

Abstract
We identified two families with an autosomal-recessive disorder manifested by severe enamel hypoplasia, delayed and failed tooth eruption, misshapen teeth, intrapulpal calcifications, and localized gingival hyperplasia. Genetic analyses identified novel FAM20A mutations associated with the disease phenotype in both families. The proband of Family 1 had an altered splice junction in Intron 1 (g.502011G>C; c.405-1G>C) and a missense mutation in Exon 8 (g.65094G>A; c.1207G>A; p.D403N). The missense mutation is notable because D403 is strictly conserved among FAM20A homologues, and the corresponding defect in FAM20C caused osteosclerotic bone dysplasia and a loss of kinase activity. The proband at age 12 yrs tested negative for nephrocalcinosis. The proband and her affected father in Family 2 were homozygous for a single nucleotide deletion that altered a splice junction in Intron 10 (g.66622del; c.1361+4del). Minigene analyses demonstrated that this alteration precluded normal splicing. Immunohistochemistry (IHC) of mouse maxillary first molars localized FAM20A in secretory-stage ameloblasts, in odontoblasts, and in the eruption pathway. IHC of kidneys localized FAM20A in the renal tubules. We conclude that FAM20A is likely a secretory pathway kinase and that loss-of-function mutations cause pathology where its phosphorylations are necessary for normal development or homeostasis.
Keywords: amelogenesis imperfecta, gingival hyperplasia, enamel, tooth, delayed eruption, nephrocalcinosis
Introduction
Recently there has been great excitement about a small family of kinases designated FAM20A, FAM20B, and FAM20C. Mutations in FAM20C cause autosomal-recessive osteosclerotic bone dysplasia (Raine Syndrome; OMIM #259775; Simpson et al., 2007, 2009). Early investigations into the function of FAM20C portrayed it as an extracellular matrix protein that bound calcium and facilitated biomineralization. However, this paradigm shifted with the seminal discovery that FAM20C is Golgi casein kinase (G-CK) (Tagliabracci et al., 2012), the enzyme that phosphorylates key extracellular matrix molecules involved in biomineralization as well as other processes. Its homologue, FAM20B, is a Golgi kinase that helps regulate glycosaminoglycan synthesis by phosphorylating the xylose in the glycosaminoglycan–protein linkage region of proteoglycans (Koike et al., 2009), which precedes and influences subsequent steps in GAG synthesis and termination (Nadanaka et al., 2013). Defects in FAM20B have not been associated with any human genetic disease. In contrast, FAM20A can cause amelogenesis imperfecta with gingival fibromatosis syndrome (AIGFS; OMIM #614253) (O’Sullivan et al., 2011; Cho et al., 2012) and enamel renal syndrome (ERS; OMIM 204690) (Jaureguiberry et al., 2013; Wang et al., 2013a), but its substrates have not been identified, so it is not even certain that FAM20A has kinase activity.
By analogy to its closest homologues, FAM20A is a kinase in the secretory pathway. Based upon the phenotype in persons with AIGFS and ERS, we suspect that FAM20A phosphorylates secreted proteins involved in dental enamel formation, root formation, tooth eruption, the dental pulp, and perhaps calcium or phosphate homeostasis in the kidneys. A better understanding of normal FAM20A expression, as well as the chronological development of pathologies in its absence, is desired. We recently identified two families with autosomal-recessive amelogenesis imperfecta associated with delayed/arrested tooth eruption, misshapen teeth, pulp stones, and minor gingival enlargement. We determined the molecular etiologies in these families and better characterized FAM20A expression in the kidneys and during tooth development and eruption.
Materials & Methods
The human study protocol/patient consents, and all procedures involving animals, were reviewed and approved by the Institution Review Boards and the Institutional Animal Care and Use Committee (IACUC) at the University of Michigan, respectively.
PCR Amplification
Genomic DNA was isolated from peripheral whole blood as previously described (Wang et al., 2013a). The 11 FAM20A exons and intron borders were amplified with genomic DNA from the two probands as template and then characterized by Sanger sequencing. Amplification and sequencing of Exons 2 and 8 in Family 1 and Exon 10 in Family 2 determined the distribution of potential disease-causing sequence variations among other recruited members of the two kindreds.
Minigene Splicing Assay
Wild-type and mutant genomic DNA from a heterozygous carrier in Family 2 was amplified with PCR primers (GGAA TATGGACCGGCACCAT; TAGTCAGCTTGGGCCAGCAG) to obtain a 2,276-bp amplicon originating at the start of FAM20A Exon 9 and extending 51 basepairs into the 5′ end of Exon 11. The amplification products were cloned into the pcDNA3.3-TOPO vector (Invitrogen, Carlsbad, CA, USA). Insert sequence and orientation were validated by Sanger sequencing.
HEK293 cells were cultured with 2 mL Dulbecco’s Modified Eagle Serum (DMEM) with 10% Fetal Bovine Serum (FBS) overnight to reach ~80% confluence and then transfected with 4 µg of mutant or wild-type minigene plasmid constructs in 10 µL Lipofectamine 2000 (Invitrogen) in 500 µL Opti-MEM I reduced serum media. The next day, the cells were harvested, RNA was extracted with an RNeasy RNA extraction kit (Qiagen Inc., Valencia, CA, USA), converted to cDNA, and amplified with the vector-specific primers (TCCAGCCTCCGGACTCTAGA and CTCATTACTAACCGGTAGGG). The amplification products were characterized by agarose gel electrophoresis and by DNA sequencing.
Immunohistochemistry
Immunohistochemistry was carried out as described in the Appendix (Wang et al., 2013b; Appendix Methods and Appendix Fig. 1).
Results
Family 1
Family 1 is of Irish and Norwegian descent. The proband first presented at age 2.5 yrs with enamel hypoplasia and was the only affected member in the family, suggesting that the disorder was either sporadic or recessive (Fig. 1A). There was no history of consanguinity. Although only recently recruited for genetic studies, the proband’s dental condition had been well-documented by dental practitioners. The initial finding (at age 2.5 yrs) was that the enamel layer was thin (hypoplastic) (Fig. 1B). All of the primary teeth had erupted into the oral cavity, but there was an anterior open bite. The gingiva was not enlarged. The secondary teeth erupted slowly and incompletely. The crowns were short, yellow-brown, and covered with little or no enamel (Fig. 1C). The anterior gingiva appeared to be mildly hyperplastic, an impression enhanced by the smallness of the teeth (due to incomplete eruption and the lack of enamel). When the child was 11.5 yrs old, a panorex showed that the secondary teeth were generally small and misshapen, and only partially erupted, with pulp stones in most of the posterior teeth (Fig. 1D). The incomplete eruption worsened posteriorly. Ultrasound analyses of the proband’s kidneys at age 12 yrs were negative for nephrocalcinosis (Appendix Fig. 2). When the child was 13 yrs old, oral photographs showed that the primary cuspids had exfoliated and exposed the tips of the permanent cuspids, with little sign of their further eruption (Fig. 1E). One first molar (#14) and all 4 second molars had not erupted. The gingiva was not noticeably enlarged.
Figure 1.

Family 1. (A) Pedigree. Dots mark the five persons who donated samples for DNA sequencing. Triangles represent stillborns. (B) Oral photographs of the proband (III:7) at age 2.5 yrs. (C) Oral photographs of the proband at age 8.5 yrs. The anterior incisors are present and exhibit severe enamel hypoplasia. The white cuspids are dental restorations. The attached gingiva is enlarged, but the impression is enhanced by the small sizes of the clinical crowns. (D) Panorex of the proband at age 11.5 yrs. Enamel is missing or does not contrast with dentin throughout. Most tooth roots are short, and eruption is less than expected. Pulp stones are observed in many teeth, particularly in the first molars. (E) Oral photographs of the proband at age 13 yrs. The gingival hyperplasia is minimal and could readily be missed in an oral examination. (F) DNA sequencing chromatograms of Family 1. Top: Sequence from the border of Exon 1 and Intron 1, revealing heterozygosity for a splice junction mutation (g.502011G>C; c.405-1G>C) that occurred in the mother and proband. Bottom: Exon 8 sequence revealing heterozygosity for a missense mutation (g.65094G>A; c.1207G>A; p.D403N) that occurred in the father and proband. The unaffected brother (III:5) and sister (III:8) had neither of these mutations (data not shown). The mutation designations are with respect to the FAM20A genomic reference sequence NG_029809.1 and cDNA reference sequence NM_017565.3 (for mRNA transcript variant 1). Key: arrowhead = mutation point; R = A or G; S = G or C.
Mutation analyses identified novel, disease-causing mutations in both FAM20A alleles (Fig. 1F). The first sequence variation altered the splice acceptor sequence in Intron 1 (g.502011G>C; c.405-1G>C). This variation was identified in the mother (II:6) and the proband (III:7). The second variation was a missense mutation in Exon 8 (g.65094G>A; c.1207G>A; p.D403N) that was identified in the father (II:5) and the proband. The proband’s unaffected brother and sister had the wild-type sequence at both positions. These findings are consistent with an autosomal-recessive pattern of inheritance and a diagnosis of amelogenesis imperfecta with gingival fibromatosis syndrome (AIGFS; OMIM #614253) (O’Sullivan et al., 2011) or ERS (OMIM 204690) (Wang et al., 2013a).
The parents of the proband in Family 1 were characterized by oral photographs and radiographs (Appendix Fig. 3). Their dentitions were within normal limits, although some dental features were suggestive of a mild dental phenotype. The enamel in the cervical third of the mandibular incisors was thin and shaded, and some roots were short or thin. On radiographs, the enamel crowns did not contrast sharply with dentin. Many teeth were restored, suggesting possible susceptibility to caries.
Family 2
Family 2 is of Mexican descent with a history of consanguinity in generations III and IV (Fig. 2A). The proband (V:1) was a 10-year-old girl presenting with dental anomalies in her permanent dentition (Fig. 2B). Her mother (IV:7) reported that her daughter’s condition did not affect her baby teeth. Inspection of the few remaining primary teeth determined that they were only mildly hypoplastic. In contrast, the enamel layer of the permanent dentition was severely hypoplastic throughout. The gingiva was mildly hyperplastic. A panorex (taken when the girl was 10 yrs old) revealed that the second molars were not erupting and had radiolucencies with sclerotic borders covering their crowns (Fig. 2C). This was also true of the right mandibular second bicuspid (#29), whose eruption pathway was blocked by a non-resorbing primary molar (#T). Bitewing and periapical radiographs taken when the girl was 9 yrs old showed almost no enamel (Fig. 2D).
Figure 2.

Family 2. (A) Pedigree. Dots mark the five persons who donated samples for DNA sequencing. Triangles represent stillborns. (B) Oral photographs of the proband (V:I) at age 10 yrs. (C) Panorex radiograph taken when the girl was 10 yrs old. (D) Bitewings and periapical radiographs taken when the girl was 9 yrs old. (E) DNA sequencing chromatograms for Family 2. Top: Sequencing chromatogram from the border of Exon 10 and Intron 10 revealing heterozygosity for a splice junction deletion (g.66622del; c.1361+4del) in the proband’s unaffected aunt (IV:2), mother (IV:7), and the sister (V:2). Bottom: Sequencing chromatogram from the border of Exon 10 and Intron 10 revealing homozygosity for a splice junction deletion (g.66622del; c.1361+4del) in the proband (V:1) and her affected father (IV:6). The mutation designations are with respect to the FAM20A genomic reference sequence NG_029809.1 and cDNA reference sequence NM_017565.3 (for mRNA transcript variant 1).
Mutation analyses identified a novel single-nucleotide deletion in a splice junction in Intron 10 (g.66622del; c.1361+4del) (Fig. 2E). This deletion was found in both FAM20A alleles in the proband (V:I) and his affected father (IV:6), while the unaffected aunt, mother, and sister were heterozygous.
The dentition of the father (IV:6) had been largely reconstructed (Appendix Fig. 4). Gingival hyperplasia was mild or absent. No enamel was discernible on the panorex. The crowns of the unerupted mandibular molars had undergone extensive resorption. Pulp stones were observed in many teeth. The dentition of the heterozygous mother was within normal limits, although pulp stones were evident and many posterior teeth had been restored (Appendix Fig. 4). The participants of Family 2 declined kidney ultrasound analyses.
FAM20A Sequence Variations are Disease-causing
The “A” nucleotide deleted in the fourth position after the exon is an “A” in 68% of splice donor sites (Lewin, 1997) and becomes a “G” in the mutant allele. To test if this change would affect splicing, we expressed wild-type and mutant minigene constructs driven by a cytomegalovirus (CMV) promoter in HEK293 cells (Fig. 3A). The wild-type minigene expressed mRNA transcripts that had been correctly spliced to remove Introns 9 and 10, whereas no correctly spliced mRNA transcripts from the mutant (c.1361+4del) minigene could be detected. The main mRNA product of the mutant minigene used a downstream cryptic splice acceptor site that extended Exon 10 by 27 nucleotides and introduced a premature stop codon after Met454 (Fig. 3B). This analysis supports the conclusion that the c.1361+4del FAM20A mutation in Family 2 is disease-causing.
Figure 3.
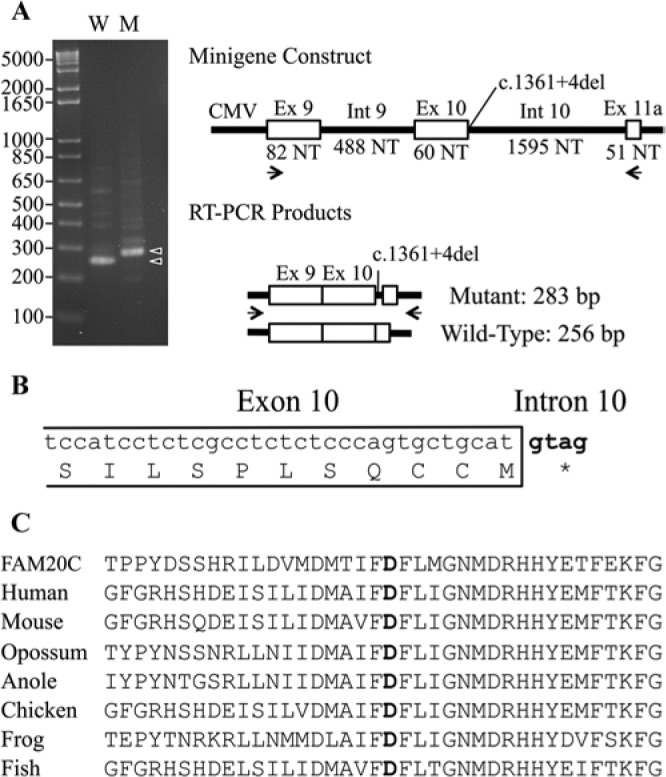
Analysis of disease-causing mutations. (A) Ethidium-bromide-stained agarose gel showing RT-PCR amplification products from the wild-type (W) and mutant (M) minigene constructs. The major amplification products (arrowheads) were 283 and 256 basepairs (bp) and differed by the inclusion of 27 bp from the 5′ end of Intron 10 in the mutated transcript. (B) Diagram showing the 3′ end of Exon 10 and how the extension of this exon into Intron 10 introduces an immediate stop codon that terminates translation after Met454. (C) Alignment of the human FAM20C amino acid sequence with FAM20A protein sequences from human (Homo sapiens), mouse (Mus musculus), opossum (Monodelphis domestica), anole (Anolis carolinensis), chicken (Gallus gallus), frog (Xenopus tropicalis), and fish (Danio rerio). The aspartic acid changed to asparagine in Family 1 is in bold.
Strong evidence supports the conclusion that the p.D403N FAM20A substitution in Family 1 is disease-causing. This position (D403) is strictly conserved among FAM20A homologues going back to fish (Fig. 3C), as is the corresponding position (D451; NM_020223.3) in the kinase domain of FAM20C (Simpson et al., 2009). A substitution exactly analogous to the p.D403N FAM20A missense mutation occurred in FAM20C (p.D451N), causing Raine syndrome (Simpson et al., 2009). Furthermore, recombinant FAM20C that carried this same amino acid substitution lacked kinase activity in vitro (Tagliabracci et al., 2012).
Immunohistochemistry
Given that severe dental malformations, eruption defects, and nephrocalcinosis are associated with FAM20A mutations, we sought to better define the expression of this protein in mouse developing teeth and kidneys by immunohistochemistry. FAM20A signal was strongest and consistently observed in the oral epithelium (Fig. 4). It was also observed in ameloblasts and odontoblasts. The immunoreactivity in ameloblasts was strongest on day 5, greatly reduced by day 8, and negative on days 11 and 14, demonstrating that FAM20A is expressed by secretory- but not maturation-stage ameloblasts. In contrast, the signal in odontoblasts was consistently observed throughout. Within the connective tissue between the oral epithelium and the developing teeth, sporadic signals were observed in morphologically distinct cells, some of which had a dendritic cell appearance. More interestingly, a signal was detected within the dental follicle just above the molar cusp tips on day 14. Furthermore, FAM20A localized in the kidneys, with stronger immunoreactivity in the cortex and less in the medulla. Specifically, the signal was detected in the renal tubules and Bowman’s capsule, but not in the glomerular capillaries.
Figure 4.

FAM20A immunohistochemistry. Top: FAM20A localization during mouse first maxillary molar development. (A1-A3) At post-natal day 5 (PN5), FAM20A signal was observed in secretory-stage ameloblasts, odontoblasts, and in oral epithelium (A3). (B1-B4) At post-natal day 8 (PN8), FAM20A signal was minimal in transition or early maturation-stage ameloblasts, but strong in odontoblasts and the oral epithelium (B2 and B4). (C1-C2) At post-natal day 11 (PN11), no FAM20A signal was observed in maturation-stage ameloblasts. FAM20A was detected in odontoblasts and in oral epithelium. A band of cells positive for FAM20A was observed in the connective tissue between the oral epithelium and the developing cusp tip. The box in C1 corresponds to the position of panel C2. (C3) On post-natal day 14 (PN14), just prior to eruption, FAM20A signal was evident in the dental follicle above the molar cusp tips. (D1-D3) FAM20A localization in four-week-old mouse kidney. Key: bc, Bowman’s capsule; c, renal cortex; m, renal medulla; oe, oral epithelium. Blue is DAPI-stained nuclei, green is FAM20A signal, solid arrowheads indicate ameloblasts, and hollow arrowheads indicate odontoblasts.
Discussion
We have identified 3 novel FAM20A mutations that caused autosomal-recessive amelogenesis imperfecta with delayed and arrested tooth eruption. Eruption failed posteriorly with slow expansion of the dental follicle over the unerupted crowns, forming coronal radiolucencies delimited by a sclerotic border. Pulp stones, particularly in the first molars, were a consistent feature. Less obvious in our patients was gingival hyperplasia, which was mild and localized. The enamel defects were mild in the primary dentition and severe, almost to the point of having no enamel, in the permanent dentition. Eruption of the primary teeth was within normal limits.
This is the fifth report of disease-causing FAM20A mutations, which describe 28 nonsense mutations, frameshifts, and splice junction alterations throughout the gene (Appendix Fig. 5) that would induce degradation of the mutated transcripts, suggesting that an absence of FAM20A protein is the usual cause of disease. Two of the FAM20A missense mutations (p.D403N and p.G331D) are at positions that coincidentally caused osteosclerotic bone dysplasia (Simpson et al., 2007, 2009) and abrogated kinase activity in vitro (Tagliabracci et al., 2012) when mutated in FAM20C (c.1135G>A, g.1366G>A, p.G379R; c.1136G>A, G1367G>A, p.G379E; c.1351G>A, g.1582, p.D451N). Such functional significance of specific amino acids strongly suggests that FAM20A, like FAM20B and FAM20C, is a kinase, although its substrates remain to be identified.
FAM20A is expressed by secretory-stage ameloblasts as they actively secrete enamel proteins, and absence of FAM20A causes severe enamel hypoplasia. This suggests that FAM20A-catalyzed phosphorylations are required for proper secretory-stage enamel formation. FAM20A is expressed in the dental follicle just above the molar cusp tips, and the absence of FAM20A causes failures in tooth eruption, suggesting that FAM20A-catalyzed phosphorylations may play a role in forming eruption pathways, although tooth eruption seems to be relatively unaffected in the human primary dentition and in Fam20a null mice. The formation of pericoronal radiolucencies with a sclerotic border over the unerupted teeth might also be related to FAM20A absence in the dental follicle. Gingival fibromatosis is another feature often reported in FAM20A mutation patients. This manifestation might be associated with FAM20A expression in selected cells within the gingival tissues, rather than in the oral epithelium. FAM20A is expressed by odontoblasts, but scanning electron microscopy (SEM) of impacted human molars from a FAM20A compound heterozygote (p.R136*/p.R478*) revealed that the dentin looked similar to dentin from wild-type teeth, with well-organized dentinal tubules (Wang et al., 2013a). Despite this finding, other observations, such as extensive calcification of pulp chambers and widespread resorption of dentin (on both external root and root canal surfaces) and its replacement by soft tissue or well-formed lamellar bone, suggest that the dentin layer was not normal in all respects.
Nephrocalcinosis is a consistent phenotype in persons with biallelic FAM20A defects (Jaureguiberry et al., 2013). The proband of Family 3, who was untested at the time of our previous publication (Wang et al., 2013a), eventually underwent kidney ultrasound analysis and tested positive for nephrocalcinosis at age 27 yrs (Appendix Fig. 6). So far, at age 12 yrs, the proband in Family 1 shows no sign of nephrocalcinosis and is the first person with bi-allelic FAM20A mutations to test negative. Not much is known about the age of onset of nephrocalcinosis in persons with ERS. One ERS patient of unknown genotype exhibited, at age 5 yrs, minimal renal calcifications that increased in density until age 14 yrs and then stabilized (Lubinsky et al., 1985). Fam20a null mice exhibited widespread and severe ectopic calcification throughout the body, but most prominently in the kidneys. Normal blood calcium and phosphorus in mutant mice and human patients suggested that the ectopic calcification might be caused by local rather than systemic effects (Vogel et al., 2012). Therefore, FAM20A absence in renal tubules might preclude the phosphorylation of proteins that normally prevent nephrocalcinosis.
Given the early and severe nature of the dental defects in patients with bi-allelic FAM20A defects, the initial diagnosis is likely to be made before nephrocalcinosis is detected. The finding of severe enamel hypoplasia with impaired tooth eruption should warrant a genetic consultation and testing for mutations in FAM20A. If bi-allelic sequence variations likely to cause a loss-of-function are found, a tentative diagnosis of ERS is warranted, and periodic kidney examinations should be made to detect nephrocalcinosis and its progression.
Supplementary Material
Acknowledgments
We thank the study participants for their contributions, Dr. Karen David and Karina Acrich for patient referrals, Drs. Thomas Wilson, Donna Hecker, Steven Giannoutsos, and the dental office of Velazquez-Montes in Woodside, New York, for providing dental records, and Stephanie Nunez for communicating with Family 2 in Spanish.
Footnotes
A supplemental appendix to this article is published electronically only at http://jdr.sagepub.com/supplemental.
This work was supported by the National Institutes of Health (NIDCR grant DE015846).
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Cho SH, Seymen F, Lee KE, Lee SK, Kweon YS, Kim KJ, et al. (2012). Novel FAM20A mutations in hypoplastic amelogenesis imperfecta. Hum Mutat 33:91-94. [DOI] [PubMed] [Google Scholar]
- Jaureguiberry G, De la Dure-Molla M, Parry D, Quentric M, Himmerkus N, Koike T, et al. (2013). Nephrocalcinosis (enamel renal syndrome) caused by autosomal recessive FAM20A mutations. Nephron Physiol 122:1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike T, Izumikawa T, Tamura J, Kitagawa H. (2009). FAM20B is a kinase that phosphorylates xylose in the glycosaminoglycan-protein linkage region. Biochem J 421:157-162. [DOI] [PubMed] [Google Scholar]
- Lewin B. (1997). Genes VI. Oxford, UK: Oxford University Press. [Google Scholar]
- Lubinsky M, Angle C, Marsh PW, Witkop CJ., Jr (1985). Syndrome of amelogenesis imperfecta, nephrocalcinosis, impaired renal concentration, and possible abnormality of calcium metabolism. Am J Med Genet 20:233-243. [DOI] [PubMed] [Google Scholar]
- Nadanaka S, Zhou S, Kagiyama S, Shoji N, Sugahara K, Sugihara K, et al. (2013). EXTL2, a member of EXT family of tumor suppressors, controls glycosaminoglycan biosynthesis in a xylose kinase-dependent manner. J Biol Chem 288:9321-9333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan J, Bitu CC, Daly SB, Urquhart JE, Barron MJ, Bhaskar SS, et al. (2011). Whole-exome sequencing identifies FAM20A mutations as a cause of Amelogenesis Imperfecta and Gingival Hyperplasia Syndrome. Am J Hum Genet 88:616-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson MA, Hsu R, Keir LS, Hao J, Sivapalan G, Ernst LM, et al. (2007). Mutations in FAM20C are associated with lethal osteosclerotic bone dysplasia (Raine syndrome), highlighting a crucial molecule in bone development. Am J Hum Genet 81:906-912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson MA, Scheuerle A, Hurst J, Patton MA, Stewart H, Crosby AH. (2009). Mutations in FAM20C also identified in non-lethal osteosclerotic bone dysplasia. Clin Genet 75:271-276. [DOI] [PubMed] [Google Scholar]
- Tagliabracci VS, Engel JL, Wen J, Wiley SE, Worby CA, Kinch LN, et al. (2012). Secreted kinase phosphorylates extracellular proteins that regulate biomineralization. Science 336:1150-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel P, Hansen GM, Read RW, Vance RB, Thiel M, Liu J, et al. (2012). Amelogenesis imperfecta and other biomineralization defects in Fam20a and Fam20c null mice. Vet Pathol 49:998-1017. [DOI] [PubMed] [Google Scholar]
- Wang SK, Aref P, Hu Y, Milkovich RN, Simmer JP, El-Khateeb M, et al. (2013a). FAM20A mutations can cause Enamel-Renal Syndrome (ERS). PLoS Genet 9:e1003302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SK, Samann AC, Hu JC, Simmer JP. (2013b). FAM20C functions intracellularly within both ameloblasts and odontoblasts in vivo. J Bone Miner Res [Epub ahead of print 5/23/2013] (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.