen loss and LV dilatation precede cardiomyocyte elongation, suggesting that collagen breakdown is the first step in the pathophysiology of LV dilatation in response to a pure volume overload.5
LV mast cell density increases as early as two weeks and persists for four to six months in isolated MR in the dog4 and the ACF rat.6 Mast cell degranulation causes the release of proteolytic enzymes tryptase and chymase. We have previously shown that mast cell chymase activity is increased in the chronic MR dog,2 and chymase has been shown to activate matrix metalloproteinases (MMPs).7,8 Thus, it has been suggested that mast cell degranulation mediates collagen degradation and adverse LV and cardiomyocyte remodeling and function in isolated MR.
Mast cell stabilizers (MCS) are commonly used in the treatment of allergic conditions. They stabilize the mast cell through blockade of calcium channels, which prevents degranulation and the release of histamine and other proteolytic enzymes.9 Treatment with a MCS in the ACF rat prevents cardiac mast cell infiltration and degranulation, and attenuates MMP activity and LV dilatation.10 Taken together, these results suggest that MCS improves LV remodeling through the preservation of extracellular matrix (ECM). Many other animal studies have implicated mast cells in the pathophysiology of vascular and cardiac remodeling.6,10–14 However, it is well appreciated that mast cell contents differ between species. For example, the mouse and rat have multiple isoforms of β chymase, whereas the dog and human possess only the α isoform.14 Thus, the dog model of MR may be more clinically relevant in studying the presumed deleterious effects of mast cells in heart disease.
In the current study of experimentally induced MR in the dog, we report the effects of the prophylactic anti-asthma drug, ketotifen, which has been shown to possess potent mast cell stabilizing effects in dogs.15,16 Ketotifen is a histamine H1 receptor and calcium entry antagonist that inhibits the release of leukotrienes and improves β-adrenoreceptor sensitivity.9 Here we show that MCS does not attenuate eccentric LV remodeling nor does it preserve interstitial collagen after four months of MR in the dog. Further, the calcium entry antagonistic effects of ketotifen further decreases cardiomyocyte function and LV function in dogs with isolated MR.
METHODS
Experimental Preparation and Protocol
Mitral valve regurgitation was induced in 15 conditioned adult mongrel dogs of either sex (19–26 kg) by chordal rupture using a fluoroscopic guided catheterization method as previously described in our laboratory.2–4 Dogs with MR were randomized to: (1) untreated (MR, n = 9; male = 5; female = 4) and (2) MCS (MR+MCS, ketotifen, 3mg/kg PO, twice daily; n = 6; male = 5; female = 1). This chronically administered dose is comparable to that used to prevent mast cell degranulation in dogs.15,16 Treatment was started 24 hours after MR induction. Only 5 of 9 untreated MR dogs (male = 3; female = 2) and 4 of 6 MR dogs treated with MCS (male = 3; female = 1) survived to four months. Normal dogs (n = 6, male = 3; female = 3) were obtained for MRI, morphometry and cell studies.
Prior to induction of MR, each dog underwent isofluorane anesthesia and hemodynamics were recorded from right heart catheterization through a left internal jugular vein cutdown. In addition, a high-fidelity catheter-tip manometer (Millar, Houston, TX) was inserted into the LV through an 8Fr sheath in the left carotid artery. Hemodynamic studies were repeated at Auburn College of Veterinary Medicine after approximately four months and animals were subsequently transported to University of Alabama at Birmingham (UAB) for terminal studies. At UAB MRI was performed prior to sacrifice on a 1.5 Tesla GE Signa Horizon (Milwaukee, WI) instrument optimized for cardiac application. The animals were allowed to recover and were sacrificed within five days. Prior to sacrifice, LV pressure volume relations were obtained with the impedance catheter in the closed chest anesthetized state. This study was approved by the Animal Services Committees at both institutions.
Magnetic resonance imaging at UAB and analysis
Left ventricular endocardial and epicardial contours were manually traced on the LV end-diastolic (ED) and end-systolic (ES) images, excluding papillary muscles. LVED and LVES volumes (V) were determined by summating serial short axis slices as previously described in our laboratory.2–3
The contour data were then transformed to a coordinate system aligned along the long-axis of the LV and converted to a prolate spheroidal coordinate system as described previously.18 The prolate spheroidal coordinate system has one radial coordinate (λ) and two angular coordinates (μ,θ). Prolate spheroidal coordinates were used because surfaces of constant λ are ellipsoids, which more closely approximate the shape of the LV wall than cylinders or spheres. Cubic B-spline surfaces, λendo(μ,θ) and λepi(μ,θ), were fit to the λ coordinates of the endocardial and epicardial contours for each time frame. Each surface used 12 control points in the circumferential (θ) direction and 10 control points in the longitudinal direction (μ). The control points of each surface were computed to minimize the following error function
where γ is a weight set to 0.1. The first term in the error function is the squared difference between the contour points, λk, and the corresponding surface points λ(μk,θk). The second term is a smoothing function, which penalizes the bending energy of the surface
where Ω is the domain of the surface. Surface curvatures were computed using standard formulas19 at the wall segments as previously defined (excluding the apex).20 Three-dimensional (3-D) wall thickness was computed at the same segments by measuring the 3-D distance from a point on the epicardial surface to the closest point on the endocardial surface along a line perpendicular to the epicardial surface. Radius of curvature to wall thickness ratio (R/T) was computed by the reciprocal of the product of the endocardial circumferential curvature (κ) and 3-D wall thickness (T).
Sacrifice Instrumentation at UAB
Animals were maintained at a deep plane of general anesthesia using 1–2% isofluorane in 100% oxygen. LV pressure volume relations were determined using a pressure/volume impedance catheter equipped with full-bridge silicone microchip transducers (12 electrodes, 8 mm spacing, CD Leycom, Zoetemeer, The Netherlands). The simultaneous measurement of LV pressure and volume from the impedance catheter were collected in a baseline state and during transient inferior vena cava occlusion via balloon catheter. Thus LV preload was altered to obtain isochronal points from the recorded LV pressure-volume loops at multiple loads and used to compute LV Emax, an index of LV contractility defined as maximal LV elastance.21,22 These data were generated using a commercially available software package (PVAN, version 3.0, Millar, Houston, Texas). Prior to inferior vena cava balloon occlusion, LV volumes were calibrated to an LV volume estimate obtained from a single plane right anterior oblique LV ventriculogram using a nonionic contrast agent (Loxilan, Guerbet). To normalize for heart size, LV Emax was multiplied by the LVEDV obtained in the baseline state measured by the impedance catheter.
After LV pressure volume analysis, the chest was opened, the heart was arrested with KCl, quickly extirpated, and placed in ice-cold Krebs solution (120.4 mM NaCl, 14.7 mM KCl, 0.6 mM KH2PO4, 0.6 mM Na2HPO4, 1.2 mM MgSO4.7H2O, 4.6 mM NaHCO3, 10 mM Na-HEPES, 30 mM taurine, 5.5 mM glucose). The coronary arteries were flushed with the same solution. The atria, RV and LV were carefully separated and weighed.
Isolated cardiomyocyte studies
After the LV was weighed, small LV wedges (from the posterior wall) were cannulated at larger branch arteries at the base then perfused with warm (36–37°C) calcium-free Krebs solution (120.4 mM NaCl, 14.7 mM KCl, 0.6 mM KH2PO4, 0.6 mM Na2HPO4, 1.2 mM MgSO4.7H2O, 4.6 mM NaHCO3, 10 mM Na-HEPES, 30 mM taurine, 5.5 mM glucose) and gassed with 100% O2 for 5–7 minutes. Cardiomyocytes were isolated from the tissue by recirculating perfusion buffer supplemented with 2mg/mL collagenase type II (Invitrogen, Carlsbad, California) for 10–20 minutes.
Intracellular calcium was measured with the fluorescent indicator fluo 3-acetoxymethyl ester (fluo 3-AM; Molecular Probes, Eugene, Oregon). Cardiomyocytes were incubated at 37°C in the dark for 30 minutes in normal HEPES-buffered solution consisting of 126 mM NaCl, 11 mM dextrose, 4.4 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 24 mM HEPES with pH adjusted to 7.4 and 5 μM fluo 3-AM added. Fluo 3-AM loaded cardiomyocytes were placed in a glass-bottom, temperature-controlled bath (37°C) that was mounted on the stage of an inverted microscope (Eclipse TE300/200, Nikon, Lewisville, Texas), as described previously.17 Cells were continuously bathed at a rate of 1–2 ml/min with the HEPES-buffered bathing solution containing 0.5 mM probenicid to help retard fluo 3-AM transport from the cells. Cardiomyocytes were allowed to equilibrate for at least 5 minutes prior to stimulation. Fluorescence emission (530 nm) was collected with a photomultiplier tube via the 20x objective during continuous excitation at 485 nm and is reported as the fluorescence normalized to baseline values, after background subtraction, F/F0. The contractile activity (fractional shortening) of field-stimulated cardiomyocytes was measured with a video-edge detector (Crescent Electronics, Sandy, Utah). Field stimulation was done with 5 ms square pulses with constant voltage at ~20% above threshold and 1000 ms cycle length. Calcium fluorescence and fractional shortening variables were obtained in the same cardiomyocyte at baseline and after 5 minutes of superfusion with 25 nM D-L isoproterenol or 50 μM ketotifen fumarate (both from Sigma, St. Louis Missouri). Signals were recorded with the Axoclamp 2B amplifier system (Axon Instruments, Inc., Foster City, California), digitized with a 12-bit A/D converter (Axon Instruments, Digidata 1200A), and recorded with a computer using pClamp 9 software (Axon Instruments).
Collagen analysis
Paraformaldehyde fixed paraffin-embedded sections (3μm) from the long-axis and short-axis epicardial and endocardial sections through the LV mid-region were stained with Picric Acid Sirius Red F3BA. Interstitial collagen was identified by light microscopy at high power (40x objective, 1600x total magnification) and collagen volume percent quantified (in 30–40 randomly selected fields per animal) with a digital-based image-analyzer system (Image-Pro Plus version 6.0, MediaCybernetics, Bethesda, Maryland).
Cardiomyocyte cross-sectional area
Formalin-fixed frozen LV mid-endocardial sections were laminin-stained for cardiomyocyte cross-sectional area analysis. Briefly, 5μm sections were subjected to Proteinase K (20μg/mL – Roche, Indianapolis, Indiana) enzymatic antigen retrieval for 30min at 37°C, blocked with 5% goat serum/1% bovine serum in phosphate buffered saline for 1hr at room temperature. Sections were incubated with rabbit α-laminin (1:200 - Abcam, Cambridge, Massachusetts) overnight at 4°C then incubated with an FITC-conjugated goat α-rabbit IgG secondary antibody (1:400 - Molecular Probes) for 1hr at room temperature, then coverslipped with DAPI-hard set mounting medium (Vector Labs, Burlingame, California). Fluorescent images in cross-section were analyzed using a digital-based image-analyzer system (Image Pro Plus) to count cardiomyocyte number per μm2 of tissue to determine cross-sectional area. A minimum of four areas were measured for each animal.
Statistics
Data are presented as mean ± standard error of the mean (SEM). One-way analysis of variance and the Holm-Sidak post-hoc test for significance was used to analyze data using Sigma Stat version 3.5 (Systat Software, Chicago, Illinois). P < 0.05 was considered significant.
RESULTS
LV hemodynamics and function
Body weight (BW) did not differ among normal (n =6), MR (n =6), and MR+MCS (n =4) groups (Table 1). LV mass:BW was increased by 25% (P = 0.002) in MR dogs and was significantly higher in MR+MCS dogs (P = 0.04). Right ventricular mass did not differ from normal in both MR groups. Mean heart rate, cardiac output, LV ejection fraction, peak +LV dP/dt and systemic or pulmonary vascular resistance did not differ from baseline values in both MR groups. However, pulmonary arterial wedge pressure was significantly elevated in MR+MCS dogs compared to MR and normal dogs (P < 0.02). LV fractional shortening was increased in MR (P < 0.05) but did not differ from normal in MR+MCS dogs, suggestive of significant LV systolic dysfunction in the MR+MCS dogs.
TABLE 1.
LV Function and Hemodynamics.
| Normal | MR | MR+MCS | |
|---|---|---|---|
| Body Weight (kg) | 20 ± 1 | 21 ± 1 | 22 ± 1 |
| LV:Body Weight (g/kg) | 3.9 ± 0.3 | 4.9 ± 0.2 * | 5.7 ± 0.3 *# |
| RV:Body Weight (g/kg) | 1.7 ± 0.1 | 1.8 ± 0.1 | 1.8 ± 0.1 |
| n | 6 | 5 | 4 |
| Hemodynamics | Baseline | ||
| Heart Rate (bpm) | 107 ± 4 | 110 ± 6 | 111 ± 10 |
| Cardiac output (L/min) | 3.9 ± 0.3 | 3.2 ± 0.3 | 3.0 ± 0.7 |
| LV ejection fraction (%) | 43 ± 4 | 51 ± 5 | 42 ± 6 |
| LV fractional shortening (%) | 17 ± 1 | 25 ± 2 * | 18 ± 5 |
| LV +dP/dtmax (mmHg/s) | 2181 ± 156 | 2423 ± 297 | 1865 ± 160 |
| Systemic vascular resistance (dyne cm sec−5) | 1972 ± 160 | 2143 ± 312 | 1815 ± 215 |
| Pulmonary artery mean pressure (mmHg) | 13 ± 1 | 12 ± 2 | 17 ± 3 |
| Pulmonary artery wedge pressure (mmHg) | 5 ± 1 | 5 ± 1 | 11 ± 4 * # |
| Pulmonary vascular resistance (dyne cm sec−5) | 177 ± 25 | 179 ± 37 | 174 ± 69 |
| n | 9 | 5 | 4 |
P < 0.05 versus normal,
P < 0.05 versus MR.
LV maximal time varying elastance (Emax) was significantly decreased in MR and MR+MCS vs. normals dogs (P < 0.05, Table 2 and Figure 1). LV end-diastolic volumes measured with LV impedance catheter correlated with LVEDV from summated serial short axis magnetic resonance images (Figure 2, left panel, R2 = 0.59, P < 0.001). LV Emax and LV Ees are always lower in larger vs. normal sized hearts, which has raised the need for normalization of LV Emax and LV Ees to heart size.21,22 Indeed, there was a curvilinear relation between LV Emax and LV impedance catheter-derived LVEDV (Figure 2, right panel). There was a mean 2-fold decrease in LV Emax normalized to LVEDV in MR (171 ± 51, P = 0.045) which did not achieve significance in MR+MCS dogs (184 ± 76, P = 0.09) compared to normal dogs (374 ± 76). However, LV preload recruitable stroke work was significantly decreased only in MR+MCS dogs vs. normals (P = 0.007). Consistent with the increase in pulmonary arterial wedge pressure, MR+MCS dogs also demonstrated a trend toward an increased LV chamber stiffness constant compared to MR dogs (Table 2).
TABLE 2.
LV function assessed by pressure/volume impedance catheter.
| Normal | MR | MR+MCS | |
|---|---|---|---|
| Diastolic Function | |||
| LV Chamber Stiffness Constant | 0.022 ± 0.006 | 0.012 ± 0.005 | 0.025 ± 0.008 |
| LV Systolic Function | |||
| LV Emax (mmHg/mL) | 5.82 ± 1.74 | 2.00 ± 0.79 * | 1.62 ± 0.54 * |
| LV Preload Recruitable Stroke Work | 61 ± 10 | 44 ± 3 | 24 ± 6 * |
| n | 7 | 5 | 4 |
P < 0.05 vs. Normal.
Emax = maximal time varying elastance
FIGURE 1. LV pressure volume relationships in (A) Normal (B) 4 month MR and (C) 4 month MR+MCS dogs.
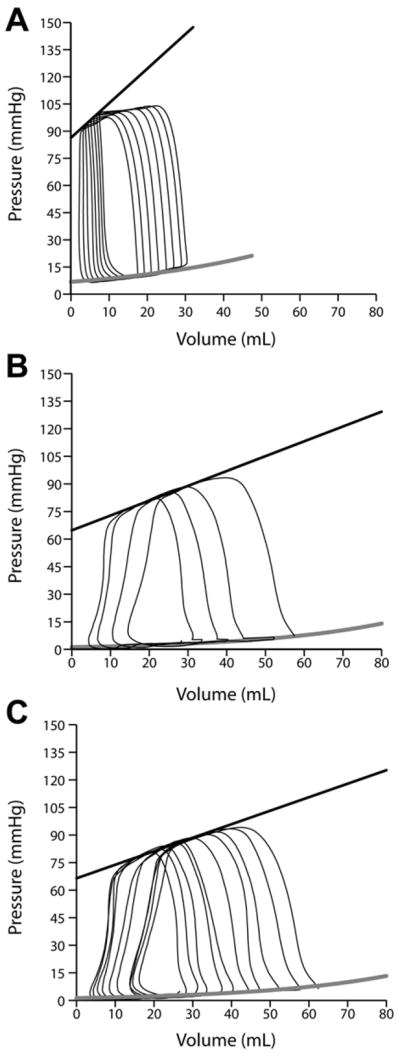
Black linear regression and gray exponential lines indicate the LV end-systolic (Ees) and end-diastolic (k1EDPVR) pressure volume relationship, respectively. Note how the end-systolic pressure volume linear regression line (black) is steeper in normal vs. MR and MR+MCS dogs.
Figure 2.
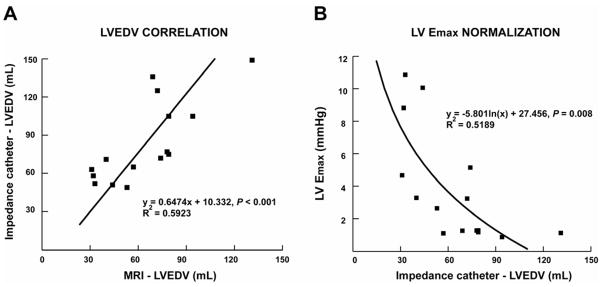
Panel A. Correlation of LV end-diastolic volume (LVEDV) by LV impedance catheter and cine-magnetic resonance imaging (MRI) in normal, MR and MR+MCS dogs. Panel B. Demonstration of the curvilinear relation of LV Emax and LVEDV.
LV remodeling by magnetic resonance imaging
LVESV and LVEDV were significantly increased in MR and MR+MCS vs. normal dogs (Table 3). In both MR groups there was a significant two-fold increase in LV stroke volume in the absence of any other valvular disease. This finding coupled with severe mitral valvular regurgitation on visual analysis of cine-MR imaging in each dog, documented a significant amount of MR in all dogs irrespective of treatment. LVEDV/mass ratio and LVED R/T increased significantly in both MR and MR+MCS vs. normals, indicative of an eccentric LV remodeling. MRI-derived LV mass correlated with the LV mass measured at sacrifice (R2 = 0.8594; P = 0.003, n = 9, MR and MR + MCS). In addition, MR+MCS had greater LV posterior wall thickness vs. untreated MR and normal dogs after four months (Fig 2, Table 3).
TABLE 3.
MRI-derived indices of LV remodeling in normal and four month MR dogs.
| Normal | MR | MR+MCS | |
|---|---|---|---|
| LV end-systolic volume, mL | 36 ± 2 | 54 ± 9 * | 59 ± 12 * |
| LV end-diastolic volume, mL | 57 ± 4 | 96 ± 15 * | 108 ± 16 * |
| LV stroke volume, mL | 21 ± 2 | 41 ± 6 * | 49 ± 8 * |
| LV end-diastolic volume: mass, mL/g | 0.88 ± 0.07 | 1.15 ± 0.10 * | 1.18 ± 0.14 * |
| LV end-diastolic radius/wall thickness | 2.5 ± 0.2 | 3.2 ± 0.2 * | 3.2 ± 0.4 * |
| LV end-diastolic posterior wall thickness, cm | 0.78 ± 0.05 | 0.74 ± 0.02 | 0.99 ± 0.06 *# |
| n | 6 | 5 | 4 |
P < 0.05 versus Normal.
P < 0.05 versus MR.
LV isolated cardiomyocyte function at baseline and in response to isoproterenol
We previously reported an increase in the length of individual isolated cardiomyocytes from chronic MR dogs.3 In the current study, analysis of LV endocardial tissue sections demonstrated a significant increase in cardiomyocyte cross-sectional area in MR (381 ± 29 μm2, P < 0.05) and MR+MCS (413 ± 35 μm2, P < 0.05) vs. normal dogs (309 ± 26 μm2). The increased cross-sectional area may be a reflection of higher LV wall stress in the LV endocardium of the MR heart.
LV cardiomyocyte fractional shortening was depressed in MR cardiomyocytes vs. normal (P < 0.05), and worsened in MCS treated dogs (P < 0.05) (Fig. 3A – white bars). Isoproterenol (ISO) induced ~ 50% increase in normal cardiomyocyte fractional shortening (P < 0.05) and this increase was blunted in both MR and MR+MCS cardiomyocytes (Fig. 3A – black vs. white bars). Calcium transients were decreased in both MR and MR+MCS cardiomyocytes vs. normals (P < 0.05) (Fig. 3B – white bars). ISO significantly increased calcium transients in normal cardiomyocytes but this effect was abolished in MR and MR+MCS cardiomyocytes (Fig. 3B – black vs. white bars).
FIGURE 3. Cine-Magnetic resonance images in MR dogs after four months.
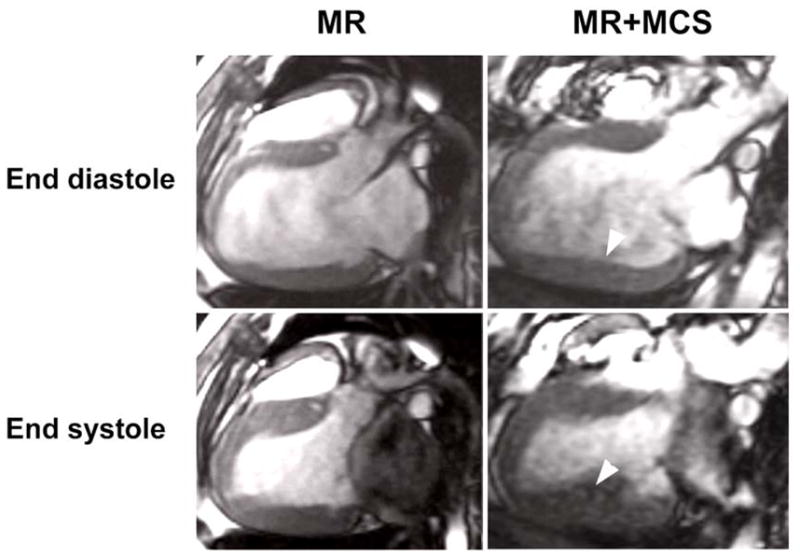
Long axis views of the LV at end diastole and end systole in dogs with MR and MR+MCS. In both groups, there is LV dilatation; however, in the MR+MCS dog there is greater LV posterior wall thickening (arrowheads – right panels) compared to the untreated MR dog (left panels).
Effect of ketotifen on isolated cardiomyocyte function in vitro
To determine whether the chronic effect of mast cell stabilization on cardiomyocyte function in MR dogs was due to inhibition of mast cell degranulation or a direct negative inotropic effect of the drug on the cardiomyocyte, we isolated cardiomyocytes from normal dogs and treated them with ketotifen (50μM). Administration of ketotifen to normal isolated cardiomyocytes caused a 67% and 11% decrease from baseline in both fractional shortening and calcium transients, respectively (P < 0.001) (Fig. 4).
FIGURE 4. Isoproterenol-induced change in fractional shortening and calcium transients in isolated cardiomyocytes.

(A) Fractional shortening and (B) Calcium transients at baseline (white bars) and after Isoproterenol stimulation (25 nmol/L) (black bars). * P < 0.05 vs. Normal baseline, # P < 0.05 vs. MR baseline, † P < 0.05 isproterenol vs. baseline of group.
Effect of mast cell stabilization on extracellular matrix
Interstitial collagen in both the endo- and epicardial region of the LV midwall were decreased in MR dogs after four months (Fig. 5). This 30% decrease in interstitial collagen was not attenuated by MCS.
FIGURE 5. Cardiomyocyte fractional shortening and calcium transients after treatment with ketotifen in normal cardiomyocytes.
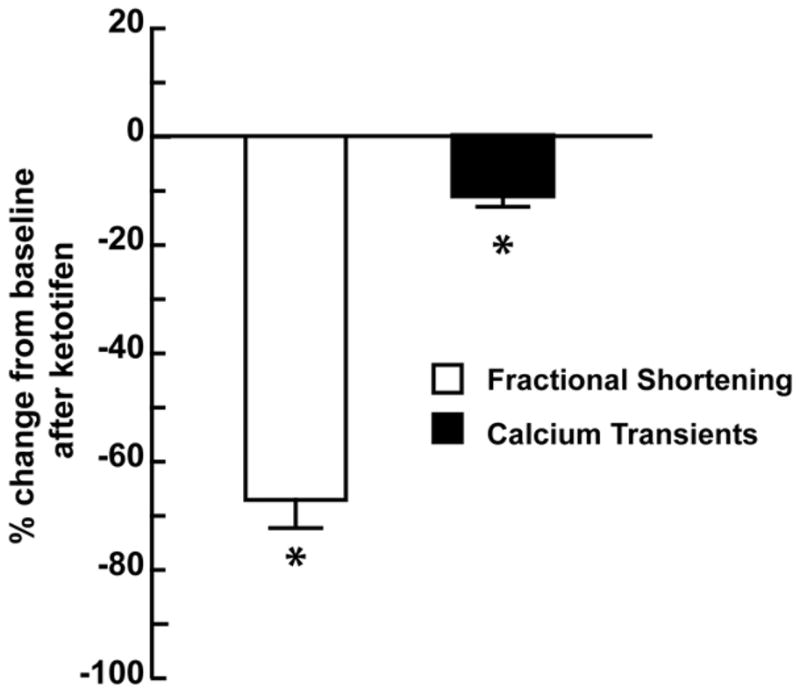
Percent change from baseline of fractional shortening and calcium transients after treatment with ketotifen (50μm). * P < 0.001 vs. baseline.
DISCUSSION
Here we show that long term stabilization of mast cells, does not attenuate eccentric LV remodeling or preserve LV interstitial collagen in isolated MR in the dog. Further, long term treatment significantly depresses cardiomyocyte and LV function. This effect is most likely caused by a heretofore unrecognized direct negative inotropic effect of ketotifen on the cardiomyocyte.
Mast cells are a source of many inflammatory mediators—cytokines, chemoattractants and proteases (tryptase and chymase)—all of which have been implicated in the progression of atherosclerosis11 and LV remodeling.12 Further, mast cells are not phenotypically static and their secretory pattern alters according to the organ and its changing microenvironment. In support of their functional versatility, treatment with MCS drugs has been reported to prevent fibrosis and LV dysfunction in the mouse with pressure overload,13 while in the rat with ACF, MCS therapy prevents matrix breakdown and attenuates LV dilatation, presumably through inhibition of MMP activation.10 Although these studies indicate that mast cells appear to be deleterious in heart failure, the divergent effects of MCS drugs on ECM homeostasis underscores the pathophysiology of the underlying condition in order to predict effect. Further, there is now emerging evidence that mast cells may have beneficial effects in host defense mechanisms in infection.14,23 It is of interest that mast cell deficient rats have been reported to develop diastolic dysfunction and perivascular fibrosis at nine months of age.24 Taken together, these studies support a multi-potential role for mast cells on ECM homeostasis and LV function that is dependent both on the type and stage of cardiac hemodynamic stress.
In the present study, MCS does not improve interstitial collagen loss and further depress cardiomyocyte fractional shortening compared to untreated MR dogs. Ketotifen has been reported to mediate changes in membrane permeability of mast cells to calcium and other ions required for mast cell degranulation.9,25 This suggests that ketotifen may also affect calcium homeostasis in the cardiomyocyte. Indeed, we show that administration of ketotifen to normal cardiomyocytes significantly decreases fractional shortening and calcium transients, indicating that the drug itself has a negative inotropic effect on the cardiomyocyte, that may be independent of mast cell mediated effects.
In addition to cardiomyocyte dysfunction, MCS treatment does not improve the decrease in LV Emax. In the volume overloaded heart, it has been suggested that LV Emax and LV Ees be normalized for increases in heart size.21,22 Indeed, after normalization there is still a persistent mean decrease in contractility; which is significantly different in MR dogs and trended towards a decrease in MR+MCS dogs compared to normals. In addition, there is a marked and significant decrease in preload recruitable stroke work (PRSW) in MR+MCS dogs. LV PRSW is an index of a functional Starling mechanism in the heart and may be a more accurate reflection of inherent contractile function in the setting of the pure volume overload of MR.26 In addition, LV fractional shortening is increased in MR dogs and did not change compared to normal dogs in MR+MCS dogs, which is further evidence of greater contractile dysfunction. Finally, the greater LV hypertrophy and posterior wall thickness in MR+MCS dogs is also consistent with greater intrinsic cardiomyocyte dysfunction and a higher pulmonary arterial wedge pressure with MCS.
The decrease in cardiomyoctye and LV function with MCS is a very interesting finding in view of the potential therapeutic implications for MCS in heart failure from previous studies.10 Studies in the eight week ACF rat demonstrate that MCS with nedocromil attenuated LV remodeling;10 however, assessments of both diastolic remodeling and systolic function were performed in the isolated heart and not in vivo as in the current investigation. The findings of the current study indicate that more extensive studies may be necessary to fully characterize the efficacy of mast cell stabilizers in the treatment of volume overload heart failure, especially given that long term administration of ketotifen in the current study caused a significant decline in cardiomyocyte and LV function in the MR dog.
FIGURE 6. Interstitial collagen volume percent in normal, MR and MR+MCS dogs.
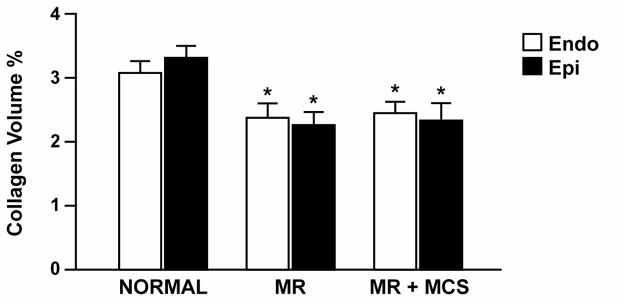
LV interstitial collagen volume percent by PASR in the endocardial and epicardial mid-wall of normal, 4 month untreated MR and MR+MCS dogs. * P < 0.05 vs. normal.
Acknowledgments
Sources of funding: This study was supported by the Office of Research and Development, Medical Service, Department of Veteran Affairs (LJD) and National Heart, Lung and Blood Institute Grant Specialized Centers of Clinically Orientated Research grant in Cardiac Dysfunction P50HL077100.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gaasch WH, Aurigemma GP. Inhibition of the renin-angiotensin system and the left ventricular adaptation to mitral regurgitation. J Am Coll Cardiol. 2002;39:1380–3. doi: 10.1016/s0735-1097(02)01766-7. [DOI] [PubMed] [Google Scholar]
- 2.Dell’Italia LJ, Meng QC, Balcells E, Straeter-Knowlen IM, Hankes GH, Dillon R, et al. Increased ACE and chymase-like activity in cardiac tissue of dogs with chronic mitral regurgitation. Am J Physiol. 1995;269:H2065–73. doi: 10.1152/ajpheart.1995.269.6.H2065. [DOI] [PubMed] [Google Scholar]
- 3.Dell’Italia LJ, Balcells E, Meng QC, Su X, Schultz D, Bishop SP, et al. Volume overload cardiac hypertrophy is unaffected by ACE inhibitor treatment in the dog. Am J Physiol. 1997;273:H961–70. doi: 10.1152/ajpheart.1997.273.2.H961. [DOI] [PubMed] [Google Scholar]
- 4.Stewart JA, Wei CC, Brower GL, Rynders PE, Hankes GH, Dillon AR, et al. Cardiac mast cell and chymase mediated matrix metalloproteinase activity and left ventricular remodeling in mitral regurgitation in the dog. J Mol Cell Cardiol. 2003;35:311–9. doi: 10.1016/s0022-2828(03)00013-0. [DOI] [PubMed] [Google Scholar]
- 5.Ryan TD, Rothstein EC, Aban I, Tallaj JA, Husain A, Lucchesi PA, et al. Left ventricular eccentric remodeling and matrix loss are mediated by bradykinin and precede cardiomyocyte elongation in rats with volume overload. J Am Coll Cardiol. 2007;49:811–21. doi: 10.1016/j.jacc.2006.06.083. [DOI] [PubMed] [Google Scholar]
- 6.Brower GL, Chancey AL, Thanigaraj S, Matsubara BB, Janicki JS. Cause and effect relationship between myocardial mast cell number and matrix metalloproteinase activity. Am J Physiol Heart Circ Physiol. 2002;283:H518–25. doi: 10.1152/ajpheart.00218.2000. [DOI] [PubMed] [Google Scholar]
- 7.Fang KC, Raymond WW, Blount JL, Caughey GH. Dog mast cell alpha-chymase activates progelatinase B by cleaving the Phe88-Gln89 and Phe91-Glu92 bonds of the catalytic domain. J Biol Chem. 1997;272:25628–35. doi: 10.1074/jbc.272.41.25628. [DOI] [PubMed] [Google Scholar]
- 8.Tchougounova E, Lundequist A, Fajardo I, Winberg JO, Abrink M, Pejler G. A key role for mast cell chymase in the activation of pro-matrix metalloprotease-9 and pro-matrix metalloprotease-2. J Biol Chem. 2005;280:9291–6. doi: 10.1074/jbc.M410396200. [DOI] [PubMed] [Google Scholar]
- 9.Craps LP, Ney UM. Ketotifen: current views on its mechanism of action and their therapeutic implications. Respiration. 1984;45:411–21. doi: 10.1159/000194648. [DOI] [PubMed] [Google Scholar]
- 10.Brower GL, Janicki JS. Pharmacologic inhibition of mast cell degranulation prevents left ventricular remodeling induced by chronic volume overload in rats. J Card Fail. 2005;11:548–56. doi: 10.1016/j.cardfail.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 11.Sun J, Sukhova GK, Wolters PJ, Yang M, Kitamoto S, Libby P, et al. Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nature Med. 2007;13:719–24. doi: 10.1038/nm1601. [DOI] [PubMed] [Google Scholar]
- 12.Shiota N, Rysä J, Kovanen PT, Ruskoaho H, Kokkonen JO, Lindstedt KA. A role for cardiac mast cells in the pathogenesis of hypertensive heart disease. J Hypertens. 2003;21:1935–44. doi: 10.1097/00004872-200310000-00022. [DOI] [PubMed] [Google Scholar]
- 13.Hara M, Ono K, Hwang MW, Iwasaki A, Okada M, Nakatani K, et al. Evidence for a role of mast cells in the evolution to congestive heart failure. J Exp Med. 2002;195:375–81. doi: 10.1084/jem.20002036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caughey GH. Mast cell tryptases and chymases in inflammation and host defense. Immunol Rev. 2007;217:141–54. doi: 10.1111/j.1600-065X.2007.00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsumoto I, Inoue Y, Tsuchiya K, Shimad T, Aikawa T. Degranulation of mast cells located in median eminence in response to compound 48/80 evokes adrenocortical secretion via histamine and CRF in dogs. Am J Physiol Regul Integr Comp Physiol. 2004;287:R969–R980. doi: 10.1152/ajpregu.00734.2003. [DOI] [PubMed] [Google Scholar]
- 16.Matsumoto I, Inoue Y, Shimada T, Matsunaga T, Aikawa T. Stimulation of brain mast cells by compound 48/80, a histamine liberator, evokes renin and vasopressin release in dogs. Am J Physiol Regul Integr Comp Physiol. 2008;294:R689–98. doi: 10.1152/ajpregu.00453.2007. [DOI] [PubMed] [Google Scholar]
- 17.Huelsing DJ, Spitzer KW, Pollard AE. Electrotonic suppression of early afterdepolarizations in isolated rabbit Purkinje myocytes. Am J Physiol Heart Circ Physiol. 2000;279:H250–9. doi: 10.1152/ajpheart.2000.279.1.H250. [DOI] [PubMed] [Google Scholar]
- 18.Young AA, Orr R, Smaill BH, Dell’Italia LJ. Three-dimensional changes in left and right ventricular geometry in chronic mitral regurgitation. Am J Physiol. 1996;271:H2689–700. doi: 10.1152/ajpheart.1996.271.6.H2689. [DOI] [PubMed] [Google Scholar]
- 19.Lipshultz M. Differential Geometry. New York, NY: McGraw-Hill; 1969. pp. 61–79. [Google Scholar]
- 20.Cerqueira MD, Weissman NJ, Dilsizian V, Jacobs AK, Kaul S, Laskey WK, et al. American Heart Association Writing Group on Myocardial Segmentation and Registration for Cardiac Imaging. Standardized myocardial segmentation and nomenclature for tomographic imaging of the heart. Int J Cardiovasc Imaging. 2002;18:539–49. [PubMed] [Google Scholar]
- 21.Berko B, Gaasch WH, Tanigawa N, Smith D, Craige E. Disparity between ejection and end-systolic indexes of left ventricular contractility in mitral regurgitation. Circulation. 1987;75:1310–19. doi: 10.1161/01.cir.75.6.1310. [DOI] [PubMed] [Google Scholar]
- 22.Sagawa K. The end-systolic pressure-volume relation of the ventricle: Definition, modifications and its use. Circulation. 1981;63:1223–27. doi: 10.1161/01.cir.63.6.1223. [DOI] [PubMed] [Google Scholar]
- 23.Galli SJ, Tsai M. Mast cells: Versatile regulators of inflammation, tissue remodeling, host defense and homeostasis. J Derm Sci. 2008;49:7–9. doi: 10.1016/j.jdermsci.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kennedy RH, Hauer-Jensen M, Joseph J. Cardiac function in hearts isolated from a rat model deficient in mast cells. Am J Physiol. 2005;288:H632–7. doi: 10.1152/ajpheart.00803.2004. [DOI] [PubMed] [Google Scholar]
- 25.Franzius D, Hoth M, Penner R. Non-specific effects of calcium entry antagonists in mast cells. Pflugers Arch. 1994;428:433–8. doi: 10.1007/BF00374562. [DOI] [PubMed] [Google Scholar]
- 26.Glower DD, Spratt JA, Snow ND, Kabas JS, Davis JW, Olsen CO, Tyson GS, Sabiston DC, Rankin JS. Linearity of the Frank-Starling relationship in the intact heart: the concept of preload recruitable stroke work. Circulation. 1985;71:994–109. doi: 10.1161/01.cir.71.5.994. [DOI] [PubMed] [Google Scholar]