

Abstract
Objective
Dual specificity phosphatase 1 (DUSP1) inhibits mitogen activated protein kinase activity, and is activated by several stimuli such as sustained hypoxia, oxidative stress, and hormones. However, the effect of intermittent hypoxia is not known. The aim of this study was to evaluate the role of intermittent hypoxia on DUSP1 expression, and to validate its role in a human model of intermittent hypoxia, as seen in obstructive sleep apnea (OSA). OSA is characterized by recurrent episodes of hypoxemia/reoxygenation and is a known risk factor for cardiovascular morbidity.
Methods
In-vitro studies using human coronary artery endothelial cells (HCAEC) and ex-vivo studies using white blood cells isolated from healthy and OSA subjects.
Results
Intermittent hypoxia induced DUSP1 expression in human coronary artery endothelial cells (HCAEC), and in granulocytes isolated from healthy human subjects. Functionally, DUSP1 increased the expression and activity of manganese superoxide dismutase (MnSOD) in HCAEC. Further, significant increases in DUSP1 mRNA from total blood, and in DUSP1 protein in mononuclear cells and granulocytes isolated from OSA subjects, was observed in the early morning hours after one night of intermittent hypoxemia due to untreated OSA. This early-morning OSA-induced augmentation of DUSP1 gene expression was attenuated by continuous positive airway pressure (CPAP) treatment of OSA.
Conclusion
Intermittent hypoxia increases MnSOD activity via increased DUSP1 expression in HCAEC. Similarly, overnight intermittent hypoxemia in patients with OSA induces expression of DUSP1, which may mediate increases of MnSOD expression and activity. This may contribute significantly to neutralizing the effects of reactive oxygen species, a consequence of the intermittent hypoxemia/reperfusion elicited by OSA.
Keywords: Dual specificity phosphatase 1, intermittent hypoxia, Obstructive sleep apnea, reactive oxygen species
Introduction
Dual specificity phosphatase 1 (DUSP1) belongs to a family of mitogen activated protein kinase (MAPK) phosphatases and is closely involved in the regulation of intracellular signaling. DUSP1 is a nuclear enzyme encoded by an immediate-early gene and its transcription is rapidly induced by stimuli such as growth factors, oxidative stresses and continuous hypoxia [1–3]. DUSP1 inactivates several members of the MAPK family including JNK, p38 and ERK through dephosphorylation of threonine and tyrosine residues within their activation site [4]. These kinases play an important regulatory role in cellular apoptosis, proliferation, and proinflammatory responses which are critical to the pathophysiology of cardiovascular disease.
Obstructive sleep apnea (OSA) is the commonest form of sleep disordered breathing, and is characterized by recurrent episodes of cessation of respiratory airflow during sleep leading to periods of intermittent hypoxia (IH) and sleep fragmentation. Apnea duration of greater than 10 seconds is commonly accompanied by significant decreases in systemic oxygen concentrations, with consequent myocardial and systemic hypoxemia. OSA has been independently linked to increased cardiovascular morbidity and mortality [5–7]. Clinical studies have shown that the association between OSA and coronary artery disease may be via nocturnal hypoxemia [8–10]. Some studies have also reported a link between apnea index and incidence of myocardial infarction, myocardial ischemia and stroke in OSA patients [11, 12]. However, while the molecular mechanisms linking OSA to cardiovascular diseases are poorly understood, oxidative stress and free radical generation in response to repetitive hypoxemic stress have been implicated [13].
DUSP1 is increased in response to sustained hypoxia [1, 2] but how DUSP1 expression changes in intermittent hypoxia (IH) is not known. Intermittent hypoxia experienced by OSA patients has been implicated in the development of cardiovascular pathophysiology in this patient population [14]. Therefore, we tested the hypothesis that IH would increase DUSP1 expression in vascular endothelial cells and granulocytes in-vitro, and that this increase in DUSP1 would also be evident in an in-vivo human model of IH, as seen in patients with OSA. These findings would provide important insights regarding the signaling mechanisms involved in OSA-related oxidative stress.
Materials and Methods
Effect of intermittent hypoxia (IH) in cultured human coronary artery endothelial cells (HCAEC) and granulocytes
Vascular endothelial cells in conjunction with cells of the immune system play an important role in the genesis of cardiovascular disease. Initiation, development, and progression of atherosclerotic lesions are orchestrated by interactions between granulocytes, lymphocytes, and the endothelium [15]. Granulocytes play an important regulatory role in the inflammatory process and are likely to be modulated by oxidative stress. Since both inflammation and oxidative stress constitute putative elements of sleep apnea pathophysiology, we studied the effects of intermittent hypoxia in coronary artery endothelial cells and granulocytes. Further, accessibility of white blood cells from humans would enable validation of our in-vitro findings in OSA subjects before and after episodes of overnight intermittent hypoxemia.
Human coronary artery endothelial cells (HCAEC; Clonetics, USA) were grown in EBM-2 media supplemented with growth factors and 2% FBS. All experiments were performed at 3–5 passages with 70–80% confluence. We optimized the time and oxygen concentrations required to achieve deoxygenation of the cell media to the level observed in severe OSA patients. pO2 was measured after each hypoxia exposure using i- STAT EG7+ (Abbott, USA). A 30–40% decreases in pO2, mimicking the degree of hypoxemia observed in severe OSA patients during sleep, were achieved in the conditioned media by 25 min of continuous exposure of cells to 0% O2 + 5% CO2. Intermittent hypoxia was achieved by repeated 25 min exposure to 21% O2 + 5% CO2 followed by 25 min exposure to 0% O2 + 5% CO2, 7 hours each day for 4 days. Cells grown in continuous 21% O2 + 5% CO2 were used as normoxic controls for these experiments. After the 4 days of IH treatment, the cells were lysed in buffer containing 50 mM sodium pyrophosphate, 50 mM sodium fluoride, 50 mM sodium chloride, 5 mM EDTA, 5 mM EGTA, 100 uM sodium vanadate, 10 mM HEPES, 0.1% Triton and 2% proteases inhibitor cocktail (Sigma, USA). Western blot analysis was done to determine the expression of DUSP1 and β-actin was used as loading control (anti-human DUSP1, Santa Cruz Biotech, USA; anti-human β-actin, Sigma, USA). Since manganese superoxide dismutase (MnSOD) is an important enzyme involved in neutralizing the effects of reactive oxygen species and is likely increased with intermittent hypoxia, cell lysates were used to determine MnSOD expression as well (anti-MnSOD, Stressgen, Canada).
To determine the role of DUSP1 in intermittent hypoxia, we inhibited DUSP1 expression by RNAi. Transfection studies were done using a mixture of DUSP1 Stealth RNAi oligo (HSS1029830 and HSS1029840), recommended Control Stealth RNAi oligo and Lipofectamine™ RNAi MAX (Invitrogen, USA). Transfection was performed according to the manufacturer’s recommendations in 6 well plates using OptiMEM I serum reduced medium and 80 pmol of each oligo. Transfected cells were exposed to intermittent hypoxia for two days and protein analyzed by western blot. Total SOD activity was measured in the experimental cell lysate using SOD assay kit (Sigma-Aldrich, Japan) according to the manufacturer’s instructions.
Similarly, to examine the effect of IH on granulocytes, cells were isolated from healthy human subjects by standard protocol using Histopaque (Sigma, USA) gradient. The isolated cells were aliquoted into two 100 mm dishes (2×106 cells/ml) in serum free RPMI media. Group 1 was the control group and incubated at 21% O2 + 5% CO2 for 48 hours, and group 2 was exposed to intermittent hypoxia (25 min exposure to 21% O2 + 5% CO2 followed by 25 min exposure to 0% O2 + 5% CO2), 7 hours each day for two days. We restricted the IH treatment for two days in these cells to balance viability and maximize the effects of IH. After 48h of intermittent hypoxia treatment, the cells were lysed and Western blot analysis was done to determine the expression of DUSP1.
Human subjects
16 subjects were enrolled in the study. We compared subjects with severe OSA (apnea hypopnea index [AHI] >30) to whose without OSA (AHI<5). The OSA patients were newly diagnosed and free of other diseases, had never been treated for OSA and were taking no medications. They were age, BMI and gender matched with 8 healthy subjects in whom occult OSA was excluded. All subjects underwent full polysomnography. Of the OSA patients, four underwent a subsequent polysomnography study with CPAP treatment. Blood was collected at three time points, before sleep (9 pm, 4 hours after last meal), immediately after arousal (at 6 am), and five hours after waking from sleep (11 am). Written informed consent was obtained from all subjects. The study was approved by the Institutional Human Subjects Review Committee.
DUSP1 mRNA quantization from total blood
RNA was isolated from the blood and quantified for the DUSP1 expression. The blood containing tubes were initially incubated at room temperature for 3 hours to stabilize cellular RNA followed by its isolation using PAX Gene RNA isolation kit (Qiagen, Chatsworth, CA) according to manufacturer’s instructions. cDNA was synthesized using total RNA (1μg/reaction) with high capacity cDNA Archive kit (Applied Biosystems, Foster City, CA). DUSP1 TaqMan probe and β-actin RNA TaqMan probe (endogenous control) (Applied Biosystems, Foster City, CA) were used in standard conditions (as determined by the manufacturer) to determine the level of DUSP1 transcription in the isolated cells. Absolute values of DUSP1 and β-actin RNA transcripts were calculated using standards. The DUSP1 value obtained from the standard curve was divided by the value for endogenous control to obtain a normalized target value ratio.
DUSP1 protein analysis in mononuclear cells and granulocytes from human subjects
The blood samples were collected at three time-points in tubes containing citrate, diluted in PBS-citrate buffer, layered with Histopaque 1077 (Sigma, USA) and centrifuged to separate the mononuclear cells and granulocytes. The mononuclear layer and the granulocyte containing pellet were collected and washed with RBC lysis buffer and PBS citrate. The cells were lysed and proteins analyzed by western blots.
Statistical analysis
The differences between the groups were assessed using Student’s T-Test and two-way analysis of variance (ANOVA) for repeated measures. All western blots and in vitro experiments were repeated at least three times.
Results
Intermittent hypoxia induces DUSP1 and MnSOD protein expression
Increased expression and nuclear localization of DUSP1 was seen in HCAEC treated with intermittent hypoxia (Figure 1A; Figure 1C, control). The effect of IH on DUSP1 expression was also determined in granulocytes isolated from healthy human subjects. An increased expression of DUSP1 was observed in cells treated with IH as compared to untreated cells (Figure 1B). Further, intermittent hypoxia increased MnSOD expression (Figure 1C, control) as well as superoxide dismutase (SOD) activity (Figure 1D, light grey bars).
Figure 1.

Effect of IH treatment in human coronary artery endothelial cells (HCAEC) and granulocytes. (A) Confocal images showing increased expression and nuclear (blue) localization of DUSP1 (red) in intermittent hypoxia treated HCEAC. (B) Representative Western blot showing increased DUSP1 expression in cultured granulocytes after IH treatment. (C) Representative Western blots showing increased expression of DUSP1 and MnSOD in control-RNAi transfected cells after IH treatment. The cells transfected with DUSP1 RNAi failed to show any increase in DUSP1 or MnSOD after IH treatment. (D) Graphical representation of SOD activity in HCAEC cells from Control RNAi (light grey bars) and DUSP1 RNAi (dark grey bars) treated cells after IH treatment. IH increased SOD activity in control, however cells transfected with DUSP1 RNAi did not show any increase in SOD activity after IH exposure. Data presented as mean ± SD of three independent experiments. Statistical significance was determined by unpaired t-tests. †is P<0.05 N vs. IH treatment in control-RNAi transfected cells. $ is P<0.005 control vs. DUSP1 RNAi transfected cells in IH condition. N: Normoxia; IH: Intermittent hypoxia.
Increased MnSOD expression and activity with IH treatment is mediated by increased DUSP1 expression
The role of DUSP1 in induction of MnSOD was determined using RNAi specific for DUSP1. Cells transfected with DUSP1 RNAi did not show any significant changes in expression of MnSOD after the IH treatment (Figure 1C, DUSP1 transfected) indicating that the increases in MnSOD expression with IH treatment were dependent on increases in DUSP1 protein expression. Similarly, IH dependent increases in total SOD activity were inhibited by DUSP1 RNAi transfection (Figure 1D, dark grey bars).
Increased DUSP1 expression in OSA subjects after sleep
The baseline characteristics and sleep profiles of the control subjects and OSA patients are shown in Table 1. All variables associated with sleep profiles differed significantly between the two groups, specifically nocturnal oxygen saturation, apnea/hypopnea index (AHI) and arousal index.
Table 1.
Anthropometrics and sleep profiles of study population.
| Variable | Controls (N=8) | OSA (N=8) | P value |
|---|---|---|---|
| Age (yrs) | 36.5 ± 4.8 | 39.7 ± 10.8 | NS |
| BMI (kg/m2) | 30.0 ± 4.07 | 33.0 ± 3.7 | NS |
| Baseline SpO2 | 96.9 ± 0.8 | 97.14 ± 1.06 | NS |
| AHI (events/hr) | 0.6 ± 0.7 | 47.9 ± 23.8 | <0.0001 |
| Mean nocturnal SpO2 | 96.0 ± 0.92 | 93.8 ± 1.86 | 0.01 |
| Lowest nocturnal SpO2 | 90.16 ± 3.68 | 67.42 ± 19.07 | 0.005 |
| % of sleep time with SpO2>90% | 99.95 ± 0.07 | 91.62 ± 9.94 | 0.03 |
| Arousal Index (events/hr) | 15.87 ± 6.02 | 45.87 ± 15.52 | <0.0001 |
Data presented as mean values ± SD. P-values for unpaired t-tests. BMI – body mass index; SpO2 – blood oxygen saturation; AHI – apnea/hypopnea index.
The effect of overnight intermittent hypoxia experienced by OSA subjects on DUSP1 expression was determined at the mRNA level in whole blood (Figure 2) and at the protein level in isolated white blood cells (Figure 3). DUSP1 gene expression and protein level did not differ between healthy controls (white bars) and OSA subjects (black bars) at baseline (before the sleep). A significant increase in DUSP1 transcripts in total blood, as determined by the quantitative RNA analysis, was seen only at the second time point of the OSA patients, namely at 6:00 am, after waking from a night of untreated OSA (Figure 2A). DUSP1 mRNA tended to decrease at the third time point in OSA patients at 11 am, after about 5 hours of normoxic wakefulness. Control subjects did not show any significant differences across the three time points. The increase in DUSP1 mRNA as seen in OSA patients during the second time point, however, was attenuated by CPAP treatment (Figure 2B, grey bars). The increased transcription of DUSP1 mRNA resulted in increased expression of DUSP1 protein during the early morning hours in the granulocytes (Figure 3A) and mononuclear cells (Figure 3B) isolated from the OSA patients. Similar to mRNA results, the healthy control subjects did not show any changes in DUSP1 protein expression at any time points.
Figure 2.
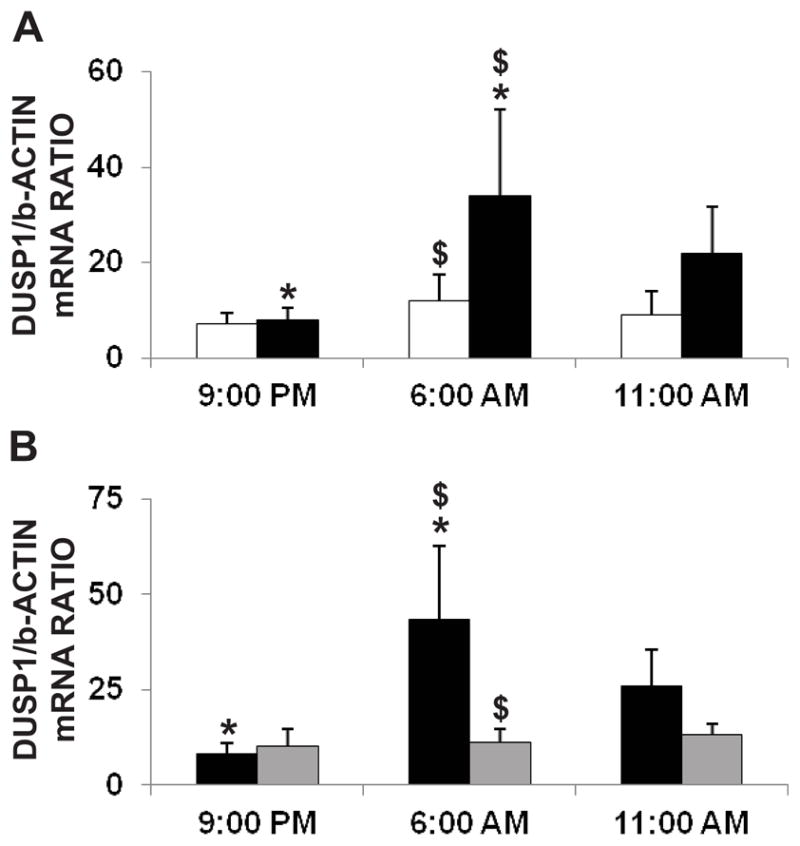
Effect of overnight IH on whole blood DUSP1 mRNA from control and OSA subjects. (A) Graphical representation of whole blood DUSP1 mRNA before sleep (9:00 pm), after sleep (6:00 am) and during day (11:00 am) in healthy controls (white bars; n=8) and OSA subjects (black bars; n=8). Increased DUSP1 mRNA was seen in OSA subjects at 6:00 am after a night of IH episodes (*is P≤ 0.05, 6:00 am vs. 9:00 pm OSA group; $ is P≤ 0.05, 6:00 am control vs. OSA group). DUSP1 mRNA showed no differences over the three time points in the control group. (B) Increases in early morning DUSP1 mRNA in OSA subjects is attenuated by nCPAP treatment. The black bars represent OSA subjects without nCPAP (n=4) and the grey bars represent OSA subjects with nCPAP treatment (n=4). No change in DUSP1 mRNA was observed in OSA subjects with nCPAP treatment in all three time points whereas patients naïve to treatment were characterized by evident DUSP1 mRNA increases at 6:00 am (*is P≤0.05, 6:00 am vs. 9:00 pm OSA group without nCPAP treatment; $ is P≤0.05, 6:00 am OSA group with vs. without nCPAP treatment). Data are presented as mean ± SD. Statistical significance was determined by unpaired student t-test for direct comparisons between groups, and ANOVA with repeated measurements for time-related assessment.
Figure 3.

Effect of overnight IH on DUSP1 protein expression in white blood cells from control and OSA subjects. Representative western blot and graph showing expression of DUSP1 protein in (A) granulocytes and (B) lymphocytes from control (white bars, n=8) and OSA subject (black bar, n=8) before sleep (9:00 pm), after sleep (6:00 am) and during day (11:00 am). DUSP1 protein expression was increased only in OSA subjects after sleep (6:00 am) in both granulocytes and lymphocytes. Statistical significance was determined by unpaired student t-test *P < 0.05, 9.00 pm vs. 6.00 am OSA group; $ p<0.04, 6:00 am Control vs. OSA group.
Discussion
The most important finding of our study is that there is a marked increase in DUSP1 gene transcription and translation in white blood cells of OSA patients. This increased expression is observed in the early morning hours immediately after waking from a night of untreated obstructive apnea, and decreases later during daytime wakefulness. The increase of DUSP1 expression seems to be an early acute response to intermittent hypoxemic episodes occurring during sleep. We also demonstrate that OSA patients undergoing CPAP treatment, and healthy subjects free of OSA, do not show any increase in DUSP1, further confirming the role of intermittent hypoxia during sleep apnea in mediating DUSP1 expression. In-vitro studies in vascular endothelial cells and granulocytes isolated from healthy subjects further validate the concept that recurrent episodes of hypoxia induce the expression of DUSP1.
OSA patients are exposed to intermittent hypoxemia which is a putative source of oxidative stress. However, objective evidence of oxidative stress in these patients has been inconsistent [16–23]. Any available evidence of oxidative stress is indirect and suggests that there exists a very effective mechanism which manages the increased reactive oxygen species (ROS) [24]. Oxidative stress is a strong stimulus for DUSP1 induction [1–3]; hence we hypothesized that over-expression of DUSP1 may itself contribute to attenuation of oxidative stress. We investigated the effect of DUSP1 in regulation of MnSOD, a key protein involved in oxidative stress management, and demonstrate that DUSP1 regulates the expression of MnSOD in HCAEC. MnSOD is one of three isoenzymes of superoxide dismutase that are present in mitochondria. These enzymes react directly with reactive oxygen species converting superoxide anion to comparatively less reactive hydrogen superoxide. MnSOD might also play an important role in the endothelial cell life cycle, such as by promoting endothelial wound healing [25]. Therefore, IH-induced DUSP1 mediated activation of MnSOD in HCAEC through anti-oxidative and anti-aging properties might protect against certain adverse processes occurring during intermittent hypoxia. It has been previously reported that MnSOD appears to act as a signaling mediator for the activation of survival genes following hypoxia/reoxygenation injury [26]. These observations, in the context of our studies, give new insight into the role of MnSOD in endothelial cells in obstructive sleep apnea, with a potential role for DUSP1 as the oxidative stress controlling agent. This role of DUSP1 in reducing oxidative stress is consistent with findings from other studies [27].
The major role of DUSP1 is inhibition of mitogen activated protein kinases (MAPK) dependent downstream cellular signal transmission. MAPKs are crucial cellular signaling mechanisms. Cellular responses to oxidative stress, hypoxia, inflammation, and other stresses are mediated via this pathway. When activated, the MAPK pathway leads to increased expression of downstream transcription factors involved in regulating the cell cycle, inflammation, apoptosis, and cell differentiation. The role of MAPK has been widely implicated in the pathophysiology of cardiovascular disease, including in cardio-protection against ischemia/reperfusion injury and ischemic preconditioning [28–30], as well as anti-apoptotic mechanisms, and activation of inflammatory processes (activation of E-selectin, cyclooxygenase COX-2, IL-6, IL-1, TNF alpha and macrophage colony stimulating factor) [31–34]. In other words, while MAPKs are crucial for sustaining the most important cell functions, hyperactivation of these molecules could disrupt normal cell cycle activities and contribute to development of pathology. DUSP1 is involved in inactivation of the MAPK pathway as part of a negative feedback mechanism which may contribute to regulating and limiting stress-induced cellular responses. Increased activity of DUSP1 in OSA might play a crucial role in adaptation to hypoxemia–reoxygenation, and may be protective in acute conditions. We have demonstrated the role of DUSP1 in induction of MnSOD in HCAEC, suggesting a protective role of DUSP1 in intermittent hypoxia and sleep apnea. Increased expression of DUSP1 in response to OSA, with consequent induction of MnSOD and attenuation of ROS, may help explain the absence of overt oxidative stress in otherwise healthy patients with OSA [16–18, 23, 27].
OSA has been linked to systemic inflammation with evidence of leukocyte activation, production of adhesion molecules, leukocyte binding to endothelial cells, and increased levels of C-reactive protein, IL-6, TNF alpha, IFN gamma [7]. White blood cells are known to be activated in OSA [35, 36]. Several studies have demonstrated that DUSP1 modulates inflammatory immune responses, acting as an anti-inflammatory agent [37–39]. Therefore the changes in DUSP1 activity in white blood cells might play a significant role in modulating activity of the immune system in OSA.
Our present findings are consistent with microarray data from OSA vs. healthy control subjects, which suggest that transcription of genes encoding several proteins involved in cellular cycle and cell proliferation are up-regulated in OSA subjects [24]. Expression of DUSP1 as the direct transcriptional target of p53 has been shown to prevent cells from entering into the cell cycle [40]. Hence further investigation of the role of DUSP1 in OSA in terms of this potentially beneficial response, protecting the most vulnerable phase of the cell cycle from the stresses associated with apnea and hypoxemia, is warranted.
However the role of DUSP1 is somewhat controversial. DUSP1, via deactivation of p38 or JNK, can contribute to adaptation to hostile conditions. Recent studies confirm a significant protective role of DUSP1 in attenuation of both acute inflammatory responses [41], and cardiac remodeling associated with chronic elevation in blood pressure [42]. On the other hand, decreased DUSP1 activity has been linked to a reduction of atherosclerosis and atherosclerotic lesion development [43–45]. DUSP1 is also activated by intermittent hypoxemia in OSA. This diversity of outcomes might depend on the severity and duration of DUSP1 activation, polymorphisms which influence its activity, co-morbidities, unique individual-specific phenotypes, or other factors regulating intracellular MAPK signaling.
Conclusion
Intermittent hypoxemia during OSA elicits increases in DUSP1 expression, an effect that is attenuated by treatment of OSA with CPAP. DUSP1 increases MnSOD, hence modulating ROS, a likely key mediator of cardiovascular pathophysiology. We suggest that acute increases in DUSP1 activity during apneic sleep in OSA patients might contribute to attenuating the potential harmful effects of hypoxemia-reoxygenation, i.e. oxidative stress and inflammation. DUSP1 activation in OSA may help explain the absence of overt increases in oxidative stress in OSA patients [16–18, 23]. However, it is not known if recurrent nightly increases of DUSP1 over a long duration, as in chronically untreated OSA, may contribute to acceleration of atherosclerosis. The response of DUSP1 to intermittent hypoxemia in OSA, as well as differential protective versus pathologic effects of DUSP1, may be implicated in the development of cardiovascular complications in some but not all OSA patients.
Highlights.
Obstructive sleep apnea is associated with increased DUSP1 expression
Treatment of sleep apnea with CPAP prevents overnight increases in DUSP1 expression
Intermittent hypoxia induces DUSP1 expression
Role of DUSP1 in increasing expression and activity of MnSOD
Molecular insight into initiation and progression of cardiovascular diseases in OSA
Acknowledgments
Funding from the following sources is acknowledged. MSH is supported by PNRF-213-AI-1/07 grant. PS is supported by AHA Scientist Development grant 11SGD7260046 and NIH R01 HL65176, KN is supported by PNRF-213-AI-1/07 grant, and VKS is supported by NIH R01 HL65176 as well as UL1 TR000135 from the National Center for Advancing Translational Sciences (NCATS). The contents of the manuscript are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
Competing interests: VKS has served as a consultant for Medtronic, Neu Pro, and Res Med, and works with Mayo Health Solutions and their industry partners on intellectual property related to sleep and cardiovascular disease. Mayo Foundation has received a gift from the Phillips-Respironics Foundation for the study of sleep apnea and cardiovascular disease.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Laderoute KR, Mendonca HL, Calaoagan JM, et al. Mitogen-activated protein kinase phosphatase-1 (MKP-1) expression is induced by low oxygen conditions found in solid tumor microenvironments. A candidate MKP for the inactivation of hypoxia-inducible stress-activated protein kinase/c-Jun N-terminal protein kinase activity. J Biol Chem. 1999;274(18):12890–97. doi: 10.1074/jbc.274.18.12890. [DOI] [PubMed] [Google Scholar]
- 2.Seta KA, Kim R, Kim HW, et al. Hypoxia-induced regulation of MAPK phosphatase-1 as identified by subtractive suppression hybridization and cDNA microarray analysis. J Biol Chem. 2001;276(48):44405–12. doi: 10.1074/jbc.M103346200. [DOI] [PubMed] [Google Scholar]
- 3.Franklin CC, Kraft AS. Conditional expression of the mitogen-activated protein kinase (MAPK) phosphatase MKP-1 preferentially inhibits p38 MAPK and stress-activated protein kinase in U937 cells. J Biol Chem. 1997;272(27):16917–23. doi: 10.1074/jbc.272.27.16917. [DOI] [PubMed] [Google Scholar]
- 4.Hirsch DD, Stork PJ. Mitogen-activated protein kinase phosphatases inactivate stress-activated protein kinase pathways in vivo. J Biol Chem. 1997;272(7):4568–75. doi: 10.1074/jbc.272.7.4568. [DOI] [PubMed] [Google Scholar]
- 5.Young T, Palta M, Dempsey J, et al. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med. 1993;328(17):1230–35. doi: 10.1056/NEJM199304293281704. [DOI] [PubMed] [Google Scholar]
- 6.Peppard PE, Young T, Palta M, et al. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med. 2000;342(19):1378–84. doi: 10.1056/NEJM200005113421901. [DOI] [PubMed] [Google Scholar]
- 7.Kuniyoshi FH, Pusalavidyasagar S, Singh P, et al. Cardiovascular consequences of obstructive sleep apnoea. The Indian journal of medical research. 2010;131:196–205. [PubMed] [Google Scholar]
- 8.Mooe T, Rabben T, Wiklund U, et al. Sleep-disordered breathing in women: occurrence and association with coronary artery disease. The American journal of medicine. 1996;101(3):251–56. doi: 10.1016/S0002-9343(96)00122-2. [DOI] [PubMed] [Google Scholar]
- 9.Mooe T, Rabben T, Wiklund U, et al. Sleep-disordered breathing in men with coronary artery disease. Chest. 1996;109(3):659–63. doi: 10.1378/chest.109.3.659. [DOI] [PubMed] [Google Scholar]
- 10.Sert Kuniyoshi FH, Singh P, Gami AS, et al. Patients with obstructive sleep apnea exhibit impaired endothelial function after myocardial infarction. Chest. 2011;140(1):62–67. doi: 10.1378/chest.10-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hung J, Whitford EG, Parsons RW, et al. Association of sleep apnoea with myocardial infarction in men. Lancet. 1990;336(8710):261–64. doi: 10.1016/0140-6736(90)91799-g. [DOI] [PubMed] [Google Scholar]
- 12.Yaggi HK, Concato J, Kernan WN, et al. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med. 2005;353(19):2034–41. doi: 10.1056/NEJMoa043104. [DOI] [PubMed] [Google Scholar]
- 13.Lavie L, Lavie P. Molecular mechanisms of cardiovascular disease in OSAHS: the oxidative stress link. The European respiratory journal. 2009;33(6):1467–84. doi: 10.1183/09031936.00086608. [DOI] [PubMed] [Google Scholar]
- 14.Punjabi NM, Sorkin JD, Katzel LI, et al. Sleep-disordered breathing and insulin resistance in middle-aged and overweight men. American journal of respiratory and critical care medicine. 2002;165(5):677–82. doi: 10.1164/ajrccm.165.5.2104087. [DOI] [PubMed] [Google Scholar]
- 15.Harrison DG, Guzik TJ, Lob HE, et al. Inflammation, immunity, and hypertension. Hypertension. 2011;57(2):132–40. doi: 10.1161/HYPERTENSIONAHA.110.163576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alzoghaibi MA, Bahammam AS. Lipid peroxides, superoxide dismutase and circulating IL-8 and GCP-2 in patients with severe obstructive sleep apnea: a pilot study. Sleep & breathing = Schlaf & Atmung. 2005;9(3):119–26. doi: 10.1007/s11325-005-0022-1. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J, Veasey S. Making sense of oxidative stress in obstructive sleep apnea: mediator or distracter? Frontiers in neurology. 2012;3:179. doi: 10.3389/fneur.2012.00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Svatikova A, Wolk R, Lerman LO, et al. Oxidative stress in obstructive sleep apnoea. Eur Heart J. 2005;26(22):2435–39. doi: 10.1093/eurheartj/ehi440. [DOI] [PubMed] [Google Scholar]
- 19.Barcelo A, Miralles C, Barbe F, et al. Abnormal lipid peroxidation in patients with sleep apnoea. The European respiratory journal : official journal of the European Society for Clinical Respiratory Physiology. 2000;16(4):644–47. doi: 10.1034/j.1399-3003.2000.16d13.x. [DOI] [PubMed] [Google Scholar]
- 20.Barcelo A, Barbe F, de la Pena M, et al. Antioxidant status in patients with sleep apnoea and impact of continuous positive airway pressure treatment. European Respiratory Journal. 2006;27(4):756–60. doi: 10.1183/09031936.06.00067605. [DOI] [PubMed] [Google Scholar]
- 21.Itzhaki S, Dorchin H, Clark G, et al. The effects of 1-year treatment with a herbst mandibular advancement splint on obstructive sleep apnea, oxidative stress, and endothelial function. Chest. 2007;131(3):740–49. doi: 10.1378/chest.06-0965. [DOI] [PubMed] [Google Scholar]
- 22.Lavie L, Vishnevsky A, Lavie P. Evidence for lipid peroxidation in obstructive sleep apnea. Sleep. 2004;27(1):123–28. [PubMed] [Google Scholar]
- 23.Gozal D, Kheirandish-Gozal L. Cardiovascular morbidity in obstructive sleep apnea: oxidative stress, inflammation, and much more. American journal of respiratory and critical care medicine. 2008;177(4):369–75. doi: 10.1164/rccm.200608-1190PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoffmann MS, Singh P, Wolk R, et al. Microarray studies of genomic oxidative stress and cell cycle responses in obstructive sleep apnea. Antioxidants & redox signaling. 2007;9(6):661–69. doi: 10.1089/ars.2007.1589. [DOI] [PubMed] [Google Scholar]
- 25.Marrotte EJ, Chen DD, Hakim JS, et al. Manganese superoxide dismutase expression in endothelial progenitor cells accelerates wound healing in diabetic mice. J Clin Invest. 2010;120(12):4207–19. doi: 10.1172/JCI36858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pardo M, Tirosh O. Protective signalling effect of manganese superoxide dismutase in hypoxia-reoxygenation of hepatocytes. Free radical research. 2009;43(12):1225–39. doi: 10.3109/10715760903271256. [DOI] [PubMed] [Google Scholar]
- 27.Xu Q, Konta T, Nakayama K, et al. Cellular defense against H2O2-induced apoptosis via MAP kinase-MKP-1 pathway. Free radical biology & medicine. 2004;36(8):985–93. doi: 10.1016/j.freeradbiomed.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 28.Bogoyevitch MA, Gillespie-Brown J, Ketterman AJ, et al. Stimulation of the stress-activated mitogen-activated protein kinase subfamilies in perfused heart. p38/RK mitogen-activated protein kinases and c-Jun N-terminal kinases are activated by ischemia/reperfusion. Circ Res. 1996;79(2):162–73. doi: 10.1161/01.res.79.2.162. [DOI] [PubMed] [Google Scholar]
- 29.Armstrong SC. Protein kinase activation and myocardial ischemia/reperfusion injury. Cardiovasc Res. 2004;61(3):427–36. doi: 10.1016/j.cardiores.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 30.Hausenloy DJ, Yellon DM. Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc Res. 2006;70(2):240–53. doi: 10.1016/j.cardiores.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 31.Dean JL, Brook M, Clark AR, et al. p38 mitogen-activated protein kinase regulates cyclooxygenase-2 mRNA stability and transcription in lipopolysaccharide-treated human monocytes. J Biol Chem. 1999;274(1):264–69. doi: 10.1074/jbc.274.1.264. [DOI] [PubMed] [Google Scholar]
- 32.Brook M, Sully G, Clark AR, et al. Regulation of tumour necrosis factor alpha mRNA stability by the mitogen-activated protein kinase p38 signalling cascade. FEBS Lett. 2000;483(1):57–61. doi: 10.1016/s0014-5793(00)02084-6. [DOI] [PubMed] [Google Scholar]
- 33.Miyazawa K, Mori A, Miyata H, et al. Regulation of interleukin-1beta-induced interleukin-6 gene expression in human fibroblast-like synoviocytes by p38 mitogen-activated protein kinase. J Biol Chem. 1998;273(38):24832–38. doi: 10.1074/jbc.273.38.24832. [DOI] [PubMed] [Google Scholar]
- 34.Lasa M, Mahtani KR, Finch A, et al. Regulation of cyclooxygenase 2 mRNA stability by the mitogen-activated protein kinase p38 signaling cascade. Mol Cell Biol. 2000;20(12):4265–74. doi: 10.1128/mcb.20.12.4265-4274.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dyugovskaya L, Lavie P, Hirsh M, et al. Activated CD8+ T-lymphocytes in obstructive sleep apnoea. The European respiratory journal : official journal of the European Society for Clinical Respiratory Physiology. 2005;25(5):820–28. doi: 10.1183/09031936.05.00103204. [DOI] [PubMed] [Google Scholar]
- 36.Dyugovskaya L, Lavie P, Lavie L. Increased adhesion molecules expression and production of reactive oxygen species in leukocytes of sleep apnea patients. American journal of respiratory and critical care medicine. 2002;165(7):934–39. doi: 10.1164/ajrccm.165.7.2104126. [DOI] [PubMed] [Google Scholar]
- 37.Hammer M, Mages J, Dietrich H, et al. Control of dual-specificity phosphatase-1 expression in activated macrophages by IL-10. Eur J Immunol. 2005;35(10):2991–3001. doi: 10.1002/eji.200526192. [DOI] [PubMed] [Google Scholar]
- 38.Abraham SM, Lawrence T, Kleiman A, et al. Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J Exp Med. 2006;203(8):1883–89. doi: 10.1084/jem.20060336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wadgaonkar R, Pierce JW, Somnay K, et al. Regulation of c-Jun N-terminal kinase and p38 kinase pathways in endothelial cells. American journal of respiratory cell and molecular biology. 2004;31(4):423–31. doi: 10.1165/rcmb.2003-0384OC. [DOI] [PubMed] [Google Scholar]
- 40.Li M, Zhou JY, Ge Y, et al. The phosphatase MKP1 is a transcriptional target of p53 involved in cell cycle regulation. J Biol Chem. 2003;278(42):41059–68. doi: 10.1074/jbc.M307149200. [DOI] [PubMed] [Google Scholar]
- 41.Korhonen R, Turpeinen T, Taimi V, et al. Attenuation of the acute inflammatory response by dual specificity phosphatase 1 by inhibition of p38 MAP kinase. Molecular immunology. 2011;48(15–16):2059–68. doi: 10.1016/j.molimm.2011.06.439. [DOI] [PubMed] [Google Scholar]
- 42.McCollum LT, Gallagher PE, Ann Tallant E. Angiotensin-(1-7) attenuates angiotensin II-induced cardiac remodeling associated with upregulation of dual-specificity phosphatase 1. American journal of physiology. Heart and circulatory physiology. 2012;302(3):H801–10. doi: 10.1152/ajpheart.00908.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Imaizumi S, Grijalva V, Priceman S, et al. Mitogen-activated protein kinase phosphatase-1 deficiency decreases atherosclerosis in apolipoprotein E null mice by reducing monocyte chemoattractant protein-1 levels. Molecular genetics and metabolism. 2010;101(1):66–75. doi: 10.1016/j.ymgme.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shen JZ, Chandrasekharan UM, Ashraf MZ, et al. Lack of Mitogen-Activated Protein Kinase Phosphatase-1 Protects ApoE-Null Mice Against Atherosclerosis. Circ Res. 2010;106(5):902–10. doi: 10.1161/CIRCRESAHA.109.198069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reddy ST, Nguyen JT, Grijalva V, et al. Potential role for mitogen-activated protein kinase phosphatase-1 in the development of atherosclerotic lesions in mouse models. Arterioscler Thromb Vasc Biol. 2004;24(9):1676–81. doi: 10.1161/01.ATV.0000138342.94314.64. [DOI] [PubMed] [Google Scholar]