

Abstract
DNA and poly(N-isopropylacrylamide) are co-assembled onto gold nanoparticles. The DNA sequences can be reversibly exposed or hidden from the polymer surface in response to temperature cues, thereby translating the temperature trigger to the on-off switching of the surface chemistry and function. When exposed by heating (~30 °C), the DNA rapidly hybridizes to complementary strands, and chain-end biotin groups become readily accessible, while at lower temperatures these activities are largely blocked.
Chemically dynamic nanoparticle systems are of great interest. These systems are capable of switching between at least two distinct structural or chemical states,1-4 which allow them to possess unusual properties such as on-demand activation5,6 and autonomous regulation.7 Indeed, many systems are being gradually realized in a number of fields spanning biomedical research,8-11 sensing,12 self-healing materials,13 and energy research,14 and can incorporate sensitivity to a variety of environmental cues, such as temperature,15 pH,16-18 light,19 redox potential,20,21 the presence or absence of small molecules,22 and mechanical stress.23 Poly(N-isoproylacrylamide) (pNIPAM) often has been utilized as a temperature-responsive material, due to its ability to undergo a reversible lower critical solution temperature (LCST) phase transition from a swollen, hydrated state to a shrunken, hydrophobic state, during which the polymer loses 90% of its volume.24 Because the LCST can be engineered through copolymerization to be near physiological temperature, pNIPAM and its derivatives have often been employed in tissue engineering and controlled drug release applications.25-27
It has been previously shown that spherical nucleic acid-gold nanoparticles (AuNPs) conjugates (termed SNAs)28 can undergo rapid endocytosis, which is facilitated by engaging with membrane-bound scavenger proteins.29-31 This cell entry mechanism appears to be general with respect to many cell types (almost all cells internalize SNAs, albeit with different rates and to different extents), and cannot effectively distinguish many diseased tissues from healthy ones.32,33 In order to achieve selective cell entry, one strategy would be to mask the DNA temporarily until an environmental trigger is applied at the region of interest. In this study, we report the synthesis of a dynamic nanoparticle system consisting of pNIPAM and DNA co-assembled onto AuNPs, and demonstrate that access to DNA is significantly retarded at temperatures below the LCST of pNIPAM. Above this temperature, PNIPAM becomes dehydrated, revealing the DNA for rapid binding with complementary strands (Scheme 1). The chain end of DNA is also exposed, as demonstrated with biotin-terminated DNA, which binds to streptavidin only at elevated temperatures. Therefore, the current study potentially paves the way for novel nucleic acid structures capable of being reversibly activated by temperature for selective and/or localized cell entry.
Scheme 1.
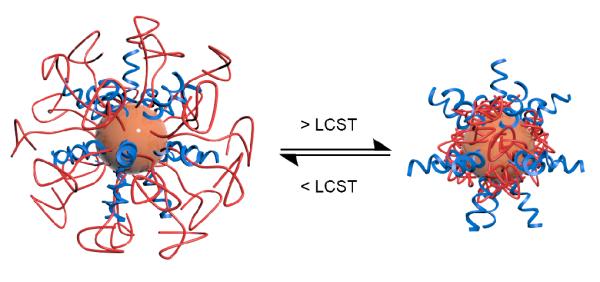
Schematic drawing of PNIPAM- and DNA-functionalized gold nanoparticles (AuNP) and response to temperature. Increasing solution temperature above the polymer LCST results in a reduction of polymer hydration and an increase in DNA accessibility.
To realize such temperature-activated DNA nanostructures, several design parameters must be established. First, the AuNP surface should have appropriate pNIPAM coverage such that all DNA molecules are adequately masked at temperatures below the LCST. Similarly, the pNIPAM should have a molecular weight high enough to offer sufficient blockage of the DNA. However, the pNIPAM molecular weight cannot be too high as to dominate the surface while in a dehydrated state above LCST, in which case hydrophobicity-induced AuNP aggregation is expected. To estimate these parameters, a molecular dynamics simulation has been implemented (see Supporting Information, Figure S1). The simulation suggested that a combination of a 10 kDa pNIPAM and a 12-mer DNA at 1:1 DNA:polymer molar ratio would be feasible.
To prepare the target structures, we first synthesized two DNA strands whose 3′ or 5′ terminus are modified with propyl thiol groups (sequence: 5′-GAG GGT AAG GAG-SH-3′ and 5′-HS-GGA AAG GTT AGT-3′). Each of the sequences was mixed with 10 kDa α-thiol ω-COOH pNIPAM (PDI: 1.1) at various DNA:polymer molar ratios (1:2, 1:1, 5:1) in Nanopure™ water. A longer pNIPAM (30 kDa, a PDI of 1.2) was used as a control. Immediately prior to mixing, the polymers and DNA strands were treated with dithiolthreitol (DTT) and dialyzed or passed through a size-exclusion column to ensure that free thiol groups were present. Thereafter, the mixture containing the thiol-modified ligands was added to citrate-stabilized AuNPs (10 nM, 13 nm, synthesized by a modified Frens-Turkevich method)34,35 with 0.01% TWEEN 20 to give a total ligand concentration of 5 μM. Over 24 h, sodium chloride was added, and its concentration was gradually brought up to 0.5 M, a process necessary for charged species (i.e. DNA) to assemble in a densely packed fashion on the AuNP surface.36 Thereafter, successive centrifugation-resuspension steps were used to remove unbound DNA and pNIPAM. Finally, AuNPs were suspended in phosphate buffered saline with 0.01% TWEEN 20 (PBST) at a particle concentration of 10 nM. Note that the DNA:polymer feed ratios do not equal their actual ratios on the nanoparticles. Using fluorophore-labeled pNIPAM (Supporting Information), we determined the actual DNA:polymer molar ratios and the total number of ligands (DNA and pNIPAM) present on the AuNPs surface (Table 1).
Table 1.
DNA:polymer molar ratio on AuNP surface.
| Feed molar ratios | 1:2 | 1:1 | 5:1 | |
|---|---|---|---|---|
| Actual ratios (Total ligand per particle) |
30 kDa | 1.4:1 (154) | 4.2:1 (112) | ~40:1 (111) |
|
| ||||
| 10 kDa | 1.1:1 (193) | 2.9:1 (164) | ~20:1 (139) | |
Once the DNA- and pNIPAM-functionalized AuNPs were prepared and purified, we examined whether they would aggregate at temperatures above the LCST due to hydrophobic interactions, or remain stable in solution. For AuNPs with 30 kDa pNIPAM (DNA:polymer 1.4:1), visible aggregation can be observed within seconds when the solution temperature is heated above 30 °C. UV-Vis spectroscopy monitoring absorption at 524 nm (AuNP plasmon resonance maximum)37 as the temperature was ramped up at 0.5 °C/min shows complete aggregation within ~1 °C (Figure 1), indicating that the AuNP surface is predominantly hydrophobic when the LCST is reached. In comparison, AuNPs with 10 kDa pNIPAM (DNA:polymer 1.1:1) only show slight reduction in absorption (5%) when heated to 50 °C, suggesting that the surface composition had switched from pNIPAM to predominantly DNA, which stabilizes the AuNPs in solution. The hydrodynamic diameter initially decreased from 21.0 ± 2.6 nm to 16.3 ± 0.8 nm as measured by dynamic light scattering (DLS) when the temperature was increased from 25 to 40 °C, consistent with pNIPAM shrinkage (Figure S2). However, after 1 h incubation at 50 °C, particle size increased to 44 nm, suggesting that a small degree of aggregation had occurred.
Figure 1.

UV-Vis measurement at 524 nm for pNIPAM-DNA AuNPs as a function of temperature. High molecular weight pNIPAM (30 kDa) leads to AuNP aggregation above LCST even for particles with low polymer content, while particles having 10 kDa pNIPAM remain stable.
To verify that the DNA is available for binding with complementary sequence once pNIPAM is dehydrated, we mixed two particles (with 10kDa pNIPAM, DNA:polymer 1.4:1) and a common complementary linker strand (sequence: 5′-CTC CTT ACC CTC ACT AAC CTT TCC-3′) in PBST buffer, and monitored AuNP absorption at 524 nm by UV-Vis spectroscopy as the temperature was increased from 25 to 80 °C at 0.5 °C/min. At temperatures below 30 °C, absorption values stay largely unchanged, suggesting effective blockage of DNA hybridization (Figure 2). As the temperature is increased, a rapid drop in absorption is observed at ~30 °C due to particle assembly, which can result from either hydrophobic interactions of pNIPAM, or DNA hybridization. As the temperature continued to increase, a sharp rise in absorption occurred at 53-55 °C, returning absorption values to nearly that before assembly. The second transition is attributed to DNA melting, which indicates that the initial aggregation is due to DNA hybridization. In contrast, DNA-AuNP conjugates without pNIPAM show immediate aggregation upon mixing, and only exhibit the DNA melting transition, and particles with pNIPAM but matched with a non-complementary linker show neither the aggregation nor the melting transition. These data indicate that the DNA strands can be blocked from hybridization below the LCST and become exposed for hybridization in response to temperature.
Figure 2.

A) A divalent linker DNA strand hybridizes with DNA-AuNPs when the temperature is raised above the LCST of pNIPAM, causing AuNP assembly. A further increase in temperature leads to the melting of double-stranded DNA and re-dispersion of the aggregates. B) UV-Vis temperature scan is performed at 0.5 °C /min, monitoring AuNP absorption at 524 nm.
To further validate that the terminus of the DNA chain is hindered from accessing the surface at lower temperatures and exposed upon heating, we designed and performed a biotin-streptavidin pull-down assay. Briefly, we synthesized a DNA sequence with 3′ biotin and 5′ propyl thiol modifications (sequence: 5′-SH-GGA AAG GTT AGT-biotin-3′). The biotin group is linked with the DNA through a triethylene glycol spacer, which minimizes the steric hindrance between the biotin and the oligonucleotide.38 It was co-assembled on the AuNP surface with pNIPAM (10 kDa) at DNA:polymer feed molar ratios ranging from 1:5 to 5:1. A DNA strand without the biotin is co-assembled with pNIPAM at 1:1 feed molar ratio and used as a control. Thereafter, the modified AuNPs were mixed with Dynabeads® coupled with streptavidin in the presence of 0.01% TWEEN 20, and incubated at either room temperature or at 40 °C. Following 5 min incubation, the beads were pulled down by a neodymium magnet, and the supernatant was analyzed for absorption at 524 nm by UV-Vis spectroscopy. If the biotin group at the chain end of the DNA is accessible, it leads to AuNP capture by the Dynabeads® and a drop in optical absorption of the supernatant. When incubated at room temperature, we found that significant amounts of particles (>85% relative to non-treated AuNPs) remained in solution for particles with DNA:polymer feed ratios of 1:5 or 1:2 (Figure 3). When a 1:1 feed ratio was used, ~60% AuNPs were bound to streptavidin. Further decreases in pNIPAM content (at 2:1 or 5:1) led to high degrees of capture by the Dynabeads® (>90%), consistent with our hypothesis that sufficient pNIPAM coverage is important for DNA blockage. On the other hand, when incubated at 40 °C, all particles containing biotin are almost completely captured, with <3% remaining in solution even for particles with low biotin-DNA contents, indicating that the DNA 3′ termini are exposed. A control involving non-biotinylated AuNP was not captured under otherwise same conditions, ruling out the possibility of non-specific binding at 40 °C. These data suggest that elevation of temperature is highly efficient in exposing DNA molecules that are blocked by the pNIPAM in its hydrated state.
Figure 3.

A) A magnetic pull-down assay for determining if DNA chain ends can be hidden/revealed by 10 kDa pNIPAM. B) UV-Vis measurements of NP solutions at 524 nm after incubation with streptavidin-coated Dynabeads® at 25 °C and 40 °C (bead removed using a neodymium magnet).
In summary, we have demonstrated a strategy to allow the surface chemistry of AuNPs to interchange between synthetic polymers and nucleic acids in response to a temperature trigger. This dynamic system allows DNA accessibility only above the LCST of pNIPAM, and therefore points to the possibility of creating novel nucleic acid constructs capable of selective activation in the vicinity of specific materials and tissues. Importantly, the general co-assembly approach to create “smart” systems reported herein can be extended to a number of biomolecules, such as peptides, small molecules targeting moieties, and proteins.
Supplementary Material
ACKNOWLEDGMENT
This material is based upon work supported by Northeastern University start-up fund, and the Northwestern University Center for Cancer Nanotechnology Excellence (CCNE) initiative of the National Institutes of Health (NIH) under Award No. U54 CA151880.
Footnotes
Supporting Information Experimental procedures, DNA sequences, additional characterization data, and details of molecular dynamics simulation. This materials is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
REFERENCES
- (1).Randolph LM, Chien MP, Gianneschi NC. Chem. Sci. 2012;3:1363. doi: 10.1039/C2SC00857B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Chien MP, Rush AM, Thompson MP, Gianneschi NC. Angew. Chem. Int. Ed. 2010;49:5076. doi: 10.1002/anie.201000265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Lee Y, Miyata K, Oba M, Ishii T, Fukushima S, Han M, Koyama H, Nishiyama N, Kataoka K. Angew. Chem. Int. Ed. 2008;47:5163. doi: 10.1002/anie.200800963. [DOI] [PubMed] [Google Scholar]
- (4).Spruell JM, Hawker CJ. Chem. Sci. 2011;2:18. [Google Scholar]
- (5).Kong SD, Zhang WZ, Lee JH, Brammer K, Lal R, Karin M, Jin SH. Nano Lett. 2010;10:5088. doi: 10.1021/nl1033733. [DOI] [PubMed] [Google Scholar]
- (6).Xiong MH, Bao Y, Yang XZ, Wang YC, Sun BL, Wang J. J. Am. Chem. Soc. 2012;134:4355. doi: 10.1021/ja211279u. [DOI] [PubMed] [Google Scholar]
- (7).He XM, Aizenberg M, Kuksenok O, Zarzar LD, Shastri A, Balazs AC, Aizenberg J. Nature. 2012;487:214. doi: 10.1038/nature11223. [DOI] [PubMed] [Google Scholar]
- (8).Wright ER, Conticello VP. Adv. Drug. Deliver. Rev. 2002;54:1057. doi: 10.1016/s0169-409x(02)00059-5. [DOI] [PubMed] [Google Scholar]
- (9).Hu SH, Chen SY, Gao XH. ACS Nano. 2012;6:2558. doi: 10.1021/nn205023w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wang YP, Byrne JD, Napier ME, DeSimone JM. Adv. Drug Deliver. Rev. 2012;64:1021. doi: 10.1016/j.addr.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Azzaroni O. J. Polym. Sci. Part A: Polym. Chem. 2012;50:3225. [Google Scholar]
- (12).Yan Q, Zhou R, Fu CK, Zhang HJ, Yin YW, Yuan JY. Angew. Chem. Int. Ed. 2011;50:4923. doi: 10.1002/anie.201100708. [DOI] [PubMed] [Google Scholar]
- (13).Chen Y, Guan Z. Polym. Chem. 2013;4:4885. [Google Scholar]
- (14).Guo XF, Xiao SX, Myers M, Miao Q, Steigerwald ML, Nuckolls C. Proc. Natl. Acad. Sci. U. S. A. 2009;106:691. doi: 10.1073/pnas.0807596106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Qian XM, Li J, Nie SM. J. Am. Chem. Soc. 2009;131:7540. doi: 10.1021/ja902226z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sahoo B, Devi KSP, Banerjee R, Maiti TK, Pramanik P, Dhara D. ACS Appl. Mater. Inter. 2013;5:3884. doi: 10.1021/am400572b. [DOI] [PubMed] [Google Scholar]
- (17).He QJ, Gao Y, Zhang LX, Zhang ZW, Gao F, Ji XF, Li YP, Shi JL. Biomaterials. 2011;32:7711. doi: 10.1016/j.biomaterials.2011.06.066. [DOI] [PubMed] [Google Scholar]
- (18).Wilson JT, Keller S, Manganiello MJ, Cheng C, Lee CC, Opara C, Convertine A, Stayton PS. ACS Nano. 2013;7:3912. doi: 10.1021/nn305466z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Angelatos AS, Radt B, Caruso F. J. Phys. Chem. B. 2005;109:3071. doi: 10.1021/jp045070x. [DOI] [PubMed] [Google Scholar]
- (20).Lee MH, Yang Z, Lim CW, Lee YH, Dongbang S, Kang C, Kim JS. Chem. Rev. 2013;113:5071. doi: 10.1021/cr300358b. [DOI] [PubMed] [Google Scholar]
- (21).Dunn SS, Tian SM, Blake S, Wang J, Galloway AL, Murphy A, Pohlhaus PD, Rolland JP, Napier ME, DeSimone JM. J. Am. Chem. Soc. 2012;134:7423. doi: 10.1021/ja300174v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wu Q, Wang L, Yu HJ, Wang JJ, Chen ZF. Chem. Rev. 2011;111:7855. doi: 10.1021/cr200027j. [DOI] [PubMed] [Google Scholar]
- (23).Kolmakov GV, Revanur R, Tangirala R, Emrick T, Russell TP, Crosby AJ, Balazs AC. ACS Nano. 2010;4:1115. doi: 10.1021/nn901296y. [DOI] [PubMed] [Google Scholar]
- (24).Schild HG. Prog. Polym. Sci. 1992;17:163. [Google Scholar]
- (25).Guan Y, Zhang YJ. Soft Matter. 2011;7:6375. [Google Scholar]
- (26).Yavuz MS, Cheng YY, Chen JY, Cobley CM, Zhang Q, Rycenga M, Xie JW, Kim C, Song KH, Schwartz AG, Wang LHV, Xia YN. Nat. Mater. 2009;8:935. doi: 10.1038/nmat2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ohya S, Nakayama Y, Matsuda T. Biomacromolecules. 2001;2:856. doi: 10.1021/bm010040a. [DOI] [PubMed] [Google Scholar]
- (28).Cutler JI, Auyeung E, Mirkin CA. J. Am. Chem. Soc. 2012;134:1376. doi: 10.1021/ja209351u. [DOI] [PubMed] [Google Scholar]
- (29).Rosi NL, Giljohann DA, Thaxton CS, Lytton-Jean AK, Han MS, Mirkin CA. Science. 2006;312:1027. doi: 10.1126/science.1125559. [DOI] [PubMed] [Google Scholar]
- (30).Giljohann DA, Seferos DS, Prigodich AE, Patel PC, Mirkin CA. J. Am. Chem. Soc. 2009;131:2072. doi: 10.1021/ja808719p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Rosi NL, Giljohann DA, Thaxton CS, JLytton-ean AKR, Han MS, Mirkin CA. Science. 2006;312:1027. doi: 10.1126/science.1125559. [DOI] [PubMed] [Google Scholar]
- (32).Zhang K, Hao LL, Hurst SJ, Mirkin CA. J. Am. Chem. Soc. 2012;134:16488. doi: 10.1021/ja306854d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Agbasi-Porter C, Ryman-Rasmussen J, Franzen S, Feldheim D. Bioconjugate Chem. 2006;17:1178. doi: 10.1021/bc060100f. [DOI] [PubMed] [Google Scholar]
- (34).Frens G. Colloid Polym. Sci. 1972;250:736. [Google Scholar]
- (35).Turkevich J, Stevenson PC, Hillier J. Discuss. Faraday Soc. 1951;11:55. [Google Scholar]
- (36).Hill HD, Mirkin CA. Nat. Protoc. 2006;1:324. doi: 10.1038/nprot.2006.51. [DOI] [PubMed] [Google Scholar]
- (37).Daniel MC, Astruc D. Chem. Rev. 2004;104:293. doi: 10.1021/cr030698+. [DOI] [PubMed] [Google Scholar]
- (38).De Greef TFA, Kade MJ, Feldman KE, Kramer E, Hawker CJ, Meijer EW. J. Polym. Sci. Part A: Polym. Chem. 2011;49:4253. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.