

Abstract
Here we review the recent applications of ion transfer (IT) at the interface between two immiscible electrolyte solutions (ITIES) for electrochemical sensing and imaging. In particular, we focus on the development and recent applications of the nanopipet-supported ITIES and double-polymer-modified electrode, which enable the dynamic electrochemical measurements of IT at nanoscopic and macroscopic ITIES, respectively. High-quality IT voltammograms are obtainable using either technique to quantitatively assess the kinetics and dynamic mechanism of IT at the ITIES. Nanopipet-supported ITIES serves as an amperometric tip for scanning electrochemical microscopy to allow for unprecedentedly high-resolution electrochemical imaging. Voltammetric ion sensing at double-polymer-modified electrodes offers high sensitivity and unique multiple-ion selectivity. The promising future applications of these dynamic approaches for bioanalysis and electrochemical imaging are also discussed.
1. Introduction
The transfer of charge (ions and electrons) at the interface between two immiscible electrolyte solutions (ITIES; also known as the liquid/liquid interface) has been extensively studied electrochemically [1]. These fundamental studies have been aimed at obtaining a greater understanding as to how charge transfer kinetics [2] is related to interfacial structure [3, 4]. In addition, these studies have laid the foundation for various electrochemical applications of charge transfer at the ITIES [5]. In particular, ion transfer (IT) at the ITIES is attractive for the electrochemical sensing of the ionic analytes that are undetectable or difficult to detect by redox electrochemistry. For instance, potentiometry of IT at the ITIES has been widely used as the basis of ion-selective electrodes (ISEs) [6–9] for biological [10], clinical [11], and environmental [12] analysis. In contrast, the sensing applications of IT voltammetry and amperometry at the macroscopic ITIES [13] had been limited by the traditional use of a four-electrode setup for the compensation of the large ohmic potential (or iR) drop in the resistive non-aqueous media [14]. To overcome this limitation, microscopic ITIES [15] was developed by supporting it at the tip of a glass micropipet [16, 17] or at the microhole drilled through a polymer film [18]. A micropipet-supported ITIES was also useful as an amperometric tip for scanning electrochemical microscopy (SECM) [19]. The macroscopic array of microhole-supported ITIES was employed for various sensing applications [20]. Alternatively, a macroscopic ITIES system with low resistance was developed by coating a solid electrode with a thin water-immiscible solution [21].
In this review, we discuss the recent applications of IT at the ITIES for electrochemical sensing and imaging. Specifically, our discussion is focused on the development and emerging applications of two important enabling technologies (i.e., nanopipet-supported ITIES and double-polymer-modified electrode) (Figure 1). We provide the brief historical overview of each technology as to how it was originally introduced for potentiometric ISEs and then adapted for voltammetry and amperometry. Then, we focus on and detail our recent work on the development and applications of the dynamic electrochemical methods for sensing and imaging.
Figure 1.

Scheme of IT voltammetry/amperometry at (A) the nanopipet-supported ITIES and (B) the double-polymer-modified electrode.
2. Electrochemical Measurement of IT at the Nanopipet-Supported ITIES
Nanopipet-supported ITIES was developed in 1970s [22] for ion-selective potentiometry in the intracellular and extracellular spaces of various cells and tissues [23]. A pair of nanopipets was readily prepared by heat-pulling a glass capillary to yield tip diameters down to ~10 nm [24]. A glass nanopipet was silanized to be hydrophobic [25, 26] and filled with a water-immiscible organic solution with high selectivity to a target ion. The ion selectivity of the inner solution was controlled by the ionophore that selectively forms a complex with an analyte ion such as H+, Li+, Na+, K+, Ca2+, and Mg2+ as the counterion of a hydrophobic anion (also known as ionic sites [8]) in the organic phase [27]. The penetration of the organic-filled nanopipet into the intracellular or extracellular space formed the nanoscopic ITIES to drive the selective and equilibrium partitioning of the analyte ion between the two phases. Subsequently, the Nernstian phase boundary potential, E, was developed across the ITIES and measured potentiometrically to determine the activity, ai, of an analyte ion with a charge z, iz, in the aqueous sample as given by [8]
| (1) |
where Ei is a constant and includes the constant sample-independent activity of the analyte ion in the organic phase. Eq 1 indicates that the size and shape of the interface is neither relevant to nor determinable by potentiometry. In practice, the tip diameter of a pipet was limited to >100 nm because the pipet was filled with the highly resistive solution based on a viscous organic solvent such as o-nitrophenyl octyl ether (oNPOE) [24].
In 1997, Shao and Mirkin reported on their pioneering work of nanopipet IT voltammetry at the ITIES [28]. In comparison to voltammetry at the ITIES supported by a tapered glass micropipet with a tip radius, a, of >0.5 μm (Figure 2A) [16], the use of nanopipets with a = 4–250 nm enabled the determination of kinetic parameters for faster IT reactions owing to higher mass transport conditions [28]. In fact, nanopipet voltammetry of the K+ transfer facilitated by dibenzo-18-crown-6 gave large standard rate constants, k0, of 1.3–1.7 cm/s as well as transfer coefficients, α, of 0.4–0.6 [28, 29]. The k0 values are still much lower than a high mass transfer coefficient of 35 cm/s based on the steady-state diffusion of the crown ether ionophore from the external 1,2-dichloroethane (DCE) solution to the nanopipet-supported ITIES with the smallest radius of 5 nm [28]. The small current at the nanoscopic ITIES minimizes the ohmic potential drop to enable the study of IT at the relatively resistive ITIES systems based on low-electrolyte [30, 31] or electrolyte-free [32–34] media. For quantitative studies, an inlaid disk-shaped ITIES was formed at the tip of a well silanized nanopipet [35] to yield a limiting current, iss, as given by
Figure 2.

(A) Scheme of a pipet electrode and (B) SEM image of a silanized nanopipet.
| (2) |
where x is a function of RG [36] (RG= rg/a; rgis the outer radius of a pipet tip), and D and c* are the diffusion coefficient and concentration of the diffusion-limiting species in the external bulk solution, respectively. The nanopipet-supported ITIES was used as an SECM tip to confirm that the nanoscale interface is flush with the surrounding surface of the pipet tip [35, 37]. A sharp nanopipet with a small RG value of ~1.5 (Figure 2B) can approach very close to a target substrate for obtaining highly quantitative kinetic data and high-resolution images.
3. Nanopipet Voltammetry of Fast IT Reactions: A Common-Ion Approach
Recently, we developed a new advanced approach for the reliable kinetic analysis of steady-state voltammograms to investigate fast IT reactions at the nanopipet-supported ITIES [38]. Unlike the conventional voltammetric protocol, this approach requires the initial addition of a transferable ion to both liquid phases, i.e., to the filling solution inside a nanopipet and the external solution. The resulting steady-state IT voltammogram comprises of two waves corresponding to the ingress of the common ion into the pipet and its egress into the external solution (Figure 1A). Both ingress and egress waves are required for the characterization of pipet geometry and the precise determination of thermodynamic and kinetic parameters for rapid IT reactions. In contrast, large uncertainties were inherent in the kinetic parameters determined from the steady-state nanopipet voltammogram of nearly reversible simple IT when the transferred ion was initially present only in one phase [35].
The new approach based on nanopipet voltammetry of a common ion was employed to reliably determine kinetic and thermodynamic parameters for the simple transfer of tetraethylammonium (TEA+) [39]. This IT reaction is a particularly important model system to check the IT theory [2]. Nanopipet voltammetry of TEA+ as a common ion gave a pair of sigmoidal waves based on ingress and egress TEA+ transfers at the DCE/water interface (Figure 3A). Both branches of the voltammogram fit very well with theoretical voltammograms, which were originally simulated by employing the finite element method and then represented by approximate analytical equations. The inner radius (9.7–33 nm) and inner angle (θ = 9°–22°) of a nanopipet were determined from the limiting currents controlled by diffusion of TEA+ in the external and internal solutions. These geometrical parameters were used for the precise determination of the unique pair of k0 and α values. Consistent kinetic parameters of k0 = 6.1 ± 0.9 cm/s and α = 0.49 ± 0.9 were obtained with various concentration ratios of TEA+ in the DCE and aqueous phases. In addition, a formal potential was determined directly from the zero-current potential of a voltammogram. More recently, this powerful common-ion approach was applied to the kinetic study of simple IT at the ionic liquid/water interface [40].
Figure 3.

(A) Steady-state CV of TEA+ transfer across the DCE/water interface as obtained using a 19 nm-radius water-filled nanopipet at 10 mV/s. The DCE and water phases contained 1.7 and 2.6 mM TEA+ as tetrakis(4-chlorophe- nyl)borate (TPBCl) and chloride salts, respectively. Supporting electrolytes in the respective phases are 9.4 mM tetrahexylammonium TPBCl and 0.1 M LiCl. Adapted with permission from ref. [39]. Copyright 2010 American Chemical Society. (B) Plot of log k0 values for TEA+ transfer reported in literature versus the year of the publication. This type of plot was reported originally by Kakiuchi and more recently by Samec. See ref. [2].
A large k0 value of 6.1 cm/s for simple TEA+ transfer was determined by our new approach to be compared with theoretical and experimental values reported in literature [2]. In contrast to other experimental values, this k0 value is free from an error due to a mass transfer limitation. This k0 value close to an approximate theoretical estimation of 1.5 cm/s as estimated by Marcus [41]. In the Marcus model, various parameters involved in the determination of the k0 value were estimated for the equilibrium association of an aqueous (or organic) TEA+ molecule with the protrusion of the organic (or aqueous) solution from the interface was followed by the diffusional passage of the TEA+ molecule through the 1 nm-thick interface as the rate-determining step. Apparently, the theoretical value is closer to a k0 value of 2.1 cm/s as determined by nanopipet voltammetry ofTEA + only in the external DCE phase [35]. This experimental value, however, was overestimated without the consideration of hindered ion diffusion in the inner solution of a nanopipet [42]. The neglect of ion diffusion in the inner solution gave an even larger k0 value of (1.1 ± 0.2) × 102 cm/s [43], which was overestimated by factor of ~10 [39].
Noticeably, all k0 values for simple TEA+ transfer at nanopipet-supported ITIES are significantly higher than those determined at macroscopic [44–51] and microscopic [52, 53] ITIES (Figure 3B). Apparent kinetic effects observed at the latter systems are likely due to the uncompensated ohmic potential drop, thereby underestimating the k0 values. In fact, reversible voltammograms of simple TEA+ transfer were obtained using the micrometer-sized ITIES supported by well silanized pipets [39, 42]. Nevertheless, one may wonder whether apparently faster IT kinetics at the nanopipet-supported ITIES may be due to its small size [1] or its confinement within the glass orifice [38]. These effects will be addressable by studying TEA+ transfer at the macroscopic ITIES using a nanopipet-supported ITIES as an SECM tip. Sun et al. employed this SECM approach to yield k0= 0.7 ± 0.3 cm/s and α = 0.56 ± 0.08 for the K+ transfer facilitated by dibenzo-18-crown-6 [54]. The authors concluded that these kinetic parameters are similar to the values determined by nanopipet voltammetry (see above), thereby indicating the minimal effect of interfacial size on the kinetics of this facilitated IT reaction. This result, however, is not directly relevant to the size effect on the simple transfer of TEA+ because the rate-determining step of facilitated IT is not simple IT but is ion–ionophore complexation at the interface [55] as discussed below.
4. Nanopipet-Supported ITIES for High-Resolution SECM Imaging
The application of nanopipet-supported ITIES as an SECM tip allows for high-resolution electrochemical imaging [37]. For instance, Laforge et al employed a nanopipet-supported ITIES tip with a radius of 106 nm for successful topographic imaging in the constant-height mode of SECM [56]. More recently, we demonstrated the quantitative imaging of ion transport through single nanopores at an unprecedentedly high spatial resolution by employing even smaller nanopipet tips [57]. In this study, we imaged a highly porous nanocrystalline silicon (pnc-Si) membrane (Figure 4A). This emerging class of ultrathin nanoporous membranes has extremely high permeability, which enables its unique practical applications such as for the efficient separation of macromolecules [58–60] and nanoparticles [61], tissue engineering [62], and cell culture [62]. The future development of a pnc-Si membrane with even higher permeability requires the quantitative characterization of single nanopore permeability by SECM as fundamental study because the permeability of the whole nanoporous membrane depends on the convolution of several structural properties of each nanopore, including its shape, size, and density [63, 64].
Figure 4.

(A) TEM image of a pnc-Si membrane and (B) scheme of SECM line scan with a nanopipet-supported ITIES tip over the impermeable and nanoporous regions of the membrane. (C) SECM image of the pnc-Si membrane based on the transfer of 10 mM TBA+ in 0.3 M KCl at a 17 nm-radius pipet as silanized and filled with a DCE solution of 0.1 M tetradodecylammonium tetrakis(pentafluorophenyl)borate. Adapted with permission from ref. [57]. Copyright 2012 American Chemical Society.
The nanoscale spatial resolution of SECM was achieved by scanning a 17 nm-radius pipet tip at a distance down to 1.3 nm from a pnc-Si membrane. In the constant-height imaging mode of SECM, the peak current response was solely based on the nanopore-mediated diffusional transport of tetrabutylammonium (TBA+), which is transferred at the nanopipet-supported ITIES tip (Figure 4B). TBA+ transport through a nanopore is not due to migration because no potential was applied across the nanoporous membrane. A 280 nm × 500 nm image resolved 13 nanopores (Figure 4C), which corresponds to a high density of 93 pores/μm2. A finite element simulation of the SECM image was performed to determine the actual size of nanopore 7, which was approximated as an elliptic cylinder with a depth of 30 nm and major and minor axes of 53 and 41 nm, respectively. These structural parameters are consistent with those determined by TEM (Figure 4A), which thereby confirms the reliability of quantitative SECM imaging at the nanoscale. The numerical simulation also indicates that two adjacent pores with an edge-edge separation of the tip diameter or larger, i.e., ≥ 34 nm, are completely resolvable. Remarkably, this spatial resolution is the highest reported for SECM-based imaging to date, with the exception of one study [65], where a nanometer-thick electrolyte solution was needed and no quantitative image analysis was shown. Moreover, this spatial resolution exceeds that of any electrochemical method employed for single pore imaging. The shortest separations between two resolvable pores were limited to > 250 nm and ~1.5 μm for SECM [66] and scanning ion conductance microscopy [67], respectively. Advantageously, no feedback distance control was needed to achieve the highest resolution of SECM under normal solution conditions. This was possible not only because the surface of a pnc-Si membrane was flat, but also because the SECM stage was isolated from the ambient environment using an isothermal chamber to suppress thermal drift (see below). The application of this simple, quantitative, and high-resolution SECM approach to the imaging of biological nanopores is envisioned.
5. Stabilizing and Monitoring Nanogap for Nanoscale SECM
The control of a nanometer-wide gap between a tip and a substrate is critical for various nanoscale applications of SECM, not only for electrochemical imaging [56, 65, 68, 69] but also for the kinetic study of heterogeneous [54, 70–76] and homogeneous [77] reactions and single molecule detection [78–81]. Recently, we reported for the first time that the stability of the nanogap in ambient conditions is significantly compromised by the thermal expansion and contraction of components of an SECM stage upon a temperature change [82]. In this study, nanopipet-supported ITIES tips were required to precisely monitor the thermal drift of the tip–substrate gap, which was dramatically suppressed down to a subnanometer level in a newly developed isothermal chamber. Importantly, the thermal drift has been recognized as the origin of vertical and lateral image distortions and unstable nanomanipulation in other scanning probe microscopy techniques [83, 84]. Currently, these problems due to thermal drift are unavoidable, unless a cryostat or fast scanning is employed, and are only correctable [85]. Importantly, thermal-drift errors in either vertical or lateral probe position can not be eliminated by the feedback control of the probe–substrate distance. Thus, the isothermal chamber will be useful for SECM and, potentially, for other scanning probe microscopes.
The sub-nanometer scale drift and fluctuation of a ~20 nm-wide gap under a 12 nm-radius nanopipet tip was achieved by accommodating an SECM stage in the isothermal chamber developed in our laboratory [82]. Air temperature in the chamber changed only at ~0.2 mK/min to remarkably and reproducibly slow down the drift of tip–substrate distance to ~0.4 nm/min (Figure 5) or even lower as seen in the constant-height imaging of single nanopores (Figure 4C). In fact, a ~10 cm-height SECM stage with a typical coefficient of linear thermal expansion in the order of 10−5 K−1 is expected to expand or contract only by 0.2 nm upon a temperature change of 0.2 mK [85]. The subnanometer fluctuation of the nanogap was mainly caused by the instability of a piezoelectric positioner. In contrast, the thermal drift of the gap width without the chamber was much more significant and strongly dependent on ambient conditions to vary in the wide range of 5–150 nm/min. Such a drift was readily detected using the 0.5 μm-radius tip of either Pt or pipet-supported ITIES. This finding explains why the holding of a tip at a constant feedback distance of <1 μm for >1 min in imaging [86, 87] and voltammetry [74] is sometimes very difficult without the chamber. Evidently, the thermal drift effect may be very different in other laboratories and with different SECM instruments. Previously, a monotonic change in tip–substrate distance was ascribed to the drift of a piezoelectric positioner [88]. In our setup, closed-loop piezoelectric positioners were employed to avoid such creeping. Moreover, the positioners were mounted on a micromanipulator stage, which does not generate heat. In contrast, the step motor of a commercial SECM instrument is significantly warmed up when it is plugged to a power supply. The motor is seriously heated during continuous or repetitive movement.
Figure 5.

Time profile of (top) the current based on the transfer of 10 mM TEA+ in 0.3 M KCl at a 12 nm-radius pipet as silanized and filled with a DCE solution of 0.1 M tetradodecylammonium tetrakis(pentafluorophenyl)borate and (bottom) the corresponding tip–substrate gap width calculated using eq 3 in ref. [82]. Chamber temperature drifted at −0.2 mK/min during the current measurement. Adapted with permission from ref. [82]. Copyright 2012 American Chemical Society.
6. Electrochemical Ion Sensing with Double-Polymer-Modified Electrodes
A double-polymer-modified electrode is the solid electrode that is successively modified with an electrochemically active polymer membrane and then with an ion-selective polymer membrane, typically, a plasticized poly(vinyl chloride) (PVC) membrane. Such a chemically modified electrode was originally introduced as a potentiometric pH electrode by Oyama et al. [89], and has been developed as “solid-state” or “solid-contact” ISEs for potentiometry [90]. In contrast to conventional potentiometric ISEs, the solid-state electrodes are free from the internal solution behind the ion-selective membrane, which facilitates the handling and miniaturization of the electrodes. Recently, significant attention has been given to potentiometric solid-state ISEs as an approach to achieve the nanomolar and subnanomolar limits of detection (LOD) by eliminating the internal solution as the contamination source of an analyte ion [91, 92]. The LOD of conventional potentiometric ISEs is limited to the micromolar level by the transmembrane flux of an analyte ion from the internal solution, which contaminates the interface between the ion-selective membrane and the sample solution [93].
In zero-current potentiometry, an electrochemically active polymer membrane stabilizes the potential of a solid-state ISE by inhibiting the formation of a water layer between the ion-selective membrane and the solid support [94]. The inner water layer is formed by the condensation of water molecules that are partitioned from the sample solution into the PVC membrane [95, 96]. The concentrations of O2, CO2, and H+ in the inner water layer affect the resultant mixed potential of the underlying electrode. In fact, “good” membranes such as poly(3-octylthiophene) (POT) [97] and poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) [98] inhibit the formation of an undesirable water layer as confirmed by neutron reflectometry, synchrotron radiation/Fourier transform-infrared microscopy, and secondary ion mass spectrometry. Noticeably, the respective conducting polymers are completely reduced or oxidized so that the stabilized potential of the solid-state ISEs is not thermodynamically well-defined. In fact, the reduced form of the POT membrane formed between a gold electrode and a PVC membrane was externally polarizable [99]. Thus, even a highly capacitive intermediate layer of nanoporous carbon with a large surface area successfully stabilizes the potential of a solid-state ISE [100, 101]. A recent review comprehensively summarizes various polymer and carbon materials that were employed as good intermediate layers for potentiometric solid-state ISEs [102].
The voltammetric and amperometric operation of a double-polymer-modified electrode was proposed by Sun et al. [103], and was experimentally demonstrated by us [99] (Figure 1B). In contrast to the potentiometric mode, an intermediate polymer film must be electrolyzed in these dynamic modes to drive the net flux of an analyte ion across the interface between the ion-selective membrane and the aqueous sample. The slow electrolysis of a redox-active osmium polymer, in contact with the viscous PVC membrane, hindered the dynamic operation [103]. We found that the intermediate membranes of polythiophenes are quickly electrolyzed with small overpotentials to voltammetrically mediate IT at the interface between the PVC membrane and the aqueous sample [104]. Specifically, the oxidation of an undoped POT membrane was coupled with the transfer of an aqueous anion into the PVC membrane [99, 105] while cation transfer was driven by the reduction of the oxidatively-doped membrane of PEDOT [106] or its lipophilic derivative (Figure 6). The transferred cations (or anions) can be stripped from the PVC membrane to the aqueous sample by applying anodic (or cathodic) potentials to the underlying electrode, which drive the electrolysis of the oxidized POT (or reduced PEDOT) membrane.
Figure 6.
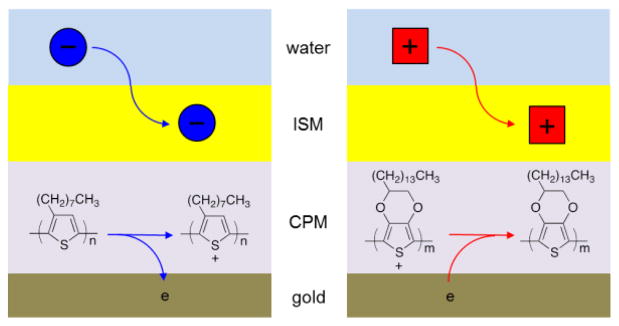
Scheme of (left) anion and (right) cation transfer at the solid electrodes (e.g., gold electrodes) modified with a conducting polymer membrane (CPM) and then with an ion-selective membrane (ISM).
For IT voltammetry and amperometry, a thin PVC membrane (0.7–20 μm) was coated on a thin conducting polymer membrane not only to minimize the ohmic potential drop across the double-polymer membrane, but also to maximize the sensitivity of IT stripping voltammetry (ITSV) based on a thin layer effect (see below). In addition, the thin membrane of a conducting polymer with a high redox density offers sufficient redox capacity. In contrast, a thicker PVC membrane is required for obtaining a sufficient redox capacity when redox-active molecules such as ferrocene are dissolved in the PVC membrane [107, 108] or covalently attached to the backbone of PVC [109]. Alternatively, a redox-active monolayer was densely adsorbed to the surface of a solid electrode to provide a high redox capacity [110–112].
7. Electrochemical Mechanism of Ion–Ionophore Recognition at the ITIES
The ionophores that were developed for potentiometric and optical ion sensors [7, 113] are attractive as selective ion-recognition elements for ITIES-based voltammetric and amperometric sensing with superior sensitivity and unqiue selectivity. As the first step toward this goal, we doped double-polymer-modified electrodes with various ionophores and voltammetrically investigated the kinetics and mechanism of ionophore-facilitated IT at the ITIES [55]. Such quantitative kinetic data have been demanded for resolving long-standing controversies on the mechanism of facilitated IT [114, 115]. The IT reaction facilitated by an ionophore, L, may proceed based on the one-step electrochemical (E) mechanism, where an 1:n ion–ionophore complex, iLnz, is formed at the very interface and then transferred into the membrane as given by
| (3) |
This reaction, however, is thermodynamically equivalent to the two-step electrochemical–chemical (EC) mechanism. In the EC mechanism, the simple transfer of an aqueous ion is followed by its complexation with ionophore in the bulk membrane as given by
| (4) |
| (5) |
Kakutani et al., proposed that these two mechanisms can be kinetically discriminated [116]. For instance, they excluded the EC mechanism for the Na+ transfer facilitated by dibenzo-18-crown-6 at the macroscopic nitrobenzene/water interface. They reasoned that, if this electrochemically reversible reaction is based on the EC mechanism, the dissociation of the stable ion–ionophore complex in the organic phase (eq 5) must be unrealistically fast. The validity of the E mechanism, however, was not addressable because of a lack of kinetic data.
We employed ionophore-doped double-polymer electrodes to reveal that facilitated IT is kinetically controlled by the E mechanism (eq 3) based on ion–ionophore recognition at the very interface [55]. In contrast to the previously reported voltammetric ISEs [117], our double-polymer-modified electrodes are free from a significant ohmic potential drop to give high-quality IT cyclic voltammograms (CVs) for quantitative kinetic study. Specifically, quasi-reversible CVs of the Ag+, K+, Ca2+, Ba2+, and Pb2+ transfers facilitated by highly selective ionophores were measured at various potential sweep rates (Figure 7A for Ca2+ and Ba2+). The CVs were analyzed numerically using the E mechanism to determine k0 values in the range of 10−2–10−3 cm/s and α values of ~0.5. Similar kinetic parameters were also obtained at the micropipet-supported interface with a much less viscous DCE solution. The little effect of the viscosity of non-aqueous media on the IT kinetics excludes the EC mechanism, where the kinetics of simple IT (eq 4) is viscosity-dependent [41]. Moreover, the fit of the experimental CVs with the theoretical CVs based on the EC mechanism required unrealistically large k0 values of >60 cm/s for simple IT (eq 4) in comparison to k0 = 6 cm/s for fast TEA+ transfer (see above). In addition, our study excludes the EC mechanism also because the dissociation of stable ion–ionophore complexes in the non-aqueous phase must be faster than a diffusion limit, as pointed out by Kakutani et al [116].
Figure 7.

(A) Background-subtracted CVs (solid lines) of the facilitated transfer of Ca2+ and Ba2+ (as 10 μM chloride salts in 10 mM CH3COOK at pH 7.1) as obtained using the glassy carbon electrode modified with a lipophilic PEDOT membrane and the oNPOE-plasticized PVC membrane doped with ETH 129. Theoretical CVs (closed circles) were obtained by the finite element simulation based on the E mechanism. (B) Scheme of the formation of 1:3 Ca2+–ionophore (ETH 129) complexes at an oNPOE-plasticized PVC membrane/water interface with a mixed solvent layer. Adapted with permission from ref. [55]. Copyright 2011 American Chemical Society.
The new kinetic data thus obtained gave molecular-level insights into the E mechanism. Microscopically, rate constants of 30–90 cm/s were estimated for a diffusion-limited collision between an ionophore molecule and an analyte ion at the interface. These rate constants are much higher than k0 values of 10−2–10−3 cm/s for the E mechanism, which is kinetically limited by ion–ionophore complexation at the interface. Interestingly, the formation of a three-dimensional complex at the two-dimensional ITIES may play an important kinetic role in the E mechanism. CVs in Figure 7A demonstrate that the formation of a 1:3 Ca2+–ionophore complex is thermodynamically more favorable, but is slower in comparison to the formation of a 1:2 Ba2+–ionophore complex. The slower kinetics of the facilitated Ca2+ transfer was ascribed to the hindered access of a lipophilic ionophore to a Ca2+ ion from its aqueous side, which is mediated only through the mixed solvent layer at the interface (Figure 7B).
8. IT Stripping Voltammetry for Trace Analysis
ITSV at the ITIES is a powerful electroanalytical method that enables the trace analysis of various ions. In comparison to traditional redox-based stripping voltammetry [118], the ITSV technique has complementary principles, as well as greater versatile applicability because target ions do not need to be reduced or oxidized [119]. In ITSV, an aqueous analyte ion is potentiostatically preconcentrated into a water-immiscible organic phase and then voltammetrically stripped to yield an enhanced current response. ITSV was successfully applied for relatively lipophilic ions such as acetylcholine [120, 121], vitamin B1 [122], various protonated amines [123], tetraalkylammoniums [124–126], anionic surfactants [126, 127], and β-blocker propranolol [128, 129]. In addition, various ionophores were employed to enable ITSV of highly hydrophilic ions such as alkaline earth metal ions [130] and oligopeptides [131]. ITSV of reducible heavy metal ions [119, 132–134] is a good candidate to replace anodic stripping voltammetry with a mercury electrode [135]. Moreover, adsorptive ITSV was developed to detect macromolecular ions such as anticoagulant/antithrombotic heparin [136], lysozyme [137], and digested proteins [138]. ITSV of neutral surfactants is also possible because these analytes form electrically-charged complexes with aqueous metal ions to be preconcentrated associatively and stripped dissociatively [127, 139, 140].
We demonstrated that double-polymer-modified electrodes massively improve the sensitivity of ITSV to low nanomolar [105] and subnanomolar [106] levels. The extremely low LODs are due to the solid-supported thin membrane, where the preconcentrated analyte ions are exhaustively stripped to maximize the resultant current response. For instance, ITSV of perchlorate in drinking water gave LODs of 0.2–0.5 nM when a double-polymer electrode was rotated at 4000 rpm to accelerate ion preconcentration [105]. These detection limits are much lower than the interim health advisory level of ~150 nM in drinking water set by the U.S. Environmental Protection Agency (EPA) and even comparable to the LODs of the most sensitive methods for perchlorate analysis, i.e., ion chromatography coupled with a suppressed conductivity detector or electrospray ionization mass spectrometry. The EPA’s recent decision to regulate perchlorate in drinking water [141] augmented the significance of our perchlorate sensor. Moreover, ITSV at the rotating double-polymer electrode gave subnanomolar LODs for more lipophilic analyte ions, which can be preconcentrated at higher concentrations in the hydrophobic PVC membrane [106]. A detection limit of 90 pM thus obtained for hexafluoroarsenate is lower than that for less lipophilic perchlorate and is comparable to the lowest LOD of 80 pM as obtained for this arsenical biocide so far using inductively coupled plasma–mass spectrometry with anion-exchange chromatography. Also, a PVC/POT-modified electrode was useful for adsorptive ITSV of heparins to achieve a very low LOD of 0.005 unit/mL in a stirred 0.12 M NaCl solution, which is equivalent to ~2.5 nM heparin with an average molecular weight of 15 kDa [99].
Recently, we reported on the first ITSV application of an ionophore-doped double-polymer electrode to demonstrate nanomolar LOD and multiple-ion detectability [142]. We developed a theoretical model for ionophore-based ITSV at the thin polymer membrane to predict that the LOD based on equilibrium preconcentration is lower when a more stable ion–ionophore complex is formed. The theoretical prediction was confirmed using valinomycin, which forms a ~60 times more stable complex with K+ than with NH4+ as confirmed by cyclic voltammetry. A LOD of 0.6 nM K+ was achieved by ITSV using commercial ultrapure water as a K+-free media, where voltammetric responses to 1–10 nM of spiked K+ are comparable to or larger than those to ~100 nM of contaminated NH4+ (Figure 8). This background concentration of NH4+ contamination in the ultrapure water was determined from the dependence of the ITSV response on the preconcentration time under the rotating electrode configuration. Advantageously, this approach does not require the background ITSV measurement of a contamination-free blank solution, which is not available for NH4+ as a target ion. The ITSV approach was also used to determine ~5 nM K+ contamination in laboratory-purified water. Importantly, the presence of easily contaminable and hardly removable K+ and NH4+ in a blank solution may be the origin of higher LODs of 5 nM [143] and 100 nM [92] K+ for potentiometry with conventional and solid-state ISEs based on valinomycin, respectively. The background contamination of a sample solution with these ions is not detectable by potentiometry, which measures only a change in the electrode potential in the sample solution with respect to that in the blank solution.
Figure 8.

(A) ITSV of 0–10 nM K+ as sulfate salt in ultrapure water at 0.1 V/s after 5 min of preconcentration as obtained using the gold electrode modified with a lipophilic PEDOT membrane and the oNPOE-plasticized PVC membrane doped with valinomycin. Aqueous and organic supporting electrolytes are H2SO4/HCl and tetradodecylammonium tetrakis(pentafluorophenyl)borate, respectively. Preconcentration potential, 0.38 V. The electrode was rotated at 4000 rpm. Adapted with permission from ref. [142]. Copyright 2012 American Chemical Society.
Finally, an inexpensive and disposable electrode for ITSV was developed by simply coating a pencil lead with a POT membrane, followed by a plasticized PVC membrane [144]. Remarkably, the simple electrode allows for the ITSV detection of perchlorate in tap water at concentrations of 100–1000 nM below the interim health advisory level set by the EPA (Figure 9). This result shows the versatility of the double-polymer approach to IT voltammetry. Moreover, the pencil lead electrode is useful as an educational tool for undergraduate instrumental laboratories. Students can fabricate and utilize their own electrodes in the 3 to 4 hour laboratory session to learn important concepts and methods of electrochemistry and also quantitatively address whether their local drinking water is contaminated with nanomolar perchlorate.
Figure 9.

Stripping voltammograms of perchlorate at a PVC/POT-modified pencil lead (see inset for its image). Aqueous and organic supporting electrolytes are Li2SO4 and tetradodecylammonium tetrakis(pentafluorophenyl)borate, respectively. Scan rate, 0.1 V/s. Adapted with permission from ref. [144]. Copyright 2012 American Chemical Society.
9. Perspectives
Electrochemical methods based on IT at the ITIES found early success in potentiometric ion analysis and adopted the concepts and methodologies of voltammetry and amperometry from redox electrochemistry at solid/liquid interfaces to be reinforced as the most advanced approaches for electrochemical ion sensing and imaging. The promising future applications of these dynamic approaches for bioanalysis and electrochemical imaging are foreseen, as discussed in this section.
The establishment of such robust and practical sensing formats as the pipet-supported ITIES and the double-polymer-modified electrode for IT voltammetry and amperometry will offer great opportunities in bioanalysis. In fact, various bioanalytical applications of dynamic electrochemical methods have been seen for redox electrochemistry, from amperometric sensors for blood glucose [145] to in-vivo voltammetry and amperometry of neurotransmitters in the brain and other tissues [146, 147]. Moreover, ion analysis in biological media such as blood samples and extracellular/intracellular spaces has been extensively explored by ion-selective potentiometry. The feasibility of IT voltammetry in undiluted blood samples was also demonstrated using the fast simple transfer of tetrapropylammonium at the micropipet-supported ITIES as a model system [136].
Voltammetric/amperometric ion analysis in complicated biological media will be challenging and require highly selective ionophores. Fortunately, >1,000 natural and synthetic ionophores have been reported for potentiometric and optical sensors during the past five decades [7, 113]. Subsequently, the ion selectivity of most of these ionophores has been determined under equilibrium conditions and has not been tested under dynamic conditions for voltammetry or amperometry at the ITIES. Interestingly, ETH 129 is thermodynamically more selective for Ca2+ against Ba2+ while the transfer of the latter ion is kinetically more facile. In addition to the appreciation of both thermodynamic and kinetic ion selectivity, these dynamic approaches allow for the exploration of multiple-ion selectivity of an ionophore in contrast to the ionophore-based potentiometric and optical sensors that respond only to the most selective ion. Moreover, IT voltammetry is useful for both thermodynamic and kinetic characterization of ionophores in contrast to the equilibrium potentiometric and optical measurements. Micropipet IT voltammetry was applied for the comprehensive characterization of new ionophores for inorganic anions [148] and heparins [149, 150]. Advantageously, pipet-supported ITIES and double-polymer-modified electrodes require a tiny amount (≪1 mg) of an expensive or elaborately synthesized ionophore, in contrast to the traditional four-electrode setup, which requires the bulk non-aqeuous solution of an ionophore.
Nanopipet-supported ITIES tips allowed us to establish a technological basis toward the future advancement of nanoscale SECM for high-resolution imaging and fast kinetic measurement. Significantly, the simple and robust operation of SECM, which has been appreciated at the micrometer-scale, is also possible at the nanoscale by minimizing thermal drift. The success of a nanopipet-based tip originates not only from its simple fabrication, but also from its design with a thin glass sheath, which facilitates tip positioning within nanometer distances from a target substrate.
Acknowledgments
This work was supported by the National Science Foundation (CHE-1213452) and the National Institutes of Health (GM073439).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Girault HH. Electrochemistry at liquid-liquid interfaces. In: Bard AJ, Zoski CG, editors. Electroanalytical chemistry. Vol. 23. Taylor & Francis; Boca Raton: 2010. p. 1. [Google Scholar]
- 2.Samec Z. Dynamic electrochemistry at the interface between two immiscible electrolytes. Electrochim Acta. 2012;84:21. [Google Scholar]
- 3.Luo GM, Malkova S, Yoon J, Schultz DG, Lin BH, Meron M, Benjamin I, Vanysek P, Schlossman ML. Ion distributions near a liquid-liquid interface. Science. 2006;311:216. doi: 10.1126/science.1120392. [DOI] [PubMed] [Google Scholar]
- 4.Laanait N, Mihaylov M, Hou B, Yu H, Vanýsek P, Meron M, Lin B, Benjamin I, Schlossman ML. Tuning ion correlations at an electrified soft interface. Proc Natl Acad Sci USA. 2012;109:20326. doi: 10.1073/pnas.1214204109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reymond F, Fermin D, Lee HJ, Girault HH. Electrochemistry at liquid/liquid interfaces: Methodology and potential applications. Electrochim Acta. 2000;45:2647. [Google Scholar]
- 6.Bakker E, Bühlmann P, Pretsch E. Carrier-based ion-selective electrodes and bulk optodes. 1. General characteristics. Chem Rev. 1997;97:3083. doi: 10.1021/cr940394a. [DOI] [PubMed] [Google Scholar]
- 7.Bühlmann P, Pretsch E, Bakker E. Carrier-based ion-selective electrodes and bulk optodes. 2. Ionophores for potentiometric and optical sensors. Chem Rev. 1998;98:1593. doi: 10.1021/cr970113+. [DOI] [PubMed] [Google Scholar]
- 8.Amemiya S. Potentiometric ion-selective electrodes. In: Zoski CG, editor. Handbook of electrochemistry. Ch 7. Elsevier; New York: 2007. p. 261. [Google Scholar]
- 9.Bobacka J, Ivaska A, Lewenstam A. Potentiometric ion sensors. Chem Rev. 2008;108:329. doi: 10.1021/cr068100w. [DOI] [PubMed] [Google Scholar]
- 10.Fry CH, Langley SEM. Ion-selective electrodes for biological systems. Harwood Academic Publishers; Amsterdam, Netherlands: 2001. [Google Scholar]
- 11.Meyerhoff ME. Electrochemistry and chemical sensors. In: Burtis CA, Ashwood ER, Bruns DE, editors. Tietz textbook of clinical chemistry and molecular diagnostics. Elsevier Saunders; St. Louis, MO: 2011. p. 259. [Google Scholar]
- 12.Zuliani C, Diamond D. Opportunities and challenges of using ion-selective electrodes in environmental monitoring and wearable sensors. Electrochim Acta. 2012;84:29. [Google Scholar]
- 13.Samec Z, Samcová E, Girault HH. Ion amperometry at the interface between two immiscible electrolyte solutions in view of realizing the amperometric ion-selective electrode. Talanta. 2004;63:21. doi: 10.1016/j.talanta.2003.11.023. [DOI] [PubMed] [Google Scholar]
- 14.Samec Z. Electrochemistry at the interface between two immiscible electrolyte solutions. Pure Appl Chem. 2004;76:2147. [Google Scholar]
- 15.Liu B, Mirkin MV. Electrochemistry at microscopic liquid-liquid interfaces. Electroanalysis. 2000;12:1433. [Google Scholar]
- 16.Taylor G, Girault HH. Ion transfer reactions across a liquid-liquid interface supported on a micropipette tip. J Electroanal Chem. 1986;208:179. [Google Scholar]
- 17.Shao Y, Mirkin MV. Voltammetry at micropipet electrodes. Anal Chem. 1998;70:3155. doi: 10.1021/ac980244q. [DOI] [PubMed] [Google Scholar]
- 18.Campbell JA, Girault HH. Steady state current for ion transfer reactions at a micro liquid/liquid interface. J Electroanal Chem. 1989;266:465. [Google Scholar]
- 19.Mirkin MV, Tsionsky M. Charge transfer processes at the liquid/liquid interface. In: Bard AJ, Mirkin MV, editors. Scanning electrochemical microscopy. Ch 8. CRC Press Taylor & Francis; Boca Raton, FL: 2012. p. 191. [Google Scholar]
- 20.Wilke S, Wang H, Muraczewska M, Müller H. Amperometric detection of heavy metal ions in ion pair chromatography at an array of water/nitrobenzene micro interfaces. Fresenius J Anal Chem. 1996;356:233. doi: 10.1007/s0021663560233. [DOI] [PubMed] [Google Scholar]
- 21.Sun L, Weber SG. Electrochemically driven ion-selective sensing and extraction based on PVC membrane-coated electrodes. Polym Mater Sci Eng. 1997;76:614. [Google Scholar]
- 22.Ammann D. Ion-selective microelectrodes. Springer-Verlag; Berlin: 1986. [Google Scholar]
- 23.Tsien RY. Intracellular measurements of ion activities. Ann Rev Biophys Bioeng. 1983;12:91. doi: 10.1146/annurev.bb.12.060183.000515. [DOI] [PubMed] [Google Scholar]
- 24.Brown KT, Flaming DG. Advanced micropipette techniques for cell physiology. Wiley; New York: 1986. [Google Scholar]
- 25.Walker JL. Ion specific liquid ion exchanger microelectrodes. Anal Chem. 1971;43:89A. [Google Scholar]
- 26.Munoz JL, Deyhimi F, Coles JA. Silanization of glass in the making of ion-sensitive microelectrodes. J Neurosci Meth. 1983;8:231. doi: 10.1016/0165-0270(83)90037-7. [DOI] [PubMed] [Google Scholar]
- 27.Bakker E, Pretsch E. Nanoscale potentiometry. Trends Anal Chem. 2008;27:612. doi: 10.1016/j.trac.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shao Y, Mirkin MV. Fast kinetic measurements with nanometer-sized pipets. Transfer of potassium ion from water into dichloroethane facilitated by dibenzo-18-crown-6. J Am Chem Soc. 1997;119:8103. [Google Scholar]
- 29.Yuan Y, Shao YH. Systematic investigation of alkali metal ion transfer across the micro- and nano-water/1,2-dichloroethane interfaces facilitated by dibenzo-18-crown-6. J Phys Chem B. 2002;106:7809. [Google Scholar]
- 30.Jing P, Zhang MQ, Hu H, Xu XD, Liang ZW, Li B, Shen L, Xie SB, Pereira CM, Shao YH. Ion-transfer reactions at the nanoscopic water/n-octanol interface. Angew Chem, Int Ed. 2006;45:6861. doi: 10.1002/anie.200601826. [DOI] [PubMed] [Google Scholar]
- 31.Li F, Chen Y, Zhang MQ, Jing P, Gao Z, Shao YH. Ion transfer reactions in media of low ionic strength. J Electroanal Chem. 2005;579:89. [Google Scholar]
- 32.Sun P, Laforge FO, Mirkin MV. Ion transfer at nanointerfaces between water and neat organic solvents. J Am Chem Soc. 2005;127:8596. doi: 10.1021/ja050999w. [DOI] [PubMed] [Google Scholar]
- 33.Laforge FO, Sun P, Mirkin MV. Shuttling mechanism of ion transfer at the interface between two immiscible liquids. J Am Chem Soc. 2006;128:15019. doi: 10.1021/ja0656090. [DOI] [PubMed] [Google Scholar]
- 34.Sun P, Laforge FO, Mirkin MV. Role of trace amounts of water in transfers of hydrophilic and hydrophobic ions to low-polarity organic solvents. J Am Chem Soc. 2007;129:12410. doi: 10.1021/ja075774v. [DOI] [PubMed] [Google Scholar]
- 35.Cai CX, Tong YH, Mirkin MV. Probing rapid ion transfer across a nanoscopic liquid-liquid interface. J Phys Chem B. 2004;108:17872. [Google Scholar]
- 36.Lefrou C. A unified new analytical approximation for positive feedback currents with a microdisk secm tip. J Electroanal Chem. 2006;592:103. [Google Scholar]
- 37.Elsamadisi P, Wang Y, Velmurugan J, Mirkin MV. Polished nanopipets: New probes for high-resolution scanning electrochemical microscopy. Anal Chem. 2011;83:671. doi: 10.1021/ac102704z. [DOI] [PubMed] [Google Scholar]
- 38.Rodgers PJ, Amemiya S, Wang Y, Mirkin MV. Nanopipet voltammetry of common ions across the liquid–liquid interface. Theory and limitations in kinetic analysis of nanoelectrode voltammograms. Anal Chem. 2010;82:84. doi: 10.1021/ac9022428. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y, Velmurugan J, Mirkin MV, Rodgers PJ, Kim J, Amemiya S. Kinetic study of rapid transfer of tetraethylammonium at the 1,2-dichloroethane/water interface by nanopipet voltammetry of common ions. Anal Chem. 2010;82:77. doi: 10.1021/ac902244s. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y, Kakiuchi T, Yasui Y, Mirkin MV. Kinetics of ion transfer at the ionic liquid/water nanointerface. J Am Chem Soc. 2010;132:16945. doi: 10.1021/ja1066948. [DOI] [PubMed] [Google Scholar]
- 41.Marcus RA. On the theory of ion transfer rates across the interface of two immiscible liquids. J Chem Phys. 2000;113:1618. [Google Scholar]
- 42.Rodgers PJ, Amemiya S. Cyclic voltammetry at micropipet electrodes for the study of ion-transfer kinetics at liquid/liquid interfaces. Anal Chem. 2007;79:9276. doi: 10.1021/ac0711642. [DOI] [PubMed] [Google Scholar]
- 43.Li Q, Xie S, Liang Z, Meng X, Liu S, Girault HH, Shao Y. Fast ion-transfer processes at nanoscopic liquid/liquid interfaces. Angew Chem, Int Ed. 2009;48:8010. doi: 10.1002/anie.200903143. [DOI] [PubMed] [Google Scholar]
- 44.Gavach C, D’Epenoux B, Henry F. Transfer of tetra-n-alkylammonium ions from water to nitrobenzene: Chronopotentiometric determination of kinetic parameters. J Electroanal Chem. 1975;64:107. [Google Scholar]
- 45.Samec Z, Marecek V. Charge transfer between two immiscible electrolyte solutions: Part X. Kinetics of tetraalkylammonium ion transfer across the water/nitrobenzene interface. J Electroanal Chem. 1986;200:17. [Google Scholar]
- 46.Wandlowski T, Marecek V, Holub K, Samec Z. Ion transfer across liquid-liquid phase boundaries: Electrochemical kinetics by faradic impedance. J Phys Chem. 1989;93:8204. [Google Scholar]
- 47.Kakiuchi T, Noguchi J, Kotani M, Senda M. Ac polarographic-determination of the rate of ion transfer for a series of alkylammonium ions at the nitrobenzene water interface. J Electronanal Chem. 1990;296:517. [Google Scholar]
- 48.Kakiuchi T, Noguchi J, Senda M. Double-layer effect on the transfer of some monovalent ions across the polarized oil–water interface. J Electroanal Chem. 1992;336:137. [Google Scholar]
- 49.Wandlowski T, Marecek V, Samec Z, Fuoco R. Effect of temperature on the ion transfer across an interface between two immiscible electrolyte solutions: Ion transfer dynamics. J Electroanal Chem. 1992;331:765. [Google Scholar]
- 50.Kakiuchi T, Teranishi Y. Effect of the viscosity of the aqueous phase on the rate of ion transfer across the nitrobenzene|water interface. J Electroanal Chem. 1995;396:401. [Google Scholar]
- 51.Rahman MA, Doe H. Ion transfer of tetraalkylammonium cations at an interface between frozen aqueous solution and 1,2-dichloroethane. J Electroanal Chem. 1997;424:159. [Google Scholar]
- 52.Marecek V, Lhotský A, Racinský S. Fluctuation analysis and faradaic impedance at micro liquid/liquid interface--I. Electrochim Acta. 1995;40:2905. [Google Scholar]
- 53.Lhotsky A, Holub K, Neuzil P, Marecek V. Ac impedance analysis of tetraethylammonium ion transfer at liquid/liquid microinterfaces. J Chem Soc, Faraday Trans. 1996;92:3851. [Google Scholar]
- 54.Sun P, Zhang ZQ, Gao Z, Shao YH. Probing fast facilitated ion transfer across an externally polarized liquid-liquid interface by scanning electrochemical microscopy. Angew Chem, Int Ed. 2002;41:3445. doi: 10.1002/1521-3773(20020916)41:18<3445::AID-ANIE3445>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 55.Ishimatsu R, Izadyar A, Kabagambe B, Kim Y, Kim J, Amemiya S. Electrochemical mechanism of ion–ionophore recognition at plasticized polymer membrane/water interfaces. J Am Chem Soc. 2011;133:16300. doi: 10.1021/ja207297q. [DOI] [PubMed] [Google Scholar]
- 56.Laforge FO, Velmurugan J, Wang Y, Mirkin MV. Nanoscale imaging of surface topography and reactivity with the scanning electrochemical microscope. Anal Chem. 2009;81:3143. doi: 10.1021/ac900335c. [DOI] [PubMed] [Google Scholar]
- 57.Shen M, Ishimatsu R, Kim J, Amemiya S. Quantitative imaging of ion transport through single nanopores by high-resolution scanning electrochemical microscopy. J Am Chem Soc. 2012;134:9856. doi: 10.1021/ja3023785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Striemer CC, Gaborski TR, McGrath JL, Fauchet PM. Charge- and size-based separation of macromolecules using ultrathin silicon membranes. Nature. 2007;445:749. doi: 10.1038/nature05532. [DOI] [PubMed] [Google Scholar]
- 59.Fang DZ, Striemer CC, Gaborski TR, McGrath JL, Fauchet PM. Pore size control of ultrathin silicon membranes by rapid thermal carbonization. Nano Lett. 2010;10:3904. doi: 10.1021/nl101602z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Snyder JL, Clark A, Fang DZ, Gaborski TR, Striemer CC, Fauchet PM, McGrath JL. An experimental and theoretical analysis of molecular separations by diffusion through ultrathin nanoporous membranes. J Membr Sci. 2011;369:119. doi: 10.1016/j.memsci.2010.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gaborski TR, Snyder JL, Striemer CC, Fang DZ, Hoffman M, Fauchet PM, McGrath JL. High-performance separation of nanoparticles with ultrathin porous nanocrystalline silicon membranes. ACS Nano. 2010;4:6973. doi: 10.1021/nn102064c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Agrawal AA, Nehilla BJ, Reisig KV, Gaborski TR, Fang DZ, Striemer CC, Fauchet PM, McGrath JL. Porous nanocrystalline silicon membranes as highly permeable and molecularly thin substrates for cell culture. Biomaterials. 2010;31:5408. doi: 10.1016/j.biomaterials.2010.03.041. [DOI] [PubMed] [Google Scholar]
- 63.Kim E, Xiong H, Striemer CC, Fang DZ, Fauchet PM, McGrath JL, Amemiya S. A structure-permeability relationship of ultrathin nanoporous silicon membrane: A comparison with the nuclear envelope. J Am Chem Soc. 2008;130:4230. doi: 10.1021/ja711258w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ishimatsu R, Kim J, Jing P, Striemer CC, Fang DZ, Fauchet PM, McGrath JL, Amemiya S. Ion-selective permeability of a ultrathn nanopore silicon membrane as probed by scanning electrochemical microscopy using micropipet-supported ITIES tips. Anal Chem. 2010;82:7127. doi: 10.1021/ac1005052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fan FRF, Bard AJ. Imaging biological macromolecules on mica in humid air by scanning electrochemical microscopy. Proc Natl Acad Soci USA. 1999;96:14222. doi: 10.1073/pnas.96.25.14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Macpherson JV, Jones CE, Barker AL, Unwin PR. Electrochemical imaging of diffusion through single nanoscale pores. Anal Chem. 2002;74:1841. doi: 10.1021/ac0157472. [DOI] [PubMed] [Google Scholar]
- 67.Zhou Y, Chen CC, Baker LA. Heterogeneity of multiple-pore membranes investigated with ion conductance microscopy. Anal Chem. 2012;84:3003. doi: 10.1021/ac300257q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sun P, Laforge FO, Abeyweera TP, Rotenberg SA, Carpino J, Mirkin MV. Nanoelectrochemistry of mammalian cells. Proc Natl Acad Soci USA. 2008;105:443. doi: 10.1073/pnas.0711075105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Takahashi Y, Shevchuk AI, Novak P, Zhang Y, Ebejer N, Macpherson JV, Unwin PR, Pollard AJ, Roy D, Clifford CA, Shiku H, Matsue T, Klenerman D, Korchev YE. Multifunctional nanoprobes for nanoscale chemical imaging and localized chemical delivery at surfaces and interfaces. Angew Chem, Int Ed. 2011;50:9638. doi: 10.1002/anie.201102796. [DOI] [PubMed] [Google Scholar]
- 70.Sun P, Mirkin MV. Kinetics of electron-transfer reactions at nanoelectrodes. Anal Chem. 2006;78:6526. doi: 10.1021/ac060924q. [DOI] [PubMed] [Google Scholar]
- 71.Velmurugan J, Sun P, Mirkin MV. Scanning electrochemical microscopy with gold nanotips: The effect of electrode material on electron transfer rates. J Phys Chem C. 2008;113:459. [Google Scholar]
- 72.Li F, Chen Y, Sun P, Zhang MQ, Gao Z, Zhan DP, Shao YH. Investigation of facilitated ion-transfer reactions at high driving force by scanning electrochemical microscopy. J Phys Chem B. 2004;108:3295. [Google Scholar]
- 73.Mirkin MV, Richards TC, Bard AJ. Scanning electrochemical microscopy. 20. Steady-state measurements of the fast heterogeneous kinetics in the ferrocene/acetonitrile system. J Phys Chem. 1993;87:7672. [Google Scholar]
- 74.Nioradze N, Kim J, Amemiya S. Quasi-steady-state voltammetry of rapid electron transfer reactions at the macroscopic substrate of the scanning electrochemical microscope. Anal Chem. 2011;83:828. doi: 10.1021/ac102352v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shen M, Bard AJ. Localized electron transfer and the effect of tunneling on the rates of Ru(bpy)32+ oxidation and reduction as measured by scanning electrochemical microscopy. J Am Chem Soc. 2011;133:15737. doi: 10.1021/ja206136h. [DOI] [PubMed] [Google Scholar]
- 76.Amemiya S, Nioradze N, Santhosh P, Deible MJ. Generalized theory for nanoscale voltammetric measurements of heterogeneous electron-transfer kinetics at macroscopic substrates by scanning electrochemical microscopy. Anal Chem. 2011;83:5928. doi: 10.1021/ac200862t. [DOI] [PubMed] [Google Scholar]
- 77.Bi S, Liu B, Fan FRF, Bard AJ. Electrochemical studies of guanosine in DMF and detection of its radical cation in a scanning electrochemical microscopy nanogap experiment. J Am Chem Soc. 2005;127:3690. doi: 10.1021/ja042433y. [DOI] [PubMed] [Google Scholar]
- 78.Fan FRF, Bard AJ. Electrochemical detection of single molecules. Science. 1995;267:871. doi: 10.1126/science.267.5199.871. [DOI] [PubMed] [Google Scholar]
- 79.Bard AJ, Fan FRF. Electrochemical detection of single molecules. Acc Chem Res. 1996;29:572. [Google Scholar]
- 80.Fan FRF, Kwak J, Bard AJ. Single molecule electrochemistry. J Am Chem Soc. 1996;118:9669. [Google Scholar]
- 81.Sun P, Mirkin MV. Electrochemistry of individual molecules in zeptoliter volumes. J Am Chem Soc. 2008;130:8241. doi: 10.1021/ja711088j. [DOI] [PubMed] [Google Scholar]
- 82.Kim J, Shen M, Nioradze N, Amemiya S. Stabilizing nanometer scale tip-to-substrate gaps in scanning electrochemical microscopy using an isothermal chamber for thermal drift suppression. Anal Chem. 2012;84:3489. doi: 10.1021/ac300564g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Moheimani SOR. Invited review article: Accurate and fast nanopositioning with piezoelectric tube scanners: Emerging trends and future challenges. Rev Sci Instrum. 2008;79 doi: 10.1063/1.2957649. [DOI] [PubMed] [Google Scholar]
- 84.Marinello F, Balcon M, Schiavuta P, Carmignato S, Savio E. Thermal drift study on different commercial scanning probe microscopes during the initial warming-up phase. Meas Sci Technol. 2011;22:094016. [Google Scholar]
- 85.Rahe P, Bechstein R, Kuhnle A. Vertical and lateral drift corrections of scanning probe microscopy images. J Vac Sci Technol B. 2010;28:C4E31. [Google Scholar]
- 86.Xiong H, Gross DA, Guo J, Amemiya S. Local feedback mode of scanning electrochemical microscopy for electrochemical characterization of one-dimensional nanostructure: Theory and experiment with nanoband electrode as model substrate. Anal Chem. 2006;78:1946. doi: 10.1021/ac051731q. [DOI] [PubMed] [Google Scholar]
- 87.Kim J, Xiong H, Hofmann M, Kong J, Amemiya S. Scanning electrochemical microscopy of individual single-walled carbon nanotubes. Anal Chem. 2010;82:1605. doi: 10.1021/ac9028032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.O’Mullane AP, Macpherson JV, Unwin PR, Cervera-Montesinos J, Manzanares JA, Frehill F, Vos JG. Measurement of lateral charge propagation in [Os(bpy)2(PVP)nCl]Cl thin films: A scanning electrochemical microscopy approach. J Phys Chem B. 2004;108:7219. [Google Scholar]
- 89.Oyama N, Hirokawa T, Yamaguchi S, Ushizawa N, Shimomura T. Hydrogen ion selective microelectrode prepared by modifying an electrode with polymers. Anal Chem. 1987;59:258. [Google Scholar]
- 90.Bobacka J. Conducting polymer-based solid-state ion-selective electrodes. Electroanalysis. 2006;18:7. [Google Scholar]
- 91.Sutter J, Lindner E, Gyurcsányi R, Pretsch E. A polypyrrole-based solid-contact pb2+-selective pvc-membrane electrode with a nanomolar detection limit. Anal Bioanal Chem. 2004;380:7. doi: 10.1007/s00216-004-2737-4. [DOI] [PubMed] [Google Scholar]
- 92.Chumbimuni-Torres KY, Rubinova N, Radu A, Kubota LT, Bakker E. Solid contact potentiometric sensors for trace level measurements. Anal Chem. 2006;78:1318. doi: 10.1021/ac050749y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sokalski T, Ceresa A, Zwickl T, Pretsch E. Large improvement of the lower detection limit of ion-selective polymer membrane electrodes. J Am Chem Soc. 1997;119:11347. [Google Scholar]
- 94.Fibbioli M, Morf WE, Badertscher M, de Rooij NF, Pretsch E. Potential drifts of solid-contacted ion-selective electrodes due to zero-current ion fluxes through the sensor membrane. Electroanalysis. 2000;12:1286. [Google Scholar]
- 95.Chan ADC, Li X, Harrison DJ. Evidence for water-rich surface region in poly(vinylchloride)-based ion-selective electrode membranes. Anal Chem. 1992;64:2512. [Google Scholar]
- 96.Lindfors T, Höfler L, Jágerszki G, Gyurcsányi RBE. Hyphenated FT-IR-attenuated total reflection and electrochemical impedance spectroscopy technique to study the water uptake and potential stability of polymeric solid-contact ion-selective electrodes. Anal Chem. 2011;83:4902. doi: 10.1021/ac200597b. [DOI] [PubMed] [Google Scholar]
- 97.Veder JP, De Marco R, Clarke G, Chester R, Nelson A, Prince K, Pretsch E, Bakkert E. Elimination of undesirable water layers in solid-contact polymeric ion-selective electrodes. Anal Chem. 2008;80:6731. doi: 10.1021/ac800823f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Veder JP, De Marco R, Clarke G, Jiang SP, Prince K, Pretsch E, Bakker E. Water uptake in the hydrophilic poly(3,4-ethylenedioxythiophene):Poly(styrene sulfonate) solid-contact of all-solid-state polymeric ion-selective electrodes. Analyst. 2011;136:3252. doi: 10.1039/c1an15267j. [DOI] [PubMed] [Google Scholar]
- 99.Guo J, Amemiya S. Voltammetric heparin-selective electrode based on thin liquid membrane with conducting polymer-modified solid support. Anal Chem. 2006;78:6893. doi: 10.1021/ac061003i. [DOI] [PubMed] [Google Scholar]
- 100.Lai CZ, Fierke MA, Stein A, Buhlmann P. Ion-selective electrodes with three-dimensionally ordered macroporous carbon as the solid contact. Anal Chem. 2007;79:4621. doi: 10.1021/ac070132b. [DOI] [PubMed] [Google Scholar]
- 101.Fierke MA, Lai CZ, Bühlmann P, Stein A. Effects of architecture and surface chemistry of three-dimensionally ordered macroporous carbon solid contacts on performance of ion-selective electrodes. Anal Chem. 2010;82:680. doi: 10.1021/ac902222n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Michalska A. All-solid-state ion selective and all-solid-state reference electrodes. Electroanalysis. 2012;24:1253. [Google Scholar]
- 103.Sun L, Jiao K, Weber SG. Charge transport through osmium-containing redox polymers in nitrophenyl-based solvents: Effect of solvent size. J Phys Chem B. 1998;102:1945. [Google Scholar]
- 104.Amemiya S, Kim Y, Ishimatsu R, Kabagambe B. Electrochemical heparin sensing at liquid/liquid interfaces and polymeric membranes. Anal Bioanal Chem. 2011;399:571. doi: 10.1007/s00216-010-4056-2. [DOI] [PubMed] [Google Scholar]
- 105.Kim Y, Amemiya S. Stripping analysis of nanomolar perchlorate in drinking water with a voltammetric ion-selective electrode based on thin-layer liquid membrane. Anal Chem. 2008;80:6056. doi: 10.1021/ac8008687. [DOI] [PubMed] [Google Scholar]
- 106.Kim Y, Rodgers PJ, Ishimatsu R, Amemiya S. Subnanomolar ion detection by stripping voltammetry with solid-supported thin liquid membrane. Anal Chem. 2009;81:7262. doi: 10.1021/ac900995a. [DOI] [PubMed] [Google Scholar]
- 107.Langmaier J, Olsak J, Samcova E, Samec Z, Trojanek A. Amperometry of heparin polyion using a rotating disk electrode coated with a plasticized PVC membrane. Electroanalysis. 2006;18:115. [Google Scholar]
- 108.Zhang J, Harris AR, Cattrall RW, Bond AM. Voltammetric ion-selective electrodes for the selective determination of cations and anions. Anal Chem. 2010;82:1624. doi: 10.1021/ac902296r. [DOI] [PubMed] [Google Scholar]
- 109.Pawlak M, Grygolowicz-Pawlak E, Bakker E. Pulsed chronopotentiometric membrane electrodes based on plasticized poly(vinyl chloride) with covalently bound ferrocene functionalities as solid contact transducer. Pure Appl Chem. 2012;84:2045. [Google Scholar]
- 110.Fibbioli M, Bandyopadhyay K, Liu S-G, Echegoyen L, Enger O, Diederich F, Bühlmann P, Pretsch E. Redox-active self-assembled monolayers as novel solid contacts for ion-selective electrodes. Chem Commun. 2000;339 [Google Scholar]
- 111.Fibbioli M, Bandyopadhyay K, Liu SG, Echegoyen L, Enger O, Diederich F, Gingery D, Bühlmann P, Persson H, Suter UW, Pretsch E. Redox-active self-assembled monolayers for solid-contact polymeric membrane ion-selective electrodes. Chem Mater. 2002;14:1721. [Google Scholar]
- 112.Sardar R, Beasley CA, Murray RW. Interfacial ion transfers between a monolayer phase of cationic Au nanoparticles and contacting organic solvent. J Am Chem Soc. 2010;132:2058. doi: 10.1021/ja909584p. [DOI] [PubMed] [Google Scholar]
- 113.Umezawa Y, Bühlmann P, Umezawa K, Tohda K, Amemiya S. Potentiometric selectivity coefficients of ion-selective electrodes part I. Inorganic cations (technical report) Pure Appl Chem. 2000;72:1851. [Google Scholar]
- 114.Shao Y, Osborne MD, Girault HH. Assisted ion transfer at micro-ITIES supported at the tip of micropipettes. J Electroanal Chem. 1991;318:101. [Google Scholar]
- 115.Liu SJ, Li Q, Shao YH. Electrochemistry at micro- and nanoscopic liquid/liquid interfaces. Chem Soc Rev. 2011;40:2236. doi: 10.1039/c0cs00168f. [DOI] [PubMed] [Google Scholar]
- 116.Kakutani T, Nishiwaki Y, Osakai T, Senda M. On the mechanism of transfer of sodium ion across the nitrobenzene/water interface facilitated by dibenzo-18-crown-6. Bull Chem Soc Jpn. 1986;59:781. [Google Scholar]
- 117.Jadhav S, Bakker E. Selectivity behavior and multianalyte detection capability of voltammetric ionophore-based plasticized polymeric membrane sensors. Anal Chem. 2001;73:80. doi: 10.1021/ac000569i. [DOI] [PubMed] [Google Scholar]
- 118.Wang J. Stripping analysis: Principles, instrumentation, and applications. VCH; Deerfield Beach, FL: 1985. [Google Scholar]
- 119.Senda M, Katano H, Kubota Y. Stripping analysis using ion-transfer voltammetry at liquid/liquid interface. Collect Czech Chem Commun. 2001;66:445. [Google Scholar]
- 120.Marecek V, Samec Z. Electrolysis at the interface between two immiscible electrolyte solutions: Determination of acetylcholine by differential pulse stripping voltammetry. Anal Lett. 1981;14:1241. [Google Scholar]
- 121.Ohkouchi T, Kakutani T, Osakai T, Senda M. Voltammetry with an ion-selective microelectrode based on polarizable oil/water interface. Anal Sci. 1991;7:371. [Google Scholar]
- 122.Huang B, Yu BZ, Li PB, Jiang MA, Bi YS, Wu SF. Vitamin-B-1 ion-selective microelectrode based on a liquid-liquid interface at the tip of a micropipette. Anal Chim Acta. 1995;312:329. [Google Scholar]
- 123.Homolka D, Marecek V, Samec Z, Base K, Wendt H. The partition of amines between water and an organic solvent phase. J Electroanal Chem. 1984;163:159. [Google Scholar]
- 124.Marecek V, Samec Z. Electrolysis at the interface between two immiscible electrolyte solutions by means of a hanging electrolyte drop electrode. Anal Chim Acta. 1982;141:65. [Google Scholar]
- 125.Strutwolf J, Scanlon MD, Arrigan DWM. The performance of differential pulse stripping voltammetry at micro-liquid-liquid interface arrays. J Electroanal Chem. 2010;641:7. [Google Scholar]
- 126.Ohtani T, Nishi N, Kakiuchi T. Differential pulse stripping voltammetry of moderately hydrophobic ions based on hydrophobic ionic liquid membranes supported on the ag/agcl electrode. J Electroanal Chem. 2011;656:102. [Google Scholar]
- 127.Katano H, Senda M. Ion-transfer stripping voltammetry of polyoxyethylene alkyl and alkylphenyl ether surfactants at the organic-gel vertical bar water interface and its application to trace analysis. J Electroanal Chem. 2001;496:103. [Google Scholar]
- 128.Collins CJ, Arrigan DWM. Ion-transfer voltammetric determination of the β-blocker propranolol in a physiological matrix at silicon membrane-based liquid|liquid microinterface arrays. Anal Chem. 2009;81:2344. doi: 10.1021/ac802644g. [DOI] [PubMed] [Google Scholar]
- 129.Collins CJ, Lyons C, Strutwolf J, Arrigan DWM. Serum-protein effects on the detection of the β-blocker propranolol by ion-transfer voltammetry at a micro-ITIES array. Talanta. 2010;80:1993. doi: 10.1016/j.talanta.2009.10.060. [DOI] [PubMed] [Google Scholar]
- 130.Marecek V, Samec Z. Determination of calcium, barium and strontium ions by differential pulse stripping voltammetry at a hanging electrolyte drop electrode. Anal Chim Acta. 1983;151:265. [Google Scholar]
- 131.Scanlon MD, Herzog G, Arrigan DWM. Electrochemical detection of oligopeptides at silicon-fabricated micro-liquid|liquid interfaces. Anal Chem. 2008;80:5743. doi: 10.1021/ac800089p. [DOI] [PubMed] [Google Scholar]
- 132.Katano H, Senda M. Stripping voltammetry of mercury(II) and lead(II) ions at liquid/liquid interface. Anal Sci. 1998;14:63. [Google Scholar]
- 133.Sherburn A, Arrigan DWM, Dryfe RAW, Boag NM. Square-wave voltammetric transfer of silver ions across the water|1,2-dichloroethane interface. Electroanalysis. 2004;16:1227. [Google Scholar]
- 134.Lee HJ, Lagger G, Pereira CM, Silva AF, Girault HH. Amperometric tape ion sensors for cadmiurn(II) ion analysis. Talanta. 2009;78:66. doi: 10.1016/j.talanta.2008.10.059. [DOI] [PubMed] [Google Scholar]
- 135.Economou A, Fielden PR. Mercury film electrodes: Developments, trends and potentialities for electroanalysis. Analyst. 2003;128:205. [PubMed] [Google Scholar]
- 136.Guo J, Yuan Y, Amemiya S. Voltammetric detection of heparin at polarized blood plasma/1,2-dichloroethane interfaces. Anal Chem. 2005;77:5711. doi: 10.1021/ac050833d. [DOI] [PubMed] [Google Scholar]
- 137.de Eulate EA, Arrigan DWM. Adsorptive stripping voltammetry of hen-egg-white-lysozyme via adsorption–desorption at an array of liquid–liquid microinterfaces. Anal Chem. 2012;84:2505. doi: 10.1021/ac203249p. [DOI] [PubMed] [Google Scholar]
- 138.Herzog G, Roger A, Sheehan D, Arrigan DWM. Ion-transfer voltammetric behavior of protein digests at liquid|liquid interfaces. Anal Chem. 2010;82:258. doi: 10.1021/ac901909j. [DOI] [PubMed] [Google Scholar]
- 139.Senda M, Katano H, Yamada M. Amperometric ion-selective electrode. Voltammetric theory and analytical applications at high concentration and trace levels. J Electroanal Chem. 1999;468:34. [Google Scholar]
- 140.Katano H, Senda M. Ion-transfer stripping voltammetry of nonionic and anionic surfactants and its application to trace analysis. Anal Sci. 2001;17:i337. [Google Scholar]
- 141. [accessed March 2013];EPA Perchlorate. http://water.epa.gov/drink/contaminants/unregulated/perchlorate.cfm.
- 142.Kabagambe B, Izadyar A, Amemiya S. Stripping voltammetry of nanomolar potassium and ammonium ions using a valinomycin-doped double-polymer electrode. Anal Chem. 2012;84:7979. doi: 10.1021/ac301773w. [DOI] [PubMed] [Google Scholar]
- 143.Qin W, Zwickl T, Pretsch E. Improved detection limits and unbiased selectivity coefficients obtained by using ion-exchange resins in the inner reference solution of ion-selective polymeric membrane electrodes. Anal Chem. 2000;72:3236. doi: 10.1021/ac000155p. [DOI] [PubMed] [Google Scholar]
- 144.Izadyar A, Kim Y, Ward MM, Amemiya S. Double-polymer-modified pencil lead for stripping voltammetry of perchlorate in drinking water. J Chem Edu. 2012;89:1323. [Google Scholar]
- 145.Heller A, Feldman B. Electrochemistry in diabetes management. Acc Chem Res. 2010;43:963. doi: 10.1021/ar9002015. [DOI] [PubMed] [Google Scholar]
- 146.Wightman RM. Probing cellular chemistry in biological systems with microelectrodes. Science. 2006;311:1570. doi: 10.1126/science.1120027. [DOI] [PubMed] [Google Scholar]
- 147.Robinson DL, Hermans A, Seipel AT, Wightman RM. Monitoring rapid chemical communication in the brain. Chem Rev. 2008;108:2554. doi: 10.1021/cr068081q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cui R, Li Q, Gross DE, Meng X, Li B, Marquez M, Yang R, Sessler JL, Shao Y. Anion transfer at a micro-water/1,2-dichloroethane interface facilitated by β-octafluoro-meso-octamethylcalix[4]pyrrole. J Am Chem Soc. 2008;130:14364. doi: 10.1021/ja804631p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Rodgers PJ, Jing P, Kim Y, Amemiya S. Electrochemical recognition of synthetic heparin mimetic at liquid/liquid microinterfaces. J Am Chem Soc. 2008;130:7436. doi: 10.1021/ja800568q. [DOI] [PubMed] [Google Scholar]
- 150.Jing P, Kim Y, Amemiya S. Voltammetric extraction of heparin and low-molecular-weight heparin across 1,2-dichloroethane/water interfaces. Langmuir. 2009;25:13653. doi: 10.1021/la902336w. [DOI] [PubMed] [Google Scholar]