

Abstract
Objective
Vascular diabetic complications are associated with abnormal extracellular matrix and dysfunction of vascular cells, which later result in aberrant angiogenesis and development of atherosclerotic lesions. The tissue and cell specificity of the effects of high glucose are well recognized, but the underlying cell type–specific molecular mechanisms controlled by glucose are still unclear. We sought to identify cell type–specific mechanisms by which high glucose regulates transcription of genes in vascular cells.
Methods and Results
Thrombospondin-1 is a potent antiangiogenic protein associated with development of several diabetic complications and regulated by high glucose in multiple cell types. We report that distinct cell type–specific mechanisms regulate thrombospondin-1 gene (THBS1) transcription in endothelial cells (ECs) and vascular smooth muscle cells (VSMCs) in response to high glucose: although a proximal fragment of 280 nucleotides is sufficient to drive transcription in ECs, THBS1 was regulated cooperatively by interaction between proximal (−272 to −275) and distal (−1016 to −1019) promoter elements in VSMCs. Transcription factors activated by high glucose in VSMCs were cell type–specific. The formation of a single complex interacting with both distal and proximal glucose-responsive elements of THBS1 promoter in VSMCs was confirmed using gel-shift assays, binding sequence decoy oligomers, and specific mutant promoter fragments.
Conclusion
Transcriptional response of vascular cells to high glucose is cell type–specific and involves activation of distinct transcription factors, providing a basis for tissue-specific changes of vasculature in diabetics.
Keywords: IRF-1, aryl hydrocarbon receptor (AhR), glucose, smooth muscle cell, thrombospondin-1
Hyperglycemia is an independent cause for the development of all vascular complications.1–4 Multiple tissues and organs respond to hyperglycemia by altering their structure and function,5,6 but the nature and consequences of hyperglycemia-induced changes are tissue-specific. Although the tissue specificity of diabetic vascular complications has been known for many years (eg, aberrant angiogenesis manifesting as tissue-specific neovascularization or tissue-specific insufficient angiogenesis in different organs of the same patient at the same time), the molecular mechanisms regulating the tissue specificity of the vascular responses to hyperglycemia are unknown.
We report a novel vascular smooth muscle cell (VSMC)-specific transcriptional response that is distinctly different from the transcriptional pathways used by endothelial cells (ECs) in response to high glucose. The difference between the 2 cell types manifests as differential activation of transcription factors (TFs), assembly of distinct cell type–specific transcriptional complexes, and use of distinct cell type–specific glucose-response elements in the promoter of thrombospondin-1 gene (THBS1).
As an example target gene, we used a potent antiangiogenic and proatherogenic protein, thrombospondin (TSP)-1. TSP-1 is a secreted matricellular protein with cell type–specific effects: eg, it induces the proliferation of VSMCs and inhibits the proliferation of ECs.7–9 TSP-1 is secreted into the extracellular matrix by multiple cell types, and elevated levels of TSP-1 in a tissue create antiangiogenic pressure by limiting the survival of ECs and affecting the proliferation of SMCs and vessel maturation. The tissue-specific regulation of TSP-1 production and secretion into the matrix may result from distinct cellular composition of organs and from the cell-specific molecular mechanisms of regulation and can determine the fates of angiogenesis and SMC proliferation in a tissue. Cell-specific response to extracellular stimuli depends on differential cell type–specific activation of signaling proteins and TFs and the formation of cell type–specific protein complexes on gene promoters. Such cell type–specific activation of transcription provides additional levels of differential regulation of protein expression, ie, different time course of expression and specific feedback mechanisms.
Recently, we identified a short promoter fragment (−280/+66) sufficient for upregulation of the TSP-1 gene (THBS1) promoter in ECs.10 We now report that the mechanism of THBS1 transcription induced by high glucose is regulated in a cell type–specific manner, and in VSMCs, this regulation depends on the interaction of 2 highly evolutionarily conserved promoter regions and formation of a protein complex between TFs binding to these regions. In contrast to ECs, where a single promoter element controls the response to glucose, a cooperative interaction between aryl hydrocarbon receptor (AhR) element and γ activation site/interferon-stimulated response element (GAS/ISRE) positively transactivates the TSP-1 gene in SMCs, apparently via a direct interaction between the transcription proteins binding to the proximal (AhR, −272 to 275) and distal (GAS/ISRE, −1016 to 1019) binding elements of the promoter.
GAS/ISRE is a common cis element that binds both STAT1 (signal transducer and activator of transcription 1) and IRF-1 (interferon regulatory factor 1). IRF-1 is an interferon-inducible TF that plays a role in signal transduction and gene-regulation events.11–13 AhR, a ligand-activated TF, is a receptor for multiple chemically unrelated xenobiotic compounds such as environmental toxins, eg, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and other polyaromatic hydrocarbons.14–17 UV light18 and modified low-density li-poproteins19 were reported to activate AhR, and we recently described glucose as a physiological and pathological activator of AhR.10 Multiple studies have established a well-documented role for IRF-1 and AhR in cardiovascular disease and diabetes,20–22 and a further understanding of their function in gene-regulation events has been obtained using the corresponding knockout mice.13,23,24
This novel VSMC-specific mechanism of cooperation of distal (GAS/ISRE-binding) and proximal (AhR-binding) elements in THBS1 promoter provides a direct evidence of a differential transcriptional regulation of gene expression by glucose in a cell-selective manner.
Methods
For a detailed description of the procedures, see the expanded Methods section in the online Data Supplement (available at http://atvb.ahajournals.org.).
Cell Culture
HASMCs were stimulated with 30 mmol/L glucose for 24 hours.
Promoter Reporter Constructs
THBS1 promoter fragments were generated by polymerase chain reaction. Core sequences of the THBS1-specific AhR- and GAS/ISRE-binding elements with nucleotide substitutions in bold were:
AhR mutant: 5′-CGGCAG-3′
Wild type: 5′-CGCGTG- 3′
GAS/ISRE mutant: 5′-ATGCGC-3′
Wild-type: 5′-ATGAAA-3′.
Antibodies Used
The following antibodies were used: anti-AhR (Novus Biologicals, Abcam); RL2 (Abcam); anti–IRF-1, anti–upstream stimulatory factor (USF)1/2, and anti–peroxisome proliferator activated receptor (PPAR)-γ (Santa Cruz Biotechnology); and anti-luciferase (Promega).
AhR- and GAS/ISRE-Binding Decoy and Mutant Oligomers
Sense and antisense strands of AhR- and GAS/ISRE-binding decoy and corresponding mismatched mutant oligonucleotides were designed based on their THBS1 putative binding element sequences as follows:
AhR mutant S: 5′-AGCCCGGCAGGCGCA-3′
AhR decoy S: 5′-AGCCCGCGTGGCGCA-3′
GAS/ISRE binding oligo-mutant S: 5′-AAAAATGCCAAGAAC-3′
GAS/ISRE binding oligo-decoy S: 5′-AAAAATGAAAAGAAC-3′.
Chromatin Immunoprecipitation
A ChIP-IT kit from Active Motif was used according to the instructions of the manufacturer. Anti-AhR and anti–IRF-1 antibodies were used to precipitate the TFs, and the coprecipitated DNA corresponding to the glucose-response elements was amplified using primers described in the online Data Supplement.
Statistical Analysis
All the experiments were performed at least 3 times. All data are presented as means±SEM, with a probability value of ≤0.05 considered statistically significant.
Results
Minimal Fragment of the Human THBS1 Promoter Responsive to High Glucose is Cell Type–Specific
We previously reported that high glucose (10 to 30 mmol/L) increases TSP-1 expression in major vascular cell types of large blood vessels,9 and this activation occurs at the level of transcription.10,25 Recently, we identified a short promoter fragment (−280/+66) sufficient for upregulation of THBS1 promoter in ECs.10 We have now determined that the transcriptional activation of THBS1 promoter by glucose is cell type–specific: the minimal glucose-responsive promoter fragment required for upregulation of the reporter gene in HASMCs is longer than that required in ECs (Figure 1). To identify the minimal THBS1 promoter fragment responsive to high glucose in HASMCs, we examined the activity of several THBS1 promoter deletion constructs driving the expression of luciferase cDNA. Unlike in ECs, a promoter region −1270/+66 was optimal for activation of the THBS1 promoter by glucose in HASMCs. A glucose concentration of 30 mmol/L produced a 7-fold increase in luciferase activity in HASMCs transfected with the −1270/+66 THBS1 promoter fragment (Figure 1A). We observed no glucose response in HASMCs transfected with a shorter promoter fragment (−280/+66) (Figure 1A), which was glucose-responsive in ECs. These data suggested the presence of a glucose-responsive element (GRE) in the longer promoter region of THBS1, which appears to be active in HASMCs, but not in ECs.10
Figure 1.

THBS1 promoter region maximally responsive to high glucose in HASMCs. A, HASMCs were transiently transfected with pGL3 vector or −1270/+66 or −280/+66 THBS1 promoter–luciferase reporter gene. After 6 hours of recovery, cells were placed in 5 or 30 mmol/L glucose. Luciferase activity was measured 42 hours later (n=6 to 7). *P<0.05 vs 5 mmol/L glucose. B, Cell-specific glucose-responsive regions of THBS1 promoter were analyzed using MatInspector 7.4.3. Top, HASMC-specific (−1270/+66). Bottom, EC-specific (−280/+66). Glucose-activated TFs are underlined; TFs coprecipitating with IRF-1 and AhR in HASMCs or with AhR in ECs are in bold.
The basal activity of −280/+66 fragment was comparable to the basal activity of −1270/+66 fragment, with net luciferase activity being normalized to total protein concentrations in lysates (≈170 642 and ≈154 634 luciferase activity/μg protein, respectively). Deletion of either the proximal or distal GRE did not result in a loss or decrease of the basal activity of −1270/+66 promoter (≈134 534 and ≈131 241 versus ≈146 345 net luciferase activity of the wild-type fragment/μg protein extract).
We analyzed the −1270/+66 THBS1 promoter fragment using MatInspector 7.4.3 (Genomatix, http://www.genomatix.de),26 which revealed putative binding sites for several TFs (PPAR, IRFs, USFs, Oct-1) close to the −1270 site (Figure 1B, top). The predicted binding sites for TFs within the distant VSMC-specific GRE, −1270/+66 (Figure 1B, top), were widely different from those in the EC-specific GRE, −280/+66 (Figure 1B, bottom).
Promoters of Homo sapiens, Bos taurus, and Mus musculus were compared using MatInspector (Figure I in the online Data Supplement). The homology in 2000-nucleotide promoters between the 3 species was 58% between human and bovine, 46% between human and mouse, and 44% between mouse and bovine promoters. However, when the regions corresponding to the 2 GREs in the human promoter were compared, a striking similarity was detected. AhR-binding site was present in all 3 promoters at a similar distance from the transcriptional site (supplemental Figure IA). In all 3 promoters, several Egr-1–binding sites were predicted in close proximity (1 always immediately next to the AhR-binding site) and at least 1 E-box and a STAT-binding site next to the AhR-binding site was found. Carbohydrate response elements and Pax-5–binding sites were predicted in human and bovine but not in mouse promoters. A human-like distant GRE was also clearly identified in all 3 promoters and contained a thoroughly conserved STAT-binding site and IRF-1–binding and Oct-1–binding sites (≥2 of each in close proximity). PPAR-binding site was found in the distal element in human but not in bovine or mouse promoters. Sequence alignment revealed even greater similarities: the above-mentioned elements and the distances between the elements were conserved among the 3 species (supplemental Figure IB), whereas the sequences surrounding the response elements were very different (data not shown).
High Glucose Activates Cell Type–Specific Set of TFs in VSMCs
Using a targeted proteomic approach, we identified TFs activated following 24-hour glucose stimulation. HASMCs and ECs were incubated with either 5 or 30 mmol/L glucose and a TranSignal Combo Protein/DNA array (Panomics), which identifies active TFs in nuclear extracts, was used. The full comparative list of activated TFs is presented in supplemental Table I. Although the lists of activated TFs identified in ECs and SMCs overlap, there is a striking difference between the 2 cell types: both SMC-specific and EC-specific TFs were found. Among the factors activated in SMCs were TFs with putative binding elements in both the non–cell-specific proximal (AhR, activator protein [AP]-2) and VSMC-specific distal GRE of the promoter (GAS/ISRE-binding proteins, IRF-1, PPARγ). Out of these TFs, we have shown here (supplemental Table I) and previously10 that only AhR and AP-2 are activated by high glucose in ECs. However, activation of TFs with predicted binding sites in the distal SMC-specific GRE was never detected in ECs (the Table and supplemental Table I). These results demonstrated that glucose-activated TFs in HASMCs are different from those in ECs, consistent with the activity of the promoter fragments, and suggest that the regulatory mechanism of THBS1 transcription in vascular cells is cell type–specific.
Table.
Cell Type–Specific Activation of Transcription Factors With Predicted Binding Sites in Proximal and Distal Glucose-Responsive Elements in THBS1 Promoter
| Probe (Panomics Protein/DNA Array) | Increase in Activity (30 mmol/L Glc/5 mmol/L Glc)
|
|
|---|---|---|
| SMCs | ECs | |
| AhR | 6.7±1.3* | 6.9±2.2* |
| AP2 (B1) | 1.3±0.1 | 3.6±1.2* |
| AP2 (B16) | 3.1±0.6* | 1.8±0.5 |
| EGR | 1.3±0.2 | 2.4±0.9* |
| USF | 1.4±0.2 | 3.3±1.4* |
| GAS-ISRE | 6.1±1.1* | 1.2±0.1 |
| IRF-1 | 3.1±0.7* | 1.5±0.5 |
| PPAR | 3.1±1.1* | 1±0.1 |
Panomics Protein/DNA array, increase in detection of binding to probe in the nuclear extracts from cells incubated in 30 mmol/L glucose vs 5 mmol/L glucose. Mean increase in probe detection is shown.
P<0.05 (n=3).
Confirmation of Activation of TFs by Electrophoretic Mobility-Shift Assay
Based on our promoter analysis implicating a glucose-responsive distal element and protein/DNA array results that detected activation of TFs with putative binding sites in both HASMC-specific distal element and a proximal fragment identified earlier in ECs,10 we focused on TFs with the maximal level of activation in glucose-stimulated HASMCs with binding sites in either distal or proximal THBS1 promoter fragments: GAS/ISRE-binding proteins (binding site predicted at −1016 to −1019) and AhR (binding site predicted at −272 to −275), respectively. To further assess activation of these TFs by glucose, we used nuclear extracts from control (5 mmol/L glucose) and 30 mmol/L glucose-treated HASMCs in electrophoretic mobility-shift assay (EMSA) with GAS/ISRE- and AhR-specific consensus oligonucleotide probes (Panomics). High glucose increased GAS/ISRE- and AhR-binding activity in these cells, and the resulting protein–DNA complex was inhibited by the addition of 66-fold molar excess of unlabeled probes (Figure 2A). Additional gel-shift experiments were performed to examine interactions between the THBS1-specific GAS/ISRE- and AhR-binding sequences with the corresponding glucose-activated nuclear proteins. A protein–DNA complex was formed in each case, confirming activation of a GAS/ISRE-binding complex and AhR by glucose in HASMCs and binding of these proteins to their putative THBS1-specific binding elements (Figure 2B).
Figure 2.

AhR and GAS/ISRE binding are activated by glucose in HASMCs. EMSA was performed with GAS/ISRE and AhR consensus probes (Panomics) (A) or THBS1-specific probes and 5 to 10 μg nuclear extract (B). In competition assays, a 66-fold molar excess of the unlabeled probe was added to the reaction mixture. Arrows indicate specific protein–DNA complexes; NS, nonspecific binding.
Glucose-Induced Association of TFs Occurs in a Cell-Specific Manner
We previously reported that in glucose-stimulated ECs, AhR precipitates in complex with other glucose-activated TFs (AP-2, Egr-1, USF2, and Pax-5), with predicted binding sites in close proximity to each other and to the AhR binding site in the glucose-responsive −280/+66-bp THBS1 promoter fragment.10 In contrast, here, we have identified a distinctly different group of glucose-activated TFs forming a complex in HASMCs, thus providing a direct support for a cell-specific mechanism of transcriptional regulation of THBS1. TFs binding to both proximal and distal promoter fragments were activated by glucose in HASMCs (Table), and the distal promoter element was required for promoter activation (Figure 1). To examine a possible interaction between TFs with putative binding sites in the proximal (eg, AhR) and distal (eg, IRF-1) promoter elements, as well as to identify other associated TFs, we performed a TranSignal TF-TF interaction array. This assay is based on precipitation of proteins complexed with the TF of interest using a specific antibody and detection of active TFs in the complex using their binding to corresponding specific probes. We used specific antibodies against both IRF-1 (Figure 3A) and AhR (Figure 3B) to precipitate and detect active TFs forming complexes with a protein interacting with distal (IRF-1) and proximal (AhR) glucose-responsive promoter fragments. We determined that in HASMCs, glucose induced formation of a complex of AhR, AP-2, GAS/ISRE-binding factor(s), PPARγ, Oct-1, and USF1/2 that is distinctly different from that formed in glucose-stimulated ECs (AhR, USF2, AP-2, Pax-5, and Egr-1).10 The same activated TFs were precipitated using both anti–IRF-1 and anti-AhR antibodies, with the exception of Egr-1, which precipitated in complex with AhR but not IRF-1, suggesting that AhR forms more than 1 transcriptional complex in response to glucose. Thus, unlike in ECs,10 the complex of TFs in glucose-stimulated HASMCs includes TFs with binding sites in both the proximal and distal ends of the −1270/+66 THBS1 promoter fragment. Neither of these proteins was precipitated with nonimmune mouse IgG (supplemental Table II). The full list of TFs that coprecipitated with anti–IRF-1 and anti-AhR antibodies is presented in supplemental Tables II and IV, respectively.
Figure 3.

TFs activated by high glucose in HASMCs form a complex on THBS1 promoter. A, Nuclear extracts from HASMCs incubated with 5 mmol/L (control) or 30 mmol/L glucose with or without 40 μmol/L DON were used for immunoprecipitation with anti–IRF-1 antibody, and associated active TFs were identified using TF-TF interaction array (Panomics). B, TF-TF interaction array was done as in A with anti-AhR antibody. C, TFs directly activated by glucose via glycosylation were analyzed by protein/DNA array. D, Hexosamine pathway mediates glucose-induced GAS/ISRE- and AhR-binding activity in HASMCs. EMSA was performed using GAS/ISRE and AhR consensus probes (Panomics) (n=3 to 4). Arrows indicate specific protein–DNA complexes.
Mechanism of Glucose-Induced Activation of GAS/ISRE–Binding Complex and AhR Complex
We previously reported that the hexosamine pathway of intracellular glucose metabolism and subsequent intracellular glycosylation mediate upregulation of THBS1 transcription in HASMCs.25 Consistent with these findings, we observed herein that activation of the DNA-binding complex in HASMCs is regulated by activity of the hexosamine pathway producing intermediates for glycosylation. HASMCs were preincubated with the glutamine:fructose 6-phosphate amido transferase (GFAT)-specific inhibitor 6-diazo-5-oxo-norleucine (DON), followed by stimulation with 30 mmol/L glucose for 24 hours. GFAT is the enzyme controlling the hexosamine pathway leading to formation of metabolites required for glycosylation. There was a dramatic decrease in the amount of active TFs in complex with IRF-1 or AhR when cells were pretreated with DON (Figure 3A and 3B). Precipitation of AhR, AP-2, PPARγ, GAS/ISRE-binding protein(s), and USF1/2 with IRF-1 was decreased, whereas interaction of Oct-1 with IRF-1 appeared unaffected by inhibition of the hexosamine pathway. Association of GAS/ISRE-binding protein(s), PPARγ, Oct-1, and USF1/2 with AhR was also dramatically reduced, whereas formation of complexes between AhR and Egr-1 or AP-2 remained unaffected by DON. Such reduced amounts of an active protein in the complex may result from either reduced activation of the specific TF or reduced association with active proteins. To address the mechanism of formation of the transcriptional complex in greater detail, we examined the effect of DON pretreatment on activation of AhR, AP-2, PPARγ, GAS/ISRE-binding protein(s), Oct-1, and USF1/2 and their binding to corresponding specific consensus probes (Figure 3C). These analyses suggest that AhR, AP-2, PPARγ, and GAS/ISRE-binding proteins(s) are activated by glucose in HASMCs through a glycosylation event, either glycosylation of TF(s) itself or signaling protein(s) involved in activation of the TF. Although there was no or little direct activation of Oct-1 and USF1/2 by glucose in HASMCs (supplemental Table I), their activity was decreased in the cells treated with DON, suggesting that their constitutive activity or expression depends on glycosylation. Additionally, USF that is forming a complex with other proteins in response to glucose was not found in the complexes in cells treated with DON (Figure 3A and 3B), further confirming that its constitutive expression or activity depends on glycosylation. Interestingly, Oct-1 binding to IRF-1 was not prevented by DON, but the binding to AhR complex was, suggesting that glycosylation of a factor other than Oct-1 regulates the binding and the complex assembly.
Thus, although it is difficult at this point to clearly understand the precise mechanism of protein activation and dynamics of the complex formation, it appears that modification of 1 or more of these TFs by glycosylation mediates their glucose-induced activation or formation of the transcriptional complex. The complete list of TFs whose precipitation in complex with IRF-1 depends on the hexosamine pathway is presented in supplemental Table III. Pretreatment with DON before glucose stimulation directly confirmed that formation of transcriptional complexes on the probes corresponding to AhR-binding site and GAS/ISRE depends on intracellular glycosylation. Both GAS/ISRE and AhR DNA-binding protein complexes were partially inhibited in glucose-stimulated HASMCs, pretreated with DON (Figure 3D). The inhibition of the complex formation was detected in array experiments detecting TFs precipitated with anti-AhR antibody (supplemental Table IV). In both precipitations with anti–IRF-1 and anti-AhR, DON prevented the activation/complex formation of AhR-, USF-, AP-2–, GAS/ISRE-, PPAR probe–binding proteins but not other proteins found in complex with IRF-1 and AhR.
Proteins in the Transcriptional Complex That Bind THBS1 Promoter in VSMCs
To further confirm and characterize proteins present in the transcriptional complex detected in glucose-stimulated HASMCs in association with THBS1 promoter, we performed chromatin immunoprecipitation using antibodies against IRF-1 and AhR and primer amplifying the promoter region corresponding to the distal or proximal GRE of THBS1 promoter (Figure 4A). The control antibody (anti-luciferase) did not precipitate either of the promoter fragments from the chromatin of nonstimulated or glucose-stimulated cells. Both anti-AhR and anti–IRF-1 antibodies precipitated both the proximal and the distal GREs from the cells stimulated with glucose.
Figure 4.
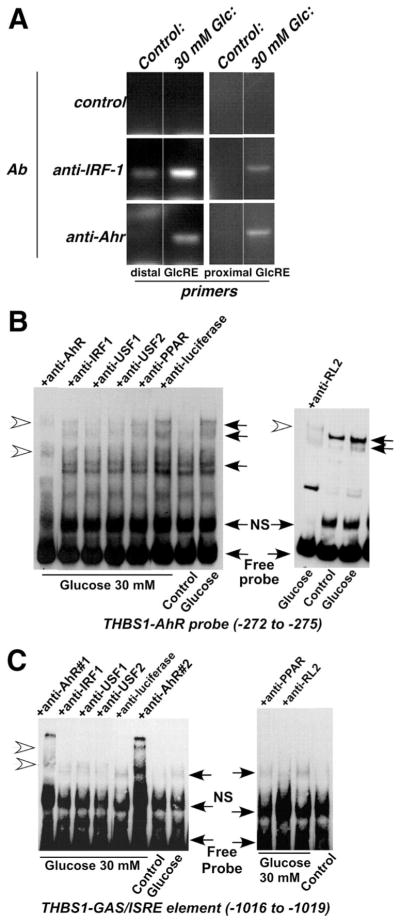
Glucose-activated TFs in HASMCs form a complex with AhR- and GAS/ISRE-binding element on THBS1 promoter. A, Chromatin precipitation using anti-AhR and anti–IRF-1 antibodies was performed as described in Methods. Approximately 200 nucleotides containing either the distal or the proximal GRE were amplified and resolved in 2% agarose gel. B and C, Nuclear extracts (5 to 10 μg) were preincubated with antibodies against AhR (1 μg) and IRF-1 (1 μg) for 30 minutes on ice, followed by further incubation with AhR-specific (B) or GAS/ISRE-specific (C) THBS1 probe for 30 minutes at room temperature. Additionally, the complex of USF1, USF2, and PPARγ with IRF-1 and AhR was confirmed with antibodies against USF1, USF2, and PPARγ (1 μg each). RL2 antibody was used to prove that at least 1 of the proteins is O-glycosylated. Formation of complex was not prevented by an unrelated antibody, anti-luciferase (1 μg) (n=3). Closed arrows indicate specific protein–DNA complexes; open arrows, specific supershifted complexes.
To confirm the formation of the single protein complex, we performed supershift experiments using oligonucleotide probes corresponding to specific binding sites in the THBS1 promoter in combination with antibodies against AhR, IRF-1, USF1 and -2, and PPARγ. Each of these antibodies prevented binding of the complex to the labeled probe, whereas an unrelated antibody (anti-luciferase) used as a negative control had no effect on the specific protein–DNA binding (Figure 4). These results suggested that, unlike in ECs, glucose stimulation of HASMCs induced an interaction of AhR with IRF-1, the latter being a GAS/ISRE-binding protein. This complex further associates with USF1/2 and PPARγ, resulting in a cooperative activation of THBS1 transcription. Specifically, glucose-stimulated HASMC nuclear extracts were incubated with either AhR- or GAS/ISRE-specific THBS1 oligonucleotide probe together with an anti-AhR or anti–IRF-1 antibody, respectively. In either case, binding of the complex to the corresponding labeled probe was either supershifted (AhR, IRF-1; Figure 4) or prevented, confirming the presence of AhR and IRF-1 in the same complex. Furthermore, anti-USF1/2 and anti-PPARγ either supershifted or prevented binding of the complex to both GAS/ISRE- and AhR-specific probes, suggesting that both USF1/2 and PPARγ are also present in association with AhR/IRF-1 on the promoter. Consistent with our previous report25 and further confirming our results in Figure 3, here we demonstrate that, similar to the transcriptional complex in ECs,10 the complex formed on THBS1 promoter in HASMCs includes a glycosylated protein(s). Thus, an RL2 antibody that recognizes O-GlcNAc (O-linked N-acetylglucosamine)–modified proteins either supershifted or inhibited formation of the complex when glucose-stimulated nuclear extracts were incubated with THBS1-specific AhR or GAS/ISRE oligonucleotide probe, respectively (Figure 4).
Glucose-Induced Upregulation of THBS1 Transcription in HASMCs Is Mediated by a Cooperative Association Between Proximal and Distal DNA-Binding Elements of the Promoter
To confirm the role of TFs binding to proximal and distal promoter fragments on THBS1 promoter activation, AhR-binding and GAS/ISRE-binding decoy oligonucleotides were cotransfected with −1270/+66 THBS1 promoter fragment, and luciferase activity was measured. In these experiments, binding of an activated TF to the decoy oligonucleotide sequence prevents its binding to and activation of the promoter, if this activation is dependent on the candidate TF. As a control, a mutant oligonucleotide that does not bind the corresponding TF was used (see online Data Supplement for specific oligonucleotide sequences). AhR-binding and GAS/ISRE-binding decoy oligomers led to a significant reduction in glucose-induced activation of the promoter (3- to 4-fold versus mutant oligonucleotides; Figure 5A). Thus, neutralizing either AhR- or GAS/ISRE-binding protein(s) resulted in a loss of promoter activation by glucose. Further mutagenesis experiments were undertaken where point mutations were introduced in either AhR-binding site or GAS/ISRE of the glucose-responsive −1270/+66 THBS1 promoter fragment, and HASMCs were transfected with these mutant plasmids. The mutant sequences were selected based on preliminary analysis of binding of the corresponding proteins where multiple mutants were tested, and sequences of the point mutations resulting in a lack of the complex formation were selected for mutagenesis (data not shown). Mutations in both AhR and GAS/ISRE elements completely prevented −1270/+66 THBS1 promoter activation by glucose in HASMCs (Figure 5B). These results directly confirmed that both AhR-binding site and GAS/ISRE are necessary but not independently sufficient for glucose-mediated activation of the THBS1 promoter in HASMCs. Our results demonstrate that in HASMCs, a cooperative association between proximal and distal binding elements mediates transcriptional upregulation of THBS1 promoter by glucose, whereas such interaction is not present or required in ECs.
Figure 5.

Glucose-mediated activation of −1270/+66 p THBS1 promoter in HASMCs is blocked by AhR- and GAS/ISRE-binding decoy oligomers, as well as mutations in AhR- and GAS/ISRE-binding sites. HASMCs were cotransfected with −1270/+66 THBS1 or pGL3 together with AhR- or GAS/ISRE-binding decoy or mismatched mutant oligomer (A) or transfected with wild-type, AhR-specific, or GAS/ISRE mutant −1270/+66 THBS1 promoter fragments or pGL3 (B). Cells were then treated with glucose, and luciferase activity was measured as in Figure 1A. Open bars indicate decoy; closed bars, mutant oligomers; n=3 to 4. *P<0.05 vs corresponding decoy (A) or 5 mmol/L glucose (B).
Discussion
The present study describes a novel cell type–specific mechanism of transcriptional regulation of TSP-1 in vascular cells in response to glucose. We report here that unlike our recently identified short promoter region (−280/+66) responsible for the increased THBS1 transcription in ECs, a longer promoter fragment (−1270/+66) is required for THBS1 regulation in HASMCs, as was described for specialized pericytes and mesangial cells.27 Interestingly, glucose responsiveness in ECs was in fact inhibited by the distal fragment of the promoter,10 suggesting the presence of an inhibitory element in this region, which is not active in either VSMCs or mesangial cells.27 The longer promoter region, −1270/+66, responsible for the increased THBS1 transcription in HASMCs contained distinctly different binding elements, as identified by MatInspector, located in the distal end of the promoter. These binding elements had no similarity to those in the EC-specific THBS1 promoter fragment, −280/+66, maximally responsive to glucose. Both regions of the promoter containing the GREs were strikingly conserved between species, both the sequence and the binding sites for TFs.
Similar to ECs,10 AhR was required for transcriptional upregulation of TSP-1 by high glucose in HASMCs, demonstrated by both decoy experiments and analysis of the activity of the longer fragment (−1270/+66) with mutated AhR-binding site at −272. However, our data clearly reveal the requirement for a distal GRE that includes the GAS/ISRE binding IRF-1, which is essential for THBS1 transcriptional activation by glucose in VSMCs. The specific role of AhR and GAS/ISRE in THBS1 promoter activation was initially established by promoter analysis studies. Such studies revealed the predicted GAS/ISRE within the long promoter region (−1270/+66) responsive to glucose in HASMCs. The activation of AhR and GAS/ISRE-binding protein(s) was detected by a targeted proteomic approach and confirmed by EMSA. Furthermore, GAS/ISRE- and AhR-binding decoy oligonucleotide (but not the corresponding mismatched oligonucleotides) completely abolished the glucose-induced activation of the −1270/+66 THBS1 promoter fragment in HASMCs. Additionally, the role of the distant GRE, GAS/ISRE, was confirmed using the −1270/+66 THBS1 promoter construct with substitutions in the core-sequence of the IRF-1–binding motif (−1016), IRF-1 being one of the TFs known to bind to the GAS/ISRE. Although the GAS/ISRE mutant promoter fragment contained an intact AhR-binding motif, it could not be activated by high glucose in VSMCs, suggesting a cooperative interaction between AhR- and GAS/ISRE-binding TFs that is specific to VSMCs. Thus, our data clearly demonstrate that, unlike in ECs,10 high glucose activates the GAS/ISRE-binding protein(s), in addition to AhR, in HASMCs and formation of a single complex between AhR- and GAS/ISRE-binding protein(s) controls the expression of TSP-1 in glucose-stimulated HASMCs.
Intracellular metabolism of glucose initiates several signaling pathways regulating gene expression in response to hyperglycemia.28,29 One of the pathways implicated in gene regulation in response to hyperglycemia is the hexosamine pathway, which produces intermediates for protein glycosylation. Many intracellular proteins, including proteases, signaling molecules, and TFs are modified by addition of O-GlcNAc and other sugar moieties, and such modification regulates the activity of these proteins.30,31 We previously reported that activation of the hexosamine pathway and subsequent protein glycosylation mediate TSP-1 upregulation by glucose in VSMCs.25 Recently, we reported a similar mechanism of THBS1 transcriptional activation in ECs.10 The present study clearly reveals that both ECs and VSMCs share a common signaling pathway that mediates upregulation of TSP-1 gene expression in response to glucose. Our results demonstrate that glycosylation of a TF(s) via generation of GlcNAc resulting from activation of the hexosamine pathway is the underlying mechanism for the glucose-mediated increase in THBS1 transcription in VSMCs. We have shown that glucose-induced activation of TFs binding to either proximal (AhR, AP-2) or distal (PPARγ, GAS/ISRE-binding proteins) THBS1 promoter fragments, or activation of the transcriptional complex depends on glycosylation and at least 1 of the proteins in the complex is glycosylated as demonstrated using RL2 antibody.
Regulation of gene transcription in response to extracellular and intracellular stimuli involves combinatorial interactions between several TFs, allowing for a cell-specific response to multiple conditions in the environment. This is largely dependent on the promoter structure and availability of binding elements in each specific cell type, as well as cell type–specific patterns of activation of transcriptional activators, cofactors, enhancers, and suppressors. In this work, we identified a HASMC-specific distinct group of TFs forming a complex on the THBS1 promoter in response to glucose; several common proteins were identified in complex with either AhR or IRF-1, as detected by TF-TF interaction studies and confirmed by chromatin immunoprecipitation, as well as supershift experiments with the corresponding antibodies. These include AhR, GAS/ISRE-binding protein IRF-1, PPARγ, and USF1/2. This complex is distinctly different from that associating with the shorter glucose-responsive fragment of the promoter (−280/+66) in ECs, where putative binding sites for all TFs in the complex were in close proximity to the AhR-binding site. In contrast, we demonstrate that the proteins forming a complex on the THBS1 promoter in HASMCs include TFs with GREs located in both the proximal (AhR) and distant segments (GAS/ISRE, PPARγ) of the promoter. Of these, the nuclear proteins that were consistently activated (>2.5-fold) by high glucose in HASMCs were AhR, AP-2, GAS/ISRE-binding proteins, IRF-1, and PPARγ. Although a glucose-mediated activation of Oct-1 and USF was previously reported in both ECs and VSMCs,32–36 our array experiments failed to detect a direct activation of these TFs. However, as revealed by immunoprecipitation and TF-TF interaction array, Oct-1 and USF were identified in the complex formed on the THBS1 promoter, in association with both AhR and IRF-1, suggesting a potential role of these proteins as coactivators required for THBS1 transcriptional activation. On the other hand, Egr-1 was detected in complex with only AhR, suggesting that it is not part of the transcriptional complex formed in response to glucose. However, an association of these proteins with the transcriptional complex suggests that even in the absence of additional activation of a TF by glucose, the basal levels of the active protein may contribute to the transcriptional regulation through association with a newly activated protein(s).
The combination of multiple complementary methods demonstrated that the activation of TFs in response to glucose is cell-specific, and the complexes formed by the TFs are also cell type–specific. In HASMCs, the activation of proteins binding to the 2 GREs (proximal AhR-binding element and distal IRF-1–binding element) is necessary to activate the THBS1 promoter. Evidence for the need of both elements and formation of a single protein complex providing the interaction between the elements was obtained from the mutant promoter activity assays (both deletions and element mutations in the context of the full promoter), decoy oligonucleotide transfections, immunoprecipitations of protein complexes and chromatin, and EMSA (summarized in supplemental Table V).
Taken together, results of the present study provide evidence for the cell type–specific differential regulation of THBS1 transcription in response to glucose. In VSMCs, such regulation is sustained by a bipartite promoter and a functional interaction between proximal and distal binding elements of the promoter, capable of binding other cooperating TFs, mediate the glucose-induced activation of TSP-1 transcription. Such cell type–specific mechanisms of cooperativity between AhR- and GAS/ISRE-dependent transcriptional machinery may provide a molecular basis for the altered tissue-specific patterns of endothelial dysfunction and SMC activation in diabetes. Differential cell type–specific mechanisms of gene regulation by high glucose provide novel mechanistic insights into the tissue-specific response to high glucose.
Supplementary Material
Acknowledgments
Sources of Funding
This work was supported by National Institutes of Health grants P50 HL077107 and R01 DK067532 (to O.I.S.) and American Heart Association Scientist Development Grant 0835190N (to P.R.).
Footnotes
Disclosures
None.
References
- 1.Nathan DM, Lachin J, Cleary P, Orchard T, Brillon DJ, Backlund JY, O’Leary DH, Genuth S. Diabetes Control and Complications Trial; Epidemiology of Diabetes Interventions and Complications Research Group. Intensive diabetes therapy and carotid intima-media thickness in type 1 diabetes mellitus. N Engl J Med. 2003;348:2294–2303. doi: 10.1056/NEJMoa022314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barzilay JI, Spiekerman CF, Kuller LH, Burke GL, Bittner V, Gottdiener JS, Brancati FL, Orchard TJ, O’Leary DH, Savage PJ. Cardiovascular Health Study. Prevalence of clinical and isolated subclinical cardiovascular disease in older adults with glucose disorders: the Cardiovascular Health Study. Diabetes Care. 2001;24:1233–1239. doi: 10.2337/diacare.24.7.1233. [DOI] [PubMed] [Google Scholar]
- 3.Ohkubo Y, Kishikawa H, Araki E, Miyata T, Isami S, Motoyoshi S, Kojima Y, Furuyoshi N, Shichiri M. Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with non-insulin-dependent diabetes mellitus: a randomized prospective 6-year study. Diabetes Res Clin Pract. 1995;28:103–117. doi: 10.1016/0168-8227(95)01064-k. [DOI] [PubMed] [Google Scholar]
- 4.Malmberg K. Prospective randomised study of intensive insulin treatment on long term survival after acute myocardial infarction in patients with diabetes mellitus. DIGAMI (Diabetes Mellitus, Insulin Glucose Infusion in Acute Myocardial Infarction) Study Group. BMJ. 1997;314:1512–1515. doi: 10.1136/bmj.314.7093.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stenina OI. Regulation of vascular genes by glucose. Curr Pharm Des. 2005;11:2367–2381. doi: 10.2174/1381612054367283. [DOI] [PubMed] [Google Scholar]
- 6.Knoll KE, Pietrusz JL, Liang M. Tissue-specific transcriptome responses in rats with early streptozotocin-induced diabetes. Physiol Genomics. 2005;21:222–229. doi: 10.1152/physiolgenomics.00231.2004. [DOI] [PubMed] [Google Scholar]
- 7.Majack RA, Goodman LV, Dixit VM. Cell surface thrombospondin is functionally essential for vascular smooth muscle cell proliferation. J Cell Biol. 1988;106:415–422. doi: 10.1083/jcb.106.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lawler J. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J Cell Mol Med. 2002;6:1–12. doi: 10.1111/j.1582-4934.2002.tb00307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stenina OI, Krukovets I, Wang K, Zhou Z, Forudi F, Penn MS, Topol EJ, Plow EF. Increased expression of thrombospondin-1 in vessel wall of diabetic Zucker rat. Circulation. 2003;107:3209–3215. doi: 10.1161/01.CIR.0000074223.56882.97. [DOI] [PubMed] [Google Scholar]
- 10.Dabir P, Marinic TE, Krukovets I, Stenina OI. Aryl hydrocarbon receptor is activated by glucose and regulates the thrombospondin-1 gene promoter in endothelial cells. Circ Res. 2008;102:1558–1565. doi: 10.1161/CIRCRESAHA.108.176990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee SH, Kim JW, Lee HW, Cho YS, Oh SH, Kim YJ, Jung CH, Zhang W, Lee JH. Interferon regulatory factor-1 (IRF-1) is a mediator for interferon-gamma induced attenuation of telomerase activity and human telomerase reverse transcriptase (hTERT) expression. Oncogene. 2003;22:381–391. doi: 10.1038/sj.onc.1206133. [DOI] [PubMed] [Google Scholar]
- 12.Kano S, Sato K, Morishita Y, Vollstedt S, Kim S, Bishop K, Honda K, Kubo M, Taniguchi T. The contribution of transcription factor IRF1 to the interferon-gamma-interleukin 12 signaling axis and TH1 versus TH-17 differentiation of CD43 T cells. Nat Immunol. 2008;9:34–41. doi: 10.1038/ni1538. [DOI] [PubMed] [Google Scholar]
- 13.Kamijo R, Harada H, Matsuyama T, Bosland M, Gerecitano J, Shapiro D, Le J, Koh SI, Kimura T, Green SJ, Mak TW, Taniguchi T, Vilcek J. Requirement for transcription factor IRF-1 in NO synthase induction in macrophages. Science. 1994;263:1612–1615. doi: 10.1126/science.7510419. [DOI] [PubMed] [Google Scholar]
- 14.Bertazzi PA, Zocchetti C, Pesatori AC, Guercilena S, Sanarico M, Radice L. Mortality in an area contaminated by TCDD following an industrial incident. Med Lav. 1989;80:316–329. [PubMed] [Google Scholar]
- 15.Muto H, Takizawa Y. Dioxins in cigarette smoke. Arch Environ Health. 1989;44:171–174. doi: 10.1080/00039896.1989.9935882. [DOI] [PubMed] [Google Scholar]
- 16.Lofroth G, Rannug A. Ah receptor ligands in tobacco smoke. Toxicol Lett. 1988;42:131–136. doi: 10.1016/0378-4274(88)90070-7. [DOI] [PubMed] [Google Scholar]
- 17.Vena J, Boffetta P, Becher H, Benn T, Bueno-de-Mesquita HB, Coggon D, Colin D, Flesch-Janys D, Green L, Kauppinen T, Littorin M, Lynge E, Mathews JD, Neuberger M, Pearce N, Pesatori AC, Saracci R, Steenland K, Kogevinas M. Exposure to dioxin and nonneoplastic mortality in the expanded IARC international cohort study of phenoxy herbicide and chlorophenol production workers and sprayers. Environ Health Perspect. 1998;106(Suppl 2):645–653. doi: 10.1289/ehp.98106645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fritsche E, Schäfer C, Calles C, Bernsmann T, Bernshausen T, Wurm M, Hübenthal U, Cline JE, Hajimiragha H, Schroeder P, Klotz LO, Rannug A, Fürst P, Hanenberg H, Abel J, Krutmann J. Lightening up the UV response by identification of the arylhydrocarbon receptor as a cyto-plasmatic target for ultraviolet B radiation. Proc Natl Acad Sci U S A. 2007;104:8851–8856. doi: 10.1073/pnas.0701764104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McMillan BJ, Bradfield CA. The aryl hydrocarbon receptor is activated by modified low-density lipoprotein. Proc Natl Acad Sci U S A. 2007;104:1412–1417. doi: 10.1073/pnas.0607296104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wessely R, Hengst L, Jaschke B, Wegener F, Richter T, Lupetti R, Paschalidis M, Schömig A, Brandl R, Neumann FJ. A central role of interferon regulatory factor-1 for the limitation of neointimal hyperplasia. Hum Mol Genet. 2003;12:177–187. doi: 10.1093/hmg/ddg018. [DOI] [PubMed] [Google Scholar]
- 21.Korashy HM, El-Kadi AO. The role of aryl hydrocarbon receptor in the pathogenesis of cardiovascular diseases. Drug Metab Rev. 2006;38:411–450. doi: 10.1080/03602530600632063. [DOI] [PubMed] [Google Scholar]
- 22.Remillard RB, Bunce NJ. Linking dioxins to diabetes: epidemiology and biologic plausibility. Environ Health Perspect. 2002;110:853–858. doi: 10.1289/ehp.02110853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thackaberry EA, Bedrick EJ, Goens MB, Danielson L, Lund AK, Gabaldon D, Smith SM, Walker MK. Insulin regulation in AhR-null mice: embryonic cardiac enlargement, neonatal macrosomia, and altered insulin regulation and response in pregnant and aging AhR-null females. Toxicol Sci. 2003;76:407–417. doi: 10.1093/toxsci/kfg229. [DOI] [PubMed] [Google Scholar]
- 24.Vasquez A, Atallah-Yunes N, Smith FC, You X, Chase SE, Silverstone AE, Vikstrom KL. A role for the aryl hydrocarbon receptor in cardiac physiology and function as demonstrated by AhR knockout mice. Cardiovasc Toxicol. 2003;3:153–163. doi: 10.1385/ct:3:2:153. [DOI] [PubMed] [Google Scholar]
- 25.Raman P, Krukovets I, Marinic TE, Bornstein P, Stenina OI. Glycosylation mediates up-regulation of a potent antiangiogenic and proatherogenic protein, thrombospondin-1, by glucose in vascular smooth muscle cells. J Biol Chem. 2007;282:5704–5714. doi: 10.1074/jbc.M610965200. [DOI] [PubMed] [Google Scholar]
- 26.Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–2942. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- 27.Wang S, Skorczewski J, Feng X, Mei L, Murphy-Ullrich JE. Glucose up-regulates thrombospondin 1 gene transcription and transforming growth factor-beta activity through antagonism of cGMP-dependent protein kinase repression via upstream stimulatory factor 2. J Biol Chem. 2004;279:34311–34322. doi: 10.1074/jbc.M401629200. [DOI] [PubMed] [Google Scholar]
- 28.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 29.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 30.Wells L, Hart GW. O-GlcNAc turns twenty: functional implications for post-translational modification of nuclear and cytosolic proteins with a sugar. FEBS Lett. 2003;546:154–158. doi: 10.1016/s0014-5793(03)00641-0. [DOI] [PubMed] [Google Scholar]
- 31.Whelan SA, Hart GW. Proteomic approaches to analyze the dynamic relationships between nucleocytoplasmic protein glycosylation and phosphorylation. Circ Res. 2003;93:1047–1058. doi: 10.1161/01.RES.0000103190.20260.37. [DOI] [PubMed] [Google Scholar]
- 32.Cuif MH, Porteu A, Kahn A, Vaulont S. Exploration of a liver-specific, glucose/insulin-responsive promoter in transgenic mice. J Biol Chem. 1993;268:13769–13772. [PubMed] [Google Scholar]
- 33.Vaulont S, Kahn A. Transcriptional control of metabolic regulation genes by carbohydrates. FASEB J. 1994;8:28–35. doi: 10.1096/fasebj.8.1.8299888. [DOI] [PubMed] [Google Scholar]
- 34.Bayraktar M, Dündar S, Kirazli S, Teletar F. Platelet factor 4, beta-thromboglobulin and thrombospondin levels in type I diabetes mellitus patients. J Int Med Res. 1994;22:90–94. doi: 10.1177/030006059402200204. [DOI] [PubMed] [Google Scholar]
- 35.Ha H, Yu MR, Choi YJ, Kitamura M, Lee HB. Role of high glucose-induced nuclear factor-kappaB activation in monocyte chemoattractant protein-1 expression by mesangial cells. J Am Soc Nephrol. 2002;13:894–902. doi: 10.1681/ASN.V134894. [DOI] [PubMed] [Google Scholar]
- 36.Ramana KV, Friedrich B, Srivastava S, Bhatnagar A, Srivastava SK. Activation of nuclear factor-kappaB by hyperglycemia in vascular smooth muscle cells is regulated by aldose reductase. Diabetes. 2004;53:2910–2920. doi: 10.2337/diabetes.53.11.2910. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.