

Abstract
To identify needed human equilibrative nucleoside transporter 4 (hENT4) inhibitors, we cloned and stably expressed the recombinant protein in PK15NTD (nucleoside transporter deficient) cells, and, investigated its interaction with a series of dipyridamole analogues synthesized in our laboratory. Compounds were tested in this newly established hENT4 expressing system as well in previous stably expressed hENT1 and hENT2 expressing systems. Of the dipyridamole analogues evaluated, about one fourth of the compounds inhibited hENT4 with higher potencies than dipyridamole. The most potent of them, Compound 30 displayed an IC50 of 74.4 nM, making it about 38 times more potent than dipyridamole (IC50 = 2.8 μM), and selectivities of about 80-fold and 20-fold relative to ENT1 and ENT2, respectively. Structure-activity relationship showed nitrogen-containing monocyclic rings and noncyclic substituents at the 4-and 8-positions of the pyrimido[5,4-d]pyrimidine were important for the inhibitory activity against hENT4. The most potent and selective hENT4 inhibitors tended to have a 2,6-di(N-monohydroxyethyl) substitution on the pyrimidopyrimidine ring system. The inhibitors of hENT4 identified in this study are the most selective and potent inhibitors of hENT4 adenosine transporter function to date, and should serve as useful pharmacological/biochemical tools and/or potential leads for ENT4-based therapeutics. Also, the new hENT4-expressing PK15 cell line established will serve as a useful screening tool for the discovery and design of hENT4 ligands.
Keywords: Equilibrative nucleoside transporters, human equilibrative nucleoside transporter 4, dipyridamole analogues, structure-activity relationships
1. Introduction
Nucleoside transporters play an important role in the disposition and physiological and pharmacological activities of nucleosides and their analogues [1, 2]. In mammalian cells, nucleoside transporters are subdivided into two major classes based on their energy requirements; Na+-dependent secondary active concentrative transporters (CNTs) and Na+-independent equilibrative transporters (ENTs) [3]. Mammalian genomes encode 4 members of the equilibrative nucleoside transporter family that share the ability to transport adenosine and a likely 11-transmembrane (TM) helix topology, but differ in their abilities to transport other nucleosides and nucleobases [4]. The best-characterized family members, ENT1 and ENT2, are cell surface proteins that can regulate adenosine signaling. hENT1 is sensitive to the nucleoside analogue nitrobenzylmercaptopurine ribonucleoside (NBMPR), whereas hENT2-mediated transport is only weakly inhibited by NBMPR [5, 6]. Human ENT1 (hENT1) transports a wide range of purine and pyrimidine nucleosides, but is unable to transport the pyrimidine base uracil [7, 8]. Human ENT2 (hENT2) exhibits similar substrate specificity to hENT1, although with a lower apparent affinity except in the case of inosine [7]. However, it differs from hENT1 in also being able to transport purine and pyrimidine nucleobases [8, 9]. ENT3 has been shown to be a lysosomal transporter and functions as a pH-dependent transporter [10]. The fourth member of the ENT family was first cloned by Wang and coworkers and found to function as a polyspecific organic cation transporter and was designated plasma membrane monoamine transporter (PMAT) [11]. Barnes et al. later showed that PMAT also transports adenosine at acidic pH. They reported that human and mouse ENT4 exhibited a novel, pH-dependent adenosine transport activity optimal at acidic pH, with apparent Km values 0.78 and 0.13 mmol/L, respectively, at pH 5.5, which was absent at pH 7.4, and also that its cardiac abundance suggests contribution to regulation of extracellular adenosine concentrations under ischemia conditions [12].
Several chemical classes have been shown to inhibit ENTs. Three classes of inhibitors are most significant. These are purine nucleoside analogues of which NBMPR is the prototype, pyrimidopyrimidine and pteridine derivatives such as dipyridamole, and flazine calcium channel blockers represented by lidoflazine [13]. NBMPR is a potent inhibitor of hENT1 (IC50 of 0.4–8 nM) while it is a moderate inhibitor of hENT2 (IC50 of 2.8 μM) [7, 14]. Likewise, dipyridamole and dilazep inhibit hENT1 more potently (Ki values of 48 nM and 19 nM, respectively) than hENT2 (Ki values of 6.2 μM and 134μM, respectively) [15]. Both NBMPR and dipyridamole are moderate inhibitors of hENT4 [12]. However, NBMPR and the flazine compounds are limited for further exploration of clinical application. NBMPR has immunosuppressive and mutagenic activities deriving from its 6-mercaptopurine metabolite [16–18]. The flazines are nosnspecific, having calcium channel antagonist activity, poor oral absorption and a short duration of action. [19] [13, 20].
Dipyridamole (DPM) possesses beneficial properties and broad pharmacological effects. It is a safe drug that has been used for a long time in humans to prevent strokes and other vascular diseases due to its antiplatelet and vasodilating activities as a nucleoside transport inhibitor and a non-selective phosphodiesterase inhibitor [21, 22]. Dipyridamole inhibits ENT1, ENT2 and ENT4, but blocks ENT1 better than ENT2 and only weakly inhibits ENT4. The ENT2 inhibitory activity of DPM, which NBMPR lacks, is good but is weak and needs improvement. We have previously reported the synthesis and flow cytometric determination of binding to hENT1 of a series of dipyridamole analogs [23]; and subsequently developed an hENT1 fluorescent flow cytometric probe with one of them as template [24]. In this study, we focused attention on hENT4, which is a recently characterized adenosine-specific transporter but has no potent and/or selective pharmacological inhibitors [12]. Thus, we cloned and stably expressed hENT4 in PK15NTD cells. This new hENT4 expressing PK15 cell line, along with previously expressed hENT1 and hENT2 PK15 cell systems, were then used to test the library of dipyridamole analogues we synthesized [23], to identify new potent and selective inhibitors of hENT4 relative to hENT1 and hENT2.
2. Materials and Methods
2.1. Materials
pCMV-3tag-1 mammalian expression vector and pfu DNA polymerase were purchased from Stratagene (La Jolla, CA). Restriction enzymes were obtained from New England Biolabs (Ipswich, MA). The transfection reagent, Lipofectamine 2000, was purchased from Invitrogen (Carlsbad, CA). FLAG antibody was obtained from Sigma-Aldrich (St. Louis, MO), and species-specific horseradish-peroxidase secondary antibody was from Santa Cruz (Santa Cruz, CA). ECL was purchase from Amersham Biosciences (Piscataway, NJ). [5-3H]Uridine (1 mCi/ml, 27.0 Ci/mmol) was supplied by GE Healthcare (Piscataway, NJ). All reagents were of either molecular biology or cell culture-tested grade.
2.2. Chemicals
All dipyridamole derivatives were synthesized in our laboratory [23]. Other compounds were purchased from Sigma-Aldrich (St. Louis, MO).
2.3. Cell Culture
Human umbilical vein endothelial cells (HUVECs) were purchased from American Type Culture Collections (ATCC) (Manassas, VA). Cells were maintained in Ham’s F12K medium (ATCC, Manassas, VA) with 0.1mg/ml heparin, 0.03mg/ml ECGs and 10% fetal bovine serum (Invitrogen, Grand Island, NY) at 37°C in a humidified atmosphere of a mixture of 5% CO2 and 95% air. The flask for growing cells was precoated with 0.1% gelatin at room temperature for 5–10 min. Porcine kidney tubular epithelium nucleoside transporter deficient (PK15NTD) cells and PK15 cells stably expressing hENT1 and hENT2 were kindly donated by Dr. Chung-Ming Tse (The Johns Hopkins University, Baltimore, MD). Cells were maintained in Eagle’s minimal essential medium/Earle’s Balanced Salt Solution with 0.1 mM nonessential amino acids, 1 mM sodium pyruvate (ATCC, Manassas, VA) and 10% fetal bovine serum (Invitrogen, Grand Island, NY).
2.4. Cloning of hENT4
Full-length hENT4 cDNA (GenBank accession number BK000627) was obtained from HUVECs by reverse transcription polymerase chain reaction (RT-PCR). Total RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, CA). The cDNA was synthesized from total RNA using SuperScript II reverse transcriptase (Invitrogen, Carlsbad, CA). PCR amplification was performed using primers flanking the hENT4 open reading frame with BamHI (sense) and SalI (Antisense) restriction sites (sense: 5′-ATAGGATCCCGAGCAGAGGCTGCCATG -3′, antisense: 5′-TATGTCGACCTCGGAGGACTTTGCAGAAC-3′). The PCR sample (50 ul) contained 2ul of cDNA template, 2.5 U of pfu DNA polymerase, 0.2 mM of dNTP and 25 pmol of both sense and antisense primers. The PCR first amplification cycle was performed at 95 °C for 5 min; this was followed by 29 cycles at 94 °C for 45 sec, 52 °C for 45 sec and 72 °C for 3.5 min; and a last PCR cycle at 72 °C for 10 min to generate a product of approximately 1.6 kb. The PCR product was cloned into the mammalian expression vector pCMV tagged with flag, pCMV-3flag-1A. A positive clone was confirmed by colony PCR and restriction analysis, and was subjected to DNA sequencing (Molecular Resource Center, University of Tennessee Health Science Center, Memphis, TN).
2.5. Selection of PK15 Cells Stably Expressing hENT4
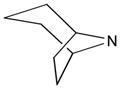
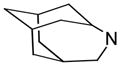
To generate hENT4 stable transfectants, 6×105 cells/well were seeded in six-well plates in modified minimal Eagle’s medium one day before transfection, which allowed them to reach about 95% confluence. The pCMV/hENT4 expression construct was transfected into PK15NTD cells using Lipofectamine 2000 according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA). Cells were transferred into a 100mm dish at 1:10 dilution in growth medium containing G418 (Invitrogen, Grand Island, NY) 48 hours after transfection. After selection for three weeks, cell colonies were picked up using cloning cylinders and screened by the successful reconstitution of [3H]adenosine uptake at acidic pH.
2.6. Preparation of Whole Cell Lysates and Western Blot Analysis
Total protein was extracted from hENT4 transfected cells by lysis in ice-cold RIPA buffer containing protease inhibitor cocktail (Santa Cruz, Santa Cruz, CA). Protein concentration was determined according to the method of BCA Protein Assay (Thermo Fisher Scientific, Rockford, IL) using bovine serum albumin as a standard. Proteins were separated on 10% SDS-polyacrylamide gels and transferred to a polyvinylidenedifluoride (PVDF) membrane (Bio-Rad, Hercules, CA). After blocking in TBST (0.1% Tween 20, Tris-buffered saline) containing 5% non-fat dry milk, membranes were incubated in blocking buffer containing mouse FLAG monoclonal antibody overnight at 4°C. The membranes were then washed three times with TBST, incubated with TBST containing anti-mouse horseradish-peroxidase secondary antibody, washed with TBST and detected by enhanced chemiluminescence.
2.7. [3H]Nucleoside Uptake and Inhibition Assay
All uptake assays were carried out in triplicate. Transport activity of cells transiently transfected with hENT4 was examined with [3H]adenosine. Transfected cells were washed twice with sodium-free buffer (choline chloride 120 mM, Tris-HCl 20 mM, K2HPO4 3mM, Glucose 10mM, MgCl2 1mM, CaCl2 1mM, pH 7.4) and incubated with transport medium containing 120 mmol/L sodium chloride at pH 6.0 for 30 min. Then 1 μM [3H]adenosine in transport buffer was added to each well and incubated for 15 min. pH dependence was examined by [3H]adenosine uptake at pH 5.5, 6.0, 7.4, and 8.0, and compared with [3H]uridine uptake. For transporter inhibition assays, PK15/hENT4 cells (PK15 cells stably expressing hENT4) were incubated with transport buffer containing test compounds at pH 6.0 for 15 min before addition of 0.2 μM [3H]adenosine, and then further incubation for 2 min. PK15/hENT1 and PK15/hENT2 cells were also incubated in sodium-free buffer (pH 7.4) with test compounds for 15 min, followed by 2 min incubation with 0.2μM [3H]uridine in sodium-free buffer (pH 7.4). Uptake was terminated by rapidly aspirating the incubation mixture and washing cells three times with ice-cold PBS. The cells were solubilized overnight in 400 μl of 5% Triton X-100 and 200 μl of cell lysate was counted using a scintillation counter (Beckman LS 3801, Fullerton, CA). Protein concentration in the cell lysate was determined using the BCA protein assay kit (Pierce, Rockford, IL). The concentrations of test compounds that caused 50 % inhibition of [3H]nucleoside uptake (IC50) were calculated using a nonlinear fitting method in the Prism 4 program (Version 4, GraphPad, San Diego, CA).
2.8. MTT Assay for Cell Viability
PK15-hENT4 cells were trypsinized, pelleted, and resuspended in 1 X MEM medium (ATCC, Manassas, VA). In 96-well plates, 100 μl 1 X MEM containing 5 × 103 cells were seeded in each well. Cells were incubated for 24 hrs and then treated with 10 μM of camptothecin, dipyridamole, compound 30 or vehicle, DMSO, in 100 μL of phenol-free 1 X MEM ((ATCC, Manassas, VA) for 24 hours. Subsequently, 10 μL of 5 mg/ml methylthiazolyldiphenyl-tetrazolium bromide (MTT) in PBS was added to each well, and then incubated for 4 hrs at 37°C. The yellow MTT was reduced to purple formazan in the mitochondria of living cells (cell viability). Following incubation, 100 μL of 0.04 N HCl in isopropanol was added for 3 hours at room temperature to solubilize the formazan crystals. The absorbance was quantified at 570 nm using a microplate reader (SpectraMax M2, Molecular Devices).
2.8. Data analysis
GraphPad Prism 5.0 was used for graphing and statistical analysis. Nucleoside uptake data are expressed as means ± S.E. for triplicate estimates of individual experiments (n=3). Statistical significance was determined by Student’s t test analysis, and a p<0.05 was considered statistically significant.
3. Results
3.1. Expression of Recombinant hENT4 in PK15NTD Cells
The full length hENT4 cDNA was cloned into the mammalian expression vector pCMV-3flag-1A. Our cDNA clone had an identical amino acid sequence with the 530-residue of hENT4 in the Genbank. The recombinant hENT4 was transiently expressed in PK15NTD cells, and the expression of hENT4 tagged with flag was verified by western blotting using FLAG antibody. An approximately 55 kDa protein, consistent with the predicted molecular mass of hENT4, was detected in the cells with recombinant hENT4 and not in the empty vector and cell controls (Fig. 1).
Fig. 1.
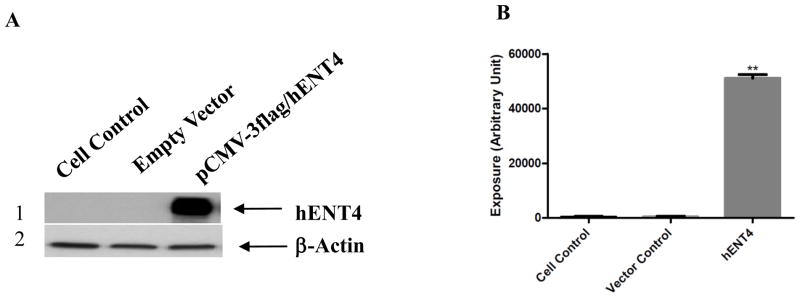
Immunoblotting detection of recombinant hENT4 in PK15NTD cells. PK15NTD cells were transiently transfected with either pCMV-3tag-1A (empty vector) or pCMV-3tag-1A/hENT4. Cell lysates were analyzed by Western blotting using FLAG antibody (A1). The blot was reprobed with β-actin for normalization (A2). The quantification of the Western blots are shown in B datapoints are mean + S.EM. for two blots. * = (p< 0.05); ** = (p <01)
3.2. Stable expression and functional characterization of hENT4
After transfection and selection of recombinant hENT4, cell colonies were screened for the successful reconstitution of [3H]adenosine uptake at acidic pH. The pH dependence of adenosine transport and specificity of hENT4 are in agreement with the literature [12]. hENT4 mediated adenosine transport, but not much of uridine transport. hENT4 activity of adenosine transport exhibited high sensitivity to pH, with much greater activity at acidic pH (pH 5.5 and 6.0) than pH 7.4 and 8.0, with optimal activity at approximately pH 6.0 (Fig. 2).
Fig. 2.

Adenosine specificity of hENT4 and pH-dependence of transport activity of stably expressed recombinant hENT4 in PK15NTD cells. pH dependence was examined at variable pH by [3H]adenosine uptake compared with [3H]uridine uptake. Data points are the mean ±S.D. for 3 samples.
3.3. Identification and IC50 determination of dipyridamole analogue inhibitors against hENT1, hENT2 and hENT4
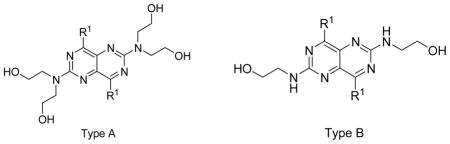
Having established the stable recombinant hENT4-expressing new transgenic cells, we used them along with previously established hENT1 and hENT2-expressing PK15 cells supplied to us by Dr. Chung-Ming Tse of Johns Hopkins University [7] to screen a series of dipyridamole analogues listed in Table 1–4. The initial screening results are presented in Figure 3. Following the initial screening, we have performed dose-response inhibition testing of selected compounds to obtain quantitative data in the form of IC50 values to use to compare the potencies and selectivities of the compounds as presented in Table 1–4. IC50 values of the compounds with very low activities or low solubility were not determined (showed as N.D. in Table 1–4). Dose response curves for the most potent and selective compounds against hENT4 are shown in Fig. 4. The compounds exhibited wide ranging inhibitory potencies and selectivities and most of them are more active against hENT1 than hENT2 and hENT4 (Fig. 3). Compounds 2, 11, 13, 15, 52, 64, 66, 74 and 79 inhibited hENT1 with IC50s less than 100 nM. Compounds 2, 11, 13, 17, 74and 79 were active against hENT2, with compound 13 (2,6-bis(diethanolamino)-4,8-diheptamethyleneimino-pyrimido[5,4d]pyrimidine) being the most potent inhibitor of both hENT1 and hENT2 with IC50 values of 0.97 nM and 90.8 nM, respectively. Compound 11, 12, 14, 16, 18, 19, 20, 25, 26, 28, 30, 52, 54, 56 and 58 showed significant activity against hENT4 with IC50 values less than 1000 nM. In fact, about one fourth of the compounds inhibited hENT4 with higher potencies than dipyridamole; and the most potent and selective hENT4 inhibitor was 2,6-diethanolamino-4,8-di-(diisobutylamino)-pyrimido[5,4-d]pyrimidine (compound 30, IC50 = 74.4 nM), which was approximately 38-fold more potent than dipyridamole (IC50 = 2,797 nM). All DPM analogues had the core structure of 2,4,6,8-tetra-substituted-pyrimido[5,4-d]pyrimidine, and maintained a symmetric feature as in the case of dipyridamole, with the exception of compound 8, which had two different substituents at the 4-, and 8-positions of the core pyrimidopyrimidine structure. Compounds listed in Table 1 are dipyridamole analogues with ring structures at the 4- and 8-positions of the pyrimidopyrimidine template; compounds listed in Table 2 are analogues with non-cyclic tertiary amines at the pyrimidopyrimidine 4- and 8-positions; compounds listed in Table 3 have primary or secondary amine substituents at the 4-and 8-positions of the core structure. Compounds listed in Table 4 are derivatives of dipyridamole [23].
Table 1.
hENT1, 2 and 4 Inhibitory Activity of Dipyridamole Analogues with different ring systems at the 4- and 8-positions
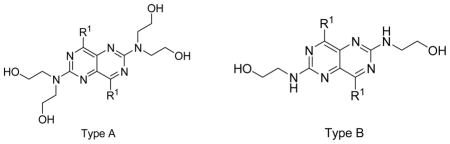
| |||||
|---|---|---|---|---|---|
| Comp. (WL) | Type | R1 | hENT1 IC50 (nM) | hENT2 IC50 (nM) | hENT4 IC50 (nM) |
| NBMPR | - | - | 0.65 | 1,350 | 2330 |
| Dipyridamole | A |
|
15.1 | 308 | 2797 |
| 1 | B | 496 | 7,440 | 2196 | |
| 2 | A |
|
47.1 | 556 | - |
| 3 | B | 115.9 | N.D.1 | 3906 | |
| 4 | A |
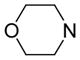
|
204.8 | 4,120 | N.D. |
| 5 | B | 5,660 | N.D. | N.D. | |
| 6 | A |
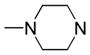
|
N.D. | N.D. | N.D. |
| 7 | A |
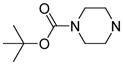
|
579.8 | 2,230 | N.D. |
| 8* | A | - | 937.1 | 3,660 | 12960 |
| 9 | A |
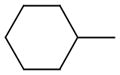
|
165.9 | N.D. | N.D. |
| 10 | A |
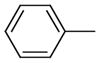
|
513.2 | 1,380 | 12000 |
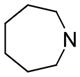
| 11 | A |
|
11.1 | 117 | 379 |
| 12 | B | 465.7 | N.D. | 595 | |
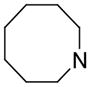
| 13 | A |
|
0.97 | 90.8 | 404 |
| 14 | B | 217 | 5,200 | 181 | |
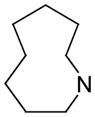
| 15 | A |
|
70.8 | 2,350 | 1320 |
| 16 | B | 167.4 | 1,390 | 513 | |
| 17 | A |
|
241.9 | 323 | - |
| 18 | B | N. D. | N.D. | 324 | |
| 19 | A |
|
393 | N.D. | 200 |
| 20 | B | 1,432 | N.D. | 602 | |
N.D.: not determined
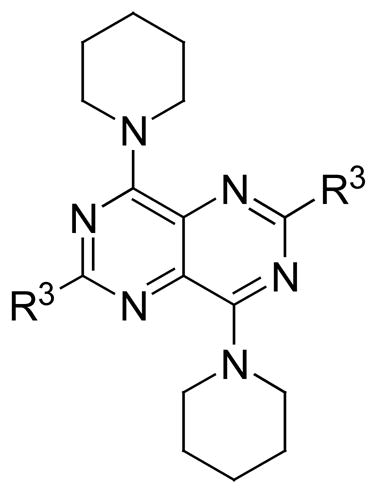
Table 4.
hENT1, 2 and 4 Inhibitory Activity of Dipyridamole Analogues with modification at hydroxyl groups of dipyridamole
| ||||
|---|---|---|---|---|
| Comp. (WL) | R3 | hENT1 IC50 (nM) | hENT2 IC50 (nM) | hENT4 IC50 (nM) |
| 72 | OCH2CH2OH | 289.6 | 1,785 | - |
| 73 |
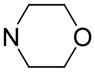
|
1,638 | N.D. | N. D. |
| 74 | N(CH2CH2OOCH)2 | 69.1 | 532 | N. D. |
| 75 | N(CH2CH2OOCCH3)2 | 238 | 708 | 14125 |
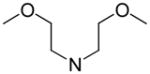
| 76 | N(CH2CH2OCH3)2 | 207.7 | 1,470 | 1820 |
| 77 | N(CH2CH2OCH2CH3)2 | 767.7 | N.D. | 29854 |
| 78 | N(CH2CH2OCH2CH2CH3)2 | N.D. | N.D. | 139637 |
| 79 |
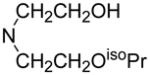
|
67.5 | 175 | N. D. |
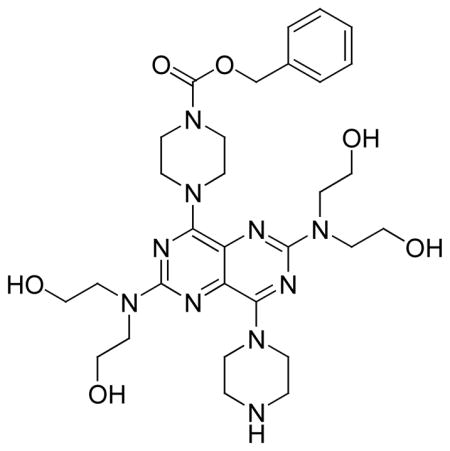
Compound 8
Fig. 3.

Inhibition profiles of dipyridamole derivatives against hENT1-, hENT2- or hENT4-mediated nucleoside uptake. The uptake of [3H]-labeled nucleoside was determined after incubation with 10 μM test compounds for 15 min. Uptake values in the presence of compounds are given as the percentage of uptake values in their absence. Data points are the mean ±S.D. for 3 determinations.
Fig. 4.

Concentration-dependent inhibition of hENT4 adenosine transport by the most potent dipyridamole analogues. Graded concentrations of test compounds were incubated with cells for 15 min prior to addition of [3H]adenosine for an uptake period of 2 min. Data are presented as percent inhibition relative to vehicle control (mean ± S.D.; n = 3). Compounds shown are the ones with IC50s ≤ 200 nM for hENT4.
Table 2.
hENT1, 2 and 4 Inhibitory Activity of Dipyridamole Analogues with opening chain analogues
| |||||
|---|---|---|---|---|---|
| Comp. (WL) | Type | R1 | hENT1 IC50 (nM) | hENT2 IC50 (nM) | hENT4 IC50 (nM) |
| 21 | A | Me2N | 1,837 | 3,250 | 59566 |
| 22 | B | N. D. | N. D. | - | |
| 23 | A | Et2N | 207.2 | 2,927 | 4121 |
| 24 | B | 612.5 | 4,223 | 3130 | |
| 25 | A | (n-Propyl)2N | 197 | 3,830 | 234 |
| 26 | B | 7,459 | N. D. | 97.7 | |
| 27 | A | (n-Butyl)2N | N. D. | 3,310 | 2960 |
| 28 | B | N. D. | 1,312 | 871 | |
| 29 | A | (iso-Butyl)2N | 2,627 | 1,962 | 1710 |
| 30 | B | 5,945 | 1,406 | 74.4 | |
| 31 | A | (n-Pentyl)2N | N. D. | N. D. | 45700 |
| 32 | B | N. D. | N. D. | N. D. | |
| 33 | A | (iso-Pentyl)2N | N. D. | N. D. | 44200 |
| 34 | B | N. D. | N. D. | - | |
| 35 | A |
|
538.4 | 2,086 | 103514 |
| 36 | B | 4,217 | 8,061 | 80352 | |
| 37 | A |
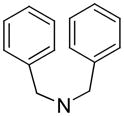
|
N.D. | N. D. | 68400 |
| 38 | B | N. D. | N. D. | N. D. | |
Table 3.
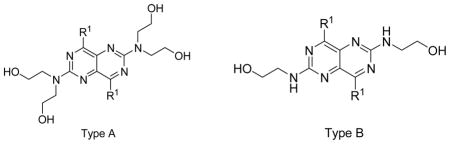
hENT1, 2 and 4 Inhibitory Activity of Dipyridamole Analogues with free hydrogen on the nitrogen of 4- and 8-position substituents
| |||||
|---|---|---|---|---|---|
| Comp. (WL) | Type | R1 | hENT1 IC50 (nM) | hENT2 IC50 (nM) | hENT4 IC50 (nM) |
| 39 | A | NH2 | N.D. | N.D. | N. D. |
| 40 | A | MeNH | 8,461 | N.D. | N. D. |
| 41 | B | N.D. | N.D. | N. D. | |
| 42 | A | EtNH | 768.3 | N.D. | N. D. |
| 43 | B | N.D. | N.D. | 38459 | |
| 44 | A | n-PropylNH | 427.7 | 3,500 | 16405 |
| 45 | B | 3,877 | N.D. | 10115 | |
| 46 | A | iso-PropylNH | 124.1 | 3,020 | 26002 |
| 47 | B | 5,873 | N.D. | 11614 | |
| 48 | A | n-ButylNH | 217 | 2,850 | 1883 |
| 49 | B | 1,069 | 2,200 | 3870 | |
| 50 | A | iso-ButylNH | 125.7 | 1,491 | 3055 |
| 51 | B | 784.2 | 1,490 | 8660 | |
| 52 | A | tert-ButylNH | 25.1 | N.D. | 767 |
| 53 | B | 6,543 | N.D. | 3273 | |
| 54 | A | n-PentylNH | 284.4 | 4,787 | 603 |
| 55 | B | N.D. | N.D. | 3067 | |
| 56 | A | iso-PentylNH | 142.6 | 1,954 | 495 |
| 57 | B | 1,173 | N.D. | 3760 | |
| 58 | A | tert-PentylNH | 78.9 | N.D. | 818 |
| 59 | B | 2,607 | N.D. | 1339 | |
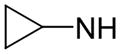
| 60 | A |
|
303.8 | 2,708 | 37600 |
| 61 | B | N. D. | N. D. | N. D. | |
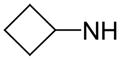
| 62 | A |
|
123.3 | 727.2 | 15668 |
| 63 | B | 1,857 | 7,754 | - | |
| 64 | A |
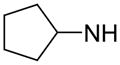
|
20.5 | 1,320 | 12162 |
| 65 | B | N.D. | N.D. | N. D. | |
| 66 | A |
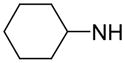
|
85.8 | N.D. | 124000 |
| 67 | B | N.D. | N.D. | 65313 | |
| 68 | A |
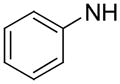
|
1,119 | N.D. | N. D. |
| 69 | B | N. D. | N. D. | N. D. | |
| 70 | A |
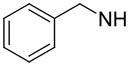
|
N. D. | N. D. | N. D. |
| 71 | B | N. D. | N. D. | 1760 | |
Compound 30 shows no cytotoxicity against PK15/hENT4 cells
To check whether the inhibition of hENT4 transporter was not due to the effect of cytotoxicity of the compound against PK15/hENT4 cells, we run an MTT assay to determine the effect of compound 30 on the viability of the cells. We compared it with the known cytotoxic agent camptothecin and also the dipyridamole. As shown in Fig. 5, the compound was significantly different from camptothecin in terms of effect on cell viability (p < 0.02). This indicates that the inhibitory effect of compound 30 against hENT4 transport function was not due to a compromise in cell viability but a true inhibition of the transporter in healthy and completely viable cells.
Fig. 5.

Cell viability testing by MTT assay. PK15/hENT cells were seeded at 5 × 103 cells were seeded in 96-well plates, treated with 10 μM test compounds or control and incubated at 37 °C in phenol-free MEM for 24 hours. Subsequently, 10 μl of 5 mg/ml methylthiazolyldiphenyl-tetrazolium bromide (MTT) in PBS was added to each well, and then incubated for 4 hrs at 37°C. Following incubation, 100 μl of 0.04N HCl in isopropanol was added for 3 hours at room temperature to solubilize the formazan produced. The absorbance was quantified at 570 nm using a microplate reader (SpectraMax M2, Molecular Devices). Data are mean ± S.E.M for 4 replicates. * = (p<0.05). Compared to the cytotoxic agent camptothecin, compound 30 was significantly non-cytotoxic at p < 0.02.
4. Discussion
In vitro studies have demonstrated that nucleoside transporters are necessary for many anti-cancer nucleoside analogues to enter cells. Antitumor nucleosides are well known to be good substrates of ENTs, and the expression of these transporters has been shown to influence the sensitivity to antitumor nucleosides [24–29]. Hence, selective inhibition of ENTs may represent a means of improving the antitumor efficacy of nucleoside drugs, in particular if these are taken up into cells by concentrative nucleoside transporters; to prevent transport out of cells as a recent example shows [30]. ENT inhibitors, by virtue of their effect on extracellular adenosine concentrations, can also modulate a variety of physiological processes, potentially leading to therapeutic benefits. For example, by inhibiting nucleoside uptake into endothelial and other cells the coronary vasodilator draflazine substantially increases and prolongs the cardiovascular effects of adenosine [31]. The beneficial effects of administering the ENT inhibitor dipyridamole during percutaneous transluminal coronary angioplasty in humans are likely to have a similar origin [32]. Transport inhibitors are also potentially of value in the context of ischaemic neuronal injury: pre-ischaemic administration of the pro-drug NBMPR-phosphate has been shown to increase brain adenosine levels and reduce ischaemia-induced loss of hippocampal CA1 neurons in the rat [33]. ENT4 expression is abundant in heart and adenosine transport pH-dependency suggests a contribution to the regulation of extracellular adenosine concentrations during myocardial ischemia. Hence, ENT4 represents a potential novel therapeutic target for coronary heart disease. Furthermore, a recent study has shown that ENT4 is transcriptionally activated by the chimeric transcription factor, EWS/WT1 expressed in desmoplastic small round cell tumor (DSRCT, [34]). ENT4 is thus a biomarker and potential diagnostic/therapeutic target for this difficult to treat cancer. The expression level and pattern of the ENTs are important determinants of the therapeutic efficacy of anticancer nucleoside analogs, suggesting that this transporter may represent an attractive pathway for targeting chemotherapeutic drugs into DSRCT. Inhibitors are acutely lacking for ENT4, thus hindering studies of the physiological functions and targeting for therapeutic purposes. Therefore, it is urgent to identify potent and selective ENT4 inhibitors. DPM has better properties for therapeutic development than NBMPR and the flazine compounds [35, 36], and has been shown to have the potential to enhance the efficacy of antimetabolites and reduce the doses of chemotherapeutic drugs and therefore the side effects of chemotherapy [37–39]. Since DPM has been shown to inhibit hENT4, albeit weakly, we envisaged that some of its analogs may possess more potent activity than the parent, and also by testing a series of 73 analogues we hoped to establish a useful SAR to guide future design of selective inhibitors of hENT4. Indeed, fortunately, we identified more potent and selective inhibitors of hENT4 than DPM.
In this study we have identified the first potent and selective inhibitors of hENT4, the best of which is compound 30 with an IC50 of 74 nM, i.e. 38 times more potent than dipyridamole (IC50 = 2,797 nM), and selectivities of about 80-fold and 20-fold relative to ENT1 and ENT2, respectively. In addition, of the 73 dipyridamole analogues evaluated against hENT4, 50 compounds were found to inhibit hENT4 with moderate to high potency (≥30%). In addition to compound 30, compounds 18, 26 and 71 are also selective inhibitors of hENT4 relative to hENT1 and hENT2.
For substituents at the 4- and 8-positions of the pyrimido[5,4-d]pyrimidine bicyclic system, nitrogen-containing cyclic ring structures gave analogues with better inhibitory activities against hENT4 than dipyridamole, as represented by compounds 11–20. The acyclic amine substituted analogues (compounds 21–38) were more active than their cyclic counterparts, which is different from their inhibitory activity SARs for hENT1 and hENT2. Of this set, the 2-, and 6-position N-(monohydroxyethyl)-substituted analogues were more potent than the 2-, and 6-position N-(dihydroxyethyl)-substituted counterparts against hENT4. Increasing the carbon chain length of the 4- and 8-postion substituents also resulted in higher inhibitory activities. The best hENT4 inhibitor was compound 30, which has a diisobutylamino substituent at the 4 and 8 positions and N-(monohydroxyethyl) substitution at the 2 and 6 positions.
In this study, the IC50s of NBMPR and dipyridamole against ENT1 and ENT2 are in agreement with the literature [40, 41]. Of the 2- and 6-position substituted dipyridamole analogues (compounds 72–79), compound 73, which has an N-(dihydroxyethyl) group at the 2 and 6 positions locked into a morpholino ring, had little activity, which indicates that this modification is detrimental to hENT4 inhibitory activity. Esterification of the dipyridamole OH groups (compounds 74 and 75) afforded relatively low activity against hENT4 compared to dipyridamole, as did ether modification of the OH groups (compounds 77 and 78). This indicates that free OH groups may be preferred, and possibly are involved in hydrogen bond donor interactions at the hENT4 inhibitor binding site. However, decreasing the carbon chain length of the ether (going from compound 78 to compound 76), increased inhibitory activity, with IC50 values going from 13, 9637 nM for compound 78 to 1, 820 nM for compound 76.
Among the three plasma membrane ENTs (1, 2 1 and 3) examined, dipyridamole analogues showed the best inhibitory activities against hENT1. Compounds with N-(di-hydroxyethyl) substituents at the 2 and 6 positions (Type A in Tables 1–3) were much more potent than the corresponding N-(monohydroxyethyl) substituted analogues (Type B in Tables 1–3) against hENT1. Also for ENT1, substituents at the 4 and 8 positions consisting of nitrogen-containing ring structures usually gave analogues with good inhibitory activities. Compound 13, the best inhibitor of hENT1 with the ring size of eight, exhibited similar inhibitory activity (IC50 = 0.97nM) as NBMPR (IC50 = 0.65 nM). Analogues which contain a primary or secondary amine at the 4 and 8 positions (compounds 39–71) and 2 and 6 positions modified dipyridamole analogues (compounds 72–79) had lower inhibitory activities than dipyridamole in general against hENT1. These results are consistent with our previous study of the hENT1 binding affinity for these dipyridamole analogues evaluated by flow cytometry [23]. In that study, hENT1 had the highest affinity for compound 13. In the present study, compound 13 exhibits about 100 times and 400 times selectivity with regard to hENT2 and hENT4, respectively, relative to hENT1. In addition, compounds 42, 64, 73 and 77 also showed selectivity towards ENT1, albeit less potent than compound 13. Compound 13 was also the best hENT2 inhibitor with an IC50 of 90.8 nM, which makes it over 3 times more potent than dipyridamole, which is the most widely used ENT2 inhibitor known hitherto. Compound 11, which has a seven-membered nitrogen containing ring substituents at the 4 and 8 positions was the second most potent hENT1 and hENT2 inhibitory compound with IC50 values of 11.1 nM and 117 nM, respectively. The SAR insights obtained from this study can be utilized in designing new potent and selective ENT1, 2 and 4 inhibitors and for modeling selective pharmacophores for these transporters.
Acknowledgments
This study was supported by a grant from the National Heart Lung and Blood Institute, NIH, grant number HL095002. We thank Dr. Chung-Ming Tse for his kind provision of PK15NTD cells and PK15/hENT1 and PK15/hENT2 cells.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cass CE, Young JD, Baldwin SA. Recent advances in the molecular biology of nucleoside transporters of mammalian cells. Biochem Cell Biol. 1998;76:761–70. doi: 10.1139/bcb-76-5-761. [DOI] [PubMed] [Google Scholar]
- 2.Balimane PV, Sinko PJ. Involvement of multiple transporters in the oral absorption of nuceoside analogues. Adv Drug Deliv Rev. 1999;39:183–209. doi: 10.1016/s0169-409x(99)00026-5. [DOI] [PubMed] [Google Scholar]
- 3.Young JD, Yao SY, Baldwin JM, Cass CE, Baldwin SA. The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol Aspects Med. 2013;34(2–3):529–47. doi: 10.1016/j.mam.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 4.Baldwin SA, Beal PR, Yao SYM, King AE, Cass CE, Young JD. The equilibrative nucleoside transporter family, SLC29. Pflug Arch Eur J Phy. 2004;447:735–43. doi: 10.1007/s00424-003-1103-2. [DOI] [PubMed] [Google Scholar]
- 5.Yao SY, Ng AM, Muzyka WR, Griffiths M, Cass CE, Baldwin SA, et al. Molecular cloning and functional characterization of nitrobenzylthioinosine (NBMPR)-sensitive (es) and NBMPR-insensitive (ei) equilibrative nucleoside transporter proteins (rENT1 and rENT2) from rat tissues. J Biol Chem. 1997;272:28423–30. doi: 10.1074/jbc.272.45.28423. [DOI] [PubMed] [Google Scholar]
- 6.Griffiths M, Beaumont N, Yao SY, Sundaram M, Boumah CE, Davies A, et al. Cloning of a human nucleoside transporter implicated in the cellular uptake of adenosine and chemotherapeutic drugs. Nat Med. 1997;3:89–93. doi: 10.1038/nm0197-89. [DOI] [PubMed] [Google Scholar]
- 7.Ward JL, Sherali A, Mo ZP, Tse CM. Kinetic and pharmacological properties of cloned human equilibrative nucleoside transporters, ENT1 and ENT2, stably expressed in nucleoside transporter-deficient PK15 cells. Ent2 exhibits a low affinity for guanosine and cytidine but a high affinity for inosine. J Biol Chem. 2000;275:8375–81. doi: 10.1074/jbc.275.12.8375. [DOI] [PubMed] [Google Scholar]
- 8.Crawford CR, Patel DH, Naeve C, Belt JA. Cloning of the human equilibrative, nitrobenzylmercaptopurine riboside (NBMPR)-insensitive nucleoside transporter ei by functional expression in a transport-deficient cell line. J Biol Chem. 1998;273:5288–93. doi: 10.1074/jbc.273.9.5288. [DOI] [PubMed] [Google Scholar]
- 9.Yao SY, Ng AM, Vickers MF, Sundaram M, Cass CE, Baldwin SA, et al. Functional and molecular characterization of nucleobase transport by recombinant human and rat equilibrative nucleoside transporters 1 and 2. Chimeric constructs reveal a role for the ENT2 helix 5–6 region in nucleobase translocation. J Biol Chem. 2002;277:24938–48. doi: 10.1074/jbc.M200966200. [DOI] [PubMed] [Google Scholar]
- 10.Baldwin SA, Yao SYM, Hyde RJ, Ng AML, Foppolo S, Barnes K, et al. Functional characterization of novel human and mouse equilibrative nucleoside transporters (hENT3 and mENT3) located in intracellular membranes. Journal of Biological Chemistry. 2005;280:15880–7. doi: 10.1074/jbc.M414337200. [DOI] [PubMed] [Google Scholar]
- 11.Engel K, Zhou M, Wang J. Identification and characterization of a novel monoamine transporter in the human brain. J Biol Chem. 2004;279:50042–9. doi: 10.1074/jbc.M407913200. [DOI] [PubMed] [Google Scholar]
- 12.Barnes K, Dobrzynski H, Foppolo S, Beal PR, Ismat F, Scullion ER, et al. Distribution and functional characterization of equilibrative nucleoside transporter-4, a novel cardiac adenosine transporter activated at acidic pH. Circ Res. 2006;99:510–9. doi: 10.1161/01.RES.0000238359.18495.42. [DOI] [PubMed] [Google Scholar]
- 13.Buolamwini JK. Nucleoside transport inhibitors: Structure-activity relationships and potential therapeutic applications. Curr Med Chem. 1997;4:35–66. [Google Scholar]
- 14.SenGupta DJ, Lum PY, Lai Y, Shubochkina E, Bakken AH, Schneider G, et al. A single glycine mutation in the equilibrative nucleoside transporter gene, hENT1, alters nucleoside transport activity and sensitivity to nitrobenzylthioinosine. Biochemistry-Us. 2002;41:1512–9. doi: 10.1021/bi015833w. [DOI] [PubMed] [Google Scholar]
- 15.Visser F, Vickers MF, Ng AM, Baldwin SA, Young JD, Cass CE. Mutation of residue 33 of human equilibrative nucleoside transporters 1 and 2 alters sensitivity to inhibition of transport by dilazep and dipyridamole. J Biol Chem. 2002;277:395–401. doi: 10.1074/jbc.M105324200. [DOI] [PubMed] [Google Scholar]
- 16.Benedict WF, Baker MS, Haroun L, Choi E, Ames BN. Mutagenicity of cancer chemotherapeutic agents in the Salmonella/microsome test. Cancer Res. 1977;37:2209–13. [PubMed] [Google Scholar]
- 17.Cass CE, Muzik H, Paterson AR. Combination therapy of mouse leukemia L1210 by 1-beta-D-arabinofuranosylcytosine and 6-[(4-nitrobenzyl)thio]-9-beta-D-ribofuranosylpurine. Cancer Res. 1975;35:1187–93. [PubMed] [Google Scholar]
- 18.Elion GB. Symposium on immunosuppressive drugs. Biochemistry and pharmacology of purine analogues. Federation proceedings. 1967;26:898–904. [PubMed] [Google Scholar]
- 19.Grover GJ, Sleph PG. The Cardioprotective Effects of R-75231 and Lidoflazine Are Not Caused by Adenosine a(1) Receptor Activation. J Pharmacol Exp Ther. 1994;268:90–6. [PubMed] [Google Scholar]
- 20.Kates RA, Dorsey LM, Kaplan JA, Hatcher CR, Jr, Guyton RA. Pretreatment with lidoflazine, a calcium-channel blocker. Useful adjunct to heterogeneous cold potassium cardioplegia. J Thorac Cardiovasc Surg. 1983;85:278–86. [PubMed] [Google Scholar]
- 21.Jones L, Griffin S, Palmer S, Main C, Orton V, Sculpher M, et al. Clinical effectiveness and cost-effectiveness of clopidogrel and modified-release dipyridamole in the secondary prevention of occlusive vascular events: a systematic review and economic evaluation. Health Technol Assess. 2004;8:iii–iv. 1–196. doi: 10.3310/hta8380. [DOI] [PubMed] [Google Scholar]
- 22.Chakrabarti S, Freedman JE. Dipyridamole, cerebrovascular disease, and the vasculature. Vascul Pharmacol. 2008;48:143–9. doi: 10.1016/j.vph.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 23.Lin WW, Buolamwini JK. Synthesis, flow cytometric evaluation, and identification of highly potent dipyridamole analogues as equilibrative nucleoside transporter 1 inhibitors. J Med Chem. 2007;50:3906–20. doi: 10.1021/jm070311l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin W, Buolamwini JK. Design, synthesis, and evaluation of 2-diethanolamino-4,8-diheptamethyleneimino-2-(N-aminoethyl-N-ethanolamino)-6-(N,N-diethanolamino)pyrimido[5,4-d]pyrimidine-fluorescein conjugate (8MDP-fluor), as a novel equilibrative nucleoside transporter probe. Bioconjug Chem. 2011;22(6):1221–7. doi: 10.1021/bc2000758. [DOI] [PubMed] [Google Scholar]
- 25.Endo Y, Obata T, Murata D, Ito M, Sakamoto K, Fukushima M, et al. Cellular localization and functional characterization of the equilibrative nucleoside transporters of antitumor nucleosides. Cancer Sci. 2007;98:1633–7. doi: 10.1111/j.1349-7006.2007.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hyde RJ, Cass CE, Young JD, Baldwin SA. The ENT family of eukaryote nucleoside and nucleobase transporters: recent advances in the investigation of structure/function relationships and the identification of novel isoforms. Mol Membr Biol. 2001;18:53–63. [PubMed] [Google Scholar]
- 27.Kong W, Engel K, Wang J. Mammalian nucleoside transporters. Curr Drug Metab. 2004;5:63–84. doi: 10.2174/1389200043489162. [DOI] [PubMed] [Google Scholar]
- 28.Pastor-Anglada M, Cano-Soldado P, Molina-Arcas M, Lostao MP, Larrayoz I, Martinez-Picado J, et al. Cell entry and export of nucleoside analogues. Virus Res. 2005;107:151–64. doi: 10.1016/j.virusres.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 29.Hillgren KM, Keppler D, Zur A, Giacomini KM, Stieger B, Cass CE, Zhang L. Emerging Transporters of Clinical Importance: An Update from the International Transporter Consortium. Clin Pharmacol Ther. 2013 Apr 8; doi: 10.1038/clpt.2013.74. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 30.Paproski RJ, Yao SY, Favis N, Evans D, Young JD, Cass CE, Zemp RJ. Human concentrative nucleoside transporter 3 transfection with ultrasound and microbubbles in nucleoside transport deficient HEK293 cells greatly increases gemcitabine uptake. PLoS One. 2013;8(2):e56423. doi: 10.1371/journal.pone.0056423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dennis DM, Raatikainen MJ, Martens JR, Belardinelli L. Modulation of atrioventricular nodal function by metabolic and allosteric regulators of endogenous adenosine in guinea pig heart. Circulation. 1996;94:2551–9. doi: 10.1161/01.cir.94.10.2551. [DOI] [PubMed] [Google Scholar]
- 32.Mubagwa K, Flameng W. Adenosine, adenosine receptors and myocardial protection: An updated overview. Cardiovasc Res. 2001;52:25–39. doi: 10.1016/s0008-6363(01)00358-3. [DOI] [PubMed] [Google Scholar]
- 33.Zhang YW, Shepel PN, Peeling J, Geiger JD, Parkinson FE. Effects of nitrobenzylthioinosine on adenosine levels and neuronal injury in rat forebrain ischemia. Neurosci Res Commun. 2002;30:83–9. doi: 10.1046/j.1471-4159.2000.0750795.x. [DOI] [PubMed] [Google Scholar]
- 34.Li HJ, Smolen GA, Beers LF, Xia L, Gerald W, Wang J, et al. Adenosine Transporter ENT4 Is a Direct Target of EWS/WT1 Translocation Product and Is Highly Expressed in Desmoplastic Small Round Cell Tumor. Plos One. 2008:3. doi: 10.1371/journal.pone.0002353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.FitzGerald GA. Dipyridamole. N Engl J Med. 1987;316:1247–57. doi: 10.1056/NEJM198705143162005. [DOI] [PubMed] [Google Scholar]
- 36.Schaper W. Dipyridamole, an underestimated vascular protective drug. Cardiovasc Drug Ther. 2005;19:357–63. doi: 10.1007/s10557-005-4659-6. [DOI] [PubMed] [Google Scholar]
- 37.Weber G, Prajda N. Targeted and non-targeted actions of anti-cancer drugs. Adv Enzyme Regul. 1994;34:71–89. doi: 10.1016/0065-2571(94)90009-4. [DOI] [PubMed] [Google Scholar]
- 38.Boyer CR, Karjian PL, Wahl GM, Pegram M, Neuteboom ST. Nucleoside transport inhibitors, dipyridamole and p-nitrobenzylthioinosine, selectively potentiate the antitumor activity of NB1011. Anticancer Drugs. 2002;13:29–36. doi: 10.1097/00001813-200201000-00003. [DOI] [PubMed] [Google Scholar]
- 39.Rodrigues M, Barbosa F, Jr, Perussi JR. Dipyridamole increases the cytotoxicity of cisplatin in human larynx cancer cells in vitro. Braz J Med Biol Res. 2004;37:591–9. doi: 10.1590/s0100-879x2004000400017. [DOI] [PubMed] [Google Scholar]
- 40.Vickers MF, Mani RS, Sundaram M, Hogue DL, Young JD, Baldwin SA, et al. Functional production and reconstitution of the human equilibrative nucleoside transporter (hENT1) in Saccharomyces cerevisiae. Interaction of inhibitors of nucleoside transport with recombinant hENT1 and a glycosylation-defective derivative (hENT1/N48Q) Biochem J. 1999;339 (Pt 1):21–32. [PMC free article] [PubMed] [Google Scholar]
- 41.SenGupta DJ, Unadkat JD. Glycine 154 of the equilibrative nucleoside transporter, hENT1, is important for nucleoside transport and for conferring sensitivity to the inhibitors nitrobenzylthioinosine, dipyridamole, and dilazep. Biochem Pharmacol. 2004;67:453–8. doi: 10.1016/j.bcp.2003.09.018. [DOI] [PubMed] [Google Scholar]