

Abstract
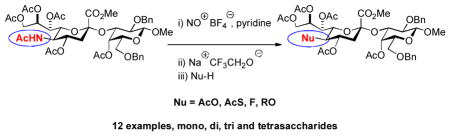
Nitrosation of peracetylated NeuAc glycosides followed by treatment with sodium trifluoroethoxide and then a suitable nucleophile enables late stage modification of sialic acid glycosides with stereospecific replacement of the acetamido functionality.
Keywords: sialic acids, deamination, diversification, glycosylation
The sialic acids are nine carbon sugars based on the 2-keto-3-deoxy-D-glycero-D-galactononulosonic acids whose α-glycosides adorn the non-reducing termini of many N-glycoproteins and gangliosides and are the monomeric units of the α-(2→8)- and α-(2→9)-oligosialic acids.[1] The recognition of these sialyl glycoconjugates by various lectins, and the trimming of sialyl residues by sialidase enzymes, play important roles in many human disease states and have stimulated interest in the synthesis of libraries of modified sialyl glycoconjugates and their deployment in the search for improved diagnostics and therapeutics.[1–2] The difficulties inherent in sialic acid chemistry and particularly in the stereocontrolled synthesis of α-sialosides[3] have, however, restricted the preparation of such libraries to enzymatic approaches which are limited by the range of substrates accepted.[4] We describe here a method for the chemical synthesis of sialyl glycoside libraries that combines recent progress in stereocontrolled α-sialoside synthesis[5] with a mild oxidative deamination process to enable late-stage modification of pre-assembled glycosides, thereby extending the range of accessible diversity.
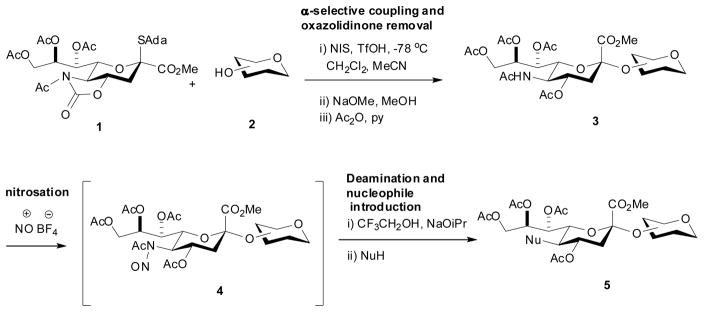
Focused libraries of specific classes of oligosaccharides and/or glycoconjugates are arguably best accessed by the late stage modification of pre-assembled substances combining synthetic efficiency with the ability to introduce targeted diversity with the minimum of synthetic effort. This strategy, which differs from the current enzymatic cascade approach to sialyl glycoside libraries which demands synthesis of a different precursor for every eventual member,[4] requires reliable, robust methodology for glycoside synthesis and mild efficient methodology for their subsequent modification. The introduction of the 4-O,5-N-oxazoldinone-protected sialyl donors and their more readily deprotected N-acetyl derivatives has removed many of the obstacles to chemical α-sialoside synthesis (Scheme 1, line 1),[5] thereby opening the door to the modification of sialyl glycosides as a means of entry into libraries, provided suitably mild chemistry for subsequent modification can be identified. We reasoned that the modified Schreiner and Zbiral oxidative deamination of neuraminic acid glycosides would be a suitable reaction for such modifications if conditions could be found to extend the range of nucleophiles beyond the acetic and and hydrazoic acid-based systems previously employed (Scheme 1, line 2).[6]
Scheme 1.
Chemical synthesis of sialoside libraries with late stage modification of preassembled glycosides.
Oxidative deamination of peractyl N-acetylneuraminic acid (NeuAc) methyl ester was achieved by Schreiner and Zbiral with nitrosyl acetate giving the N-nitroso adduct,[6a] whereas in our laboratory we prefer the more convenient, commercial nitrosyl tetrafluoroborate for this purpose.[6b] Subsequent steps involve selective removal of the acetyl group from the N-nitrosoacetamide with sodium trifluoroethoxide to give a diazo derivative of NeuAc that is then substituted by the incoming nucleophile. Participation by the 4-O-acetate is invoked to explain both the regio- and stereoselectivity of the process.[6a] As nitrosyl tetrafluoroborate is known to activate disarmed thioglycosides toward glycosylation,[7] we tested the compatibility of the oxidative deamination thioglycosides. In the event, treatment of a dichloromethane solution of thioglycoside 6 in the presence of pyridine with NOBF4 at −10 °C gave a solution of the presumed N-nitroso derivative 7 that was briefly exposed to trifluoroethanol (TFE) and sodium isopropoxide in isopropanol at the same temperature before addition of either acetic acid, thioacetic acid, or the hydrogen fluoride-pyridine complex resulting in the formation of the 5-desacetamido acetoxy, acetylthio, and fluoro NeuAc derivatives 8, 9, and 10, respectively, in good yield. albeit the fluoride 10 was accompanied by 11% of the isopropoxy analogue 11 (Scheme 2).[8] The survival of the thioglycoside moiety is due to the use of pyridine, which captures the nitrosonium ion as the N-nitrosopyridinium complex[9] 12 (Scheme 2) and moderates its reactivity. While the formation of the 2-keto-3-deoxy-D-glycero-D-galactononulosonic acid (KDN) derivative 8 recalls the earlier studies[6] with acetic acid as nucleophile, the formation of the acetylthio and fluoro derivatives demonstrate the ability to incorporate a wider range of nucleophiles. The formation of the isopropyl ether 11 as byproduct alongside fluoride 10 could be avoided by using preformed sodium trifluoroethoxide as base in conjunction with 18-crown-6 and excluding isopropanol from the reaction mixture, when 63% of the fluoride 10 was obtained. The isolation of 11 as byproduct also suggested that conditions for the use of simple alcohols as nucleophiles in the oxidative deamination protocol. Thus, after some experimentation it was found that deacetylation with sodium trifluoroethoxide, followed by the addition of propargyl alcohol and tetrafluoroboric acid, to protonate the intermediate diazo compound, yielded the propargyl ether 13 in 66% yield (Scheme 2). Next a series of α-sialosides was prepared from the donor 1 on activation with NIS/TfOH in a mixture of dichloromethane and acetonitrile at −78 °C[5c] as reported in Table 1, entries 1–5. Alternatively, for more complex acceptors based on the thioglycoside motif the Wong sialyl phosphate type donor[5e] 14 was employed with activation by trimethylsilyl trifluoromethanesulfonate at −78 °C in dichloromethane and acetonitrile (Table 1, entries 6 and 7). In each case the oxazolidinone then was removed under Zemplen conditions and any acetate esters reinstalled with acetic anhydride and pyridine (Scheme 1, and Table 1). Nitrosylation was achieved with nitrosyl tetrafluoroborate in the presence of pyridine and the intermediate N-nitrosoamides substituted under the conditions reported in Table 1.
Scheme 2.
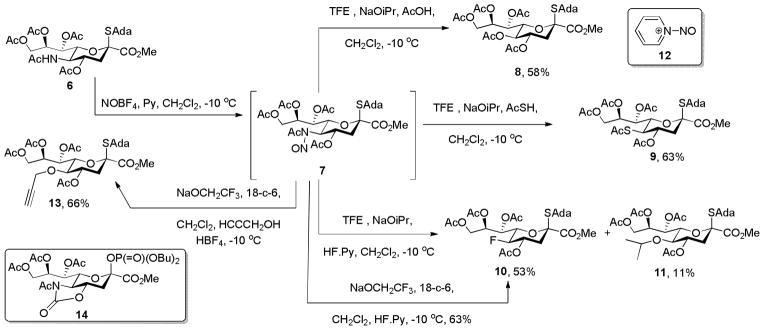
Compatibility with a thioglycoside and introduction of acetoxy, acetylthio, and fluoride groups.
Table 1.
Stereoselective α-sialoside synthesis and oxidative deamination.
| Glycosylationa | Oxazolidinonea Removal | Nucleophile, conditions | Substitution Productsa | |
|---|---|---|---|---|
| 1 |
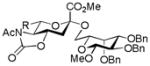 15, 81%, α- only from 1 |
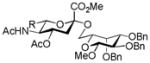 20, 77% |
AcOH, TFE, NaOiPr |
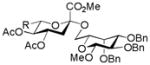 25, 51% |
| 2 |
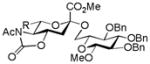 16, 84%, α- only from 1 |
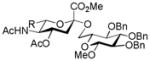 21, 88% |
AcOH, TFE, NaOiPr |
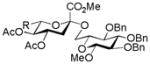 26, 48% |
| 3 |
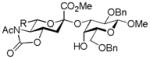 17, 83%, 10:1 β:α, from 1 |
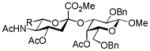 22, 76% |
AcOH, TFE, NaOiPr |
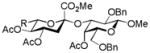 27, 54% |
| 4 | 17 | 22 | AcSH, TFE, NaOiPr |
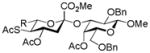 28, 67% |
| 5 | 17 | 22 | HF.NEt3, NaTFE, 18-c-6, |
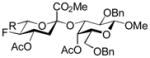 29, 63% |
| 6 |
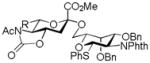 18, 86%, α- only, from 14 |
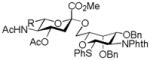 23, 91% |
AcOH, TFE, NaOiPr |
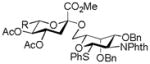 30, 49% |
| 7 |
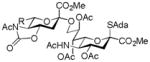 19, -b, from 14 |
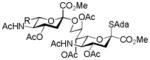 24, 74%, α- onlyc |
AcOH, Bu4NOAc, TFE, NaOiPr |
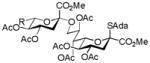 31, 42% |
R = CH(OAc)CH(OAc)CH2OAc;
not isolated;
overall yield from 14.
Entries 1 – 3 of Table 1 illustrate the application of the protocol to the synthesis of KDN glycosides incorporating α(1→6)-galactopyranoside, α(1→6)-glucopyranoside, and α(1→3)-galactopyranoside linkages and demonstrate the compatibility of the protocol with glycosidic bonds and with benzyl ethers. Entry 4 of Table 1 employs thioacetic acid as nucleophile and provides the novel disaccharide 28, which may be viewed either as a 5-acetylthio-5-desacetamido derivative of a NeuAc glycoside or as a 5-acetylthio-5-deoxy analogue of a KDN glycoside. As selective deacetylation of thioacetates in the presence of acetates is facile, thioacetate derivatives such as 28 enable further regioselective diversification at the 5-position by alkylation of thiols and disulfide formation, or by the thiol-ene and yne click[10] and other processes.[11] As 5-mercapto analogues of NeuAc and/or KDN have not previously been accessible through the use of sialyl transferases,[4b, 4c] this example extends the range of accessible α-sialoside derivatives modified at the 5-position and demonstrates the versatility of the chemical approach. Entry 5 (Table 1) illustrates the formation of a 5-fluoro derivative of NeuAc and/or KDN. Entries 6 and 7 (Table 1) reiterate the applicability of the method to thioglycoside containing substrates, whether of the arylthio or alkylthio classes. Table 1, entry 6 introduces the possibility of diversification at the 5-position of the sTn antigen, and is especially noteworthy as it has been previously demonstrated that modification of this antigen at the amide can lead to analogues that display improved antigenicity.[12] Entry 7 (Table 1) demonstrates how the oxidative deamination process may be applied concomitantly to two sialic acid residues in an α(2→9)-linked disaccharide and represents the first chemical synthesis of a KDN disaccharide of this class. Such modifications of polysialic acids are of interest in view of recent approaches to α(2→9)-linked polysialic acids because of their potential as antibacterial agents and synthetic vaccines.[13]
Application of the oxidative deamination protocol to a GM3 trisaccharide and a branched tetrasaccharide is demonstrated beginning from trisaccharide 32, acetylation of which gave the derivative 33, while glucosylation afforded the tetrasaccharide 34 (Scheme 3). Both substances were treated with nitrosyl tetrafluoroborate and pyridine and then exposed to sodium trifluoroethoxide followed by acetic acid giving the KDN analogues 35 and 36, respectively, in good yield.
Scheme 3.

Application of the oxidative deamination reaction to a GM3 trisaccharide and a branched tetrasaccharide.
The combination of N-acetyloxazolidinone-directed α-sialidation with oxidative deamination provides rapid entry into NeuAc and/or KDN analogues modified at the 5-position. Suitable nucleophiles for this process include acetate, thioacetate, fluoride and simple alcohols. The ability to chemically synthesize sialic acid glycosides in this manner and then affect late stage modification is potentially very flexible and should enable access to many derivatives with a minimum of synthetic effort. Modifications are not limited to those incorporated in substrates for sialyl transferases.
Experimental Section
General procedure for nitrosation of sialosides
A stirred 0.1 M solution of sialoside in anhydrous CH2Cl2 under Ar was treated with anhydrous pyridine (10 equiv) and cooled to −10 °C. After stirring for 10 min, powdered nitrosyl tetrafluoroborate (4 equiv) was added in one portion to the mixture. The resultant light green solution was stirred at −10 °C for 3 h then was diluted with CH2Cl2 and washed sequentially with cold 1N HCl, cold saturated aqueous NaHCO3, and brine. Drying (Na2SO4) and concentration below 10 °C gave the nitrosated sialosides which were carried forward without further purification.
General procedure for deamination with acetic or thioacetic acid as nucleophile
A 0.1 M solution of the nitrosyl sialoside and trifluoroethanol (1.5 equiv) in anhydrous CH2Cl2 under Ar was treated at −10 °C with freshly prepared 0.2 N sodium isopropoxide in isopropanol (1.2 equiv). The resulting mixture was stirred for exactly 2 min then was treated with a cold solution of glacial acetic acid (20 equiv) or thioacetic acid (20 equiv) in CH2Cl2 (0.1M). After stirring for 5 min the reaction mixture was warmed to 0 °C and was quenched with saturated aqueous NaHCO3. The organic layer was washed with cold brine, dried (Na2SO4), concentrated and chromatographed over silica gel to afford the deaminated sialosides.
General procedure for deamination with HF and alcohol nucleophiles
18-Crown-6 (0.22 mmol) and sodium 2,2,2-trifluoroethoxide (54 mg, 0.2 mmol) were dissolved in anhydrous CH2Cl2 (0.5 mL) under Ar, cooled to −10 °C, and added to the nitrosyl sialoside (0.1 mmol) in anhydrous CH2Cl2 (1 mL) at −10 °C under Ar. After 2 min, either the alcohol (2 mmol in 2 mL CH2Cl2) immediately followed by HBF4.Et2O (0.4 mmol) or HF.pyridine (2 mmol) was added to the reddish reaction mixture. The mixture was stirred for 5 min, diluted with CH2Cl2 (5 mL) then quenched with saturated aqueous NaHCO3 (5 mL). The organic layer was washed with brine (5 mL), dried, concentrated and purified by chromatography over silica gel to afford the deaminated sialosides.
Supplementary Material
Acknowledgments
We thank the NIH (GM 62160) for support of this work, and Dr. P. K. Kancharla for experimental assistance.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author
References
- 1.Miyagi T, Yamaguchi K. In: Comprehensive Glycoscience. Kamerling JP, editor. Vol. 3. Elsevier; Amsterdam: 2007. pp. 297–323. [Google Scholar]
- 2.a) Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Essentials of Glycobiology. 2. Cold Spring Harbor Lab Press; Cold Spring Harbor: 2009. p. 784. [PubMed] [Google Scholar]; b) Gabius HJ, editor. Fundamentals of Glycosciences. Wiley-VCH; Weinheim: 2009. The Sugar Code; p. 569. [Google Scholar]
- 3.a) De Meo C, Boons GJ, Demchenko AV. In: Comprehensive Glycoscience. Kamerling J, editor. Vol. 1. Elsevier; Amsterdam: 2007. pp. 583–604. [Google Scholar]; b) De Meo C, Priyadarshani U. Carbohydr Res. 2008;343:1540–1552. doi: 10.1016/j.carres.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 4.a) Rillahan CD, Schwartz E, McBride R, Fokin VV, Paulson JC. Angew Chem Int Ed. 2012;51:11014–11018. doi: 10.1002/anie.201205831. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sugiarto G, Lau K, Qu J, Li Y, Lim S, Mu S, Ames JB, Fisher AJ, Chen X. ACS Chem Biol. 2012;7:1232–1240. doi: 10.1021/cb300125k. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Song X, Yu H, Chen X, Lasanajak Y, Tappert MM, Air GM, Tiwari VK, Cao H, Chokhawala HA, Zheng H, Cummings RD, Smith DF. J Biol Chem. 2011;286:31610–31622. doi: 10.1074/jbc.M111.274217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Tanaka H, Nishiura Y, Takahashi T. J Am Chem Soc. 2006;128:7124–7125. doi: 10.1021/ja0613613. [DOI] [PubMed] [Google Scholar]; b) Crich D, Li W. J Org Chem. 2007;72:2387–2391. doi: 10.1021/jo062431r. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Crich D, Li W. J Org Chem. 2007;72:7794–7797. doi: 10.1021/jo7012912. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Farris MD, De Meo C. Tetrahedron Lett. 2007;48:1225–1227. [Google Scholar]; e) Hsu CH, Chu KC, Lin YS, Han JL, Peng YS, Ren CT, Wu CY, Wong CH. Chem Eur J. 2010;16:1754–1760. doi: 10.1002/chem.200903035. [DOI] [PubMed] [Google Scholar]
- 6.a) Schreiner E, Zbiral E. Liebigs Annalen der Chemie. 1990:581–586. [Google Scholar]; b) Crich D, Navuluri C. Angew Chem Int Ed. 2010;49:3049–3052. doi: 10.1002/anie.200907178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pozsgay V, JHJ J Org Chem. 1987;52:4635. [Google Scholar]
- 8.The stereochemistry at C5 of all deaminated compounds follows from the two trans-diaxial couplings to H5. See the supporting information.
- 9.Olah GA, Olah JA, Overchuk NA. J Org Chem. 1965;30:3373–3376. [Google Scholar]
- 10.Dondoni A. Angew Chem Int Ed. 2008;47:8995–8997. doi: 10.1002/anie.200802516. [DOI] [PubMed] [Google Scholar]
- 11.Subramanian V, Moumé-Pymbock M, Hu T, Crich D. J Org Chem. 2011;76:3691–3709. doi: 10.1021/jo102411j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu JC, Guo Z. Bioconjug J. 2006;17:1537–1544. doi: 10.1021/bc060103s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Tanaka H, Nishiura Y, Takahashi T. J Org Chem. 2009;74:4383–4386. doi: 10.1021/jo900176e. [DOI] [PubMed] [Google Scholar]; b) Liang CF, Yan MC, Chang TC, Lin CC. J Am Chem Soc. 2009;131:3138–3139. doi: 10.1021/ja808353m. [DOI] [PubMed] [Google Scholar]; c) Chu KC, Ren CT, Lu CP, Hsu CH, Sun TH, Han JL, Pal B, Chao TA, Lin YF, Wu SH, Wong CH, Wu CY. Angew Chem Int Ed. 2011;50:9391–9395. doi: 10.1002/anie.201101794. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.