

Abstract
F-box proteins are the substrate-recognition subunits of SCF (Skp1/Cul1/F-box protein) ubiquitin ligase complexes. Purification of the F-box protein FBXL2 identified the PI(3)K regulatory subunit p85β and tyrosine phosphatase PTPL1 as interacting proteins. FBXL2 interacts with the pool of p85β that is free of p110 PI(3)K catalytic subunits and targets this pool for ubiquitylation and subsequent proteasomal degradation. FBXL2-mediated degradation of p85β is dependent on the integrity of its CaaX motif. Whereas most SCF substrates require phosphorylation to interact with their F-box proteins, phosphorylation of p85β on Tyr 655, which is adjacent to the degron, inhibits p85β binding to FBXL2. Dephosphorylation of phospho-Tyr-655 by PTPL1 stimulates p85β binding to and degradation through FBXL2. Finally, defects in the FBXL2-mediated degradation of p85β inhibit the binding of p110 subunits to IRS1, attenuate the PI(3)K signalling cascade and promote autophagy. We propose that FBXL2 and PTPL1 suppress p85β levels, preventing the inhibition of PI(3)K by an excess of free p85 that could compete with p85–p110 heterodimers for IRS1.
Degradation of regulatory proteins by the ubiquitin–proteasome system is an important control mechanism to ensure that a cell’s molecular machines are turned off or on at the right time and in the proper subcellular compartment. The specificity of the ubiquitin–proteasome system relies on a large number of ubiquitin ligases, which can be subdivided into single-subunit and multi-subunit ligases1.
SCFs are multimeric ubiquitin ligase complexes composed of four main subunits: SKP1, CUL1, RBX1 and one of many F-box proteins, the substrate receptors that define the specificity of each SCF ligase2,3. The F-box motif is a stretch of ~40 amino acids that is necessary for F-box proteins to bind SKP1 and, therefore, the other SCF subunits. The human genome encodes 69 F-box proteins, of which only a few are well characterized. FBXL2 (F-box and LLR (leucine-rich repeats) containing protein 2), also known as FBL2, is a highly conserved F-box protein that was originally identified in a screen for proteins required for the replication of the hepatitis C virus4–6. Interestingly, FBXL2 has a unique CaaX motif that is required for its geranylgeranylation and membrane localization, and the requirement for both the F-box and CaaX domains in hepatitis C virus replication suggested that SCFFBXL2 targets its substrates at the cell membranes. However, no membrane-associated substrates have been identified for FBXL2. To this end, we coupled FBXL2 tandem affinity purification with multidimensional protein identification technology (MudPIT) analysis and identified the p85α and p85β regulatory subunits of phosphatidylinositol-3-OH kinases (PI(3)Ks) as FBXL2 interacting proteins.
PI(3)Ks are conserved intracellular enzymes that modify lipids at membranes7–9. On the basis of their homology and substrate specificity, PI(3)Ks are grouped into three classes. Class I PI(3)K activity on phosphatidylinositol-4-5-biphosphate (PtdIns(4, 5)P2) generates phosphatidylinositol-3-4-5-triphosphate (PtdIns(3,4,5)P3), which acts as a docking site to recruit downstream targets, such as the PDK1, AKT and mTOR kinases, to the cell membrane to regulate cell growth, proliferation, survival and motility10–14. Class I is divided in two subclasses (IA and IB) and, of these, class IA PI(3)Ks are heterodimeric lipid kinases composed of a p110 catalytic subunit and a p85 regulatory subunit. In vertebrates, there are three catalytic p110 subunits (encoded by PIK3CA, PIK3CB and PIK3CD) and three regulatory subunits (encoded by PIK3R1, PIK3R2 and PIK3R3). The mechanism(s) by which the regulatory subunits (p85α, p85β and p55γ), which lack intrinsic kinase activity, modulate PI(3)K activity are not completely understood. In particular, genetic and biochemical studies suggest that, whereas a stoichiometric amount of p85 is necessary for PI(3)K to bind phospho-Tyr (p-Tyr) docking sites at the cell membrane, excess free p85 could compete with p85–p110 heterodimers for these sites, resulting in PI(3)K inhibition15–19.
This study provides an FBXL2-dependent molecular mechanism to maintain the balance between free p85β and p110–p85 heterodimers, to regulate PI(3)K signalling on stimulation with mitogens.
RESULTS
FBXL2 specifically binds p85α and p85β, but not p110α and p110β
To identify FBXL2 interactors, FLAG–HA-tagged FBXL2 was transiently expressed in HeLa or HEK293T cells and immunopurified for analysis by MudPIT (ref. 20). Purifications of FLAG–HA–FBXO1, a different F-box protein, were used as a control. Peptides corresponding to p85α and p85β were specifically identified in FBXL2, but not in FBXO1 immunoprecipitates (Supplementary Table S1). To investigate whether the binding between p85 proteins and FBXL2 is specific, we screened a panel of F-box proteins for interactions with endogenous p85α and p85β by transient expression in HEK293T cells and immunoprecipitations. We found that only FBXL2 was able to co-immunoprecipitate endogenous p85α and p85β(Fig. 1a). Interestingly, the catalytic subunits of PI(3)K (p110α and p110β) were not present in the FBXL2 complex, as detected by immunoblotting or mass spectrometry (Fig. 1b and Supplementary Fig. S1a and Table S1). We also found that the interaction with p85α and p85β depends on the integrity of the CaaX motif in FBXL2 because FBXL2(CaaX/SaaX), a mutant in which Cys 420 of the CaaX motif is mutated to serine, did not interact with either p85α or p85β(Fig. 1c–d). Moreover, FBXL2, but not FBXL2(CaaX/SaaX), interacted with endogenous, neddylated CUL1 (Fig. 1c–d), the active form of CUL1 that is covalently linked to Nedd8, whose formation is promoted by the binding of the substrate to an SCF (ref. 21). This result suggests that FBXL2 localization to cell membranes facilitates substrate binding, which in turn stimulates CUL1 neddylation and activation of ubiquitin ligase activity.
Figure 1.

FBXL2 binds p110-free p85 regulatory subunits. (a) FBXL2 binds p85α and p85β. HEK293T cells were transfected with either an empty vector (EV) or the indicated FLAG-tagged F-box proteins (FBPs). 24 h post-transfection, cells were treated with MG132 for 3 h before collection for immunoprecipitation (IP) and immunoblotting as indicated. WCL, whole-cell lysate. (b) FBXL2 does not bind p110α and p110β. HEK293T and NIH3T3 cells were transfected with either an empty vector (EV) or the indicated FLAG-tagged proteins. The experiment was performed as described in a. (c) The CaaX motif of FBXL2 is required to bind p85α and p85β. HEK293T cells were transfected with either an empty vector (EV) or the indicated FLAG-tagged F-box proteins. The experiment was performed as described in a. (d) Table summarizing the results presented in a–c and Supplementary Fig. S1a. NT, not tested. Uncropped images of blots are shown in Supplementary Fig. S8.
p85β is targeted for degradation by SCFFBXL2
Next, we investigated whether p85α and p85β are targeted for proteolysis by FBXL2. Expression of wild-type FBXL2, but not FBXL2(CaaX/SaaX), resulted in a reduction of endogenous p85β levels, as detected using immunoblotting or immunofluorescence microscopy (Fig. 2a and Supplementary Fig. S1b). This reduction in protein levels was rescued by the addition of the proteasome inhibitor MG132, demonstrating that the decrease in p85β levels was due to enhanced proteolysis (Fig. 2a). In contrast, p85α levels remained unchanged despite expression of FBXL2 (Fig. 2a).
Figure 2.
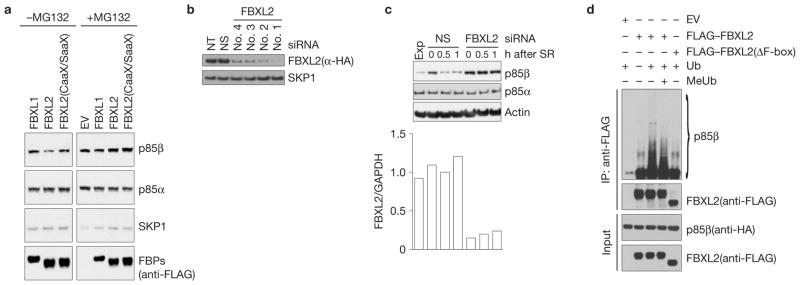
p85β is targeted for ubiquitylation and degradation by SCFFBXL2. (a) HEK293T cells were transfected with an empty vector (EV) or the indicated FLAG-tagged constructs. 24 h after transfection, cells were treated with either MG132 or solvent for 3 h before collection for immunoblotting as indicated. (b) HeLa cells stably expressing FLAG–HA-tagged FBXL2 were transfected with either siRNAs targeting FBXL2 (four different siRNAs) or a non-silencing siRNA (NS). Cells were lysed, and proteins were immunoblotted as indicated. NT, non-transfected. (c) During a 72-h serum starvation, NHFs were transfected with either an siRNA targeting FBXL2 (no. 1) or a non-silencing siRNA (NS). Cells were subsequently stimulated with media containing serum and collected at the indicated time points for immunoblotting. The graph shows FBXL2 mRNA levels in the different samples analysed using real-time PCR in triplicate measurements. The values represent the ratios between FBXL2 and GAPDH mRNAs. SR, serum re-addition. (d) HEK293T cells were transfected with HA-tagged p85β, FLAG-tagged FBXL2, FLAG-tagged FBXL2(ΔF-box) or an empty vector (EV) as indicated. After immunopurification with anti-FLAG resin, in vitro ubiquitylation of p85β was performed in the presence of E1, E2s and ubiquitin (Ub). Where indicated, an excess of methylated ubiquitin (MeUb) was also added. Samples were analysed by immunoblotting with the indicated antibodies. The ladder of bands with a relative molecular mass of >85,000 (lane 3) corresponds to ubiquitylated p85β. Immunoblots of whole-cell lysates (WCL) are shown at the bottom. Uncropped images of blots are shown in Supplementary Fig. S8
Two other mutants of FBXL2 that disrupt binding to SKP1 (and through SKP1, CUL1) also suggested that p85β is a substrate for FBXL2-mediated degradation. FBXL2(LPK-W/AAA-A) (in which the first three amino acids and the conserved tryptophan in position 28 of the F-box domain were mutated to alanine) and FBXL2(ΔF-box) (in which the entire F-box domain was deleted) bound less SKP1 and more p85β than wild-type FBXL2 (Supplementary Fig. S2a). As these mutants are unable to form an active SCF ubiquitin ligase, they should sequester FBXL2 substrates, as observed for p85β.
To further confirm that FBXL2 regulates the degradation of p85β, we used four different short interfering RNAs (siRNAs) to reduce its expression in cells (Fig. 2b). As PI(3)K activity is stimulated by mitogens, we examined the impact of FBXL2 silencing on the abundance of p85β. Normal human fibroblasts (NHFs) were deprived of serum for 72 h and then reactivated by addition of serum. In mitogen-deprived cells, the overall amount of p85β increased, but after mitogen stimulation, it rapidly decreased (Fig. 2c). Depletion of FBXL2 with four different siR-NAs (used individually) inhibited the degradation of p85β after serum addition, with no effects on p85α(Fig. 2c and Supplementary Fig. S2b).
Finally, we reconstituted the ubiquitylation of p85β in vitro. Immunopurified FBXL2, but not FBXL2(ΔF-box), promoted the in vitro ubiquitylation of p85β(Fig. 2d). Notably, methylated ubiquitin inhibited the formation of the highest-molecular-weight forms of p85β, demonstrating that the high-molecular-weight forms of p85β are indeed polyubiquitylated. These results support the hypothesis that FBXL2 directly controls the ubiquitin-mediated degradation of p85β.
Together, the results shown in Figs 1 and 2 and Supplementary Figs S1 and S2 demonstrate that FBXL2 mediates the ubiquitylation and degradation of p85β on cell membranes. The binding of p85α to FBXL2 is probably indirect and may be due to the fact that p85β forms heterodimers with p85α(Supplementary Fig. S1a and ref. 22).
Phosphorylation of p85β on Tyr 655 inhibits its binding to FBXL2
Subsequently, we mapped the FBXL2-binding motif in p85β using deletion mutants, which identified a binding motif in the SH2C domain of p85β that was necessary and sufficient to interact with FBXL2 (Supplementary Fig. S3 and Fig. 3a). Interestingly, the SH3 and GAP domains of p85β seemed to inhibit binding to FBXL2, as shown by the increase in the amount of FBXL2 co-immunoprecipitated with p85β mutants lacking these domains (Supplementary Fig. S3 and Fig. 3a). Further deletion mutants within the SH2C domain mapped the FBXL2-binding motif to a region between amino acids 640 and 660 of human p85β (Fig. 3a–b). Finally, we performed alanine scanning mutagenesis of this region and found that Gln 651 and Arg 652 are necessary for efficient binding of p85β to FBXL2 (Supplementary Fig. S4a). The Gln-Arg motif and surrounding amino acids may represent a degron for FBXL2 substrates. This region is highly conserved in p85β orthologues (Supplementary Fig. S4b), but it is not present in p85α and p55γ (Supplementary Fig. S4c). Significantly, a p85β mutant missing the SH2C domain and p85β (QR/AA), a mutant in which Gln 651 and Arg 652 are mutated to alanine, exhibited a longer half-life than wild-type p85β(Fig. 3c–d).
Figure 3.

Identification of p85β degron. (a) Schematic representation of p85β mutants. Binding of p85β to FBXL2 and p110α is indicated with the symbol +; NT, not tested. ‘++’ denotes enhanced binding. (b) HEK293T cells were transfected with GFP-tagged FBXL2 and the indicated FLAG-tagged p85β mutants. Whole-cell lysates (WCL) were immunoprecipitated (IP) with anti-FLAG resin, and immunocomplexes were probed with antibodies against the indicated proteins. (c) p85β(ΔSH2C) is more stable than wild-type p85β. RPE1-hTERT cells were infected with either a retrovirus expressing wild-type p85β or p85β (ΔSH2C). Cells were incubated with cycloheximide (CHX) for the indicated times, collected and analysed by immunoblotting as indicated. In the graph, the amount of p85β (wild-type or mutant) is represented relative to the amount at time 0. (d) p85β (QR/AA) is more stable than wild-type p85β. HEK293T cells were infected with either a retrovirus expressing wild-type p85β or p85β (QR/AA). Cells were incubated with cycloheximide (CHX) for the indicated times, collected and analysed by immunoblotting as indicated. In the graph, the amount of p85β (wild-type or mutant) is represented relative to the amount at time 0. (e) p85β (Y655A) is less stable than wild-type p85β. The experiment was performed as described in c except that p85β(Y655A) was used. In the graph, the amount of p85β(wild-type or mutant) is represented relative to the amount at time 0. Uncropped images of blots are shown in Supplementary Fig. S8.
Interestingly, in contrast to mutations in Gln 651 and Arg 652, mutation of Tyr 655 to alanine (both in the context of full-length p85β and p85β-deletion mutants) stabilized the p85β–FBXL2 interaction (Supplementary Fig. S5). Moreover, the half-life of p85β(Y655A) was shorter than the half-life of wild-type p85β(Fig. 3e). These two results suggest that phosphorylation of this residue may inhibit binding between these two proteins. To investigate whether phosphorylation of Tyr 655 affects this interaction, we used immobilized, synthetic peptides containing the candidate binding sequence (amino acids 640–660 in human) to test binding to FBXL2. Whereas non-phosphorylated peptide was able to bind FBXL2 (but not FBXL1 or FBXL3), the corresponding peptide containing phosphorylated Tyr 655 did not bind FBXL2 efficiently (Fig. 4a).
Figure 4.
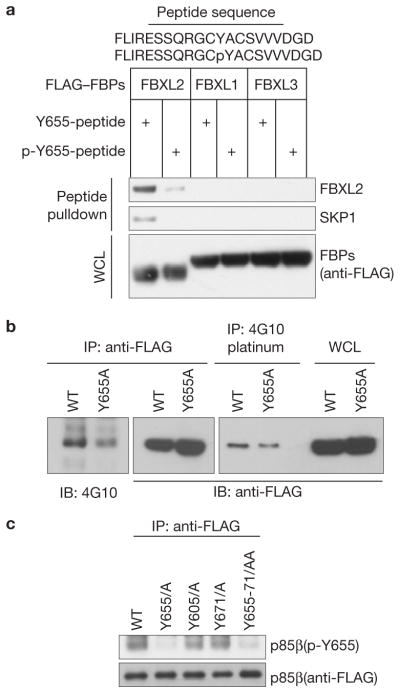
p85β interaction with FBXL2 is negatively regulated by Tyr phosphorylation. (a) Lysates from cells expressing the indicated FLAG-tagged F-box proteins (FBPs) were used in binding reactions with beads coupled to either a peptide or a phospho-peptide (sequences shown on top of the panel). Beads were washed with lysis buffer, and bound proteins were eluted and subjected to SDS–PAGE and immunoblotting. (b) Denatured extracts from HEK293T cells stably expressing either FLAG-tagged wild-type p85β or FLAG-tagged p85β(Y655A) were immunoprecipitated (IP) with either an anti-FLAG resin or a 4G10 platinum resin and immunoblotted (IB) as indicated. Immunoblots of whole-cell lysates (WCL) are also shown in the last two lanes. (c) Denatured extracts from HEK293T cells expressing either FLAG-tagged wild-type p85β or the indicated FLAG-tagged p85β mutants were immunoprecipitated (IP) with an anti-FLAG resin and immunoblotted as indicated. Uncropped images of blots are shown in Supplementary Fig. S8.
We also verified that Tyr 655 was phosphorylated in vivo. First, FLAG-tagged p85β and p85β(Y655A) were expressed in HEK293T cells, immunopurified under denaturing conditions with an anti-FLAG antibody or a mix of anti-p-Tyr antibodies, and immunoblotted. In both cases, the experiments showed that the level of phosphorylation on Tyr was decreased in p85β(Y655A) when compared with wild-type p85β (Fig. 4b). Moreover, we generated a phospho-specific antibody against a peptide containing p-Tyr at position 655. This antibody recognized wild-type p85β, as well as p85β mutants on Tyr 605 and Tyr 671, but it did not recognize a p85β mutant in which Tyr 655 was substituted to alanine (Fig. 4c), demonstrating that Tyr 655 is phosphorylated in vivo.
PTPL1 promotes the degradation of p85β
The role of Tyr 655 prompted us to search for Tyr kinases and/or p-Tyr phosphatases in the list of FBXL2 interactors that we identified. Peptides corresponding to PTPL1 (protein Tyr phosphatase L1; also known as PTPN13, FAP-1, PTP-BAS and PTP1E) were specifically identified in the FBXL2 immunoprecipitates, but not in control purifications (Supplementary Table S1). PTPL1 is a non-receptor type p-Tyr phosphatase23 (PTP). In addition to confirming that FBXL2 was able to bind endogenous PTPL1 (Fig. 5a), we observed that p85β was able to co-immunoprecipitate both exogenous and endogenous PTPL1 and that, vice versa, PTPL1 co-immunoprecipitated both exogenous and endogenous p85β (Supplementary Fig. S6a–d). Like FBXL2, PTPL1 interacted with amino acids 640–660 of p85β, and the SH2C domain of p85β was necessary and sufficient for the interaction with PTPL1 (Supplementary Fig. S6e–g and Fig. 5b). Moreover, both FBXL2 and PTPL1 interacted with p85β, but not p110α (Supplementary Fig. S6d and Fig. 1b). We then expressed tagged constructs of PTPL1, FBXL2 and p85β in HEK293T cells in different combinations. As expected, FLAG–p85β co-immunopurified both HA-tagged PTPL1 and GFP-tagged FBXL2 (Supplementary Fig. S7a). Parallel anti-FLAG immunoprecipitates were also eluted with FLAG peptide and re-precipitated with an anti-GFP antibody. Again, all three proteins were detected by immunoblotting of the second immunoprecipitation (Supplementary Fig. S7a), indicating that PTPL1, FBXL2 and p85β form a trimeric complex. However, PTPL1 could also bind FBXL2 independently of p85β, because the CaaX domain of FBXL2 was necessary to bind p85β, but not PTPL1 (Fig. 5a and Fig. 1c). These results suggest that FBXL2 brings PTPL1 to the membrane to ensure p85β dephosphorylation. In agreement with this interpretation, expression of increasing amounts of FBXL2 promoted the interaction between PTPL1 and p85β (Supplementary Fig. S7b).
Figure 5.

PTPL1 dephosphorylates p85β, promoting its binding to FBXL2 and degradation. (a) The CaaX motif of FBXL2 is not required to bind PTPL1. HEK293T cells were transfected with either FLAG-tagged wild-type FBXL2 or FLAG-tagged FBXL2(CaaX/SaaX). 24 h post-transfection, cells were collected and whole-cell lysates (WCL) were immunoprecipitated (IP) and immunoblotted as indicated. (b) Schematic representation of p85β mutants. Binding of p85β to PTPL1 and p110α is indicated with the symbol +. (c) PTPL1 stimulates the binding of FBXL2 to wild-type p85β, but not p85β(Y655A). HEK293T cells were transfected with GFP-tagged FBXL2 and either FLAG-tagged p85β or FLAG-tagged p85β(Y655A). Where indicated, HA-tagged PTPL1 or HA-tagged PTPL1(C/S) were also transfected. The experiment was performed as described in a. (d) PTPL1 silencing inhibits the FBXL2–p85β interaction. HeLa cells stably expressing FLAG-tagged FBXL2 were transfected with either an siRNA targeting PTPL1 or a non-silencing siRNA (NS). Forty-eight hours post-transfection, cells were collected and whole-cell lysates (WCL) were immunoprecipitated (IP) and immunoblotted as indicated. (e) During a 72-h serum starvation, NHFs were transfected twice with either siRNAs targeting FBXL2 or PTPL1, or a non-silencing siRNA (NS). Cells were subsequently re-stimulated with media containing serum and collected at the indicated time points for immunoblotting. SR, serum re-addition. (f) HEK293T cells were transfected twice with either siRNAs targeting FBXL2 or PTPL1, or a non-silencing siRNA (NS). Cells were transfected with p85β(Y655A) and 16 h after cells were incubated with cycloheximide (CHX) for the indicated times, collected and analysed by immunoblotting as indicated. (g) During a 48-h serum starvation, U2OS cells stably transfected with a doxycycline-inducible p85β construct were transfected twice with either an siRNA targeting PTPL1 or a non-silencing siRNA (NS). During the last 16 h before collection, p85β expression was stimulated with doxycycline. Cells were subsequently re-stimulated with media containing serum and collected 30 min later. Denatured cell lysates were immunoprecipitated with an anti-FLAG resin and immunoblotted as indicated. The asterisk indicates a non-specific band. Uncropped images of blots are shown in Supplementary Fig. S8.
As phosphorylation of p85β on Tyr 655 inhibited its binding to FBXL2, we examined whether PTPL1 could regulate this interaction. We found that the expression of wild-type PTPL1, but not an inactive PTPL1(C/S) mutant, increased the binding of FBXL2 to wild-type p85β, whereas binding to the p85β(Tyr 655) mutant was unaffected (Fig. 5c). Similarly, PTPL1 silencing inhibited FBXL2–p85β interaction (Fig. 5d). These results suggest that PTPL1 dephosphorylates p-Tyr-655 in p85β, promoting its interaction with and degradation through FBXL2. Accordingly, PTPL1 silencing inhibited the serum-induced degradation of p85β in a manner similar to FBXL2 knockdown (Fig. 5e and Supplementary Fig. S7c). Moreover, FBXL2 silencing did not alter PTPL1 levels, indicating that FBXL2 does not control the abundance of PTPL1. Significantly, p85β(Y655A) was stabilized by FBXL2 knockdown, but not PTPL1 depletion (Fig. 5f), further indicating that PTPL1 de-phosphorylates p-Tyr-655. In agreement with this hypothesis, PTPL1 silencing resulted in an increased level of phosphorylation of p85β on Tyr 655 (Fig. 5g).
Inhibition of the FBXL2-mediated degradation of p85β leads to a decrease in the interaction of p110 with IRS1 and attenuation of the PI(3)K signalling
To investigate the biological significance of the FBXL2–p85β interaction, NHFs were transfected with a non-silencing siRNA or an FBXL2 siRNA and serum starved. Following stimulation with serum, p85β levels decreased in control cells, but in the FBXL2 siRNA cells, they were elevated at time 0 and remained elevated even after serum stimulation (Fig. 6a). In parallel, we observed that the induction of phosphorylated AKT (on Thr 308 and Ser 473), S6K1 (on Thr 389), GSK3α(on Ser 21) and GSK3β(on Ser 9) was severely blunted in the FBXL2-knockdown samples (Fig. 6a). Similar results were obtained using a different siRNA to FBXL2 (Supplementary Fig. S7d) or when cells were stimulated with insulin rather than serum (Supplementary Fig. S7e).
Figure 6.

Failure to degrade p85β results in PI(3)K activation defects. (a) During a 72-h serum starvation, NHFs were transfected with either siRNAs targeting FBXL2, p85β, both FBXL2 and p85β, or a non-silencing siRNA (NS). Cells were subsequently stimulated with media containing serum and collected at the indicated time points for immunoblotting. The graph at the right shows FBXL2 mRNA levels in the different samples analysed using real-time PCR in triplicate measurements. The values represent the ratios between FBXL2 and GAPDH mRNAs. SR, serum re-addition. (b) Cells were treated as in a, except that 3 h after serum re-addition, cells were lysed, immunoprecipitated (IP) with an anti-IRS1 antibody and immunoblotted as indicated. Uncropped images of blots are shown in Supplementary Fig. S8.
To determine whether the effects of FBXL2 knockdown on the PI(3)K signalling cascade are dependent on the stabilization of p85β, we silenced p85β expression either by itself or in combination with FBXL2. No substantial differences in phosphorylation of AKT, S6K1 and GSK3 were observed in the double p85β/FBXL2 knockdown NHFs (Fig. 6a and Supplementary Fig. S7d). Significantly, when p85β was silenced (either alone or in combination with FBXL2), the levels of AKT phosphorylation on Thr 308 and S6K phosphorylation on Thr 389 were higher than controls, particularly one hour after serum stimulation.
In activated cells, p85 subunits bind p-Tyr residues of receptor Tyr kinases (for example, PDGFR) and associated binding proteins (for example, IRS1 (insulin receptor substrate 1)), recruiting p110 subunits to the membrane, where PI(3)K can phosphorylate its lipid substrates. As downregulation of FBXL2 results in the accumulation of p85β, but not p110α and p110β, we investigated the binding of PI(3)K subunits to IRS1 and PDGFR. We found that, when FBXL2 expression was silenced in NHFs, endogenous IRS1 bound more endogenous p85β, but less p110α and p110β (Fig. 6b). These results suggested that the attenuation of PI(3)K signalling observed in FBXL2-knockdown NHFs (Fig. 6a and Supplementary Fig. S7d) may be due, at least partially, to increased membrane localization of p85β monomers and/or p85β–p85β homodimers at the expense of p85–p110 complexes. (Notably, in contrast to other cell types, IRS did not co-immunoprecipitate with p85α in NHF lysates (Fig. 6b and Supplementary Fig. S7f).)
FBXL2-mediated degradation of p85β inhibits autophagy
The PI(3)K–AKT–mTOR signalling pathway negatively regulates autophagy24–27. We found that, when FBXL2 was downregulated (using two different siRNAs) in serum-starved NHFs, more autophagy was induced than in control cells, as indicated by the increased levels of the fast-migrating, lipidated species of the LC3 autophagy marker (referred to as LC3-II) and a concomitant decrease in p62 levels (Fig. 7a). Depletion of FBXL2 resulted in increased LC3-II levels even in favourable nutrient conditions (that is, three hours after reactivation of NHFs with serum; Fig. 7a). Importantly, co-depletion of p85β almost completely rescued the autophagy phenotype, as indicated by a reduction in the level of LC3-II and an increase in p62, compared with cells in which only FBXL2 was silenced (Fig. 7b). To further confirm that autophagy was induced by the accumulation of p85β, we engineered U2OS cells to express p85β in an inducible fashion at levels slightly higher than endogenous and just enough to overwhelm p85β degradation. Cells were serum starved for 48 h and reactivated with serum for various times. Expression of p85β during the last 16 h in culture led to an increase in the level of LC3-II and a decrease in p62 both in serum-starved and reactivated cells (Fig. 7c). Moreover, inducible expression of p85β resulted in a decrease of AKT, S6K and GSK3 phosphorylation in response to serum, in line with the effects of FBXL2 knockdown. Finally, forced expression of p85β led to an increase in GFP–LC3-II vesicles (Fig. 7d), consistent with an activation of autophagy.
Figure 7.

p85β degradation regulates cell autophagy. (a) During a 72-h serum starvation, NHFs were transfected with either siRNAs targeting FBXL2 (no. 1 or 2) or a non-silencing siRNA (NS). Cells were subsequently stimulated with media containing serum and collected at the indicated time points for immunoblotting. SR, serum re-addition. (b) During a 72-h serum starvation, NHFs were transfected with either siRNAs targeting FBXL2, both FBXL2 and p85β, or a non-silencing siRNA (NS). Cells were subsequently stimulated with media containing serum and collected at the indicated time points for immunoblotting. SR, serum re-addition. (c) U2OS cells stably transfected with a doxycycline-inducible p85β construct were serum starved for 48 h. During the last 16 h before collection, p85β expression was stimulated with doxycycline. Cells were subsequently stimulated with media containing serum and collected at the indicated time points for immunoblotting. SR, serum re-addition. (d) Cells constitutively expressing GFP–LC3 were transfected with HA-tagged p85β, fixed and incubated with an anti-HA p85β antibody (red). Arrows in the top panels point to the enhanced autophagic response (LC3-II vesicles) in cells expressing high levels of p85β, as shown in the bottom panels. Uncropped images of blots are shown in Supplementary Fig. S8. Scale bars, 10 μm.
DISCUSSION
The SH2 domains of p85 subunits bind p-Tyr residues in pYxxM motifs of IRS1 and receptor tyrosine kinases to mediate the recruitment of active p110–p85 heterodimers to cell membranes28. However, many studies suggest that the SH2 domains of p85 subunits form direct inhibitory contacts with the catalytic site of p110 subunits, leading to attenuation of PI(3)K activity29–32, and these observations are in agreement with the increase of insulin-mediated PI(3)K signalling in mice lacking various p85 regulatory isoforms16,18,33–35. Biochemical and structural studies suggest that, in resting cells, p85 subunits have a closed conformation, in which their SH2 domains bind and inhibit PI(3)K (refs 31,32,36). On mitogenic stimuli, p-Tyr-containing proteins favour an open, non-inhibitory conformation, in which SH2 domains bind to p-Tyr docking sites and no longer inhibit PI(3)K. Moreover, it has been suggested that the outcome of PI(3)K signalling also depends on the equilibrium between p110-free p85 monomers (or p110-free p85–p85 homodimers) and p85–p110 heterodimers. In this model, excess p85 would directly inhibit p110 catalytic activity and/or compete with p85–p110 heterodimers for p-Tyr docking sites15,19. However, there is no consensus about the existence of p110-free p85 subunits, and how this p85 pool may be regulated is not understood17,37,38. We have shown that SCFFBXL2 interacts with and promotes the proteasome-mediated degradation of a pool of p85β that is free of p110 subunits. Accumulation of p85β due to the inhibition of its degradation causes a decrease in the level of binding of p110 subunits to IRS1, attenuation of the PI(3)K signalling cascade (that is, phosphorylation of AKT, S6K and GSK3) and persistent autophagy under favourable nutrient conditions (see the model in Fig. 8). These results are in agreement with experiments in which expression of p85 subunits in skeletal muscle cells resulted in the inhibition of AKT phosphorylation39,40.
Figure 8.

A model of the FBXL2- and PTPL1-dependent regulation of the PI(3)K pathway. SCFFBXL2 interacts with and promotes the proteasome-mediated degradation of a pool of p85β that is free of p110 subunits. Phosphorylation of p85β on Tyr 655 inhibits its interaction with FBXL2 and is counteracted by the phosphatase PTPL1. Together, SCFFBXL2 and PTPL1 ensure that free p85β does not accumulate, which would result in the inhibition of the catalytic activity of p110 and/or competition with p85–p110 heterodimers for p-Tyr docking sites (for example, in IRS1).
Our results underscore the differential molecular mechanism of regulation of p85α and p85β subunits. Moreover, as both the SH2N and SH2C domains of p85β are able to bind p-Tyr residues, it has been proposed that the presence of two SH2 domains in p85 subunits increases their affinity for IRS1 and receptor Tyr kinases28,41,42. Whereas previous studies do not distinguish between the functions of the SH2N and SH2C domains, we found that FBXL2 specifically binds the SH2C domain of p85β, making it possible that FBXL2 targets p85β homodimers in which one p85β subunit binds FBXL2 and the other subunit binds IRS1 or a receptor. However, it is also possible that FBXL2 binds the SH2C domains of p85β monomers, which in turn bind IRS1 or receptor Tyr kinases through their SH2N domains.
Recent studies suggest that the SH2C domain of p85α in complex with p110β is not exposed because in quiescent cells SH2C binds p110β and in activated cells binds p-Tyr docking sites32,36. If this model is confirmed for p85β, it may explain why FBXL2 targets only p110-free p85β.
Most of the known substrates of SCF ubiquitin ligases are recognized by the F-box protein subunit only when they are phosphorylated on threonines or serines in their degrons. Therefore, it is unique that FBXL2 binding to p85β is inhibited by phosphorylation and that this regulation occurs through tyrosine phosphorylation. Indeed, whereas other SCF ligases cooperate with Ser/Thr kinases to target substrates, SCFFBXL2 requires the p-Tyr phosphatase PTPL1. Interestingly, PTPL1 levels increase 2–3-fold on mitogenic stimulation of serum-starved cells and inversely correlate with p85β levels (Fig. 5f), suggesting that increased PTPL1 expression may be at least partially responsible for enhanced degradation of p85β. However, PTPL1 silencing results in only a slight decrease in AKT phosphorylation (Supplementary Fig. S7c), probably because the combined actions of PTPL1 on p85β and IRS1 (ref. 43) balance themselves, resulting in only a minor effect on AKT activity.
PI(3)K signalling is important for cell growth, proliferation and survival, and when improperly regulated, the pathway contributes to a variety of diseases. A further understanding of the regulation of this pathway could lead to new therapies. Our study suggests that small-molecule inhibitors of FBXL2 or PTPL1 could inhibit the PI(3)K signalling cascade, impeding the growth of cancer cells that are addicted to PI(3)K pathway activity.
METHODS
Tandem affinity purification and mass spectrometry
HEK293T cells were transiently transfected with FLAG–HA-tagged FBXL2 and control plasmids using polyethylenimine, and 24 h after transfection cells were treated with MG132 for 3 h before collection. Lysis of cell pellets was carried out with lysis buffer (50 mM Tris, at pH 8.0, 150 mM NaCl, glycerol 10%, 1 mM EDTA, 5 mM MgCl2, 50 mM NaF and NP-40 0.1%) supplemented with protease and phosphatase inhibitors. The first immunoprecipitation of the soluble fraction was carried out with anti-FLAG antibodies (2 h at 4 °C, rocking) conjugated with agarose resin. FLAG-immunoprecipitates were incubated with 3X-FLAG-peptide (50 μg ml−1) for 30 min at room temperature to elute the immunoprecipitated proteins that were then subjected to a second immunoprecipitation with anti-HA antibodies (1 h at 4 °C, rocking) conjugated to agarose resin. HA-immunoprecipitates were eluted with HA-peptide (50 μg ml−1) for 30 min at room temperature. Two independent affinity purifications were performed. Both single and double immunoprecipitations were analysed by MudPIT. Eluted fractions were TCA precipitated and the pellets were solubilized in Tris–HCl at pH 8.5 and 8 M urea; TCEP (tris-(2-carboxylethyl)-phosphine hydrochloride, Pierce) and CAM (chloroacetamide, Sigma) were added to a final concentration of 5 and 10 mM, respectively. Protein suspensions were digested overnight at 37 °C using endoproteinase Lys-C at 1:50 wt/wt (Roche). Samples were brought to a final concentration of 2 M urea and 2 mM CaCl2 before performing a second overnight digestion at 37 °C using trypsin (Promega) at 1:100 wt/wt. Formic acid (5% final) was added to stop the reactions. Samples were loaded on split-triple-phase fused-silica micro-capillary columns44 and placed in-line with linear ion trap mass spectrometers (LTQ, ThermoScientific), coupled with quaternary Agilent 1100 or 1200 series HPLCs. A fully automated 10-step chromatography run (for a total of 20 h) was carried out for each sample, as described in ref. 20, enabling dynamic exclusion for 120 s. The MS/MS data sets were searched using SEQUEST (ref. 45) against a database of 58,614 sequences, consisting of 29,147 Homo sapiens non-redundant proteins (downloaded from NCBI on 16 June 2011), 177 usual contaminants (such as human keratins, IgGs and proteolytic enzymes), and, to estimate false-discovery rates, 29,307 randomized amino-acid sequences derived from each non-redundant protein entry. Peptide/spectrum matches were sorted, selected and compared using DTASelect/CONTRAST (ref. 46). Combining all runs, proteins had to be detected by at least 2 peptides, leading to false-discovery rates at the protein and spectral levels of 1.02 and 0.16%, respectively. To estimate relative protein levels, normalized spectral abundance factors were calculated as described47.
Antibodies and plasmids
Antibodies used were: p85β (1:1,000, Abcam cat. no. ab28356 and GeneTex cat. no. GTX11593), p85α (1:1,000, Abcam cat. no. ab22653 and Cell Signaling cat. no. 4257S), FLAG (1:5,000, Sigma cat. no. A2220, F7425), PTPL1 (1:1,000, Santa Cruz Biotechnology cat. no. SC-15,356), SKP1 (1:1,000, home generated), AKT and p-AKT (1:1,000, Cell Signaling cat. nos 2,920, 4058S and 9275S), S6K and p-S6K (1:1,000, Cell Signaling cat. nos 9202S and 9206S), p-GSK3α/β (1:1,000, Cell Signaling cat. no. 8566S), p110α (1:1,000, Cell Signaling cat. no. 4249S), p110β (1:1,000, Cell Signaling cat. no. 3011S), PDGFR (1:1,000, Cell Signaling cat. no. 3166S), IRS1 (1:1,000, Cell Signaling cat. no. 2,383), p62 (1:5,000, MBL cat. no. PM045), LC3B (1:7,000, Invitrogen cat. no. L10382), HA (1:3,000, Covance cat. no. MMS-101R), CUL1 (1:1,000, Invitrogen cat. no. 32-2,400), GFP (1:1,000, Cell Signaling cat. no. 2956S), β-actin (1:10,000, Sigma cat. no. A5441) and tubulin (1:5,000, Sigma cat. no. T5168). FBXL2 polyclonal rabbit antibody was generated using a peptide to the amino terminus of FBXL2. Polyclonal rabbit phospho-specific antibody against p85β was generated using the KFLIRESSQRGCpYACSVVVDGD phosphopetide. FBXL2, p85β and PTPL1 complementary DNAs were inserted into pcDNA3 either by sub-cloning or site-directed mutagenesis48,49. Where specified, we also used pEGFPC1, retroviral (pBabe) and lentiviral (pTRIPz) vectors. Specific details will be provided on request.
Retro and lentivirus-mediated gene transfer
GP-293 (Clontech) packaging cells were transiently co-transfected with retro viral(pBabe) vectors containing VSVG and the gene of interest using calcium phosphate. Lentiviruses for doxycycline-inducible p85β were prepared in HEK293 cells by co-transfecting the pTRIPz vector (p85β) and vectors containing VSVG and pΔ8.2 using calcium phosphate. Retro- or lentivirus-containing medium, 48 h after transfection, was collected and supplemented with 8 mg ml−1 Polybrene (Sigma). RPE, U2OS, HEK293T and HeLa cells were infected by replacing the cell culture medium with the viral supernatant for 5 h. Selection of stable clones was carried out using puromycin.
Immunoprecipitation and immunoblotting
HEK293T cells were transiently transfected using polyethylenimine. Sixteen hours after transfection HEK293T cells were incubated with or without MG132 for 3 h before collection. NHFs, NIH3T3, RPE and U2OS cells were subjected to experimental conditions (serum-starvation before re-addition of serum or siRNA) for various times before collection. Cell lysis was carried out with lysis buffer (50 mM Tris, at pH 8.0, 150 mM NaCl, glycerol 10%, 1 mM EDTA, 50 mM NaF and NP-40 0.1%) supplemented with protease and phosphatase inhibitors. For denaturating conditions, cells were lysed in lysis buffer containing 1% SDS, 5 mM dithiothreitol, 1 mM sodium vanadate and 50 mM Tris at pH 7.4 and boiled for 10 min before immunoprecipitation. Immunoprecipitation and immunoblotting were performed as previously described49,50.
Gene silencing with siRNA and real-time quantitative PCR
ON TARGET siRNA oligonucleotides (Dharmacon) against various messenger RNAs were used at 5 nM final concentration in serum-free medium for 24–48 h using HiPerfect (Qiagen). For real-time quantitative PCR, total RNA was isolated using Qiagen’s RNeasy kit. Reverse transcription reaction was carried out for 5.0 μg total RNA using Oligo-dT primers with Superscript III RT polymerase (Invitrogen) according to the manufacturer’s instructions. Real-time quantitative PCR was carried out for 250 ng cDNA with FBXL2 and GAPDH primers using the SYBR Green method with a Roche Light Cycler 480II machine in a 96-well format. Data were analysed using second-derivative Max with high-confidence software for RT values according to the manufacturer’s guidelines (Roche). ON-TARGETplus p85β siRNA pool: 5′-GGAAAGGCGGGAACAAUAA-3′, 5′-GGACAAGAGCCGCGAGUAU-3′, 5′-GGAACGCACUUGGUACGUG-3′, 5′-GCGCCCAGCUUAAGGUCUA-3′. ON-TARGETplus FBXL2 siRNA: 5′-GCACAGAACUGCCGAAACA-3′, 5′-GCUCGGAAUUGCCACGAAU-3′, ON-TARGETplus PTPL1 siRNA (L-008065-00-0005, ON-TARGETNon-targeting siRNA #1 (D-001810-01-05). RT-primers for FBXL2 (forward: 5′-AT TTG ACT GAC GCA GGT TT-3′, reverse: 5′-GAGCTGGATGAGTGTGCTGT-3′) and human GAPDH (forward: 5′-TGCACCACCAACTGCTTAGC-3′, reverse: 5′-GGCATGGACTGTGGTCATGAG-3′).
In vitro ubiquitylation assay
In vitro ubiquitylation assays were preformed as described previously51. Briefly, HA-tagged p85β was co-transfected with either FLAG-tagged FBXL2 or an F-box-deleted FBXL2 mutant into HEK293T cells. 24 h after transfection, cells were incubated with MG132 for 3 h before collection. FBXL2 (wild-type or mutant) was immunoprecipitated with anti-FLAG M2 agarose beads. The beads were washed four times in lysis buffer and twice in ubiquitylation reaction buffer (10 mM Tris–HCl, at pH 7.5, 100 mM NaCl and 0.5 mM dithiothreitol). In vitro ubiquitylation assays were performed on immunoprecipitated beads in a volume of 30 μl, containing 0.1 μM E1 (Boston Biochem), 0.25 μM Ubch3, 0.25 μM Ubch5c, 1 μM ubiquitin aldehyde, 2.5 μg μl−1 ubiquitin and 1× magnesium/ATP cocktail. Samples were incubated for 2 h at 30 °C and analysed by immunoblotting.
Autophagy
NHFs and U20S (expressing inducible p85β) were cultured in serum-free medium for 48–72 h to monitor the autophagic response. Recovery from the autophagic response was studied by replacing the serum-free medium with medium containing 10% fetal bovine serum for up to 3 h. Cell lysates were the subjected to SDS–PAGE and western blotting using antibodies against LC3-II and p62. U2OS cells stably expressing GFP–LC3-II were grown in serum-free medium for 1–3 h to monitor GFP–LC3-II using an inverted microscope, as described below.
Immunofluorescence microscopy
Cells were cultured on glass coverslips in 24-well plates and transiently transfected using Xtremegene transfection reagent (Roche). Cells were fixed with 4% paraformaldehyde (wt/vol) for 30 min, washed 3 times in PBS-T (PBS-Tween 0.1%) and permeabilized with 0.1% Triton X-100 in PBS before blocking with 10% normal goat serum (NGS) in PBS-T for 30 min. Primary antibodies were incubated for 1 h, and secondary antibody conjugated to Alexa-Fluor 555 was incubated for 30 min at room temperature in 10% NGS. DAPI staining was carried out for 10 min in PBS. After washing, coverslips were dried and mounted on glass slides. Fluorescence microscopy was carried out using an Axiovert 200M-ZEISS fluorescence inverted microscope. Images were acquired using the CCD (charge-coupled device) camera (RETIGA 2000R FAST) attached to the microscope with MetaMorph 6.2r6 software.
In vitro peptide binding assay
Phosphorylated and non-phosphorylated peptides of p85β (amino acids 644–664) were synthesized (YenZyme) and analysed by mass spectroscopy. Five milligrams of each peptides was coupled to Sepharose beads using an N-terminal lysine linker. Lysates from HEK293T cells expressing FLAG-tagged proteins were prepared in lysis buffer supplemented with protease and phosphatase inhibitors. Lysates were incubated for 1 h at 4 °C with either phosphorylated or non-phosphorylated peptides conjugated to Sepharose 4B beads. After five washes, pulldown was carried out at 1,000 r.p.m. Bound proteins were subjected to SDS–PAGE and western blotting.
Supplementary Material
Figure S1 The CaaX motif of FBXL2 is necessary to induce the degradation of p85β. (a) FBXL2 does not bind p110α and p110β. U2OS cells were stably transfected with either an empty vector (EV) or the indicated FLAG-tagged proteins. Cells were treated with MG132 for three hours prior to harvesting for immunoprecipitations and immunoblotting as indicated. (WCL, whole cell lysate). (b) HeLa cells were transfected with either GFP-tagged FBXL2 or GFP-tagged FBXL2(Caax/SaaX) constructs. Twenty-four hours after transfection, cells were fixed and incubated with an anti-FLAG antibody (green) and anti-p85β antibody (red). In merged images, yellow shows colocalization of FBXL2(Caax/SaaX) and p85β. Arrows in the middle panels point to the position of GFP-positive cells shown in the top panels. The graphs show the comparison of mean fluorescence between FBXL2-positive and FBXL2-negative cells (top), FBXL2(Caax/SaaX)-positive and FBXL2(Caax/ SaaX)-negative cells (middle), and FBXL2-positive and FBXL2(Caax/SaaX)-positive cells (bottom). Fluorescence intensity was measured by a Nikon Eclipse TE2000-E fluorescence microscope using Metamorph software.
Figure S2 p85β is targeted by FBXL2 for degradation. (a) FBXL2 mutants impaired in their binding to SKP1 form more stable complexes with p85β. HEK-293T cells were transfected with either an empty vector (EV) or the indicated FLAG-tagged FBXL2 constructs (wild type and mutants). Twenty-four hours post-transfection, cells were treated with either MG132 or solvent for three hours prior to harvesting for immunoprecipitations and immunoblotting as indicated. (b) During a 72-hour serum starvation, NHFs were transfected with either an siRNA targeting FBXL2 (#2) or a non-silencing siRNA (NS). Cells were subsequently stimulated with media containing serum and harvested at the indicated time points for immunoblotting. (AS, asynchronous growing cells; SR, serum re-addition).
Figure S3 Mapping of the FBXL2 binding domain on p85β. (a–e) HEK-293T cells were transfected with GFP-tagged FBXL2 and the indicated FLAG-tagged p85β mutants. Whole cell extracts (WCL) were immunoprecipitated (IP) with either anti-FLAG resin or anti-GFP resin as indicated, and immunocomplexes were probed with antibodies to the indicated proteins.
Figure S4 Identification of p85β degron. (a) HEK-293T cells were transfected with GFP-tagged FBXL2 and the indicated FLAG-tagged p85β mutants. Whole cell lysates (WCL) were immunoprecipitated (IP) with anti-FLAG resin, and immunocomplexes were probed with antibodies to the indicated proteins. (b) Alignment of the amino acid regions containing the FBXL2 binding motif in p85β orthologs. (c) Alignment of the amino acid regions containing the FBXL2 binding motif in human p85β with corresponding region in p85α and p85γ from different species.
Figure S5 Tyr655 inhibits p85β binding to FBXL2. (a–b) HEK-293T cells were transfected with GFP-tagged FBXL2 and the indicated FLAG-tagged p85β mutants. Whole cell lysates (WCL) were immunoprecipitated (IP) with anti-FLAG resin, and immunocomplexes were probed with antibodies to the indicated proteins. (c) Schematic representation of p85β mutants used in (ab). Binding of p85β to FBXL2 is indicated with symbol (+).
Figure S6 The binding domain on p85β for PTPL1 and FBXL2 overlap. (a) HEK-293T cells were transfected with HA-tagged PTPL1 in combination with an empty vector (EV) or FLAG-tagged p85β. Whole cell lysates (WCL) were immunoprecipitated (IP) with anti-FLAG resin, and immunocomplexes were probed with antibodies to the indicated proteins. (b) The experiment was performed as in (a), except that the IP was performed with an anti-HA antibody. (c) HEK-293T cells were transfected with an empty vector (EV), FLAG-tagged p85β, or FLAG-tagged p85β(ΔSH2C). Whole cell lysates (WCL) were immunoprecipitated (IP) with anti-FLAG resin, and immunocomplexes were probed with antibodies to the indicated proteins. (d) HEK-293T cells were transfected with GFP-tagged FBXL2 in combination with an empty vector (EV), HA-tagged PTPL1, or HA-tagged p85β. Whole cell lysates (WCL) were immunoprecipitated (IP) with anti-HA antibody, and immunocomplexes were probed with antibodies to the indicated proteins. (e) HEK-293T cells were transfected with GFP-tagged FBXL2 together with either wild type p85β or the indicated FLAG-tagged p85β mutants. Whole cell lysates (WCL) were immunoprecipitated (IP) with anti-FLAG resin, and immunocomplexes were probed with antibodies to the indicated proteins. (f) HEK-293T cells were transfected with wild type p85β or the indicated FLAG-tagged p85β mutants. Whole cell lysates (WCL) were immunoprecipitated (IP) with anti-FLAG resin, and immunocomplexes were probed with antibodies to the indicated proteins. (g) Table summarizing the results presented in (a–f) and Fig. 4a. (NT, not tested).
Figure S7 FBXL2, p85β and PTPL1 form a trimeric complex. (a) HEK-293T cells were transfected with different combinations of HA-tagged PTPL1, FLAG-tagged p85β, and GFP-tagged FBXL2 constructs. Whole cell lysates (WCL) were immunoprecipitated (IP) with anti-FLAG resin, and immunocomplexes were split in two. One fraction was probed with antibodies to the indicated proteins. The second was subjected to elution with an excess of FLAG peptide and re-immunoprecipitated with an anti-GFP antibody followed by immunoblotting as indicated. (b) HEK-293T cells were transfected with HA-tagged p85β and increasing amounts of a plasmid encoding FLAG-tagged FBXL2. Whole cell lysates (WCL) were immunoprecipitated (IP) with an anti-HA resin, and immunoblotting was performed as indicated. The first lane shows cells transfected with an empty vector (EV). (c) During a 72-hour serum starvation, NHFs were transfected twice with either an siRNA targeting PTPL1 or a non-silencing siRNA (NS). Cells were subsequently re-stimulated with media containing serum for 30 minutes and harvested for immunoblotting. (SR, serum re-addition). (d) During a 72-hour serum starvation, NHFs were transfected with either siRNAs targeting FBXL2, both FBXL2 and p85β or a non-silencing siRNA (NS). Cells were subsequently stimulated with media containing serum and harvested at the indicated time points for immunoblotting. The graph in the bottom shows FBXL2 mRNA levels in the different samples analyzed using real-time PCR in triplicate measurements. The values represent the ratios between FBXL2 and GAPDH mRNAs. (SR, serum re-addition). (e) Cells were treated as in (d), except that cells were stimulated with media containing only insulin. (f) Cells (different lines as indicated) were serum starved for 48 hours, then, one hour after serum re-addition, cells were lysed, immunoprecipitated with an anti-IRS1 antibody, and immunoblotted as indicated.
Figure S8 Full scans of immunoblots used in the main figures.
Table S1 MudPIT analysis of FBXL2 immunopurification. Two biological replicate FLAG-affinity purifications were analyzed for both FBXL2 and FBX01 (control). Distributed normalized spectral abundance factors (dNSAF) were averaged, while unique peptides and total spectral counts were merged from the replicate analyses.
Acknowledgments
The authors thank J. Backer, J. H. Lee and R. K. Mallampalli for reagents, and J. Backer, J. R. Skaar and E. Skolnik for critical reading of the manuscript. M.P. is grateful to T. M. Thor for continuous support. This work was financially supported by grants from the National Institutes of Health (R01-GM057587, R37-CA076584 and R21-CA161108) to M.P. and a grant from Susan G. Komen for the Cure to S.D. A.S., L.F. and M.P.W. are supported by the Stowers Institute for Medical Research. M.P. is an Investigator with the Howard Hughes Medical Institute.
Footnotes
Note: Supplementary Information is available in the online version of the paper
AUTHOR CONTRIBUTIONS
S.K. planned and performed most experiments and helped to write the manuscript. M.P. coordinated the study, oversaw the results, and wrote the manuscript. S.D., E.S. and A.P. helped with some experiments. A.S., L.F. and M.P.W. performed the mass spectrometry analysis of the FBXL2 complex purified by S.K. All authors discussed the results and commented on the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reprints and permissions information is available online at www.nature.com/reprints
References
- 1.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 2.Skaar JR, Pagan JK, Pagano M. SnapShot: F box proteins I. Cell. 2009;137:1160–1161. doi: 10.1016/j.cell.2009.05.039. [DOI] [PubMed] [Google Scholar]
- 3.Skaar JR, D’Angiolella V, Pagan JK, Pagano M, SnapShot F. Box Proteins II. Cell. 2009;137:1358. doi: 10.1016/j.cell.2009.05.040. [DOI] [PubMed] [Google Scholar]
- 4.Wang C, et al. Identification of FBL2 as a geranylgeranylated cellular protein required for hepatitis C virus RNA replication. Mol Cell. 2005;18:425–434. doi: 10.1016/j.molcel.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 5.Chen BB, Coon TA, Glasser JR, Mallampalli RK. Calmodulin antagonizes a calcium-activated SCF ubiquitin E3 ligase subunit, FBXL2, to regulate surfactant homeostasis. Mol Cell Biol. 2011;31:1905–1920. doi: 10.1128/MCB.00723-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen BB, et al. F-box protein FBXL2 targets cyclin D2 for ubiquitination and degradation to inhibit leukemic cell proliferation. Blood. 2012;119:3132–3141. doi: 10.1182/blood-2011-06-358911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 8.Katso R, et al. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001;17:615–675. doi: 10.1146/annurev.cellbio.17.1.615. [DOI] [PubMed] [Google Scholar]
- 9.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11:329–341. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 10.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 11.Klippel A, Kavanaugh WM, Pot D, Williams LT. A specific product of phosphatidylinositol 3-kinase directly activates the protein kinase Akt through its pleckstrin homology domain. Mol Cell Biol. 1997;17:338–344. doi: 10.1128/mcb.17.1.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997;275:665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- 13.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 14.Mora A, Komander D, van Aalten DM, Alessi DR. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol. 2004;15:161–170. doi: 10.1016/j.semcdb.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 15.Luo J, Field SJ, Lee JY, Engelman JA, Cantley LC. The p85 regulatory subunit of phosphoinositide 3-kinase down-regulates IRS-1 signalling via the formation of a sequestration complex. J Cell Biol. 2005;170:455–464. doi: 10.1083/jcb.200503088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mauvais-Jarvis F, et al. Reduced expression of the murine p85α subunit of phosphoinositide 3-kinase improves insulin signalling and ameliorates diabetes. J Clin Invest. 2002;109:141–149. doi: 10.1172/JCI13305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geering B, Cutillas PR, Vanhaesebroeck B. Regulation of class IA PI3Ks: is there a role for monomeric PI3K subunits? Biochem Soc Trans. 2007;35:199–203. doi: 10.1042/BST0350199. [DOI] [PubMed] [Google Scholar]
- 18.Fruman DA, et al. Hypoglycaemia, liver necrosis and perinatal death in mice lacking all isoforms of phosphoinositide 3-kinase p85α. Nat Genet. 2000;26:379–382. doi: 10.1038/81715. [DOI] [PubMed] [Google Scholar]
- 19.Ueki K, et al. Molecular balance between the regulatory and catalytic subunits of phosphoinositide 3-kinase regulates cell signalling and survival. Mol Cell Biol. 2002;22:965–977. doi: 10.1128/MCB.22.3.965-977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Florens L, Washburn MP. Proteomic analysis by multidimensional protein identification technology. Methods Mol Biol. 2006;328:159–175. doi: 10.1385/1-59745-026-X:159. [DOI] [PubMed] [Google Scholar]
- 21.Emberley ED, Mosadeghi R, Deshaies RJ. Deconjugation of Nedd8 from Cul1 is directly regulated by Skp1-Fbox and substrate, and CSN inhibits deneddylated SCF by a non-catalytic mechanism. J Biol Chem. 2012;287:29679–29689. doi: 10.1074/jbc.M112.352484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park SW, et al. The regulatory subunits of PI3K, p85α and p85β, interact with XBP-1 and increase its nuclear translocation. Nat Med. 2010;16:429–437. doi: 10.1038/nm.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abaan OD, Toretsky JA. PTPL1: a large phosphatase with a split personality. Cancer Metast Rev. 2008;27:205–214. doi: 10.1007/s10555-008-9114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans. 2009;37:217–222. doi: 10.1042/BST0370217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005;15:702–713. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 26.Zhang H, et al. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signalling through downregulation of PDGFR. J Clin Invest. 2003;112:1223–1233. doi: 10.1172/JCI17222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Songyang Z, et al. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- 29.Yu J, Wjasow C, Backer JM. Regulation of the p85/p110α phosphatidylinositol 3′-kinase. Distinct roles for the n-terminal and c-terminal SH2 domains. J Biol Chem. 1998;273:30199–30203. doi: 10.1074/jbc.273.46.30199. [DOI] [PubMed] [Google Scholar]
- 30.Wu H, et al. Regulation of Class IA PI 3-kinases: C2 domain-iSH2 domain contacts inhibit p85/p110α and are disrupted in oncogenic p85 mutants. Proc Natl Acad Sci USA. 2009;106:20258–20263. doi: 10.1073/pnas.0902369106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burke JE, et al. Dynamics of the phosphoinositide 3-kinase p110delta interaction with p85α and membranes reveals aspects of regulation distinct from p110α. Structure. 2011;19:1127–1137. doi: 10.1016/j.str.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang X, et al. Structure of lipid kinase p110β/p85β elucidates an unusual SH2-domain-mediated inhibitory mechanism. Mol Cell. 2011;41:567–578. doi: 10.1016/j.molcel.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen D, et al. p50α/p55α phosphoinositide 3-kinase knockout mice exhibit enhanced insulin sensitivity. Mol Cell Biol. 2004;24:320–329. doi: 10.1128/MCB.24.1.320-329.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Terauchi Y, et al. Increased insulin sensitivity and hypoglycaemia in mice lacking the p85α subunit of phosphoinositide 3-kinase. Nat Genet. 1999;21:230–235. doi: 10.1038/6023. [DOI] [PubMed] [Google Scholar]
- 35.Ueki K, et al. Increased insulin sensitivity in mice lacking p85β subunit of phosphoinositide 3-kinase. Proc Natl Acad Sci USA. 2002;99:419–424. doi: 10.1073/pnas.012581799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burke JE, Williams RL. Dynamic steps in receptor tyrosine kinase mediated activation of class IA phosphoinositide 3-kinases (PI3K) captured by H/D exchange (HDX-MS) Adv Biol Regul. 2013;53:97–110. doi: 10.1016/j.jbior.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kok K, Geering B, Vanhaesebroeck B. Regulation of phosphoinositide 3-kinase expression in health and disease. Trends Biochem Sci. 2009;34:115–127. doi: 10.1016/j.tibs.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 38.Chagpar RB, et al. Direct positive regulation of PTEN by the p85 subunit of phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. 2010;107:5471–5476. doi: 10.1073/pnas.0908899107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bandyopadhyay GK, Yu JG, Ofrecio J, Olefsky JM. Increased p85/55/50 expression and decreased phosphotidylinositol 3-kinase activity in insulin-resistant human skeletal muscle. Diabetes. 2005;54:2351–2359. doi: 10.2337/diabetes.54.8.2351. [DOI] [PubMed] [Google Scholar]
- 40.Ueki K, Algenstaedt P, Mauvais-Jarvis F, Kahn CR. Positive and negative regulation of phosphoinositide 3-kinase-dependent signalling pathways by three different gene products of the p85α regulatory subunit. Mol Cell Biol. 2000;20:8035–8046. doi: 10.1128/mcb.20.21.8035-8046.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Myers MG, Jr, et al. IRS-1 activates phosphatidylinositol 3′-kinase by associating with src homology 2 domains of p85. Proc Natl Acad Sci USA. 1992;89:10350–10354. doi: 10.1073/pnas.89.21.10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Otsu M, et al. Characterization of two 85 kd proteins that associate with receptor tyrosine kinases, middle-T/pp60c-src complexes, and PI3-kinase. Cell. 1991;65:91–104. doi: 10.1016/0092-8674(91)90411-q. [DOI] [PubMed] [Google Scholar]
- 43.Dromard M, et al. The putative tumour suppressor gene PTPN13/PTPL1 induces apoptosis through insulin receptor substrate-1 dephosphorylation. Cancer Res. 2007;67:6806–6813. doi: 10.1158/0008-5472.CAN-07-0513. [DOI] [PubMed] [Google Scholar]
- 44.McDonald WH, et al. Comparison of three directly coupled HPLC MS/MS strategies for identification of proteins from complex mixtures: single-dimension LCMS/MS, 2-phase MudPIT, and 3-phase MudPIT. Mass Spectrom. 2002;219:245–251. [Google Scholar]
- 45.Eng J, et al. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Amer Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 46.Tabb DL, McDonald WH, Yates JR. 3rd DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res. 2002;1:21–26. doi: 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y, et al. Refinements to label free proteome quantitation: how to deal with peptides shared by multiple proteins. Anal Chem. 2010;82:2272–2281. doi: 10.1021/ac9023999. [DOI] [PubMed] [Google Scholar]
- 48.D’Angiolella V, et al. SCF(Cyclin F) controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature. 2010;466:138–142. doi: 10.1038/nature09140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duan S, et al. mTOR generates an auto-amplification loop by triggering the β TrCP- and CK1α-dependent degradation of DEPTOR. Mol Cell. 2011;44:317–324. doi: 10.1016/j.molcel.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.D’Angiolella V, et al. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell. 2012;149:1023–1034. doi: 10.1016/j.cell.2012.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Duan S, et al. FBXO11 targets BCL6 for degradation and is inactivated in diffuse large B-cell lymphomas. Nature. 2012;481:90–93. doi: 10.1038/nature10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 The CaaX motif of FBXL2 is necessary to induce the degradation of p85β. (a) FBXL2 does not bind p110α and p110β. U2OS cells were stably transfected with either an empty vector (EV) or the indicated FLAG-tagged proteins. Cells were treated with MG132 for three hours prior to harvesting for immunoprecipitations and immunoblotting as indicated. (WCL, whole cell lysate). (b) HeLa cells were transfected with either GFP-tagged FBXL2 or GFP-tagged FBXL2(Caax/SaaX) constructs. Twenty-four hours after transfection, cells were fixed and incubated with an anti-FLAG antibody (green) and anti-p85β antibody (red). In merged images, yellow shows colocalization of FBXL2(Caax/SaaX) and p85β. Arrows in the middle panels point to the position of GFP-positive cells shown in the top panels. The graphs show the comparison of mean fluorescence between FBXL2-positive and FBXL2-negative cells (top), FBXL2(Caax/SaaX)-positive and FBXL2(Caax/ SaaX)-negative cells (middle), and FBXL2-positive and FBXL2(Caax/SaaX)-positive cells (bottom). Fluorescence intensity was measured by a Nikon Eclipse TE2000-E fluorescence microscope using Metamorph software.
Figure S2 p85β is targeted by FBXL2 for degradation. (a) FBXL2 mutants impaired in their binding to SKP1 form more stable complexes with p85β. HEK-293T cells were transfected with either an empty vector (EV) or the indicated FLAG-tagged FBXL2 constructs (wild type and mutants). Twenty-four hours post-transfection, cells were treated with either MG132 or solvent for three hours prior to harvesting for immunoprecipitations and immunoblotting as indicated. (b) During a 72-hour serum starvation, NHFs were transfected with either an siRNA targeting FBXL2 (#2) or a non-silencing siRNA (NS). Cells were subsequently stimulated with media containing serum and harvested at the indicated time points for immunoblotting. (AS, asynchronous growing cells; SR, serum re-addition).
Figure S3 Mapping of the FBXL2 binding domain on p85β. (a–e) HEK-293T cells were transfected with GFP-tagged FBXL2 and the indicated FLAG-tagged p85β mutants. Whole cell extracts (WCL) were immunoprecipitated (IP) with either anti-FLAG resin or anti-GFP resin as indicated, and immunocomplexes were probed with antibodies to the indicated proteins.
Figure S4 Identification of p85β degron. (a) HEK-293T cells were transfected with GFP-tagged FBXL2 and the indicated FLAG-tagged p85β mutants. Whole cell lysates (WCL) were immunoprecipitated (IP) with anti-FLAG resin, and immunocomplexes were probed with antibodies to the indicated proteins. (b) Alignment of the amino acid regions containing the FBXL2 binding motif in p85β orthologs. (c) Alignment of the amino acid regions containing the FBXL2 binding motif in human p85β with corresponding region in p85α and p85γ from different species.
Figure S5 Tyr655 inhibits p85β binding to FBXL2. (a–b) HEK-293T cells were transfected with GFP-tagged FBXL2 and the indicated FLAG-tagged p85β mutants. Whole cell lysates (WCL) were immunoprecipitated (IP) with anti-FLAG resin, and immunocomplexes were probed with antibodies to the indicated proteins. (c) Schematic representation of p85β mutants used in (ab). Binding of p85β to FBXL2 is indicated with symbol (+).
Figure S6 The binding domain on p85β for PTPL1 and FBXL2 overlap. (a) HEK-293T cells were transfected with HA-tagged PTPL1 in combination with an empty vector (EV) or FLAG-tagged p85β. Whole cell lysates (WCL) were immunoprecipitated (IP) with anti-FLAG resin, and immunocomplexes were probed with antibodies to the indicated proteins. (b) The experiment was performed as in (a), except that the IP was performed with an anti-HA antibody. (c) HEK-293T cells were transfected with an empty vector (EV), FLAG-tagged p85β, or FLAG-tagged p85β(ΔSH2C). Whole cell lysates (WCL) were immunoprecipitated (IP) with anti-FLAG resin, and immunocomplexes were probed with antibodies to the indicated proteins. (d) HEK-293T cells were transfected with GFP-tagged FBXL2 in combination with an empty vector (EV), HA-tagged PTPL1, or HA-tagged p85β. Whole cell lysates (WCL) were immunoprecipitated (IP) with anti-HA antibody, and immunocomplexes were probed with antibodies to the indicated proteins. (e) HEK-293T cells were transfected with GFP-tagged FBXL2 together with either wild type p85β or the indicated FLAG-tagged p85β mutants. Whole cell lysates (WCL) were immunoprecipitated (IP) with anti-FLAG resin, and immunocomplexes were probed with antibodies to the indicated proteins. (f) HEK-293T cells were transfected with wild type p85β or the indicated FLAG-tagged p85β mutants. Whole cell lysates (WCL) were immunoprecipitated (IP) with anti-FLAG resin, and immunocomplexes were probed with antibodies to the indicated proteins. (g) Table summarizing the results presented in (a–f) and Fig. 4a. (NT, not tested).
Figure S7 FBXL2, p85β and PTPL1 form a trimeric complex. (a) HEK-293T cells were transfected with different combinations of HA-tagged PTPL1, FLAG-tagged p85β, and GFP-tagged FBXL2 constructs. Whole cell lysates (WCL) were immunoprecipitated (IP) with anti-FLAG resin, and immunocomplexes were split in two. One fraction was probed with antibodies to the indicated proteins. The second was subjected to elution with an excess of FLAG peptide and re-immunoprecipitated with an anti-GFP antibody followed by immunoblotting as indicated. (b) HEK-293T cells were transfected with HA-tagged p85β and increasing amounts of a plasmid encoding FLAG-tagged FBXL2. Whole cell lysates (WCL) were immunoprecipitated (IP) with an anti-HA resin, and immunoblotting was performed as indicated. The first lane shows cells transfected with an empty vector (EV). (c) During a 72-hour serum starvation, NHFs were transfected twice with either an siRNA targeting PTPL1 or a non-silencing siRNA (NS). Cells were subsequently re-stimulated with media containing serum for 30 minutes and harvested for immunoblotting. (SR, serum re-addition). (d) During a 72-hour serum starvation, NHFs were transfected with either siRNAs targeting FBXL2, both FBXL2 and p85β or a non-silencing siRNA (NS). Cells were subsequently stimulated with media containing serum and harvested at the indicated time points for immunoblotting. The graph in the bottom shows FBXL2 mRNA levels in the different samples analyzed using real-time PCR in triplicate measurements. The values represent the ratios between FBXL2 and GAPDH mRNAs. (SR, serum re-addition). (e) Cells were treated as in (d), except that cells were stimulated with media containing only insulin. (f) Cells (different lines as indicated) were serum starved for 48 hours, then, one hour after serum re-addition, cells were lysed, immunoprecipitated with an anti-IRS1 antibody, and immunoblotted as indicated.
Figure S8 Full scans of immunoblots used in the main figures.
Table S1 MudPIT analysis of FBXL2 immunopurification. Two biological replicate FLAG-affinity purifications were analyzed for both FBXL2 and FBX01 (control). Distributed normalized spectral abundance factors (dNSAF) were averaged, while unique peptides and total spectral counts were merged from the replicate analyses.