

1. Introduction
Acquired immunodeficiency syndrome (AIDS) has been a devastating pandemic with around 33 million people infected with it worldwide. The human immunodeficiency virus (HIV) is implicated in the etiology of AIDS. The pol gene of HIV-1 encodes three enzymes that are required for viral replication viz., reverse transcriptase (RT), integrase (IN) and protease (PR).1 Currently FDA approved antiretroviral drugs target mainly RT2 and PR3 and are widely used in combination as highly active antiretroviral therapy (HAART). Entry inhibitors e.g. Maraviroc4 and fusion inhibitors such as Enfuvirtide5 have also recently been approved as anti-HIV agents. Though, HAART has been effective in reducing morbidity and mortality, it does not eliminate the virus from patients.6 Furthermore, the emergence of multidrug resistant viral strains along with severe side effect7–8 associated with existing drugs and the life-long chronic nature of the infection, demand the development of novel drugs that target other crucial steps of viral replication and survival.
The integration of viral cDNA into host chromosomes is an essential step in the viral replication process9 and is responsible for stable infection. Moreover, IN has no known structural analogs in human cells.10–13 Inhibition of this enzyme therefore provides an attractive strategy for antiretroviral drug development.
The function of IN is to integrate viral cDNA into the host genome. This process occurs in two steps: 3′-end processing (cleavage; 3′-P) and joining reaction (strand transfer, ST). In the processing reaction which occurs in the cytoplasm of an infected cell, a site specific endonucleolytic activity removes the highly conserved 3′ terminal CA dinucleotide from the 3′-end of the of viral DNA to generate a reactive nucleophilic 3′-hydroxyl group.14–15 In the strand transfer reaction, the processed 3′-hydroxyl end of the viral DNA does a nucleophilic attack at the phosphodiester bond of the host DNA to effect integration. β-Diketo acid derivatives as represented by S-136016 was the first integrase inhibitor class to have entered clinical trials. Naphthyridine carboxamides such as L-870,81017 have also advanced to clinical trials as have novel mono keto quinolone carboxylic acids18,19 such as GS-9137 which is currently undergoing Phase III clinical trials. Inspite of over two decades of extensive research leading to promising IN inhibitors,20–25 hitherto only one IN inhibitor, raltegravir,26 a pyrimidinone carboxamide, has been approved for clinical use. Moreover, emergence of resistance against raltegravir due to viral mutations mostly usually T66I, N155H, Q148H/R and E92Q)27, demands exploration of novel scaffolds for the treatment of HIV infection.
We have previously reported novel DKA IN inhibitors with aryl moiety modifications that were derived from a phenanthridinone scaffold.28 In continuing our efforts, we report herein the synthesis and biological evaluation of chalcone derivatives having 3-keto salicylic acid moieties as a new class of IN inhibitors. We have undertaken substantial analog synthesis and applied the PHASE pharmacophore analysis program in conjunction with three dimensional structure-activity relationship (3D-QSAR) studies to gain insights into their binding at the IN active site. CoMFA and CoMSIA 3D-QSAR models were derived to study the influence of stereoelectronic, hydrophobic and hydrogen bonding interactions on activity; and to establish predictive tools for design of novel inhibitors.
2. Results and discussion
2.1. Chemistry
In the present study, a novel series of IN inhibitors was discovered based on modification of the diketo acid pharmacophoric group as represented by structure III (Fig. 1). The 2-position OH and β-postion keto groups together mimic the keto-enol tautomer of the β-diketo group. Attachment of the carboxylic acid at the 1-position gave the 3-keto salicylic acid moiety of III, intended to interact with IN similar to the β-diketo acids. A styryl group was attached to the 3-position keto group to create chalcones (IV) (Fig. 1), the type of which have not yet been investigated as HIV IN inhibitors to our knowledge. The chalcone moiety was also reduced to give an ethylene linker as shown in compound 60. Furthermore, replacement of the enone group of the chalcones with an amide group was also investigated.
Figure 1.
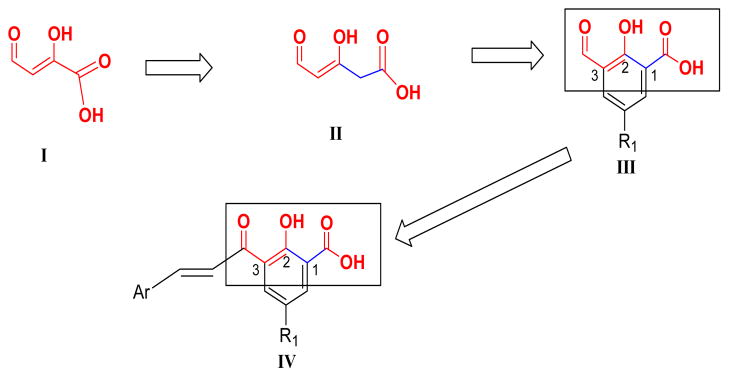
Design of 3-keto salicylic acid pharmacophore of synthesized chalcones.
The synthesis of target chalcones was performed according to reaction Scheme 1. The 5-substituted salicylic acid (X = COOH) starting material 4 was first acetylated with acetic anhydride and concentrated sulfuric acid. The acetylated intermediate 5 was then subjected to Fries rearrangement reaction using anhydrous aluminum chloride and heating at 160 °C to transfer the acetyl group from the 2-position oxygen to the 3-position to yield 3-acetyl salicylic acid intermediate 6.29 The next step involved the Claisen-Schmidt condensation reaction of intermediate 6 with various aryl aldehydes at room temperature using NaOH to obtain the target chalcones (10–57).30 To determine the importance of the α,β-unsaturated bond (chalcone scaffold) to IN inhibitory activity, the synthesis of compound 60 was undertaken by reduction of compound 36 by Pd/C-catalyzed hydrogenation (Scheme 2). The amide 61 was synthesized according to reaction Scheme 3 such that, commercially available 5-bromo salicylic acid (2) was first formylated with chloroform at the 2-position (ortho) by Reimer-Tiemann reaction to yield the aldehyde intermediate 7. However, the low yield of the above reaction, prompted to employ and alternative synthetic route. Thus, 3-formyl salicylaldehyde 1 was brominated by NBS in acetonitrile to afford intermediate 7 in higher yields. In the next step, compound 7 was condensed with 4-fluorobenzyl amine to afford intermediate 8 which was subsequently subjected to oxidation with KMnO4 to yield 61. For SAR purposes, compounds 62 and 63 were synthesized by coupling with p-fluorobenzyl amine following step c of reaction Scheme 3, from the commercially available starting materials 3a and 3b.
Scheme 1a.
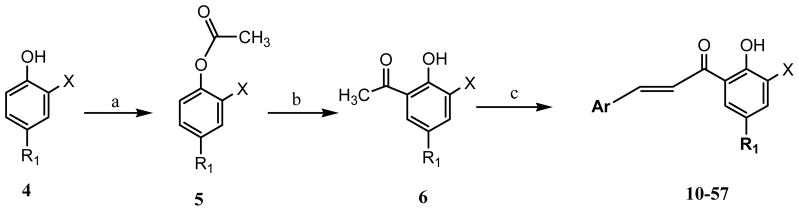
a Reagents and conditions: (a) Ac2O, H2SO4; (b) AlCl3, 160 °C; (c) ArCHO, NaOH, EtOH.
Scheme 2a.
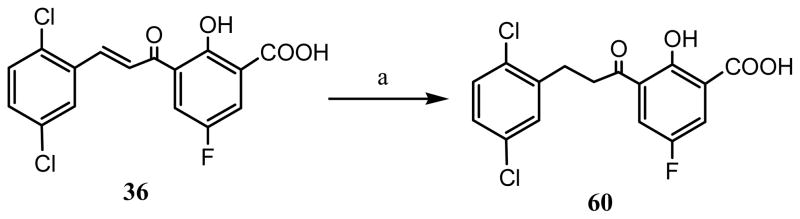
aReagents and conditions: (a) H2, Pd/C (10 mol%), methanol.
Scheme 3a. Reagents and conditions.

(a) CHCl3, NaOH, H2O; (b) NBS, CH3CN, 2 h (c) 4-F benzylamine, EDCI, HOBT, Et3N; (d) KMnO4, H2O/Pyridine, 90 °C.
2.2. HIV-1 integrase inhibition structure–activity relationship (SAR)
After synthesis, the target compounds were tested for integrase inhibitory activity to assess their potency and study the SAR. The intergrase 3′-cleavage and strand transfer (ST) inhibitory activity data are presented in Table 1 along with compound structures. In general, the compounds behaved as typical diketo acid IN inhibitors, being selective against the strand transfer (ST) step compared to the 3′-end processing (3′-P).
Table 1.
Structure and HIV-1 integrase inhibitory activities of chalcones
| |||||
|---|---|---|---|---|---|
| IC50(μM) | Activity
|
||||
| Comp. | Ar | X | R1 | 3′-Pa | STb |
| 10 | Ph | COOH | Br | 75 ± 14 | 25 ± 5 |
| 11 | 2-Cl-Ph | COOH | Br | 52 ± 13 | 14 ± 4 |
| 12 | 2-F-Ph | COOH | Br | 74 ± 25 | 32 ± 3 |
| 13 | 3-Cl-Ph | COOH | Br | 45 ± 10 | 9 ± 3 |
| 14 | 4-Cl-Ph | COOH | Br | 81 ± 28 | 12 ± 1 |
| 15 | 4-Br-Ph | COOH | Br | 11 ± 4 | 5 ± 2 |
| 16 | 4-I-Ph | COOH | Br | 69 | 35 |
| 17 | 2,4-di-Cl-Ph | COOH | Br | 28 ± 12 | 7 ± 4 |
| 18 | 2,3-di-Cl-Ph | COOH | Br | 58 ± 13 | 19 ± 11 |
| 19 | 2,3,di-MeO-Ph | COOH | Br | 62 ± 14 | 34 ± 6 |
| 20 | 2,3,4-tri-MeO-Ph | COOH | Br | 85 ± 20 | 51 ± 16 |
| 21 | 2,5-di-Cl-Ph | COOH | Br | 47 ± 14 | 18 ± 4 |
| 22 | 2,6-di-Cl-Ph | COOH | Br | 52 ± 12 | 16 ± 8 |
| 23 | 3,4-di-Cl-Ph | COOH | Br | 27 ± 11 | 14 ± 4 |
| 24 | 2,3,5-tri-Cl-Ph | COOH | Br | 20 ± 12 | 13 ± 5 |
| 25 | 2,3,6-tri-Cl-Ph | COOH | Br | 23 ± 12 | <3.7 |
| 26 | 4-Cl-Ph | COOH | Cl | 85 ± 13 | 53 ± 21 |
| 27 | 2,4-di-Cl-Ph | COOH | Cl | > 100 | > 100 |
| 28 | 2,3-di-Cl-Ph | COOH | Cl | 100 | 92 ± 14 |
| 29 | 2,5-di-Cl-Ph | COOH | Cl | 75 | 25 |
| 30 | 2,3,5-tri-Cl-Ph | COOH | Cl | 55 ± 40 | 17 ± 3 |
| 31 | 2,3,6-tri-Cl-Ph | COOH | Cl | 77 ± 20 | 30 ± 5 |
| 32 | 3,4-di-Cl-Ph | COOH | Cl | 87 ± 13 | 41 ± 9 |
| 33 | 4-Cl-Ph | COOH | F | 36 ± 13 | 17 ± 3 |
| 34 | 2,4-di-Cl-Ph | COOH | F | 52 ± 13 | 24 ± 9 |
| 35 | 2,3-di-Cl-Ph | COOH | F | 55 ± 18 | 30 ± 9 |
| 36 | 2,5-di-Cl-Ph | COOH | F | 50 | 29 ± 6 |
| 37 | 2,3,5-tri-Cl-Ph | COOH | F | 33 ± 14 | 15 ± 4 |
| 38 | 2,3,6-tri-Cl-Ph | COOH | F | 82 ± 16 | 39 ± 6 |
| 39 | 3,5-di-Me-Ph | COOH | Br | >100 | 58 |
| 40 | 2-Cl-3,4-di-MeO-Ph | COOH | Br | >100 | 76 |
| 41 | 6-F-3,4-di-MeO-Ph | COOH | Br | >100 | 76 |
| 42 | 2-BzO-Ph | COOH | Br | 21 | 9 |
| 43 | 3-(4-Cl-PhO)-Ph | COOH | Br | 25 | 18 |
| 44 | 4-(4-Cl-PhO)-Ph | COOH | Br | 22 | 16 |
| 45 | 3-thienyl | COOH | Br | 30 ± 12 | 18 ± 1 |
| 46 | 3-(cyclopentyloxy)-Ph | COOH | Br | 100 | 55 |
| 47 | 3-(2-OEt-naphthyl) | COOH | Br | 92 | 46 |
| 48 | 5-MeO-naphthyl | COOH | Br | 54 | 54 ± 25 |
| 49 | 5-benzothiophene | COOH | Br | 23 ± 3 | 11 ± 1 |
| 50 | 2-furan-2-yl-Ph | COOH | Br | >100 | 16 ± 2 |
| 51 | 3-Cl-4,5-Methylenedioxy-Ph | COOH | Br | 79 | 63 |
| 52 | 3-OCF3-Ph | COOH | Br | >100 | 52 |
| 53 | 3-NO2-Ph | COOH | Br | 49 ± 8 | 32 ± 6 |
| 54 | 3-Br-Ph | COOH | Br | 19 ± 4 | 18 ± 1 |
| 55 | 3-F-Ph | COOH | Br | 71 ± 1 | 59 ± 2 |
| 56 | 2,3,6-tri-Cl-Ph | CN | Br | >100 | 44 ± 2 |
| 57 | 2,3,6-tri-Cl-Ph | COOH | CH3 | >100 | 50 ± 9 |
| 58 | 2,3,5-tri-Cl-Ph | NO2 | Br | >333 | 68 ± 5 |
| 59 | 2,3,6-tri-Cl-Ph | NO2 | Br | 36 ± 4 | 21 ± 3 |
| S1360c | N/A | N/A | N/A | 11 ± 2 | 0.6 ± 0.1 |
3-P = 3′-processing,
ST = strand transfer,
S1360 = standard
2.2.1. Substitution on aromatic ring A
Compound 9, 2-hydroxy-3-(3-phenyl-propyl)-benzoic acid, in which salicylic acid is connected to an unsubstituted phenyl ring B by a propylene linker exhibited only weak inhibitory activity with IC50 of >100 μM for ST and 91 μM for 3′-end processing. Modification to a 3-keto salicylic acid along with substituion with Br at R1 and an α,β-unsaturated linker (chalcone) in compound 10 (used as reference) demonstrated significant increase in both activity and selectivity (IC50 25 μM for ST vs IC50 75 μM for 3′-end processing). With the presence of a bromine substituent at the R1 on aromatic ring A being shown to be important for activity, chloroine and fluorine substituents were also investigated. Compounds with a Br substituent were the most active followed by F which did a little better than the corresponding Cl substituted analogs. To determine the effect of hydrophobic interaction, a methyl group was substituted for the halogens at R1 (see compound 57). Compound 57 was found to be 13 times less active than the corresponding Br-substituted compound 25. Since Br and a methyl group are similar in steric terms, this implies that it is not the hydrophobic effect of the halogen, but possibly the electronegativity and/or polarizability that might be dominant. The 3-keto salicylic acid pharmacophore was designed to form coordinate bonds with Mg2+. To test this hypothesis, the carboxylic acid was replaced with NO2 functionality (58, 59) and this modification diminished HIV inhibitory activity. Replacement of the carboxyl group with another polar group but with weaker coordination ability, CN (compound 56) also resulted in reduced inhibitory activity. This probably indicates that the formal negative charge of the free ionizable acidic moiety, that would chelate better with the metal ion, is required for potent activity, supporting the hypothesis. Of course a chelation study will have to be done to confirm this.
2.2.2. Substitution on aromatic ring B
Various groups were substituted on aromatic B to further explore the SAR. The presence of halogens on phenyl ring B produced noticeable improvements in potency based on the type, number and position on the aromatic ring. Fluorine substitution in the ortho and meta positions (compounds 12 and 55) diminished activity, as did iodine substitution at the para position (compound 16). However, substitution with Cl enhanced activity specifically for ST in all the three positions (compounds 11, 13 and 14). Interestingly, compounds 54 and 15 with m-Br and p-Br substituent, respectively, were potent against both catalytic steps (compound 15 showed IC50 of 5 μM against ST activity and 11 μM against 3′-processing). The 2,3,6-trichloro-substituted compound (25) was the most active, with IC50 of about ~ 4 μM against the ST reaction. This suggests a binding site pocket with high affinity for halogenated phenyl rings. This might be the site that binds the fluorophenyl group common in potent IN inhibitors like the clinically used drug raltegravir.
We next turned our attention to examining the following. (1) The effect of a bulky aryl B groups: benzyloxy and chloro substituted phenoxy in ortho, meta and para positions of phenyl ring B enhanced activity for both 3′-P and ST steps; whereas an ortho furan substituent (compound 50) increased the inhibitory potency against ST but decreased potency against 3′-P; the increase in activity was most pronounced for the o-benzyloxy substituent (compound 42), with the 3′-P and ST IC50 values lowered to 21 and 9 μM, respectively, compared to corresponding IC50 values of 75 and 25 μM for the parent phenyl compound 10. (2) The effects of electron donating or withdrawing groups: introduction of electron donating groups such as methoxy, methyl and cyclopentoxy (compounds 19, 39 and 46) decreased integrase inhibitory activity; and, increasing the number of electron donating groups resulted in further decrease in activity (compare compounds 19 and 20). Introduction of strong electron withdrawing groups such as trifluoromethyl and nitro also diminished activity as exemplified by compounds 52 and 53, respectively; (3) Boisosteric replacement of phenyl ring B with polycyclic and heteroaromatic rings: replacement of phenyl with thiophene or benzothiophene (compounds 45 and 49, respectively) displayed improved activity. Naphthalene derivatives on the other hand decreased activity (compounds 47 and 48). It thus appears that heteroaromatic B rings may improve integrase inhibitory activity.
2.2.3. Reduction of the α,β-unsaturated double bond of the chalcones or replacement with an amide moiety
To explore the importance of the α,β-unsaturated bond of the chalcones on inhibitory activity, we reduced it to a single bond. Our first attempt at this was with compound 25, but reduction also led to a loss of the Br substituent. However, reduction (Scheme 2) of analog 36, having a fluorine substituent, which is not as good a leaving group as Br, was successful in achieving our objective. The reduction product, compound 60, was less active than unsaturated analog, showing that the reduction of the double bond in the chalcone template is detrimental to activity. Bioisosteric replacement of the α,β-unsaturated carbonyl system in the chalcone with an amide moiety (compound 61, see Table 2) was well tolerated with retention of comparative inhibitory activity but an increase in ST selectivity. Interestingly, the aldehyde intermediate compound 8, which was obtained during the synthesis of the amide analog compound 61, was found to be more active than the carboxylic acid product 61, indicating that an aldehyde group can substitute for the carboxylic acid group in these compounds, opening an opportunity for exploring other bioisosteric substitutions.
Table 2.
Structure and HIV-1 integrase inhibitory activities of synthesized compounds
| ||||||
|---|---|---|---|---|---|---|
| Comp. | Ar | R2 | R1 | X | Activity (IC50μM) | |
| 3′-Pa | STb | |||||
| 8 | 4-F-Ph | NH | Br | CHO | >100 | 8 ± 1 |
| 60 | 2,5-di-Cl-Ph | CH2 | F | COOH | >100 | 57 ± 5 |
| 61 | 4-F-Ph | NH | Br | COOH | >100 | 15 ± 3 |
| 62 | 4-F-Ph | NH | H | CH3 | >100 | >100 |
| 63 | 4-F-Ph | NH | Br | H | >100 | >100 |
| S1360c | N/A | N/A | N/A | N/A | 11 ± 2 | 0.6 ± 0.1 |
3-P = 3′-processing,
ST = strand transfer,
S1360 = standard
2.3. Inhibition of HIV replication in cell culture
To evaluate the potential of these new IN inhibitors as antiviral agents, five potent analogs were tested for the ability to inhibit HIV-1 replication in primary human peripheral blood mononuclear cells (PBMCs) infected with the virus. The toxicity of the compounds was also tested against PBMC and human CEM lymphoblastic leukemia cells. All five compounds tested inhibited of HIV replication (Table 3). The most potent conpounds, 25 and 15 showed comparable EC50 values of 7.3 and 8.3 μM, repectively. Compound 49 (integrase ST IC50 = 11 μM) exhibited an EC50 of 10 μM, while analog 17 (integrase ST IC50 = 7.0 μM) displayed an EC50 of 13.9 μM. Thus, in general the biological activity correlated well with the IN inhibitory potency. Moreover, the two most potent IN inhibitory compounds of the set tested, 15 and 25, were also found to have the highest activity against HIV replication, suggesting that IN might indeed be their antiviral target. These results demonstrate moderate anti-viral activity, which is not very different from early members of the diketo acid IN inhibitors, which served as important lead compounds such as L-706,908 which had an EC50 of 5.7 ± 4.7 μM for inhibition of HIV replication.31 The reason that compound 8 (integrase ST IC50 = 8.0 μM), in which the carboxylic acid is replaced by an aldehyde group showed a lower antiviral activity (EC50 = 30.8 μM) than expected is not apparent, but may have to do with solubility, absorption, binding to serum proteins in cell culture media and/or cellular distribution factors. The compounds showed mild cytotoxicity, with selectivity index up to 4-fold for compound 49. It may be noteworthy that compound 49 is the only compound among the antiviral test set that had a 5-benzothiophene for ring B; this may offer clues for improving selectivity index. These results demonstrate reasonable anti-viral activity when compared to the anti-viral activity of reported diketo acid inhibitors like L-706,908 which has an EC50 of 5.7 ± 4.7 μM31 for inhibition of HIV replication. To put data in perspective with regard to AZT’s activity, in the same assay, AZT had an EC50 of 0.001 μM.31
Table 3.
Anti-HIV activity of selected novel potent derivatives
| Comp. in | Anti-HIV-1 activity in PBMCsa | Slope | R | Cytotoxicity (IC50, μM) | ||
|---|---|---|---|---|---|---|
| EC50 (μM)b | EC90 (μM)b | PBMCs | CEM | |||
| 15 | 8.7 | 21.0 | 2.5±0.61 | 0.97 | 24.4 | 27.5 |
| 17 | 13.9 | 26.0 | 3.5 | 1.0 | 22.1 | 23.8 |
| 25 | 7.3 | 20.3 | 2.2±0.96 | 0.91 | 22.7 | 19.0 |
| 49 | 10.1 | 24.3 | 2.5±0.89 | 0.94 | 39.1 | 20.7 |
| 8 | 30.8 | 52.7 | 4.1 | 1.0 | 44.0 | 40.2 |
| AZT | 0.0025 | 0.01 | 1.6±0.30 | 0.96 | >100 | 56.1 |
| L-706,908 | 5.7±4.7c | N/A | N/A | N/A | N/A | N/A |
Human peripheral blood mononuclear cells(PBMCs),
EC50 and EC90 are the effective concentrations inhibiting 50% and 90% HIV replication, respectively.
In the human leukemic T–cell line MOLT -4; in that assay the EC50 of AZT was 0.001 μM.
3. Molecular Modeling
3.1. Generation of a Pharmacophore Model Using the PHASE program
Knowledge of the structure of target protein inhibitor interations is limited as no crystal structure of human integrase-ligand-DNA complex is available. Therefore, to explore the bioactive conformation of these novel IN inhibitors, the PHASE32 (v3.0) pharmacophore mapping program implemented in Maestro modeling program package (v8.5; Shrodinger Inc.) was used. The 3D structures of molecules were constructed in and imported from the SYBYL modeling program (v8.3, Tripos).
Conformers of each molecule were generated using the OPLS_2005 force field in the macromodel conformational analysis program of PHASE. A set of pharmacophoric sites based on features defined in PHASE were assigned to the molecules. These included H-bond acceptor (A), H-bond donor (D), hydrophobic group (H), negatively charged group (N) and aromatic ring (R). The three most potent compounds of the series, compound 15, 17 and 25 were selected as actives for use in identifying a common pharmacophore hypothesis using a tree-based partitioning technique of PHASE. The resulting pharmacophores were then scored and ranked.
The top twenty pharmacophore hypotheses were analyzed and validated by partial least square (PLS) regression-based PHASE 3D-QSAR. Random external test sets were generated automatically to determine the predictive ability of the models. The hypothesis that afforded the best PLS statistics (Table 4) and the highest predicted ability consisted of six features viz., two H-bond acceptors, two hydrophobic groups, a negative group and one aromatic ring (Fig. 2a). Two H-bond acceptor features mapped onto the oxygen of the hydroxyl and the 3-keto moiety and negative feature mapped onto the carboxylate group of the salicylic acid pharmacophore. The two hydrophobic features mapped to the halogen substituent of ring A and the para substituted halogen in aryl ring B. The aromatic pharmacophoric feature mapped onto aromatic ring A. Residuals of the training and the test set are listed in Tables 5 and 6, respectively. The prediction curve for the test set is shown in Fig. 2b. As can be seen, the hypothesis gave a good predictive model with a predictive r2 of 0.57 for an external test set of ten compounds. It was also able to distinguish active and inactive compounds, with the latter group not aligning well to the features (Fig. 2c and 2d).
Table 4.
PLS Statistics of PHASE 3D-QSAR model
| q2 | 0.57 |
| r2 | 0.74 |
| SD | 0.17 |
| F | 28.3 |
| PLS Factors | 4 |
| Pearson R | 0.75 |
Figure 2.

The best pharmacophore hypothesis (AAHHNR) mapped onto (a) compound 17 (b) alignment of actives and (c) inactives from PHASE pharmacophore mapping. Pharmacophore features are: red spheres and vectors for H-bond acceptor (A), brown rings for aromatic groups (R), dark red spheres refer to a negative feature (N) and green spheres indicate hydrophobic groups (H). Atoms in the ligands are represented as O, red; Br, wine red; Cl, dark green; H, white; C, gray. (d) PHASE 3D-QSAR predictions of the test set.
Table 5.
Residuals of training set by PHASE 3D-QSAR
| Comp. | Actual ST pIC50 | Predicted ST pIC50 | Residuals |
|---|---|---|---|
| 10 | 4.60 | 4.85 | −0.25 |
| 11 | 4.85 | 4.59 | 0.26 |
| 12 | 4.49 | 4.53 | −0.04 |
| 14 | 4.92 | 4.84 | 0.08 |
| 15 | 5.30 | 4.94 | 0.36 |
| 16 | 4.45 | 4.85 | −0.4 |
| 17 | 5.15 | 5.02 | 0.13 |
| 18 | 4.72 | 4.66 | 0.06 |
| 19 | 4.46 | 4.59 | −0.13 |
| 20 | 4.29 | 4.29 | 0.00 |
| 21 | 4.74 | 4.87 | −0.13 |
| 22 | 4.79 | 4.83 | −0.04 |
| 24 | 4.88 | 4.82 | 0.06 |
| 25 | 5.40 | 5.40 | 0.00 |
| 26 | 4.27 | 4.49 | −0.22 |
| 27 | 4.00 | 4.21 | −0.21 |
| 28 | 4.03 | 4.48 | −0.45 |
| 32 | 4.39 | 4.44 | −0.05 |
| 33 | 4.76 | 4.63 | 0.13 |
| 34 | 4.62 | 4.33 | 0.29 |
| 35 | 4.52 | 4.44 | 0.08 |
| 36 | 4.54 | 4.63 | −0.09 |
| 37 | 4.82 | 4.59 | 0.23 |
| 39 | 4.23 | 4.42 | −0.19 |
| 40 | 4.12 | 3.94 | 0.18 |
| 41 | 4.12 | 4.15 | − 0.03 |
| 42 | 5.04 | 4.99 | 0.05 |
| 43 | 4.74 | 4.79 | −0.05 |
| 44 | 4.79 | 4.79 | 0.00 |
| 45 | 4.74 | 4.85 | −0.11 |
| 46 | 4.26 | 4.32 | −0.06 |
| 47 | 4.33 | 4.16 | 0.17 |
| 48 | 4.26 | 4.28 | −0.02 |
| 49 | 4.96 | 4.86 | 0.10 |
| 53 | 4.49 | 4.46 | 0.03 |
| 54 | 4.74 | 4.60 | 0.14 |
| 55 | 4.23 | 4.32 | −0.09 |
| 56 | 4.40 | 4.24 | 0.16 |
| 57 | 4.30 | 4.37 | −0.07 |
| 58 | 4.16 | 4.10 | 0.06 |
| 59 | 4.68 | 4.80 | −0.12 |
| 61 | 4.82 | 4.83 | −0.01 |
| 63 | 4.00 | 4.26 | −0.26 |
Table 6.
Residuals of test set by PHASE 3D-QSAR
| Comp. | Actual ST pIC50 | Predicted ST pIC50 | Residual |
|---|---|---|---|
| 13 | 5.04 | 4.86 | 0.18 |
| 23 | 4.85 | 4.78 | 0.07 |
| 29 | 4.60 | 4.71 | −0.11 |
| 30 | 4.77 | 4.63 | 0.14 |
| 31 | 4.52 | 4.25 | 0.27 |
| 38 | 4.41 | 4.28 | 0.13 |
| 50 | 4.80 | 4.91 | −0.11 |
| 51 | 4.20 | 4.51 | −0.31 |
| 52 | 4.28 | 4.46 | −0.18 |
| 62 | 4.00 | 4.35 | −0.35 |
3.2. 3D-QSAR Analysis
3.2.1. CoMFA and CoMSIA 3D QSAR Models
Statistical analysis by PLS 33,34 was done using CoMFA and CoMSIA descriptors as independent variables and biological activity in the form of pIC50 values as dependent variable. The molecular alignment used for CoMFA ans CoMSIA 3D-QSAR modeling was derived from PHASE pharmacophore mapping (Shrodinger, Inc.). PHASE derived pharmacaphore hypotheses have been successfully used to derive “bioactive conformations” for 3D-QSAR modeling in previous work from our laboratory35 and elsewhere (Telvekar et al.)36. Thus, “bioactive conformations” of all the molecules from PHASE conformational analysis were superimposed using the MATCH alignment tool in SYBYL (Fig. 3). Fifty three (53) synthesized target compounds were used for the 3D-QSAR analyses. The Leave-One-Out (LOO) method of cross validation was used for intial assessment of the predictive abilities of the models with the training sets. The optimal number of components used in the final QSAR models was that which gave the smallest standard error of prediction. Reliability of the models was tested by prediction of 10 compounds selected as an external test set using factor analysis. The PLS analysis results from both the CoMFA and CoMSIA models are summarised in Table 7. Cross-validated
Figure 3.

Superimposition of the conformation of molecules obtained from PHASE pharmacophore analysis. Atoms are represented as O, red; Br, dark green; Cl, green; F, bright green; H, cyan; C, white; S, yellow; N, blue.
Table 7.
PLS Statistics of CoMFA and CoMSIA 3D QSAR
| PLS statistics | CoMFA | CoMSIA |
|---|---|---|
| q2 | 0.43 | 0.54 |
| r2 | 0.94 | 0.94 |
| s | 0.09 | 0.09 |
| F | 78.32 | 78.69 |
| PLS components | 6 | 6 |
| Field contribution | ||
| Steric | 0.474 | 0.100 |
| Electrostatic | 0.526 | 0.428 |
| Hydrophobic | 0.283 | |
| Donor | 0.146 | |
| Acceptor | 0.044 | |
| Bootstrapping r2 (20 runs) | 0.96±0.01 | 0.96±0.01 |
| Randomization q2 (20 runs) | 0.33 | 0.39 |
| Cross-validation q2 (20 runs) | 0.44 | 0.50 |
q2 values of 0.43 and 0.54 were obtained for CoMFA and CoMSIA, respectively. Results of rigorous statistical testing using group cross-validation (20 groups), bootstrapping and randomization of activity data demonstrated the robustness of the QSAR models. Both CoMSIA and CoMFA identified six outliers, which were excluded on the basis of their extreme residual values (Table 8). Out of these, five outliers were common to both CoMFA and CoMSIA. These were compounds 16, 27, 28, 55 and 57. The reasons for compound 55 being an outlier are not apparent. With compound 16 (iodo substituted) and compound 57 (X = CH3), their outlier stsus may stem from structural uniqueness, whereas the outlier stus of compounds 27 and 28 (least active analogs, X = Cl) may drive from different binding modes. The CoMFA and CoMSIA predictions for the training and the test sets are shown in Figs. 4a and 4b, respectively. The CoMFA and CoMSIA residual values for the training set are given in Table 8 and the corresponding values for the test set in Table 9. In concordance with the higher q2 value, the CoMSIA model performed better than CoMFA mpdel, and predicted all test set compounds within 0.56 log unit of the actual pIC50 values.
Table 8.
Residuals of training set of CoMFA and CoMSIA models
| Residuals
|
|||
|---|---|---|---|
| Comp. | Actual ST pIC50 | CoMFA | CoMSIA |
| 10 | 4.60 | −0.13 | −0.12 |
| 11 | 4.85 | 0.12 | 0.12 |
| 12 | 4.49 | −0.18 | −0.19 |
| 13 | 5.04 | 0.05 | 0.03 |
| 14 | 4.92 | −0.15 | −0.08 |
| 15 | 5.30 | 0.16 | 0.11 |
| 16a | 4.45 | −0.62 | −0.96 |
| 17 | 5.15 | 0.21 | 0.19 |
| 18 | 4.72 | 0.10 | −0.07 |
| 19 | 4.46 | 0.06 | −0.04 |
| 20 | 4.29 | −0.05 | −0.003 |
| 21 | 4.74 | 0.01 | 0.04 |
| 22 | 4.79 | −0.03 | −0.03 |
| 23 | 4.85 | −0.13 | −0.03 |
| 24 | 4.88 | 0.07 | 0.11 |
| 25 | 5.40 | −0.05 | −0.05 |
| 26 | 4.27 | −0.02 | 0.09 |
| 27a | 4.00 | −0.62 | −0.89 |
| 28a | 4.03 | −0.79 | −0.74 |
| 29 | 4.60 | 0.02 | 0.005 |
| 30 | 4.77 | 0.01 | 0.11 |
| 31 | 4.52 | 0.03 | 0.06 |
| 33 | 4.76 | −0.009 | 0.04 |
| 34 | 4.62 | −0.02 | −0.13 |
| 35 | 4.52 | −0.03 | −0.12 |
| 36 | 4.54 | 0.06 | 0.04 |
| 38 | 4.41 | −0.001 | −0.04 |
| 39 | 4.23 | −0.05 | − 0.05 |
| 40 | 4.12 | − 0.02 | −0.02 |
| 43 | 4.74 | −0.07 | − 0.01 |
| 44c | 4.79 | −0.07 | 0.56 |
| 46 | 4.26 | −0.03 | 0.00 |
| 47 | 4.33 | 0.07 | 0.09 |
| 48 | 4.26 | 0.002 | −0.03 |
| 49 | 4.96 | 0.10 | 0.04 |
| 51 | 4.20 | 0.01 | −0.07 |
| 52b | 4.28 | −0.84 | −0.09 |
| 55a | 4.23 | −0.41 | −0.31 |
| 56 | 4.35 | −0.05 | −0.01 |
| 57a | 4.30 | −1.03 | −1.03 |
| 58 | 4.16 | −0.01 | 0.11 |
| 62 | 4.00 | 0.07 | 0.04 |
| 63 | 4.00 | −0.047 | −0.05 |
Represents common outliers,
outlier only for CoMFA,
Outlier only for CoMSIA
Figure 4.
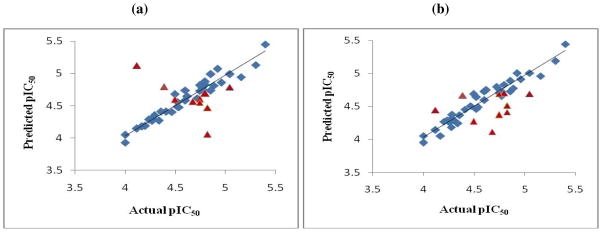
Predictions of ST activities of the training (blue diamonds) and test set (red triangles) compounds by (a) CoMFA and (b) CoMSIA
Table 9.
Residuals of the test set predictions given by CoMFA and CoMSIA models
| Comp. | Actual ST pIC50 | Residuals
|
|
|---|---|---|---|
| CoMFA | CoMSIA | ||
| 32 | 4.39 | −0.41 | −0.28 |
| 37 | 4.82 | 0.35 | 0.30 |
| 41 | 4.12 | −1.00 | −0.32 |
| 42 | 5.04 | 0.25 | 0.35 |
| 45 | 4.74 | 0.14 | 0.37 |
| 50 | 4.79 | 0.09 | 0.09 |
| 53 | 4.49 | −0.10 | 0.22 |
| 54 | 4.74 | 0.19 | 0.05 |
| 59 | 4.67 | 0.11 | 0.56 |
| 61 | 4.82 | 0.76 | 0.40 |
3.2.2. CoMFA and CoMSIA Contour Maps
The CoMFA and CoMSIA contour maps derived fro the PLS coefficients are displayed in Fig. 5. Steric CoMFA maps (Fig. 5a) show green contours around the ortho and para positions on aryl ring B indicating that bulky groups are favored at these positions. This is validated by the fact that a benzyl group substitution at the ortho position resulted in higher activity (compound 42). Compounds 44 and 49, which have a bulky chloro phenoxy and thiophene groups around the green contour at the para position also displayed better inhibitory activities. A large yellow contour surrounds a small green contour at the meta position on aryl ring B suggesting that only a limited bulk is favourable for activity. This is apparent by the loss of inhibitory activity observed with compound 46, the bulky cyclopentyl ring of which occupies the sterically disfavored yellow conour region. There is also a green contour evident at the R1 position on the aromatic ring A. This may be the reason why among the three halogens at that position, bromine, which is the bulkiest, afforded the highest activity. At the aryl ring B, CoMFA electrostatic map reveals a red contour at the 3′-position suggesting that electronegative groups could increase activity at this position. This is most exemplified by the 3′-Cl substitution in compound 13 which had an IC50 value of 9 μM. However, for compound 55, a decrease in activity was observed. Red contours are also present at the 5′- and the 6′-positions. The dichloro-substituted compounds 21 and 22 having Cl at the 5′- and 6′-positions showed improved activities. There are also distant blue contours around aryl ring B, which predicts that electropositive groups should increase activity at these positions. Electronegative oxygen atom of the methoxy substituents in compounds 20, 40 and 41 points towards the blue contours and thus may explain the loss of activity observed by the presence of methoxy groups in these compounds. The red contours around the salicylic acid moiety in aryl ring A suggest that a negative charge is important for activity in this region, possibly for chelation with the active site Mg+2 ions. This is in agreement with the proposed mechanism of action of these compounds as HIV-1 integrase inhibitors. There is a red contour around the 4′-position and both blue and red contours at the 5′-position (R1). This suggests that polarizability effect might be important at R1 and could explain the increased activity conferred by bromine substitution over other halogens and the similar-sized hydrophobic methyl group at this position.
Figure 5.

(a) CoMFA and (b) CoMSIA steric and electrostatic contour plots. The green contours indicate regions where bulky groups increase activity, whereas yellow contours indicate regions where bulky groups decrease activity. Blue contours indicate regions where electropositive groups increase activity, whereas red contours depict regions where electronegative groups increase activity. (c) CoMSIA hydrophobic, H-bond donor and H-bond acceptor contour plots; white contours indicate regions where hydrophilic groups increase activity, whereas yellow contours indicate regions where hydrophobic groups increase activity, cyan indicates region where H-donors increase activity and magenta contour indicate region where H-bond acceptor groups increase activity.
The CoMSIA steric contour map (Fig. 5b) also shows a large green contour around aryl ring B. Like in the CoMFA, the electrostatic contours of CoMSIA also show red contours at the 3′ and the 4′-positions on ring B, and at the 5′-position (R1) on ring A. There is a blue contour located at the position of the hydrogen atom of the hydroxyl group of the salicylic acid moiety. The hydophobic, H-bond donor and acceptor contours are shown in Fig. 5c. There is a white contour located near the R1 substituent position which suggests hydrophophilic groups increase activity at this region. This might be the reason why compound 57 with a hydrophobic methyl group at this position was less active than the corresponding halogen compounds. The improved activity observed with bromine at R1 position may be the result because of a combination of polarizability effects. White contours are also evident at the 2′ and the 5′-positions on the B ring, indicating that hydrophilic groups are favored for activity. This might be the explanation, or at least in part, for the decrease in potency of compounds 19 and 35, which have hydrophobic methoxy and methyl groups at these regions. Another white contour appears at the 3-keto oxygen again signifying the need for hydrophilic groups at this position. There is a yellow contour close to the 4′-position on ring B, suggesting that hydrophobic character is favored for activity. The 3′-keto functionality perfectly fits the magenta contour within its vicinity indicating that they may act as H-bond acceptors. A cyan contour appears close to, and merges into the magenta contour, suggesting that the OH group is a H-bond donor at this position. It may be involved in H-bonding interactions with the acceptor carboxylate groups of the acidic catalytic triad residues of intergase active site. The general agreement between the maps and the observed biological activity trends suggests that these models could aid in the design of more active compounds.
4. Conclusion
In summary, we have successfully synthesized and evaluated the HIV-1 integrase inhibitory activities of a series of chalcone derivatives and related amides harboring a 3-keto salicylic acid moiety as a diketo acid isostere. Consistent with the DKA class of IN inhibitors, these series of compounds were also generally selective against the IN strand transfer catalytic step. Introduction of a Br substituent at the R1 position and an α,β-unsturated carbonyl linker connecting aryl rings A and B resulted in significant enhancement of inhibitory activity. The most active compound has 2,3,6-trichloro substitution on aryl ring B. In the absence of a crystal structure of the complete human integrase, inhibitor and DNA complex, bioactive conformational hypothesis was obtained and validated by PHASE pharmacophore mapping and 3D-QSAR analysis. Statistically significant CoMFA and CoMSIA 3D-QSAR models were obtained using an alignment derived from the pharmacophore analysis, and may be useful for the design and prediction of the activities of novel related compounds as HIV-1 integrase inhibitors. Last but not the least, most of the potent compounds also inhibited HIV replication in cell culture with moderate antiviral activity, which supports further development of the series.
5. Experimental Section
All reagents and solvents were purchased from the Aldrich Chemical Company and used without further purification. Progress of reactions was monitored by TLC on silica gel GHLF-250 micron plates (Analtech, Inc.). Fisher scientific Da visil grade 1740 (170–400 mesh) silica gel was used for flash column chromatography. 1H NMR spectra were recorded on Brucker AR, 300 or 500-MHz spectrometer: chemical shifts are expressed in δ values (ppm) and coupling constants (J) in Hertz. Mass spectral data were determined on a Brucker-HP Esquire-LC spectrometer (ESI-MS). Melting points of final products were determined using a Fisher-Johns melting point apparatus and are reported uncorrected. HPLC analysis of final compounds was carried out using a reverse phase SUPELCOSIL 5 μm C-18 column of dimensions 25 cm × 4.6 cm. Area % purity was detected at 254 nm. An initial isocratic method comprising 40% water (solvent A) and 60% methanol (solvent B) for 20 min at a flow rate of 1.2 mL/min was optimized to a linear gradient elution with water (A) and methanol (B) at a flow-rate of 1.7 mL/min. Gradient elution of the mobile phase was either (a) 80 % A to 35 % A from 0–5 min, 35 % A, 65% B for 5–12 min, and 35 % A to 80 % A from 12–15 min or (b) 60 % A to 20 % A from 0–4 min, 20 % A and 80% B for 4–12 min and 20 % A to 60 % A from 12–15 min. The purity of all, but two, compounds was found to be ≥ 95%.
5.1. General procedure for the synthesis of acetylated intermediates (5a–5e)
5-Halogenated salicylic acids (46 mmol) were stirred with acetic anhydride (20 mL) and 80 μL of conc. H2SO4 was then added. After a few (20) min the reaction mixture solidified and was poured into cold water. The solid so obtained was filtered and extracted with ethyl acetate, washed with brine and dried over Na2SO4. The solvent was evaporated under reduced pressure to obtain a crude solid which was recrystalized from ethyl acetate/hexane to afford white crystals (5a–5c).
5.1.1. 2-Acetoxy-5-bromobenzoic acid (5a)
Yield 88%; mp 155–157 °C; 1H NMR (300 MHz, DMSO-d6): δ 13.44 (br s, 1H, COOH), 8.0 (d, 1H, J = 2.4 Hz, ArH), 7.85 (dd, 1H, J1 = 2.7 Hz, J2 = 8.7 Hz, ArH), 7.20 (d, 1H, J = 8.7 Hz, ArH), 2.2 (s, 3H, OCOCH3); MS (ESI): m/z 216.6 [M−COCH3]−. 6 mol
5.1.2. 2-Acetoxy-5-fluorobenzoic acid (5b)
Yield 85%; mp 133–134 °C; 1H NMR (300 MHz, DMSO-d6): δ 13.42 (br s, 1H, COOH), 7.67 (dd, 1H, J1 = 3.3 Hz, J2 = 9.0 Hz, ArH), 7.51 (ddd, 1H, J1 = 8.7 Hz, J2 = 8.1 Hz, J3 = 3.3 Hz, ArH), 7.26 (dd, 1H, J1 = 4.8 Hz, J2 = 8.7 Hz, ArH), 2.24 (s, 3H, OCOCH3); MS (ESI): m/z 154.7 [M− COCH3]−.
5.1.3. Acetoxy-5-chlorobenzoic acid (5c)
Yield 85%; mp 150–152 °C; 1H NMR (300 MHz, DMSO-d6): δ 7.88 (d, 1H, J = 2.7 Hz, ArH), 7.72 (dd, 1H, J1 = 2.7 Hz, J2 = 9.0 Hz, ArH), 7.27 (d, 1H, J = 8.4 Hz, ArH), 2.24 (s, 3H, OCOCH3); MS (ESI): m/z 170.7 [M− COCH3]−.
5.1.4. Acetic acid-4-bromo-2-cyanophenyl ester (5d)
Yeild 76%; mp 60–61 °C; 1H NMR (300 MHz, DMSO-d6) : δ 8.26 (d, 1H, J = 2.7 Hz, ArH), 8.0 (dd, 1H, J1 = 2.4 Hz, J2 = 8.7 Hz, ArH), 7.44 (d, 1H, J = 8.7 Hz, ArH), 2.29 (s, 3H, OCOCH3); MS (ESI): m/z 197.5 [M− COCH3]−.
5.1.5. 2-Acetoxy-5-methylbenzoic acid (5e)
Yeild 75%; mp 158–160 °C; 1H NMR (300 MHz, DMSO-d6) : δ 8.01 (d, 1H, J = 1.5 Hz, ArH), 7.50 (dd, 1H, J1 = 1.8 Hz, J2 = 8.4 Hz, ArH), 7.12 (d, 1H, J = 8.1 Hz, ArH), 2.43 (s, 3H, OCOCH3), 2.27 (s, 3H, CH3); MS (ESI): m/z 150.7 [M− COCH3]−.
5.2. General Procedure for the synthesis of 3-acetyl-5-halo salicylic acids (6a–6e)
The acetylated intermediates (38.61 mmol) and AlCl3 (120 mmol) were mixed in a three-necked flask and heated to 160 °C under mechanical stirring. After 3 h, the reaction mixture was cooled to room temperature and poured into ice containing 20 mL concentrated HCl. The slurry was extracted with ethyl acetate, acidified with 1 M HCl, washed with brine and dried over Na2SO4. The solvent was evaporated to give the crude product, which was washed with dichloromethane for removal of impurities, filteredand dried to give 3-acetyl salicylic acids as pale brown powders.
5.2.1. 2-Acetoxy-3-acetyl-5-bromobenzoic acid (6a)
Yield 34%; mp 182–185 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.06 (d, 1H, J = 2.7 Hz, ArH), 7.97 (d, 1H, J = 2.7 Hz, ArH), 2.60 (s, 3H, COCH3); MS (ESI): m/z 258.7 [M−H]−.
5.2.2. 2-Acetoxy-3-acetyl-5-fluorobenzoic acid (6b)
Yield 31%; mp 135–136 °C; 1H NMR (300 MHz, DMSO-d6): δ 7.78 (dd, 1H, J1 = 3.3 Hz, J2 = 8.1 Hz, ArH), 7.72 (dd, 1H, J1 = 3.3 Hz, J2 = 8.7 Hz, ArH), 2.62 (s, 3H, COCH3); MS (ESI): m/z 196.7 [M−H]−.
5.2.3. 2-Acetoxy-3-acetyl-5-chlorobenzoic acid (6c)
Yield 32 %; mp 160–163 °C; 1H NMR (300 MHz, DMSO-d6): δ 7.95 (d, 1H, J = 3.0 Hz, ArH), 7.85 (d, 1H J = 2.7 Hz, ArH), 2.61 (s, 3H, COCH3); MS (ESI): m/z 212.7 [M−H]−.
5.2.4. 3-Acetyl-5-bromo-2-hydroxybenzonitrile (6d)
Yield 26%; mp 75–77°C; 1H NMR (300 MHz, DMSO-d6): δ 13.17 (s, 1H, OH), 8.41 (d, 1H, J = 2.4 Hz, ArH), 8.32 (d, 1H J = 2.4 Hz, ArH), 2.68 (s, 3H, COCH3); MS (ESI): m/z 239.6 [M−H]−.
5.2.5. 3-Acetyl-2-hydroxy-5-methylbenzoic acid (6e)
Yield 28%; mp >180 °C (dec); 1H NMR (300 MHz, DMSO-d6): δ 7.80 (s, 1H, ArH), 7.64 (s, 1H, ArH), 2.59 (s, 3H, COCH3), 2.24 (s, 3H, CH3); MS (ESI): m/z 192.7 [M−H]−.
5.3. General procedure for the synthesis of chalcones (10–57)
Equimolar amounts of aromatic aldehydes and intermediates 6a–6e were dissolved in 10 mL of ethanol, and 5 mL of 25% aq NaOH was added. More solvent was added to help stirring in cases of excessive precipitation. The reaction mixture was stirred at rt for 1–5 days or until completion of reaction (6 h for compound 56). The mixture was then poured into ice and acidified with 3 N HCl to pH 4. The yellow precipitate formed was filtered and washed with water. The crude solid was purified by column chromatography (10–25% ethylacetate/hexane) and/or recrystalized with ethanol or THF/water to yield pure chalcones.
5.3.1. 5-Bromo-2-hydroxy-3-(3-phenyl-acryloyl)benzoic acid (10)
Yellow solid, yield 58%; mp 210–212 °C; 1H NMR (300 MHz, MeOH-d4): δ 8.08 (d, 1H, J = 2.4 Hz, ArH), 8.04 (d, 1H, J = 2.7 Hz, ArH), 7.81 (dd, 2H, J1 = 2.1 Hz, J2 = 7.5 Hz, ArH), 7.68 (s, 2H, H-α and H-β), 7.42–7.44 (m, 3H, ArH); MS (ESI): m/z 346.6 [M−H]− ; HPLC: tR 7.71 min, purity 99.10%.
5.3.2. 5-Bromo-3-[3-(2-chlorophenyl)-acryloyl]-2-hydroxybenzoic acid (11)
Yellow solid, yield 60%; mp 211–214 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.08 (d, 1H, J = 2.7 Hz, ArH), 8.03 (d, 1H, J = 2.7 Hz, ArH), 8.01 (dd, 1H, J1 = 1.5 Hz, J2 = 7.5 Hz, ArH), 7.94 (d, 1H, J = 15.9 Hz, H-β), 7.78 (d, 1H, J = 15.9 Hz, H-α), 7.59 (dd, 1H, J1 = 1.8 Hz, J2 = 8.4 Hz, ArH), 7.46 (ddd, 2H, J1 = 1.8 Hz, J2 = 7.2 Hz, J3 = 7.5 Hz, ArH); MS (ESI): m/z 378.6, 380.6 [M−H]− HPLC: tR 8.05 min, purity 98.82%.
5.3.3. 5-Bromo-3-[3-(2-fluorophenyl)-acryloyl]-2-hydroxybenzoic acid (12)
Yellow solid, yield 47%; mp 185–187 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.05 (d, 1H, J = 2.1 Hz, ArH), 8.0 (s, 1H, H-β), 7.93 (t, 1H, J = 7.2 Hz, ArH), 7.74 (s, 1H, H-α), 7.73 (d, 1H, J = 2.7 Hz, ArH), 7.51 (d, 1H, J = 6.9 Hz, ArH), 7.31 (dd, 1H, J1 = 7.2 Hz, J2 = 8.7 Hz, ArH), 7.29 (t, 1H, J = 7.2, ArH); MS (ESI): m/z 362.7, 364.7 [M−H]− HPLC: tR 7.24 min, purity 99.39%.
5.3.4. 5-Bromo-3-[3-(3-chlorophenyl)-acryloyl]-2-hydroxybenzoic acid (13)
Yellow solid, yield 56%; mp 234–236 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.06 (d, 1H, J = 2.7 Hz, ArH), 8.01 (d, 1H, J = 2.7 Hz, ArH), 7.92 (s, 1H, ArH), 7.76 (d, 1H, J = 15.9 Hz, H-β), 7.75 (t, 1H, J = 7.8 Hz ArH), 7.63 (d, 1H, J = 15.9 Hz, H-α), 7.50 (dd, 2H, J1 = 1.5 Hz, J2 = 8.7 Hz, ArH); MS (ESI): m/z 378.6, 380.6 [M−H]−. HPLC: tR 7.67 min, purity 99.30%.
5.3.5. 5-Bromo-3-[3-(4-chlorophenyl)-acryloyl]-2-hydroxybenzoic acid (14)
Yellow solid, yield 62%; mp 238–241 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.04 (d, 1H, J = 15.3 Hz, H-β), 7.91 (d, 1H, J = 2.4 Hz, ArH), 7.74 (d, 2H, J = 8.4 Hz, ArH), 7.71 (d, 1H, J = 3.0 Hz, ArH), 7.58 (d, 1H, J = 15.3 Hz, H-α), 7.50 (d, 2H, J = 8.4 Hz, ArH); MS (ESI): m/z 380.6 [M−H]−. HPLC: tR 8.1 min, purity 99.10%.
5.3.6. 5-Bromo-3-[3-(4-bromophenyl)-acryloyl]-2-hydroxybenzoic acid (15)
Yellow solid, yield 34%; mp 236–238 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.06 (d, 1H, J = 2.7 Hz, ArH), 8.02 (d, 1H, J = 2.7 Hz, ArH), 7.74 (d, 2H, J = 8.7 Hz, ArH), 7.69 (s, 1H, H-β), 7.67 (d, 2H, J = 8.7 Hz, ArH), 7.65 (s, 1H, H-α); MS (ESI): m/z 424.6 [M−H]−. HPLC: tR 6.05 min, purity: 99.02%.
5.3.7. 5-Bromo-2-hydroxy-3-[3-(4-iodophenyl)-acryloyl]benzoic acid (16)
Yellow solid, yield 48%; mp 237–239 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.06 (d, 1H, J = 2.7 Hz, ArH), 8.01 (d, 1H, J = 2.7 Hz, ArH), 7.83 (d, 1H, J = 8.4 Hz, ArH), 7.73 (d, 1H, J = 15.9 Hz, H-β), 7.69 (d, 1H, J = 8.4 Hz, ArH), 7.60 (d, 1H, J = 15.3 Hz, H-α), 7.59 (d, 2H J = 8.4 Hz, ArH); MS (ESI): m/z 472.6 [M−H]−. HPLC: tR 6.46 min, purity 94.70%.
5.3.8. 5-Bromo-3-[3-(2,4-dichlorophenyl)-acryloyl]-2-hydroxybenzoic acid (17)
Yellow solid, yield 54%; mp 234–235 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.08 (d, 1H, J = 2.7 Hz, ArH), 8.05 (d, 1H, J = 8.4 Hz, ArH), 8.05 (d, 1H, J = 2.7 Hz, ArH), 7.87 (d, 1H, J = 15.9 Hz, H-β), 7.76 (d, 1H, J = 15.9 Hz, H-α), 7.76 (d, 1H, J = 2.1 Hz, ArH), 7.52 (dd, 1H, J1 = 1.5 Hz, J2 = 8.4 Hz, ArH); MS (ESI): m/z 414.8 [M−H]−. HPLC: tR 10.91 min, purity 98.23%.
5.3.9. 5-Bromo-3-[3-(2,3-dichlorophenyl)-acryloyl]-2-hydroxybenzoic acid (18)
Yellow solid, yield 50%; mp 226–228 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.08 (d, 1H, J = 2.7 Hz, ArH), 8.03 (d, 1H, J = 2.4 Hz, ArH), 7.98 (d, 1H, J = 8.1 Hz, ArH), 7.91 (s, 1H, H-β), 7.77 (d, 1H, J = 15.6 Hz, H-α), 7.74 (d, 1H, J = 7.8 Hz, ArH), 7.46 (t, 1H, J = 7.8 Hz, ArH); MS (ESI): m/z 414.8 [M−H]−. HPLC: tR 6.46 min, purity: 99.08%.
5.3.10. 5-Bromo-3-[3-(2,3-dimethoxyphenyl)-acryloyl]-2-hydroxybenzoic acid (19)
Yellow solid, yield 51%; mp 195–197 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.02 (s, 1H, ArH), 7.92 (s, 1H, ArH), 7.86 (d, 1H, J = 16.2 Hz, H-β), 7.75 (d, 1H, J = 15.9 Hz, H-α), 7.40 (dd, 1H, J1 = 3.0 Hz, J2 = 5.7 Hz, ArH), 7.13–7.15 (m, 2H, ArH), 3.83 (s, 3H, OCH3), 3.77 (s, 3H, OCH3); MS (ESI): m/z 406.8 [M−H]−. HPLC: tR 7.03 min, purity: 99.7%.
5.3.11. 5-Bromo-2-hydroxy-3-[3-(2,3,4-trimethoxyphenyl)-acryloyl]-benzoic acid (20)
Yellow solid, yield 49%; mp 194–195 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.05 (d, 1H, J = 1.8 Hz, ArH), 8.02 (d, 1H, J = 1.8 Hz ArH), 7.80 (d, 1H, J = 15.9 Hz, H-β), 7.63 (d, 1H, J = 8.7 Hz, ArH), 7.10 (d, 1H, J = 15.9 Hz, H-α), 6.92 (d, 1H, J = 8.7 Hz, ArH), 3.87 (s, 1H, OCH3), 3.84 (s, 1H, OCH3), 3.77 (s, 1H, OCH3); MS (ESI): m/z 436.8 [M–H]−. HPLC: tR 6.88 min, purity: 99.36%.
5.3.12. 5-Bromo-3-[3-(2,5-dichlorophenyl)-acryloyl]-2-hydroxybenzoic acid (21)
Yellow solid, yield 51%; mp 232–234 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.16 (d, 1H, J = 2.4 Hz, ArH), 8.11 (d, 1H, J = 2.7 Hz, ArH), 8.0 (d, 1H, J = 2.7 Hz, ArH), 7.87 (s, 2H, H-α and H-β), 7.65 (d, 1H, J = 8.4 Hz, ArH), 7.57 (dd, 1H, J1 = 2.4 Hz, J2 = 8.4 Hz, ArH); MS (ESI): m/z 414.7 [M−H]−. HPLC: tR 8.03 min, purity 97.30%.
5.3.13. 5-Bromo-3-[3-(2,6-dichlorophenyl)-acryloyl]-2-hydroxybenzoic acid (22)
Yellow solid, yield 54%; mp 235–237 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.08 (d, 1H, J = 2.7 Hz, ArH), 7.96 (d, 1H, J = 2.7 Hz, ArH), 7.79 (d, 1H, J = 16.5 Hz, H-β), 7.67 (d, 1H, J = 16.5 Hz, H-α), 7.60 (d, 2H, J = 8.1 Hz, ArH), 7.41–7.46 (t, 1H, J = 8.7 Hz, ArH); MS (ESI): m/z 414.6 [M−H]−. HPLC: tR 7.96 min, purity 96.60%.
5.3.14. 5-Bromo-3-[3-(3,4-dichlorophenyl)-acryloyl]-2-hydroxybenzoic acid (23)
Yellow solid, yield 48%; mp 218–221 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.07 (d, 1H, J = 15.9 Hz, H-β), 8.0 (s, 1H, ArH), 7.93 (d, 1H, J = 2.7 Hz, ArH), 7.72 (d, 1H, J = 2.7 Hz, ArH), 7.70 (d, 2H, J = 7.5 Hz), 7.56 (d, 1H, J = 15.9 Hz, H-α); MS (ESI): m/z 414.6 [M−H]−. HPLC: tR 7.25 min, purity 97.0 %.
5.3.15. 5-Bromo-2-hydroxy-3-[3-(2,3,5-trichlorophenyl)-acryloyl]benzoic acid (24)
Yellow solid, yield 54%; mp 212–214 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.10 (d, 1H, J = 2.4 Hz, ArH), 8.08 (d, 1H, J = 2.7 Hz, ArH), 8.05 (d, 1H, J = 2.7 Hz, ArH), 7.96 (d, 1H, J = 2.4 Hz, ArH), 7.84 (br s, 2H, H-α and H-β); MS (ESI): m/z 448.6 [M−H]−. HPLC: tR 7.41 min, purity 98.21%.
5.3.16. 5-Bromo-2-hydroxy-3-[3-(2,3,6-trichlorophenyl)-acryloyl]benzoic acid (25)
Yellow solid, yield 57%; mp 220–223 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.07 (d, 1H, J = 2.7 Hz, ArH), 7.97 (d, 1H, J = 2.4 Hz, ArH), 7.73 (d, 1H, J = 8.7 Hz, ArH), 7.71 (d, 1H, J = 16.2 Hz, H-β), 7.61–7.64 (m, 2H, H-α and ArH); MS (ESI): m/z 448.6 [M−H]−. HPLC: tR 7.13 min, purity 99.20%.
5.3.17. 5-Chloro-3-[3-(4-chlorophenyl)-acryloyl]-2-hydroxybenzoic acid (26)
Yellow solid, yield 43%; mp 213–215 °C; 1H NMR (300 MHz, DMSO-d6): δ 7.95 (d, 1H, J = 2.1 Hz, ArH), 7.93 (d, 1H, J = 2.7 Hz, ArH), 7.84 (dd, 2H, J1 = 1.5 Hz, J2 = 6.9 Hz, ArH), 7.66 (s, 2H, H-β and H-α), 7.51 (dd, 2H, J1 = 1.5 Hz, J2 = 6.9 Hz, ArH); MS (ESI): m/z 336.9 [M−H]−. HPLC: tR 6.13 min, purity 99.55%.
5.3.18. 5-Chloro-3-[3-(2,4-dichlorophenyl)-acryloyl]-2-hydroxybenzoic acid (27)
Yellow solid, yield 48%; mp 248–250 °C; 1H NMR (300 MHz, MeOH-d4): δ 8.05 (d, 1H, J = 15.9 Hz, H-β), 7.93 (d, 1H, J = 8.7 Hz, ArH), 7.84 (d, 1H, J = 2.4 Hz, ArH), 7.80 (d, 1H, J = 15.3 Hz, H-α), 7.73 (d, 1H, J = 2.1 Hz, ArH), 7.65 (d, 1H, J = 2.7 Hz, ArH), 7.52 (dd, 1H, J1 = 2.1 Hz, J2 = 8.4 Hz, ArH); MS (ESI): m/z 370.8 [M−H]−. HPLC: tR 6.76 min, purity 99.14%.
5.3.19. 5-Chloro-3-[3-(2,3-dichlorophenyl)-acryloyl]-2-hydroxybenzoic acid (28)
Yellow solid, yield 41%; mp 250–252 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.10 (d, 1H, J = 15.3 Hz, H-β), 7.85–7.89 (m, 3H, H-α and ArH), 7.70 (d, 1H, J = 7.2 Hz, ArH), 7.62 (s, 1H, ArH), 7.45 (t, 1H, J = 7.5 Hz, ArH); MS (ESI): m/z 370.8 [M−H]−. HPLC: tR 6.06 min, purity 96.61%.
5.3.20. 5-Chloro-3-[3-(2,5-dichlorophenyl)-acryloyl]-2-hydroxybenzoic acid (29)
Yellow solid, yield 42%; mp 247–249 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.20 (d, 1H, J = 1.2 Hz, ArH), 8.04 (s, 2H, H-β and ArH), 7.90 (s, 2H, H-α and ArH), 7.67 (d, 1H, J = 8.4 Hz, ArH), 7.61 (dd, 1H, J1 = 1.5 Hz, J2 = 8.4 Hz, ArH); MS (ESI): m/z 370.7 [M−H]−. HPLC: tR 8.39 min, purity 99.0%.
5.3.21. 5-Chloro-2-hydroxy-3-[3-(2,3,5-trichlorophenyl)-acryloyl]benzoic acid (30)
Yellow solid, yield 37%; mp 235–237 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.05 (d, 1H, J = 15.9 Hz, H-β), 7.91 (d, 2H, J = 1.2 Hz, ArH), 7.82 (d, 1H, J = 3.0 Hz, ArH), 7.75 (d, 1H, J = 15.6 Hz, H-α), 7.60 (d, 1H, J = 2.7 Hz, ArH); MS (ESI): m/z 404.6 [M−H]−. HPLC: tR 7.18 min, purity 98.81%.
5.3.22. 5-Chloro-2-hydroxy-3-[3-(2,3,6-trichlorophenyl)-acryloyl]benzoic acid (31)
Yellow solid, yield 49%; mp 229–231 °C; 1H NMR (300 MHz, DMSO-d6): δ 7.99 (d, 1H, J = 16.2 Hz, H-β), 7.86 (d, 1H, J = 2.1 Hz, ArH), 7.71 (d, 1H, J = 2.7 Hz, ArH), 7.69 (d, 1H, J = 7.8 Hz, ArH), 7.65 (d, 1H, J = 8.4 Hz, ArH), 7.60 (d, 1H, J = 16.2 Hz, H-α); MS (ESI): m/z 404.6 [M−H]−. HPLC: tR 6.58 min, purity 95.66%.
5.3.23. 5-Chloro-3-[3-(3,4-dichlorophenyl)-acryloyl]-2-hydroxybenzoic acid (32)
Yellow solid, yield 54%; mp >240 °C (dec); 1H NMR (300 MHz, DMSO-d6): δ 8.05 (d, 1H, J = 15.9 Hz, H-β), 7.99 (s, 1H, ArH), 7.80 (d, 1H, J = 3.0 Hz, ArH), 7.71 (d, 2H, J = 6.0 Hz, ArH), 7.58 (d, 1H, J = 3.0 Hz, ArH), 7.56 (d, 1H, J = 15.9 Hz, H-α); MS (ESI): m/z 370.8 [M−H]−. HPLC: tR 6.78 min, purity 99.60%.
5.3.24. 3-[3-(4-Chlorophenyl)-acryloyl]-5-fluoro-2-hydroxybenzoic acid (33)
Yellow solid, yield 56%; mp 198–200 °C; 1H NMR (300 MHz, DMSO-d6): δ 7.91 (d, 1H, J = 15.9 Hz, H-β), 7.60 (d, 1H, J = 15.9 Hz, H-α), 7.68 (dd, 1H, J1 = 3.3 Hz, J2 = 8.4 Hz, ArH), 7.76 (d, 2H, J = 8.4 Hz, ArH), 7.49 (d, 2H, J = 8.7 Hz, ArH), 7.54 (dd, 1H, J1 = 3.3 Hz, J2 = 9.3 Hz, ArH); MS (ESI): m/z 318.8 [M−H]−. HPLC: tR 7.11 min, purity 99.11%.
5.3.25. 3-[3-(2,4-Dichlorophenyl)-acryloyl]-5-fluoro-2-hydroxybenzoic acid (34)
Yellow solid, yield 41%; mp 260–262 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.12 (d, 1H, J = 15.6 Hz, H-β), 7.93 (d, 1H, J = 8.4 Hz, ArH), 7.81 (d, 1H, J = 15.9 Hz, H-α), 7.74 (d, 1H, J = 2.1 Hz, ArH), 7.65 (dd, 1H, J1 = 3.3 Hz, J2 = 8.7 Hz, ArH), 7.52 (dd, 1H, J1 = 2.1 Hz, J2 = 8.4 Hz, ArH), 7.43 (dd, 1H, J1 = 3.3 Hz, J2 = 9.3 Hz, ArH); MS (ESI): m/z 352.8 [M−H]−. HPLC: tR 6.51 min, purity 99.5%.
5.3.26. 3-[3-(2,3-Dichlorophenyl)-acryloyl]-5-fluoro-2-hydroxybenzoic acid (35)
Yellow solid, yield 47%; mp 222–224 °C; 1H NMR (500 MHz, DMSO-d6): δ 7.94 (d, 1H, J = 7.8 Hz, ArH), 7.90 (s, 2H, H-α and H-β), 7.77 (dd, 1H, J1 = 3.0 Hz, J2 = 8.5 Hz, ArH), 7.73 (d, 1H, J = 8.0 Hz, ArH), 7.46 (t, 1H, J = 8.0 Hz, ArH), 7.66 (dd, 1H, J1 = 2.5 Hz, J2 = 9.0 Hz, ArH); MS (ESI): m/z 352.8 [M−H]−. HPLC: tR 5.68 min, purity 97.20%.
5.3.27. 3-[3-(2,5-Dichlorophenyl)-acryloyl]-5-fluoro-2-hydroxybenzoic acid (36)
Yellow solid, yield 59%; mp 238–240 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.07 (d, 1H, J = 15.9 Hz, H-β), 7.93 (d, 1H, J = 2.4 Hz, ArH), 7.76 (d, 1H, J = 15.9 Hz, H-α), 7.66 (dd, 1H, J1 = 3.6 Hz, J2 = 8.4 Hz, ArH), 7.60 (d, 1H J = 8.7 Hz, ArH), 7.52 (dd, 1H, J1 = 2.4 Hz, J2 = 8.7 Hz, ArH), 7.42 (dd, 1H, J1 = 3.6 Hz, J2 = 9.3 Hz, ArH); MS (ESI): m/z 352.8 [M−H]−. HPLC: tR 6.02 min, purity 98.43%.
5.3.28. 5-Fluoro-2-hydroxy-3-[3-(2,3,5-trichlorophenyl)-acryloyl]benzoic acid (37)
Yellow solid, yield 53%; mp 231–233 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.02 (d, 1H, J = 2.4 Hz, ArH), 7.95 (d, 1H, J = 15.9 Hz, H-β), 7.92 (d, 1H, J = 2.4 Hz, ArH), 7.79 (d, 1H, J = 15.6 Hz, H-α), 7.74 (dd, 1H, J1 = 3.3 Hz, J2 = 8.4 Hz, ArH), 7.64 (dd, 1H, J1 = 3.3 Hz, J2 = 9 Hz, ArH); MS (ESI): m/z 388.6 [M−H]−. HPLC: tR 8.11 min, purity 97.86%.
5.3.29. 5-Fluoro-2-hydroxy-3-[3-(2,3,6-trichlorophenyl)acryloyl]benzoic acid (38)
Yellow solid, yield 46%; mp 235–237 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.11 (d, 1H, J = 16.2 Hz, H-β), 7.70 (d, 1H, J = 8.7 Hz, ArH), 7.62 (d, 1H, J = 8.7 Hz, ArH), 7.58 (d, 1H, J = 16.2 Hz, H-α), 7.66 (dd, 1H, J1 = 3.6 Hz, J2 = 8.4 Hz, ArH), 7.44 (dd, 1H, J1 = 3.6 Hz, J2 = 9.3 Hz, ArH); MS (ESI): m/z 388.6 [M−H]−. HPLC: tR 7.57 min, purity 97.57%.
5.3.30. 5-Bromo-3-[3-(3,5-dimethylphenyl)-acryloyl]-2-hydroxybenzoic acid (39)
Yellow solid, yield 51%; mp 223–225 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.06 (d, 1H, J = 2.7 Hz, ArH), 7.99 (d, 1H, J = 1.8 Hz, ArH), 7.61 (d, 1H, J = 16.5 Hz, H-β), 7.58 (d, 1H, J = 16.5 Hz, H-α), 7.40 (s, 2H, Hz, ArH), 7.10 (s, 1H, ArH), 2.32 (s, 6H, CH3); MS (ESI): m/z 374.9 [M−H]−. HPLC: tR 6.59 min, purity 99.0%.
5.3.31. 5-Bromo-3-[3-(2-chloro-3,4-dimethoxyphenyl)-acryloyl]-2-hydroxybenzoic acid (40)
Yellow solid, yield 21%; mp 217–220 °C; 1H 1H NMR (300 MHz, DMSO-d6): δ 8.05 (d, 1H, J = 2.7 Hz, ArH), 8.00 (d, 1H, J = 2.7 Hz, ArH), 7.90 (d, 1H, J = 15.6 Hz, H-β), 7.81 (d, 1H, J = 8.7 Hz, ArH), 7.68 (d, 1H, J = 15.6 Hz, H-α), 7.16 (d, 1H, J = 8.7 Hz, ArH), 3.90 (s, 3H, OCH3), 3.77 (s, 3H, OCH3); MS (ESI): m/z 440.8 [M−H]−. HPLC: tR 6.46 min, purity 99.0%.
5.3.32. 5-Bromo-3-[3-(2-fluoro-4,5-dimethoxyphenyl)-acryloyl]-2-hydroxybenzoic acid (41)
Yellow solid, yield 32%; mp 193–195 °C; 1H NMR (300 Hz, DMSO-d6): δ 8.03 (d, 1H, J = 2.7 Hz, ArH), 7.93 (d, 1H, J = 2.7 Hz, ArH), 7.66 (s, 2H, H-β and H-α), 7.38 (d, 1H, J = 7.2 Hz, ArH), 7.00 (d, 1H, J = 12.3 Hz, ArH), 3.84 (s, 3H, OCH3), 3.82 (s, 3H, OCH3); MS (ESI): m/z 424.8 [M−H]−. HPLC: tR 7.19 min, purity 97.51%.
5.3.33. 3-[3-(2-Benzyloxyphenyl)-acryloyl]-5-bromo-2-hydroxybenzoic acid (42)
Yellow solid, yield 26%; mp > 165 °C (dec); 1H NMR (300 MHz, MeOH-d4): δ 7.92–7.95 (m, 2H, H-β and ArH), 7.72 (d, 1H, J = 7.5 Hz, ArH), 7.65 (d, 1H, J = 2.7 Hz, ArH), 7.47 (d, 2H, J = 7.2 Hz, ArH), 7.30–7.42 (m, 5H, H-α, and ArH), 7.20 (d, 1H, J = 8.1 Hz, ArH), 7.0 (t, 1H, J = 7.5 Hz, ArH), 5.22 (s, 2H, CH2),; MS (ESI): m/z 452.8 [M−H]−. HPLC: tR 7.83 min, purity 95.65%.
5.3.34. 5-Bromo-3-{3-[3-(4-chlorophenoxy)-phenyl]acryloyl}-2-hydroxybenzoic acid (43)
Yellow solid, yield 42%; mp 162–165 °C; 1H NMR (300 MHz, DMSO-d6): δ 7.94 (s, 2H, ArH), 7.74 (d, 1H, J = 6.9 Hz, ArH), 7.70 (d, 1H, J = 15.9 Hz, H-β), 7.40–7.46 (m, 6H, H- α and ArH), 7.20 (d, 1H, J = 7.8 Hz, ArH), 7.0 (t, 1H, J = 6.9 Hz, ArH); (ESI): m/z 472.6 [M−H]−. HPLC: tR 5.86 min, purity 96.8%.
5.3.35. 5-Bromo-3-{3-[4-(4-chlorophenoxy)phenyl]acryloyl}-2-hydroxybenzoic acid (44)
Yellow solid, yield 35%; mp 219–220 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.06 (d, 1H, J = 2.1 Hz, ArH), 8.02 (d, 1H, J = 2.1 Hz, ArH), 7.84 (d, 2H, J = 8.4 Hz, ArH), 7.67 (d, 1H, J = 15.9 Hz, H-β), 7.59 (d, 1H, J = 15.9 Hz, H-α), 7.48 (d, 2H, J = 8.7 Hz, ArH), 7.13 (d, 2H, J = 8.7 Hz, ArH), 7.07 (d, 2H, J = 8.7 Hz, ArH); MS (ESI): m/z 472.6 [M−H]−. HPLC: tR 7.23 min, purity 99.44%.
5.3.36. 5-Bromo-2-hydroxy-3-(3-thiophen-3-yl-acryloyl)benzoic acid (45)
Yellow solid, yield 54%; mp 207–210 °C; 1H NMR (300 MHz, DMSO-d6): δ 7.97 (s, 2H, ArH), 7.75 (d, 1H, J = 15.9 Hz, H-β), 7.65 (s, 1H, ArH), 7.61 (s, 1H, ArH), 7.58 (d, 1H, J = 15.6 Hz, H-α), 7.48 (d, 1H, J = 4.2 Hz, ArH); MS (ESI): m/z 352.6 [M−H]−. HPLC: tR 6.91 min, purity 99.71%.
5.3.37. 5-Bromo-3-[3-(3-cyclopentyloxy-4-methoxyphenyl)-acryloyl]-2-hydroxybenzoic acid (46)
Yellow solid, yield 31%; mp 233–235 °C; 1H NMR (300 MHz, MeOH-d4): δ 7.91 (d, 1H, J = 2.7 Hz, ArH), 7.82 (d, 1H, J = 15.6 Hz, H-β), 7.67 (d, 1H, J = 3.0 Hz, ArH), 7.59 (d, 1H, J = 15.9 Hz, H-α), 7.26 (d, 1H, J = 8.7 Hz, ArH), 7.25 (s, 1H, ArH), 6.99 (d, 1H, J = 8.1 Hz, ArH), 4.86 (p, 1H, J = 5.7 Hz, cyclopentyl), 3.79 (s, 3H, CH3) 1.87–1.92 (m, 2H, cyclopentyl), 1.66–1.72 (m, 4H, cyclopentyl), 1.57–1.60 (m, 2H, cyclopentyl); MS (ESI): m/z 460.8 [M−H]−. HPLC: tR 7.19 min, purity 97.51%.
5.3.38. 5-Bromo-3-[3-(2-ethoxynaphthalen-1-yl)acryloyl]-2-hydroxybenzoic acid (47)
Yellow solid, yield 23%; mp 216–218 °C; 1H NMR (300 MHz, MeOH-d4): δ 8.48 (d, 1H, J = 15.9 Hz, H-β), 8.30 (d, 1H, J = 8.4 Hz, ArH), 8.19 (d, 1H, J = 2.7 Hz, ArH), 8.17 (d, 1H, J = 15.9 Hz, H-α), 7.92 (d, 1H, J = 9.0 Hz, ArH), 7.88 (d, 1H, J = 2.7 Hz, ArH), 7.84 (d, 1H, J = 8.4 Hz, ArH), 7.55 (t, 1H, J = 7.2 Hz, ArH), 7.37–7.44 (m, 2H, ArH), 4.33 (q, 2H, J = 6.9 Hz, CH2), 1.55 (t, 3H, J = 6.9 Hz, CH3); MS (ESI): m/z 440.9 [M−H]−. HPLC: tR 8.53 min, purity 95.0%.
5.3.39. 5-Bromo-2-hydroxy-3-[3-(6-methoxynaphthalen-2-yl)-acryloyl]benzoic acid (48)
Yellow solid, yield 15%; mp 239–241 °C; 1H NMR (300 Hz, MeOH-d4): δ 8.17 (d, 1H, J = 2.4 Hz, ArH), 8.01 (s, 1H, H-β), 7.86 (d, 1H, J = 7.9 Hz, ArH), 7.84 (d, 2H, J = 8.1 Hz, ArH), 7.83 (d, 1H, J = 2.4 Hz, ArH), 7.80 (s, 2H, H-α, and ArH), 7.26 (d, 1H, J = 2.4 Hz, ArH), 7.16 (dd, 1H, J1 = 8.7 Hz, J2 = 2.4 Hz, ArH), 3.93 (s, 3H, OCH3); MS (ESI): m/z 426.8 [M−H]−. HPLC: tR 6.07 min, purity 98.93%.
5.3.40. 3-(3-Benzo[b]thiophen-5-yl-acryloyl)-5-bromo-2-hydroxybenzoic acid (49)
Yellow solid, yield 21%; mp 235–237 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.21 (s, 1H, ArH), 8.1 (d, 1H, J = 15.9 Hz, H-β), 8.0 (s, 1H, ArH), 7.92 (d, 1H, J = 2.7 Hz, ArH), 7.82 (d, 1H, J = 5.4 Hz, ArH), 7.73 (d, 1H, J = 15.9, H-α), 7.72 (d, 1H, J = 3 Hz, ArH), 7.72 (s, 1H, ArH), 7.53 (d, 1H, J = 5.1 Hz, ArH); MS (ESI): m/z 402.8 [M−H]−. HPLC: tR 5.53 min, purity 98.31%.
5.3.41. 5-Bromo-3-[3-(2-furan-2-yl-phenyl)-acryloyl]-2-hydroxybenzoic acid (50)
Yellow solid, yield 36%; mp 208–209 °C; 1H NMR (300 MHz, DMSO-d6): δ 7.92 (m, 3H, H-β and ArH), 7.87 (s, 1H, H-α), 7.81 (d, 1H, J = 2.7 Hz, ArH), 7.70 (s, 1H, ArH), 7.68 (d, 1H, 9.6 Hz, ArH), 7.48 (t, 2H, J = 7.5 Hz, ArH), 6.66 (s, 1H, ArH), 6.63 (s,1H, ArH); MS (ESI): m/z 412.8 [M−H]−. HPLC: tR 5.77 min, purity 95.02%.
5.3.42. 5-Bromo-3-[3-(6-chlorobenzo[1,3]dioxol-5-yl)-acryloyl]-2-hydroxybenzoic acid (51)
Yellow solid, yield 18%; mp 228–231 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.10 (s, 1H, ArH), 8.07 (s, 1H, ArH), 7.90 (d, 1H, J = 15 Hz, H-β), 7.66 (s, 1H, ArH), 7.63 (d, 1H, J = 14.7 Hz, H-α), 7.21 (s, 1H, ArH), 6.16 (s, 2H, CH2); MS (ESI): m/z 424.6 [M−H]−. HPLC: tR 7.82 min, purity 99.0%.
5.3.43. 5-Bromo-2-hydroxy-3-[3-(3-trifluoromethoxyphenyl)-acryloyl]benzoic acid (52)
Yellow solid, yield 44 %; mp 217–219 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.07 (d, 1H, J = 2.7 Hz, ArH), 8.03 (d, 1H, J = 2.7 Hz, ArH), 7.80–7.84 (br s, 2H, H-α and H-β), 7.72 ( s, 1H, ArH), 7.70 (s, 1H, J = 4.0 Hz, ArH), 7.58 (t, 1H, J = 7.8 Hz, ArH), 7.45 (d, 1H, J = 7.5 Hz, ArH); MS (ESI): m/z 430.8 [M−H]−. HPLC: tR 6.06 min, purity 99.30%.
5.3.44. 5-Bromo-2-hydroxy-3-[3-(3-nitrophenyl)-acryloyl]benzoic acid (53)
Yellow solid, yield 62%; mp 200–202 °C; 1H NMR (500 MHz, DMSO-d6): δ 8.62 (s, 1H, ArH), 8.26 (s, 2H, ArH), 8.07 (s, 1H, H-β), 8.02 (s, 1H, H-α), 7.81 (d, 2H, J = 7.0 Hz, ArH), 7.75 (t, 1H, J = 7.0 Hz, ArH); MS (ESI): m/z 391.8 [M−H]−. HPLC: tR 11.47 min, purity 95.59%.
5.3.45. 5-Bromo-3-[3-(3-bromophenyl)-acryloyl]-2-hydroxybenzoic acid (54)
Yellow solid, yield 31%; mp 235–237 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.0 (d, 1H, J = 15.9 Hz, H-β), 7.92 (d, 1H, J = 2.7 Hz, ArH), 7.90 (s, 1H, ArH), 7.72 (d, 1H, J = 6.6 Hz, ArH), 7.70 (d, 1H, J = 2.4 Hz, ArH), 7.59 (d, 1H, J = 8.4 Hz, ArH), 7.55 (d, 1H, J = 15.9 Hz, H-α), 7.39 (t, 1H, J = 7.8 Hz, ArH); MS (ESI): m/z 424.6 [M−H]−. HPLC: tR 6.14 min, purity 98.81%.
5.3.46. 5-Bromo-3-[3-(3-fluorophenyl)-acryloyl]-2-hydroxybenzoic acid (55)
Yellow solid, yield 59%; mp 190–193 °C; 1H NMR (500 MHz, DMSO-d6): δ 8.0 (d, 1H, J = 2.0 Hz, ArH), 7.93 (s, 1H, ArH), 7.80 (d, 1H, J = 16.0 Hz, H-β), 7.60–7.77 (m, 3H, H-α and ArH), 7.48 (d, 1H, J = 6.5 Hz, ArH), 7.28 (d, 1H, J = 8.0 Hz, ArH); MS (ESI): m/z 364.7 [M−H]−. HPLC: tR 5.39 min, purity 92.5%.
5.3.47. 5-Bromo-2-hydroxy-3-[3-(2,3,6--trichlorophenyl)-acryloyl]benzonitrile (56)
Orange solid, yield 74%; mp 139–141 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.34 (d, 1H, J = 16.2 Hz, H-β), 7.69 (d, 1H, J = 3.0 Hz, ArH), 7.66 (d, 1H, J = 9.0 Hz, ArH), 7.58 (d, 1H, J = 8.7 Hz, ArH), 7.46 (d, 1H, J = 2.4 Hz, ArH), 7.43 (d, 1H, J = 16.2 Hz, H-α); MS (ESI): m/z 431.6 [M−H]−. HPLC: tR 6.37 min, purity: 95.69%.
5.3.48. 2-Hydroxy-5-methyl-3-[3-(2,3,6-trichlorophenyl)-acryloyl]benzoic acid (57)
Yellow solid, yield 58.5%; mp 205–208 °C; 1H NMR (300 MHz, DMSO-d6): δ 7.87 (s, 1H, ArH), 7.74 (s, 1H, ArH), 7.70 (d, 1H, J = 8.7 Hz, ArH), 7.59–7.64 (m, 3H, H-α, H-β and ArH), 2.29 (s, 3H, CH3); MS (ESI): m/z 384.8 [M−H]−. HPLC: tR 6.01 min, purity 97.84%.
5.4. General procedure for the synthesis of chalcones (58–59)
A similar procedure to the one mentioned above was followed with appropriate aromatic aldehydes and commercially available 1-(5-Bromo-2-hydroxy-3-nitro-phenyl)-ethanone starting material.
5.4.1. 1-(5-Bromo-2-hydroxy-3-nitrophenyl)-3-(2,3,5-trichlorophenyl)propenone (58)
Yellow solid, yield 65%; mp 175–178 °C; 1H NMR (300 MHz, DMSO-d6): δ 8.13 (d, 1H, J = 15.6 Hz, H-β), 7.76 (d, 1H, J= 15.9 Hz, H-α), 8.05 (d, 1H, J = 2.7 Hz, ArH), 8.0 (d, 2H, J = 2.4 Hz, ArH), 7.92 (d, 1H, J = 2.4 Hz, ArH); MS (ESI): m/z 451.8 [M−H]−. HPLC: tR 5.46 min, purity 97.30 %.
5.4.2. 1-(5-Bromo-2-hydroxy-3-nitrophenyl)-3-(2,3,6-trichlorophenyl)propenone (59)
Yellow solid, yield 58%; mp 153–154 °C; 1H NMR (500 MHz, DMSO-d6): δ 8.32 (s, 1H, ArH), 8.24 (s, 1H, ArH), 7.77 (d, 1H, J = 16.5 Hz, H-β), 7.75 (d, 1H, J = 9.0 Hz, ArH), 7.65 (d, 1H, J = 15.0 Hz, H-α), 7.63 (d, 1H, J = 8.0 Hz, ArH); MS (ESI): m/z 451.8 [M−H]−. HPLC: tR 6.53 min, purity 98.0%.
5.5. General procedure for the synthesis of 3-[3-(2,5-dichlorophenyl)propionyl]-5-fluoro-2-hydroxybenzoic acid (60)
Compound 36 (0.2 g, 0.563 mmol) was dissolved in methanol (10 mL) and Pd/C (10 mol %, 20 mg) was added. The resulting solution was stirred under hydrogen at atmospheric pressure for 2 h at room temperature. After the reaction was complete, the reaction mixture was filtered through celite, and evaporated to dryness. The resulting crude solid was recrystalized from ethanol to give a white solid, yield 43%; mp 163–164 °C; 1H NMR (300 MHz, DMSO-d6): δ 7.79 (d, 1H, J = 8.4 Hz, ArH), 7.74 (d, 1H, J = 9.0, ArH), 7.49 (s, 1H, ArH), 7.45 (d, 1H, J = 8.7 Hz, ArH), 7.31 (d, 1H, J = 8.1 Hz, ArH), 3.39 (t, 2H, J = 6.9 Hz, CH2), 3.02 (t, 2H, J = 6.9 Hz, CH2); MS (ESI): m/z 356.9 [M−H]−. HPLC: tR 8.34 min, purity 96.36%.
5.6. General procedure for the synthesis of 5-bromo-N-(4-fluorobenzyl)-2-hydroxyisophthalamic acid (61)
5.6.1. 5-Bromo-3-formyl-2-hydroxybenzoic acid (7)
(Method a) NaOH (5.53 g, 0.138 mol) was added to a solution of 5-bromosalicylic acid (5 g, 23.0 mmol) in CHCl3 (45 mL) and water (1 mL, 55.55 mmol) at room temperature. The reaction mixture was refluxed for 48 h, diluted with water and acidified with 6 N HCl to pH 1.0. The resulting solution was extracted with ethyl acetate washed with brine and dried over Na2SO4. The organic layer was evaporated in vacuo to yield a crude product which was purified by column chromatography (25–35% methanol/dichloromethane) to give a white solid, yield 13%.
(Method b) 3-Formyl salicylic acid hydrate ( 1 g, 6.0 mmol) was dissolved in dry acetonitrile (20 mL). N-Bromo succinimide (1.02 g, 5.73 mmol) was added and the reaction mixture stirred at rt for 3 h. The white crystals formed were filtered and washed with water. The filtrate was extracted with ethyl acetate, and washed with brine. The organic layer was dried over Na2SO4 and evaporated to dryness. The two fractions were combined to give intermediate 7 as white crystals, yield 90%; mp 172–174 °C; 1H NMR (300 MHz, DMSO-d6): δ 10.27 (s, 1H, CHO), 8.12 (d, 1H, J = 2.7, ArH), 7.93 (d, 1H, J = 2.7 ArH); MS (ESI): m/z 244.7 [M−H]−.
5.6.2. 5-Bromo-N-(4-fluorobenzyl)-3-formyl-2-hydroxybenzamide (8)
To a solution of intermediate 7(0.4 g, 1.63 mmol), HOBt (0.441 g, 3.26 mmol) and Et3N (0.45 mL, 3.26 mmol) in DCM (20 mL) was added EDCI (0.344 g, 1.8 mmol) at 0 °C. 4-Fluorobenzylamine (0.20 mL, 1.8 mmol) was then added and the reaction mixture stirred overnight at room temperature. The reaction mixture was extracted with ethyl acetate, washed with dilute hydrochloric acid and sodium bicarbonate solution. The organic layer was further washed with brine and dried over sodium sulfate. The resulting crude was purified by recrystalization with ethylacetate/hexane to give a yellow solid. Yield 41%; mp 135–137 °C; 1H NMR (300 MHz, DMSO-d6): δ 10.27 (s,1H, CHO), 9.90 (s, 1H, NH), 8.41 (s, 1H, ArH), 7.87 (s, 1H, ArH), 7.40 (d, 2H, J = 8.1 Hz, ArH), 7.18 (t, 2H, J = 8.1 Hz, ArH), 4.52 (s, 2H, CH2); MS (ESI): m/z 351.9 [M−H]−.
5.6.3. 5-Bromo-N-(4-fluorobenzyl)-2-hydroxyisophthalamic acid (61)
Intermediate 8 (0.2 g, 0.568 mmol) was added to a mixture of water (2.5 mL) and pyridine (2.5 mL). The reaction mixture was heated to 90 °C, KMnO4 (0.18 g, 1.13 mmol) was added to the hot solution was and the heating continued overnight at 90 °C. After completion, the mixture was filtered through celite and evaporated to dryness. The resulting residue was purified by recrystalization from ethylacetate/hexane. Yield 80 %; mp 208–210 °C; 1H NMR (300 MHz, DMSO-d6): δ 10.20 (s, 1H, NH), 7.98 (s, 1H, ArH), 7.85 (s, 1H, ArH), 7.13–7.34 (m, 4H, ArH), 4.50 (s, 2H, CH2); MS (ESI): m/z 367.9 [M−H]−. HPLC: tR 4.21 min, purity 97.7%.
5.7. General procedure for the synthesis of N-(4-fluoro-benzyl)-2-hydroxy-3-methylbenzamide (62) and 5-bromo-N-(4-fluoro-benzyl)-2-hydroxybenzamide (63)
A similar procedure to the one described for the synthesis of intermediate 8 was followed by condensing 4-fluorobenzyl amine and commercially available 2-hydroxy-3-methylbenzoic acid (3a) and 5-bromosalicylic acid (3b) to give compounds 62 and 63, respectively.
5.7.1. N-(4-Fluoro-benzyl)-2-hydroxy-3-methylbenzamide (62)
Yield 76%; mp 165–166 °C; 1H NMR (300 MHz, DMSO-d6): δ 13.10 (s, 1H, OH), 9.43 (t, 1H, J = 6.0 Hz, NH), 7.72 (d, 1H, J = 7.2 Hz, ArH), 7.37 (d, 1H, J = 8.7 Hz, ArH), 7.35 (d, 1H, J = 8.4 Hz, ArH), 7.31 (d, 1H, J = 7.2 Hz, ArH), 7.15 (t, 2H, J = 9.0 Hz, ArH), 6.78 (t, 1H, J = 7.5 Hz, ArH), 4.48 (d, 1H, J = 6.0 Hz, CH2), 2.15 (s, 3H, CH3); MS (ESI): m/z 257.9 [M−H]−. HPLC: tR 8.63 min, purity 99.11%.
5.7.2. 5-Bromo-N-(4-fluoro-benzyl)-2-hydroxybenzamide (63)
Yield 62%; mp 83–84 °C; 1H NMR (300 MHz, DMSO-d6): δ 12.47 (s,1H, OH), 9.37 (t, 1H, J = 5.7 Hz, NH), 8.07 (d, 1H, J = 2.4 Hz, ArH), 7.55 (dd, 1H, J1 = 2.4 Hz, J2 = 8.7 Hz, ArH), 7.36 (dd, 2H, J1 = 5.7 Hz, J2 = 8.1, ArH), 7.15 (t, 2H, J = 9.0, ArH), 6.89 (d, 1H, J = 8.7 Hz, ArH), 4.47 (d, 2H, J = 5.7 Hz, CH2); MS (ESI): m/z 323.8 [M−H]−. HPLC: tR 9.7 min, purity 98.66%.
5.8. General procedure for the synthesis of 2-hydroxy-3-(3-phenylpropyl)benzoic acid (9)
Compound 10 (0.05 g, 0.195 mmol) was dissolved in ethanol (10 mL) and Pd/C (10 mol %, 5 mg) was added. The resulting solution was stirred under hydrogen at atmospheric pressure for 10 h at room temperature. The reaction mixture was then filtered through celite, and evaporated to dryness. The resulting crude solid was purified using preparative chromatography to give a white solid, yield 72%; mp 125–127 °C; 1H NMR (300 MHz, DMSO-d6): δ 11.66 (s,1H, OH), 7.67 (dd, 1H, J1 = 1.5 Hz, J2 = 7.8 Hz, ArH), 7.39 (dd, 1H, J1 = 1.5 Hz, J2 = 7.2 Hz, ArH), 7.16–7.30 (m, 5H, ArH), 6.85 (t, 1H, J = 7.8 Hz, ArH), 2.62 (t, 4H, J = 7.2 Hz, CH2), 1.81–1.91 (m, 2H, CH2); MS (ESI): m/z 254.8 [M−H]−. HPLC: tR 7.61 min, purity 98.05 %.
5.9. CoMFA and CoMSIA Studies
Three-dimensional structure building and CoMFA and CoMSIA studies were performed on a Dell computer workstation workstation with the Red Hat Linux operating system using the SYBYL program, version 8.0 ( Tripos Associates Inc). Partial atomic charges were assigned with MOPAC. The CoMFA descriptors, steric (Lennard-Jones 6–12 potential) and electrostatic (Coulombic potential) field energies, were calculated using Tripos force field with a distance-dependent dielectric and employing the SYBYL default parameters: 2 A grid points spacing, an sp3 carbon probe atom with +1 charge asnd vdW radius of 1.52 A, and energy cut-off of 30 kcal/mol. The five similarity indices in CoMSIA, i.e., steric, electrostatic, hydrophobic, H-bond donor and H-bond acceptor descriptors, were calculated37 using a C1+ probe atom with a radius of 1.0 A placed at regular grid spacing of 2 A. A Gaussian type distance dependence was used between the grid point q and each atom i of the molecule. The default value of 0.3 was used as the attenuation factor (R). Column filtering was set to 2.0 kcal/mol. CoMSIA steric indices are related to the third power of the atomic radii, electrostatic descriptors were derived from atomic partial charges, hydrophobic fields were derived from atom-based parameters,38 and H-bond donor and acceptor indices were obtained by a rule-based method based on experimental results.39 The CoMFA and CoMSIA descriptors were used as independent variables, and pIC50 (−log IC50) values were used as the dependent variable in PLS regression analyses to derive 3D-QSAR models. The predictive value of the models was evaluated by leave-one-out (LOO) cross-validation. The cross validated coefficient, q2, was calculated using eq 1:
| (1) |
where Ypredicted, Yactual, and Ymean are predicted, actual and mean values of the target property (pIC50), respectively. Σ(Ypredicted − Yactual)2 is the predictive sum of squares (PRESS). Conventional correlation coefficient r2 and its standard error, s, were also computed for the final PLS models. CoMFA and CoMSIA coefficient maps were generated by interpolation of the pair wise products between the PLS coefficients and the standard deviations of the corresponding CoMFA or CoMSIA descriptor values.
5.10. Biological assays
5.10.1. Materials, chemicals, and enzymes
All compounds were dissolved in DMSO and the stock solutions were stored at −20 °C. γ[32P]-ATP was purchased from either Amersham Biosciences or ICN. The expression systems for wild-type IN were a generous gift from Dr. Robert Craigie, Laboratory of Molecular Biology, NIDDK, NIH, Bethesda, MD. Cell lines were obtained from American Type Culture Collection (ATCC, Rockville, MD).
5.10.2. Preparation of oligonucleotide substrates
The oligonucleotides 21top, 5′-GTGTGGAAAATCTCTAGCAGT-3′, and 21bot, 5′-ACTGCTAGAGATTTTCCACAC-3′, were purchased from the Norris Cancer Center Microsequencing Core Facility (University of Southern California) and purified by UV shadowing on polyacrylamide gel. To analyze the extent of 3′-end processing and strand transfer using 5′-end labeled substrates, 21top was 5′-end labeled using T4 polynucleotide kinase (Epicentre, Madison, WI) and γ[32P]-ATP (Amersham Biosciences or ICN). The kinase was heat-inactivated and 21-bot was added in 1.5 M excess. The mixture was heated at 95 °C, allowed to cool slowly to room temperature, and run through a G25 mini spin column (USA Scientific) to separate double-stranded oligonucleotide from unincorporated material.
5.10.3. Integrase assays
To determine the extent of 3′-processing (3′-P) and strand transfer (ST), wild-type IN was preincubated at a final concentration of 200 nM with the inhibitor in reaction buffer (50 mM NaCl, 1mM HEPES, pH 7.5, 50 μM EDTA, 50 μM dithiothreitol, 10% glycerol (w/v), 7.5 mM MnCl2, 0.1 mg/mL bovine serum albumin, 10 mM 2-mercaptoethanol, 10% dimethylsulfoxide, and 25 mM MOPS, pH 7.2) at 30 °C for 30 min. An aliquot (5μL) was electrophoresed on a denaturing 20% polyacrylamide gel (0.09 M Tris- borate, pH 8.3, 2 mM EDTA, 20% acrylamide, and 8M urea). Gels were dried, exposed in a PhosphorImager cassette, and analyzed using a Typhoon 8610 Variable Mode Imager (Amersham Biosciences) and quantitated using ImageQuant 5.2. Percent inhibition (%I) was calculated using the following equation:
, where C, N, and D are the fractions of 21-mer substrate converted to 19-mer (3′-P product) or strand transfer products for DNA alone, DNA plus IN, and IN plus compound, respectively. The IC50 values were determined by plotting the logarithm of compound concentration versus percent inhibition to obtain the concentration that produced 50% inhibition.
5.11. Anti-HIV-1 activity assay
The testing of the ability of potent compounds to inhibit HIV replication in cell culture was done according to a previously reported procedure.40 PBMC (107 cells/T25flask) were stimulated with phytohemagglutinin for 3 days and infected with a wild-type HIV-1 strain (strain LAI) at 100 50% tissue culture infective doses, as described previously.40 The cultures were kept for 5 days in the presence of test compounds at serial 1-log dilutions. Subsequently, human PBMC were removed from the culture supernatant by centrifugation (400g, 10 min, 4 °C). This clarified supernatant was tested by a reverse transcriptase assay.
5.12. Cytotoxicity assays
The cytotoxicity of compounds was evaluated using uninfected PBMC and CEM leukemia cells according to a previous method.41 PBMC were obtained from whole blood of healthy individuals, while CEM were obtained from the ATCC (Rockville, MD). The PBMC and CEM cells were cultured in the presence or absence of compound for 6 days. After this time period, cells were stained with Trypan blue dye, and counted for cell proliferation and viability according to the previously reported procedure.42
Acknowledgments
Financial support from the National Institute of Allergy and Infectious Diseases (NIAID), NIH grant No. AI084710 and the Department of Pharmaceutical Sciences is greatfully acknowledged.
Abbreviations
- HIV
Human Immunodeficiency Virus
- ST
Strand Transfer
- FDA
Food and Drug Administration
- 3D-QSAR
Three Dimensional Quantitative Structure Activity Relationship
- CoMFA
Comparitive Molecular Field Analysis
- CoMSIA
Comparative Molecular Similarity Analysis
- VdW
Van der Waals
- NMR
Nuclear Magnetic Resonance
- HPLC
High Performace Liquid Chromatography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Coffin JC, Hughes SH, Vermus HE, editors. RetroViruses. Cold Spring Harbor Laboratory Press; Plainview, NY: 1999. [PubMed] [Google Scholar]
- 2.Sweeney ZK, Klumpp K. Curr Opin Drug Discovery Dev. 2008;11:458–470. [PubMed] [Google Scholar]
- 3.Von Hentig N. Drugs Today. 2008;44:103–132. doi: 10.1358/dot.2008.44.2.1137107. [DOI] [PubMed] [Google Scholar]
- 4.MacArthur RD, Novak RM. Clin Infect Dis. 2008;47:236–241. doi: 10.1086/589289. [DOI] [PubMed] [Google Scholar]
- 5.Manfredi R, Sabbatani S. Curr Med Chem. 2006;13:2369–2384. doi: 10.2174/092986706777935069. [DOI] [PubMed] [Google Scholar]
- 6.De Clercq E. J Med Chem. 1995;38:2491–2517. doi: 10.1021/jm00014a001. [DOI] [PubMed] [Google Scholar]
- 7.Zhang L, Ramratnam B, Tenner-Racz K, He Y, Vesanen M, Lewin S, Talal A, Racz P, Perelson AS, Korber BT, Markowitz M, Ho DD. N Eng J Med. 1999;340:1605–1613. doi: 10.1056/NEJM199905273402101. [DOI] [PubMed] [Google Scholar]
- 8.Richman DD. Nature. 2001;410:995–1001. doi: 10.1038/35073673. [DOI] [PubMed] [Google Scholar]
- 9.Esposito D, Craigie R. Adv Virus Res. 1999;52:319–333. doi: 10.1016/s0065-3527(08)60304-8. [DOI] [PubMed] [Google Scholar]
- 10.Katz RA, Skalka AM. Annu Rev Biochem. 1994;63:133–173. doi: 10.1146/annurev.bi.63.070194.001025. [DOI] [PubMed] [Google Scholar]
- 11.La Femina RL, Schnider CL, Robbins HL, Callahan PL, Legro K, Roth E, Schleif WA, Emini EA. J Virol. 1992;66:7414–7419. doi: 10.1128/jvi.66.12.7414-7419.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakai H, Kawamura M, Sakuragi S, Shibata R, Ishimoto A, Ono N, Ueda S, Adachi A. J Virol. 1993;67:1169–1174. doi: 10.1128/jvi.67.3.1169-1174.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farnet CM, Wang B, Lipford JR, Bushman FD. Proc Natl Acad Sci USA. 1996;93:9742–9747. doi: 10.1073/pnas.93.18.9742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Asante-Appiah E, Skalka AM. Adv Virus Res. 1999;52:351–369. doi: 10.1016/s0065-3527(08)60306-1. [DOI] [PubMed] [Google Scholar]
- 15.Brown PO. Integration. Cold Spring Harbor Press; Cold Spring Harbor, NY: 1999. [PubMed] [Google Scholar]
- 16.Billich A. Curr Opin Invest Drugs. 2003;4:206–209. [PubMed] [Google Scholar]
- 17.Hazuda DJ, Anthony NJ, Gomez RP, Jolly SM, Wai JS, Zhuang L, Fisher TE, Embrey M, Guare JP, Jr, Egbertson MS, Vacca JP, Huff JR, Felock PJ, Witmer MV, Stillmock KA, Danovich R, Grobler J, Miller MD, Espeseth AS, Jin L, Chen IW, Lin JH, Kassahun K, Ellis JD, Wong BK, Xu W, Pearson PG, Schleif WA, Cortese R, Emini E, Summa V, Holloway MK, Young SD. Proc Natl Acad Sci USA. 2004;101:11233–11238. doi: 10.1073/pnas.0402357101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato M, Kawakami H, Motomura T, Aramaki H, Matsuda T, Yamashita M, Ito Y, Matsuzaki Y, Yamataka K, Ikeda S, Shinkai H. J Med Chem. 2009;52:4869–4882. doi: 10.1021/jm900460z. [DOI] [PubMed] [Google Scholar]
- 19.Sorbera LA, Serradell N. Drugs Future. 2006;31:310–313. [Google Scholar]
- 20.Boros EE, Edwards CE, Foster SA, Fuji M, Fujiwara T, Garvey EP, Golden PL, Hazen RJ, Jeffrey JL, Johns BA, Kawasuji T, Kiyama R, Koble CS, Kurose N, Miller WH, Mote AL, Murai H, Sato A, Thompson JB, Woodward MC, Yoshinaga T. J Med Chem. 2009;52:2754–2761. doi: 10.1021/jm801404b. [DOI] [PubMed] [Google Scholar]
- 21.Dubois M, Bailly F, Mbemba G, Mouscadet JF, Debyser Z, Witvrouw M, Cotelle P. J Med Chem. 2008;51:2575–2579. doi: 10.1021/jm7011134. [DOI] [PubMed] [Google Scholar]
- 22.Pace P, Francesco MED, Gardelli C, Harper S, Muraglia E, Nizi E, Orvieto F, Petrocchi A, Poma M, Rowley M, Scarpelli R, Laufer R, Gonzalez P, Monteagudo E, Bonelli F, Hazuda DJ, Stillmock KA, Summa V. J Med Chem. 2007;50:2225–2239. doi: 10.1021/jm070027u. [DOI] [PubMed] [Google Scholar]
- 23.Pasquini S, Mugnaini C, Tintori C, Botta M, Trejos A, Arvela RK, Larhed M, Witvrouw M, Michiels M, Christ F, Debyser Z, Corelli F. J Med Chem. 2008;51:5125–5129. doi: 10.1021/jm8003784. [DOI] [PubMed] [Google Scholar]
- 24.Plewe MB, Butler SL, Dress KR, Hu Q, Johnson TW, Kuehler JE, Kuki A, Lam H, Liu W, Nowlin D, Peng Q, Rahavendran SV, Tanis SP, Tran KT, Wang H, Yang A, Zhang J. J Med Chem. 2009;52:7211–7219. doi: 10.1021/jm900862n. [DOI] [PubMed] [Google Scholar]
- 25.Ramkumar K, Serrao E, Odde S, Neamati N. Med Res Rev. 2010;30:890–954. doi: 10.1002/med.20194. [DOI] [PubMed] [Google Scholar]
- 26.Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, Fiore F, Gardelli C, Pazz GO, Hazuda DJ, Jones P, Kinzel O, Laufer R, Monteagudo E, Muraglia E, Nizi E, Orvieto F, Pace P, Pescatore G, Scarpelli R, Stillmock KA, Witmer MV, Rowley M. J Med Chem. 2008;51:5843–5855. doi: 10.1021/jm800245z. [DOI] [PubMed] [Google Scholar]
- 27.Al-Mawasawi LQ, Al-Safi RI, Neamati N. Expert Opin Emerging Drugs. 2008;13:213–225. doi: 10.1517/14728214.13.2.213. [DOI] [PubMed] [Google Scholar]
- 28.Patil S, Kamath S, Sanchez T, Neamati N, Schinazi RF, Buolamwini JK. Bioorg Med Chem. 2007;15:1212–1228. doi: 10.1016/j.bmc.2006.11.026. [DOI] [PubMed] [Google Scholar]
- 29.Zwaagstra ME, Timmerman H, Abdoelgafoer RS, Zhang MQ. Eur J Med Chem. 1996;31:861–874. [Google Scholar]
- 30.Zwaagstra ME, Timmerman H, Tamura M, Tohma T, Wada Y, Onogi K, Zhang MQ. J Med Chem. 1997;40:1075–1089. doi: 10.1021/jm960628d. [DOI] [PubMed] [Google Scholar]
- 31.Pannecouque C, Pluymers W, Van Maele B, Tetz V, Cherepanov P, De Clercq E, Witvrouw M, Debyser Z. Curr Biol. 2002;12:1169–1177. doi: 10.1016/s0960-9822(02)00952-1. [DOI] [PubMed] [Google Scholar]
- 32.Dixon S, Smondyrev A, Knoll E, Rao S, Shaw D, Friesner R. J Comput Aided Mol Des. 2006;20:647–671. doi: 10.1007/s10822-006-9087-6. [DOI] [PubMed] [Google Scholar]
- 33.Kuo CL, Assefa H, Kamath S, Brzozowski Z, Slawinski J, Saczewski F, Buolamwini JK, Neamati N. J Med Chem. 2004;47:385–399. doi: 10.1021/jm030378i. [DOI] [PubMed] [Google Scholar]
- 34.Clark M, Cramer IRD. Quant Struct Act Relat. 1993;12:137–145. [Google Scholar]
- 35.Zhu Z, Buolamwini JK. Bioorg Med Chem. 2008;16:3848–3865. doi: 10.1016/j.bmc.2008.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Telvekar V, Kundaikar H, Patel K, Chaudhari H. QSAR & Combinatorial Science. 2008;27:1193–1203. [Google Scholar]
- 37.Klebe G, Abraham U, Mietzner T. J Med Chem. 1994;37:4130–4146. doi: 10.1021/jm00050a010. [DOI] [PubMed] [Google Scholar]
- 38.Viswanadhan VN, Ghose AK, Revenkar GR, Robins R. J Chem Inf Comput Sci. 1989;29:163–172. [Google Scholar]
- 39.Klebe G. J Mol Biol. 1994;237:212–235. doi: 10.1006/jmbi.1994.1223. [DOI] [PubMed] [Google Scholar]
- 40.Stuyver LJ, Lostia S, Adams M, Mathew JS, Pai BS, Grier J, Tharnish PM, Choi Y, Chong Y, Choo H, Chu CK, Otto MJ, Schinazi RF. Antimicrob Agents Chemother. 2002;46:3854–3860. doi: 10.1128/AAC.46.12.3854-3860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schinazi RF, Sommadossi JP, Saalmann V, Cannon DL, Xie MY, Hart GC, Smith GA, Hahn EF. Antimicrob Agents Chemother. 1990;34:1061–1067. doi: 10.1128/aac.34.6.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sommadossi JP, Carlisle R, Schinazi RF, Zhou Z. Antimicrob Agents Chemother. 1988;32:997–1001. doi: 10.1128/aac.32.7.997. [DOI] [PMC free article] [PubMed] [Google Scholar]