

The bacterial heat shock transcription factor, σ32, maintains proper protein homeostasis only after it is targeted to the inner membrane by the signal recognition particle (SRP), thereby enabling integration of protein folding information from both the cytoplasm and cell membrane.
Abstract
All cells must adapt to rapidly changing conditions. The heat shock response (HSR) is an intracellular signaling pathway that maintains proteostasis (protein folding homeostasis), a process critical for survival in all organisms exposed to heat stress or other conditions that alter the folding of the proteome. Yet despite decades of study, the circuitry described for responding to altered protein status in the best-studied bacterium, E. coli, does not faithfully recapitulate the range of cellular responses in response to this stress. Here, we report the discovery of the missing link. Surprisingly, we found that σ32, the central transcription factor driving the HSR, must be localized to the membrane rather than dispersed in the cytoplasm as previously assumed. Genetic analyses indicate that σ32 localization results from a protein targeting reaction facilitated by the signal recognition particle (SRP) and its receptor (SR), which together comprise a conserved protein targeting machine and mediate the cotranslational targeting of inner membrane proteins to the membrane. SRP interacts with σ32 directly and transports it to the inner membrane. Our results show that σ32 must be membrane-associated to be properly regulated in response to the protein folding status in the cell, explaining how the HSR integrates information from both the cytoplasm and bacterial cell membrane.
Author Summary
All cells have to adjust to frequent changes in their environmental conditions. The heat shock response is a signaling pathway critical for survival of all organisms exposed to elevated temperatures. Under such conditions, the heat shock response maintains enzymes and other proteins in a properly folded state. The mechanisms for sensing temperature and the subsequent induction of the appropriate transcriptional response have been extensively studied. Prior to this work, however, the circuitry described in the best studied bacterium E. coli could not fully explain the range of cellular responses that are observed following heat shock. We report the discovery of this missing link. Surprisingly, we find that σ32, a transcription factor that induces gene expression during heat shock, needs to be localized to the membrane, rather than being active as a soluble cytoplasmic protein as previously thought. We show that, equally surprisingly, σ32 is targeted to the membrane by the signal recognition particle (SRP) and its receptor (SR). SRP and SR constitute a conserved protein targeting machine that normally only operates on membrane and periplasmic proteins that contain identifiable signal sequences. Intriguingly, σ32 does not have any canonical signal sequence for export or membrane-integration. Our results indicate that membrane-associated σ32, not soluble cytoplasmic σ32, is the preferred target of regulatory control in response to heat shock. Our new model thus explains how protein folding status from both the cytoplasm and bacterial cell membrane can be integrated to control the heat shock response.
Introduction
The heat shock response (HSR) maintains protein homeostasis (proteostasis) in all organisms. The HSR responds to protein unfolding, aggregation, and damage by the rapid and transient production of heat shock proteins (HSPs) and by triggering other cellular protective pathways that help mitigate the stress. Although the specific HSR is tailored to each organism, chaperones that mediate protein folding and proteases that degrade misfolded proteins are almost always included in the core repertoire of induced protein and are among the most conserved proteins in the cell. These HSPs maintain optimal states of protein folding and turnover during normal growth, while decreasing cellular damage from stress-induced protein misfolding and aggregation. Malfunction of the HSR pathway reduces lifespan and is implicated in the onset of neurodegenerative diseases in higher organisms [1]–[3].
In E. coli and other proteobacteria, σ32 mediates the HSR by directing RNA polymerase to promoters of HSR target genes [4]–[9]. Given the importance of this response and the necessity for a rapid but transient increase in expression of HSPs, it is not surprising that regulation of the HSR across organisms is complex. σ32 is positively regulated by a feed-forward mechanism in which exposure to heat melts an inhibitory mRNA structure enabling high translation of σ32 mRNA [10],[11] and is negatively regulated by two feedback loops [12] mediated through members of the σ32 regulon (Figure 1A). σ32 activity is coupled to the cellular protein folding state via a negative feedback loop executed by the two major chaperone systems, DnaK/J/GrpE and GroEL/S. There is extensive support for the model that free chaperones directly inactivate σ32 and that these chaperones are titrated by unfolded proteins that accumulate and bind chaperones during a HSR. Depletion of either chaperone system or overexpression of chaperone substrates leads to an increase in σ32 activity, and conversely, overexpression of either chaperone system decreases σ32 activity [13],[14]. Inhibition is likely direct, as DnaK/J and GroEL/S bind σ32 in vitro and inhibit its activity in a purified in vitro transcription system [13],[15]–[17]. σ32 stability is controlled by the inner membrane (IM) protease FtsH: deletion of the protease stabilizes σ32 [18]–[20], and FtsH degrades σ32 in vitro, albeit slowly [18],[20]. DnaK/J and GroEL/S also regulate stability, as their depletion leads to σ32 stabilization in vivo [13],[14],[21], although this finding has not yet been recapitulated in vitro [22].
Figure 1. Homeostatic control circuits of σ32.

(A) Current and (B) revised model for activity and degradation control of σ32. The revised model incorporates SRP-mediated trafficking of σ32 to the membrane. Interactions validated in vitro are shown as solid lines; those inferred from in vivo data are shown as dashed lines. Newly identified interactions are shown in red.
Despite the regulatory complexity of the current model, it inadequately addresses two issues that are central to our understanding of the circuitry controlling the HSR, motivating us to search for additional players in the response: (1) Exhaustive genetic screens for mutations in σ32 that result in misregulation have identified a small cluster of four closely spaced amino acid residues (Leu47, Ala50, Lys51, and Ile54), of which three are surface exposed, as well as a somewhat distant fifth residue that abuts this patch in the folded σ32 structure. When these residues are mutated, cells have both increased level and activity of σ32, indicating that this region is involved in a central process required for operation of the negative feedback loops that control both the activity and degradation of σ32 (Figure 1A) [23]–[25]. However, the phenotypes of these mutants are not recapitulated in vitro, where both FtsH degradation and chaperone-mediated inactivation of mutant and WT σ32 are experimentally indistinguishable [25],[26]. Thus, we do not understand how this “homeostatic control region” of σ32 functions. (2) σ32 is thought to monitor the folding status of IM proteins as well as cytoplasmic proteins, but the mechanism for this additional surveillance is unknown. Their close connection is indicated because (1) the IM protease, FtsH, not only degrades σ32, but also maintains quality control in the IM by degrading unassembled IM proteins; (2) induction of the HSR is a very early response to perturbations in the co-translational membrane-trafficking system that brings ribosomes translating IM proteins to the membrane [27]–[29]; and (3) IM proteins are significantly overrepresented both in the σ32 regulon [30] and in an unbiased overexpression screen for HSR inducers [30].
In this report, we identify the co-translational protein targeting machinery, comprised of the Signal Recognition Particle (SRP; Ffh protein in complex with 4.5S RNA; Figure 2A) and the SRP Receptor (SR; FtsY), as a regulator of σ32. We show that SRP preferentially binds to WTσ32 compared to a mutant σ32 with a defective homeostatic control region. We further show that a fraction of σ32 is associated with the cell membrane and that both the SRP-dependent machinery and the homeostatic control region of σ32 are important for this localization. Lastly, the regulatory defects in HSR circuitry caused by mutation of either the σ32 homeostatic control region or the co-translational targeting machinery are circumvented by artificially tethering σ32 to the IM. We propose that SRP-dependent membrane localization is a critical step in the control circuitry that governs the activity and stability of σ32. Membrane localization is widely used to control σ factors, but this is the first case where the IM-localized state is used for dynamic regulation rather than as a repository for an inactive protein.
Figure 2. σ32 binds to Ffh.

(A) Schematic representation of E. coli SRP (Ffh+4.5S RNA), indicating experimentally confirmed functions associated with each domain. (B) σ32 co-immunoprecipitates with Ffh and FtsY in vivo, but σ70 does not. Immunoprecipitations of Ffh or FtsY were carried out on lysates of Δσ32 and ΔftsH cells grown to exponential phase. Immunocomplexes were isolated, analyzed by SDS-PAGE, and immunoblotted with anti-σ32 and anti-σ70 antibodies. Proteins from approximately 15-fold more cells were loaded onto the gel for the immunoprecipitated samples against σ32 and σ70 as compared with the lysate samples. (C) Protein–protein interaction analysis indicates that σ32 binds to Ffh, but not FtsY. Purified FtsY and Ffh were run on a 10% SDS-PAGE gel, transferred to nitrocellulose, re-natured, and incubated with purified WTσ32. The Coomassie-stained gel (left) and the nitrocellulose blot probed with polyclonal anti-σ32 antibodies (right) are shown. (D) σ32 binds to the M-domain of Ffh. Ffh, partially digested by endopeptidase V8, was resolved on a 10% SDS-PAGE gel, transferred to nitrocellulose, and incubated with σ32. The Coomassie-stained gel (left) and the nitrocellulose membrane, containing transferred Ffh fragments, probed against σ32 (right) are shown.
Results
A Transposon Insertion Mutant at the ftsY Promoter Region Is Defective in Feedback Control
To identify additional players involved in activity control of σ32, we carried out a genetic screen for transposon mutants with increased σ32 activity under conditions that inactivate σ32 in wild-type cells (see Methods). To impose a condition that mimics the negative feedback control of σ32, the DnaK/J chaperones were overexpressed from an inducible promoter at their chromosomal locus. Under these conditions, a σ32-regulated lacZ chromosomal reporter (PhtpG-lacZ) is expressed so poorly that cells do not make sufficient β-galactosidase to turn colonies blue on X-gal indicator plates. We screened for blue colonies, indicative of a defect in σ32 inactivation. A conceptually similar screen previously identified mutations in the DnaK/J chaperones—key negative regulators of the σ32 response [31]. In addition to re-identifying these components, we found an insertion in the promoter region of ftsY (pftsY::Tn5), located 39 bp upstream of the ftsY open reading frame. The pftsY::Tn5 strain had a 3- to 4-fold reduction in the level of FtsY, the SR, and a ∼7-fold increase in the activity and amount of σ32 relative to WT (Table 1). Defects were complemented by a plasmid carrying ftsY. Unlike WT, in the pftsY::Tn5 strain σ32 activity did not respond to increased chaperone expression. Upon chaperone overexpression in WT cells, the specific activity (S.A.) of σ32 fell to 0.3, relative to that in cells growing without chaperone overexpression. In contrast, upon chaperone overexpression in pftsY::Tn5 cells, the S.A. of σ32 did not change, suggesting a defect in chaperone-mediated activity control in that strain (Table 1). This finding raised the possibility that the high activity of σ32 in pftsY::Tn5 resulted from disruption of activity control of σ32, rather than reflecting a cellular response to accumulation of unassembled membrane proteins.
Table 1. The altered σ32 phenotypes of the Tn5 insertion mutant (pftsY::Tn5) are significantly complemented by an ftsY + plasmid.
| Relative S.A. of σ32 | |||||
| Chaperone Overexpression | |||||
| Strain | FtsY Level | σ32 Level | σ32 Activity | None | +DnaKJ/GrpE |
| Wild-type | 1.0±0.2 | 1.0±0.1 | 1.0±0.1 | 1.0 | 0.3 |
| pftsY::Tn5 | 0.3±0.1 | 6.8±0.6 | 7.7±1.0 | 1.1 | 1.1 |
| pftsY::Tn5 +pftsY + * | 0.9±0.1 | 2.5±0.4 | 2.1±0.5 | 0.8 | 0.3 |
In this and all other experiments, protein levels were determined by SDS-PAGE followed by quantitative immunoblotting. σ32 activity was determined from a chromosomal β-galactosidase reporter (calculated as a differential rate of synthesis); values presented are from ≥3 experiments. Relative S.A. of σ32 is defined as: [(σ32 activity/σ32 level) normalized to σ32 S.A. of WT cells grown at 30°C].
The ftsY + plasmid inhibits growth of the cells by ∼30%.
σ32 Directly Interacts with SRP
We tested whether σ32 binds to either FtsY (SR) or to Ffh, the protein component of SRP. Ffh is a two-domain protein, comprised of an M-domain that binds the signal sequence and 4.5S RNA, and an NG-domain that binds to SR, the ribosome, and GTP (Figure 2A). We first used co-immunoprecipitation analysis. Interacting proteins were immunoprecipitated with antibodies against either FtsY or Ffh and, following resolution on SDS-PAGE, antibodies against σ32 or σ70 were used to probe for the presence of these proteins. σ32 was detected in the immunoprecipitations (Figure 2B, lanes 7 and 8), and this signal was dependent on the presence of σ32 in the strain (Figure 2B, lanes 1–4). By contrast, σ70, although much more abundant than σ32 in the cell, did not interact with either SRP or SR (Figure 2B, Lanes 3,4 and 7,8), indicating that interaction with SRP is not a general property of σs. It was not surprising that σ32 was co-immunoprecipitated with both SRP and SR, as the latter two components interact in vivo. To determine the direct binding partner of σ32, purified Ffh and FtsY were resolved on SDS-PAGE, transferred to nitrocellulose, and incubated with purified σ32. Antibodies against σ32 detected σ32 present at the molecular weight corresponding to Ffh but not SR (Figure 2C). In a reciprocal experiment, purified σ32 was resolved on SDS-PAGE, transferred to nitrocellulose, and incubated with purified Ffh or SR. Ffh, but not SR, bound σ32 (unpublished data). Similar studies did not reveal an interaction between σ70 and either Ffh or SR (unpublished data). We determined which Ffh domain binds σ32 by partially-proteolyzing Ffh to produce an 18 kDa M-domain and a 38 kDa NG-domain, resolving the mixture by SDS-PAGE, transferring to nitrocellulose, and probing with σ32. σ32 was detected at the position of full-length Ffh and the M-domain, but not at the position of the NG-domain (Figure 2D), indicating that the M-domain contains the determinants mediating the σ32-interaction.
We used in vivo crosslinking to validate the direct interaction of SRP (Ffh+4.5S RNA) and σ32. We created a σ32 derivative with an N-terminal 6×HIS-tag and a photoreactive amino acid analog (pBPA) at amino acid position 52 (6×HIS-σ32T52pBPA; see Methods), which is active as WTσ32 in expression of the σ32 reporter PhtpG-lacZ (activity is 150% that of WT; within the range of the variability of the assay; unpublished data). Following UV irradiation of whole cells, anti-Ffh immunoblotting of the whole cell lysate detected one predominant crosslinked product, which was dependent on UV-irradiation (Figure 3A, lanes 1 and 2) and pBPA at position 52 (Figure 3A, lanes 2 and 4). This UV- and pBPA-dependent product was also detected with anti-σ32 immunoblotting (Figure 3A, lane 6). To determine whether the crosslinked product represented 6×HIS-σ32T52pBPA-Ffh, we determined whether this product was identified both by co-immunoprecipitation with anti-Ffh antisera (Figure 3B) and by affinity purification of 6×HIS-σ32T52pBPA on a TALON resin (Figure 3C). Upon immunoprecipitation with anti-Ffh antisera, we detected a single higher molecular mass band, which reacted with both anti-Ffh (Figure 3B, lane 2) and -σ32 (Figure 3B, lane 6). Upon affinity purification on a TALON resin, anti-Ffh identified the same predominant UV- and pBPA-dependent Ffh-containing crosslinked product (compare Figure 3B and 3C, lane 2). Importantly, no free Ffh was recovered following TALON purification, indicating that the recovery of the Ffh conjugate was mediated by the covalently linked 6×HIS-σ32, rather than interaction with either the TALON resin or another protein. These results strongly suggest that σ32 directly interacts with Ffh in vivo. Although only a faint band was seen at the same position using anti-σ32 immunoblotting, this was likely a result of high background in this area of the gel, possibly because of extensive interaction between chaperones and σ32 (Figure 3C, lanes 5–8).
Figure 3. In vivo cross-linking between σ32 and Ffh.

(A) Detection of a cross-linked product following UV irradiation in whole cells. Cells of CAG48238/pEVOL-pBpF/p6XH-rpoHT52amber (lanes 1, 2, 5, and 6) and CAG48238/pEVOL-pBpF/p6XH-rpoH (lanes 3, 4, 7, and 8) were grown at 30°C in L-medium supplemented with 0.02% arabinose, induced with 1 mM IPTG for 1 h, and UV-irradiated for 0 or 10 min as indicated. Total cellular proteins were acid-precipitated and analyzed by SDS-PAGE and immunoblotting with anti-Ffh and anti-σ32 antibodies. (B) Immunoprecipitation with anti-Ffh reveals a unique cross-linked product that interacts with anti-σ32. Supernatants of sonically disrupted UV-irradiated cells were subjected to immunoprecipitation with anti-Ffh antibodies. Immunocomplexes were solubilized in SDS sample buffer, analyzed by SDS-PAGE, and immunoblotted with anti-Ffh and anti-σ32 antibodies. Proteins from approximately 4.4-fold more cells were loaded onto the gel for the immunoprecipitated samples as compared with the whole cell samples. (C) Purification of 6×H-σ32 from UV-irradiated cells reveals a band that interacts with anti-Ffh. Supernatants of sonically disrupted UV-irradiated cells were subjected to TALON affinity chromatography, and bound proteins were eluted with 300 mM imidazole. Proteins in the eluate were acid-precipitated and analyzed by SDS-PAGE and immunoblotting with anti-Ffh antibodies. Proteins form approximately 20-fold more cells were loaded onto the gel for the TALON-affinity isolated samples as compared with the whole cell samples.
I54Nσ32 Is Defective in Interacting with SRP
The function of the homeostatic control region of σ32 is not known [25]. I54Nσ32 is a mutation located in this region is severely compromised in both activity and degradation control, but the mechanism responsible for this phenotype had not yet been determined [25]. We therefore compared the binding of WTσ32 and I54Nσ32 to SRP using gel filtration. We incubated WTσ32 or I54Nσ32 either alone or in combination with SRP and subjected the mixture to gel filtration. Analysis of the elution profiles demonstrated that most WTσ32 was shifted towards the higher molecular weight region in the presence of SRP, and additionally, a fraction of σ32 eluted at a higher molecular weight than that of SRP alone, indicative of an SRP–σ32 complex [compare A280 profiles of σ32, SRP, and SRP-σ32 (Figure 4A) with immunoblotting for σ32 (Figure 4B; rows 1,2)]. σ32 present at a molecular weight between σ32 and SRP likely represents transient forms of the σ32–SRP complex. In sharp contrast, an interaction between I54Nσ32 with SRP was almost undetectable [compare A280 profiles of I54Nσ32 and SRP (Figure 4A) with immunoblotting for I54Nσ32 (Figure 4B; rows 3,4)], indicating that I54Nσ32 bound more weakly to SRP than WTσ32. Neither WTσ32 nor I54Nσ32 interacted detectably with Ffh, indicating that differential binding is dependent on the formation of SRP (Ffh+4.5S RNA), the biologically relevant cellular species of Ffh.
Figure 4. SRP (Ffh+4.5S RNA) preferentially interacts with WTσ32.
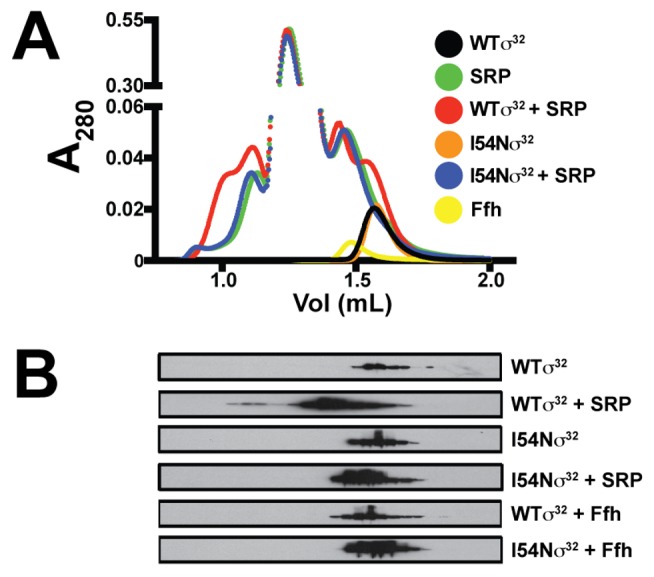
(A) A280 elution profiles of WTσ32, I54Nσ32, Ffh, and SRP alone or in complex. WTσ32 or I54Nσ32 was incubated with a 10-fold molar excess of purified SRP on ice for 10 min, and complexes were analyzed by gel filtration on a Superdex 200 PC3.2/30 column. Protein elution was monitored by A280. Gel filtration of purified WTσ32, I54Nσ32, and SRP alone was carried out to determine the migration of each individual protein on the column. (B) Eluted fractions were separated on SDS-PAGE and probed with polyclonal antibodies against Ffh and σ32; Western blots of σ32 are shown. Experiments were performed at least four times, and a representative experiment is shown.
σ32 Is Partially Membrane Associated in an SRP-Dependent Process
The biological function of SRP is co-translational protein targeting, leading us to test whether σ32 may be targeted to the IM through an SRP-dependent mechanism. Rapid degradation by FtsH normally keeps σ32 levels very close to the detection limit (∼20–50 molecules/cell; [8]), making reproducible detection following fractionation very difficult. Therefore, we performed fractionation experiments (Figure 5), either in cells expressing an enzymatically inactive mutant of the FtsH protease (FtsH E415A) or in cells lacking FtsH altogether (ΔftsH). Approximately 44% of σ32 fractionated to the membrane in a ΔftsH strain, and this fraction was increased to ∼58% in the FtsH E415A strain, raising the possibility that FtsH itself may participate in retention of σ32 at the IM. As the β′ subunit of RNA polymerase, a known interaction partner of σ32, also fractionated with the membrane, we next tested whether σ32 association with the IM was dependent on its association with RNA polymerase. To this end, we used σ32Δ21aa, which is defective in interacting with RNA polymerase [32]. We confirmed that σ32Δ21aa did not detectably interact with RNA polymerase (Figure S1A,B). Yet endogenous WTσ32 and ectopically expressed σ32Δ21aa fractionated equivalently to the IM both in ΔftsH cells (∼39%) and in FtsH E415A cells (∼58%) (Figure S2), indicating that σ32 transited to the membrane independent of RNA polymerase.
Figure 5. σ32 is partially membrane associated.

The extent of association of σ32 and the β′ subunit of RNA polymerase with the membrane fraction was determined by quantitative immunoblotting of the soluble and nonsoluble fractions. Membrane association of σ32 and β′ was assessed in several relevant strain backgrounds. In addition to endogenous σ32, all strains contained a plasmid-encoded variant of σ32 lacking its 21 C-terminal amino acids (σ32Δ21aa). Ectopically expressed σ32Δ21aa or I54Nσ32Δ21aa were present at levels comparable to native σ32 and were distinguished from endogenous σ32 on a 12% SDS-PAGE gel. All fractionation experiments were performed ≥8 times, and % fractionation was calculated from experiments where probed cytoplasmic (RuvB) and membrane (RseA) proteins separated properly.
We next tested whether the pftsY::Tn5 mutation or the homeostatic control region mutation of σ32 disrupted membrane partitioning of σ32. Both WTσ32 and ectopically expressed σ32Δ21aa were defective in partitioning to the IM in pftsY::Tn5 cells (Figure 5). To look at the effect of disrupting the homeostatic control region on membrane fractionation, we expressed I54Nσ32 as a σ32Δ21aa variant (I54Nσ32Δ21aa). The size difference allowed us to compare I54Nσ32Δ21aa and WTσ32 in the same cells (Figure S2). Whereas WTσ32 exhibited normal fractionation, I54Nσ32Δ21aa showed a severe localization defect, comparable to that of pftsY::Tn5 cells (Figure 5). We conclude that σ32 targeting to the IM is dependent on both SRP/SR and the σ32 homeostatic control region.
Both SecA and SecY Are Important for Membrane Association of σ32
SecA is an ATP-fueled motor protein that recognizes signal peptides, drives the translocation of secreted proteins through the Sec translocon [33]–[37], and collaborates with the SRP/SR for integration of a subset of IM proteins into the membrane [33],[38]. We previously found that σ32 activity is increased in a SecA(ts) strain [39]. This observation motivated us to explore the relationship of SecA to IM trafficking of σ32. Indeed, using a SecA(ts) mutant with general defects in protein export (SecAL43P) [40],[41], we observed that cells displayed a significant defect in membrane localization of σ32 (Figure 5), as well as increased σ32 activity ([39] and unpublished data). In addition, purified SecA, resolved on SDS-PAGE and transferred to nitrocellulose, showed binding affinity for σ32, suggesting that these two proteins interact (Figure S3). We conclude that SecA participates in trafficking of σ32 to the IM.
SecY forms the core of the SecYEG IM translocon. This multidomain protein has a large cytoplasmic domain (C5) that functionally interacts with SR [42], SecA, and the ribosome [43]–[50] (Figure 6A). We tested whether 10 previously described secY mutations located in various domains of SecY (Figure 6A) [51] perturb chaperone-mediated control of σ32 activity and trafficking of σ32 to the IM (Figure 6B). All mutants had enhanced σ32 activity. This result was not surprising as secY mutants are expected to accumulate secretory protein precursors that titrate chaperones [52]. Importantly, four mutants (secY124, secY351, secY40, secY129) were also defective in chaperone-mediated control of σ32 activity (Figure 6B), as indicated by a lack of down-regulation of σ32 activity in response to overexpression of one or both of the chaperone systems. We examined the secY351 mutant, which had both high σ32 activity and a significant defect in chaperone-mediated inactivation, and found it to be defective in IM trafficking of σ32 (Figure 5). secY40 and secY351 affect domain C5 (Figure 6A), implicated in the interaction of SecY with SR, raising the possibility that this interaction is important for both homeostatic control and IM targeting of σ32.
Figure 6. The SecY translocon plays a role in chaperone-mediated activity control of σ32.

(A) Schematic of SecY topology in the IM by highlighting in yellow the locations/allele names of the mutated residues used in this study [51]. The region that interacts with FtsY (Domain C5) is boxed in green. (B) Mutations in secY show higher σ32 activity and affect chaperone-mediated activity control of σ32. The activity of σ32 was measured in WT and secY mutant cells growing at 30°C in LB medium (column 1) or in the same cells following induction of DnaK/J (column 2) or GroEL/S (column 3). Activity is calculated as the differential rate of β-galactosidase synthesis from a chromosomal PtpG-lacZ reporter in each cell type relative to that of WT cells.
An Independent Methodology Indicates Association of σ32 with the IM
Alkaline phosphatase is active only in the periplasm, where it forms the disulfide bonds necessary for its activity. Therefore, translational fusions to alkaline phosphatase (PhoA) lacking its own export signal are commonly used as an indicator of membrane targeting by the appended N-terminal sequence [53]. If the appended N-terminal sequence has either an export or insertion sequence, the fusion protein will exhibit alkaline phosphatase activity in vivo because it is partly transported to the periplasmic side of the membrane through the SecYEG translocon. Although σ32 has neither a membrane insertion nor an export sequence, it may contain a sequence that targets it to the cytosolic face of the IM. There is some evidence that the secretory apparatus can recognize the mature domains of exported proteins at low efficiency [54]. If so, proximity of PhoA to the translocon resulting from the IM targeting signal might enable transit of some fraction of PhoA to localize to the periplasmic side of the membrane, where it is active. By random insertion of the transposon probe TnphoA into rpoH, encoding σ32 (see Materials and Methods), we found that a phoA fusion to the first 52 amino acids of σ32 (N52-σ32-PhoA) showed ∼10-fold greater PhoA activity than signal-less PhoA itself, indicating that the N-terminus of σ32 facilitates PhoA export (Table 2). Moreover, PhoA activity enhancement is dependent both on the SRP/SR-dependent trafficking system and on SecY, as both pftsY::Tn5 and secY351 decreased the PhoA activity ∼2-fold, whereas leaderless PhoA exhibited little response to these perturbations (Table 2). Thus, this assay is consistent with the idea that the N-terminus of σ32 carries an IM-trafficking sequence and that the targeting process is dependent on SRP and SecY.
Table 2. N-terminal segment of σ32 directs activation of PhoA protein through the SRP-Sec pathway.
| N52-σ32-PhoA | Signal-Less PhoA | |||
| Strain | PhoA Activity | Protein Level | PhoA Activity | Protein Level |
| Wild-type | 1.00±0.11* | 1.0 | 0.11±0.03 | 0.15 |
| pftsY::Tn5 | 0.43±0.05 | 0.8 | 0.14±0.03 | 0.2 |
| secY351 | 0.46±0.07 | 1.5 | 0.32±0.05 | 0.2 |
Wild-type strain (MG1655) and its derivatives carrying the mutation as indicated were transformed by the plasmid containing each PhoA construct. The resulting transformants were grown to log phase in LB medium at 30°C. PhoA activity and protein levels were determined by standard procedures (see Materials and Methods).
The activity of N52-σ32-PhoA was set to 1.00 in the wild-type strain.
Membrane-Tethering of Otherwise Deregulated σ32 Restores Homeostatic Control
The I54Nσ32 mutant and mutants in the IM-targeting machinery (pftsY::Tn5, secA(ts), secY351) were both defective in proper regulation of σ32 and in σ32 association with the IM. This convergence motivated us to test whether artificially tethering σ32 to the IM could restore homeostatic control. To this end, we exploited the bacteriophage Pf3 coat protein. With the addition of three leucine residues in its membrane-spanning region, 3L-Pf3 translocates spontaneously in an orientation-specific manner to the IM, where it inserts in an N-out/C-in orientation [55]. We modified rpoH (encoding σ32) at its chromosomal locus to encode a σ32 variant with the 3L-Pf3 membrane-insertion signal attached to its N-terminus (schematized in Figure S4A). Strains carrying 3L-Pf3-σ32 (IM-WTσ32) or 3L-Pf3-I54Nσ32 (IM-I54Nσ32) as their sole source of σ32 were viable, even though 99% of IM-WTσ32 was inserted in the membrane as judged by fractionation studies (Figure S4B). Thus, σ32 functions when it is tethered to the IM.
We determined whether IM-WTσ32 was subject to homeostatic control circuitry exhibited by WTσ32. σ32 is maintained at a low level by FtsH degradation, and its activity is decreased by chaperone-mediated inactivation. Both phenotypes are evident by comparing the amount and activity of σ32 in a WT versus a ΔftsH strain. In a ΔftsH strain, the level of WTσ32 increases ∼30-fold because the major protease degrading σ32 is removed (Table 3; Figure S5 [compare lanes 1 and 3]; and [25]). However, the activity of σ32 increases only 3-fold as a consequence of chaperone-mediated activity control, leading to a 10-fold reduction in the S.A. of σ32 in ΔftsH cells relative to that in WT cells (Table 3 and [56]). Both the level and S.A. of WTσ32 and IM-WTσ32 were closely similar in a ΔftsH strain, indicating that the chaperone-mediated activity control circuit is active in IM-WTσ32 (Table 3 and Figure S5 [compare lanes 3 and 4]). Additionally, the level of IM-WTσ32 was significantly lower in ftsH + than in a ΔftsH strain, indicating that IM-WTσ32 was efficiently degraded by FtsH (Table 3 and Figure S5 [compare lanes 2 and 4]). The presence of a contaminating band prevented absolute quantification of IM-WTσ32 levels via Western blot analysis (Figure S5). However, if the relative S.A. of IM-WTσ32 and WTσ32 are equivalent in the ftsH + strain as we found in the ΔftsH strain, then the 2-fold decrease in activity of IM-WTσ32 relative to WTσ32 implies a slight increase in the rate of degradation of IM-WTσ32 relative to WTσ32. Note that the 3L-Pf3 membrane-insertion tag itself is not a signal for FtsH degradation, as the stability of the FliA σ factor, which is closely related to σ32, was unchanged when expressed as 3L-Pf3-FliA (Figure S6). In summary, both the chaperone-mediated activity control circuit and the FtsH-mediated degradation control circuit are active on IM-tethered σ32.
Table 3. IM-insertion of σ32 significantly restores homeostatic control to mutant cells.
| σ32 Level | σ32 Activity | Relative S.A. of σ32 | ||||
| σ32 Variant | WT | ΔftsH | WT | ΔftsH | WT | ΔftsH |
| σ32 | 1.0±0.1 | 32.1±5.0 | 1.0±0.1 | 3.1±0.1 | 1.0 | 0.1 |
| IM-σ32 | a | 26.7±4.5 | 0.7±0.1 | 2.0±0.1 | N.D. | 0.1 |
| I54Nσ32 | 11.4±2.1 | b | 6.7±0.5 | b | 0.6 | N.D. |
| IM-I54Nσ32 | a | 29.5±6.0 | 1.3±0.6 | 4.3±0.8 | N.D. | 0.1 |
| pftsY::Tn5 | 6.8±0.6 | 7.7±1.0 | 1.1 | |||
| pftsY::Tn5; IM-σ32 | a | 0.7±0.1 | N.D. | |||
Protein levels were determined by SDS-PAGE followed by immunoblotting, and averages from 3–4 expts are presented. Relative S.A., relative Specific Activity, is calculated as described in Table 1. N.D., not determined, denotes that values could not be determined because of a and b.
Levels of the σ32 variants could not be measured accurately.
An I54Nσ32ΔftsH strain is inviable.
Next, we asked whether the forced and stable tethering of σ32 to the IM bypassed the regulatory defects of I54Nσ32 and the reduced-level SR mutant pftsY:::Tn5. I54Nσ32 is degraded poorly by FtsH as its level was 11-fold higher than that of WTσ32 (Table 3; Figure S5 [compare lanes 1 and 6] and [25]). I54Nσ32 also had compromised chaperone-mediated activity control as the high chaperone levels in this strain did not reduce the S.A. of I54Nσ32 (Table 3; and [25]). In stark contrast, both degradation and activity control were restored when I54Nσ32 was converted to IM-I54Nσ32. FtsH efficiently degraded the membrane-tethered variant: IM-I54Nσ32 was undetectable in ftsH + cells but present at a high level in ΔftsH cells (Table 3 and Figure S5 [compare lanes 5 and 7]). Additionally, IM-I54Nσ32 and IM-WTσ32 exhibited comparable reductions in relative S.A. of σ32 in ΔftsH cells (Table 3). Stable tethering of σ32 to the IM also bypassed the regulatory defects of pftsY::Tn5 as IM-WTσ32 in the reduced-level SR background was degraded and subject to chaperone-mediated activity control. Indeed, IM-WTσ32 behaved identically in WT and pftsY::Tn5 strains, exhibiting comparable σ32 activity at a protein level below detection (Table 3 and Figure S5 [compare lanes 8 and 9]). Finally, IM-tethering relieved the growth defects of both I54Nσ32 (Figure S7A and C) and of pftsY::Tn5 (Figure S7B, C, and D). In summary, stable tethering of σ32 to the IM restored both homeostatic control and normal growth to cells with a defective σ32 homeostatic control region and to cells with a compromised SRP/SR co-translational targeting apparatus.
Discussion
Our work has led to a revised model of the HSR circuitry (Figure 1B). σ32 first transits to the IM via an SRP/SR-dependent process and is then subjected to the chaperone-mediated activity control and FtsH-mediated degradation control that have been previously described. This revised model enables the homeostatic control circuit to integrate information on both cytosolic and IM status. Importantly, the efficiency of co-translational protein targeting depends on the cumulative effect of multiple SRP checkpoints including differences in cargo binding affinities, kinetics of SRP-SR complex assembly, and GTP hydrolysis [57]. Multiple checkpoints and the fact that SRP is sub-stoichiometric relative to translating ribosomes (∼1∶100; SRP molecules to translating ribosomes [58]) may allow SRP to modulate the extent of IM-localization of σ32 during times of stress and/or increased protein flux. Thus, σ32 down-regulation through its localization to the membrane could be alleviated when the IM is disturbed or SRP is overloaded in assisting membrane protein biogenesis. This feed-forward mechanism allows the σ32 homeostatic control to sense the state of cytosolic and IM proteostasis before unfolded proteins accumulate to a significant extent. Interestingly, ffh (encoding the protein subunit of the SRP) is a σ32 regulon member as its expression increases at least 3-fold following induction of σ32 either by heat shock or by deletion of dnaK/J ([30] and unpublished data). This could provide an additional connection between σ32 and protein flux to the IM. Finally, and more speculatively, given the demonstrated involvement of SecA in IM targeting of σ32 and its direct interaction with σ32, the σ32 homeostatic control circuit may also monitor protein flux through SecA to the periplasm and outer membrane.
The idea that the high activity of σ32 in the I54Nσ32 homeostatic control mutant and in SRP/SR mutants (eg. pftsY::Tn5) results from σ32 mislocalization to the cytosol and consequent homeostatic dysregulation, rather than from chaperone titration by a buildup of unfolded proteins, is supported by our data. First, forced IM-tethering overcomes the inviability of the I54Nσ32 mutation in the ΔftsH strain background (Table 3), as well as the growth defects of I54Nσ32 and pftsY::Tn5 (Figure S7), suggesting that high expression of σ32 is aberrant and deleterious to cells, rather than required to remodel misfolded proteins. This is reminiscent of previous findings that reduced-function σ32 mutants suppress physiological defects of a ΔdnaK strain [59] and that overexpression of HSPs was deleterious to growth [13],[60]. Second, secY mutants dysregulated in chaperone-mediated activity control were not distinguished by their extent of σ32 induction. This is contrary to the prediction of the chaperone titration model, which posits that secY mutants with the highest σ32 induction would have the highest level of unfolded proteins. These mutants would then be refractory to activity control because the additional chaperones resulting from chaperone overexpression would actually be needed to remodel the misfolded protein burden. We conclude that homeostatic dysregulation of σ32 results from σ32 mislocalization, rather than from the buildup of unfolded proteins.
The molecular nature of IM-localized σ32 remains unclear. Prediction programs [61],[62] do not detect either a signal peptide-like or transmembrane sequence in σ32. We favor the idea that following transit to the IM, σ32 is maintained at the membrane via interactions with other proteins and/or lipid head groups during its short half-life in the cell (30–60″). Indeed, we have already demonstrated interactions between σ32 and several membrane-associated or IM proteins, including SRP, SecA, and FtsH itself. Moreover, the chaperone systems regulating σ32 (DnaK/J/GrpE and GroEL/S) show partial distribution to the membrane [63]–[68], whereas other potential membrane-associated protein partners have not yet been tested for σ32 interaction (e.g., SecY and additional members of the Sec machinery). Each of these proteins could result in partial membrane localization of σ32, as was shown for FtsH where deletion of the protein decreased localization relative to cells with the protease-dead mutation FtsH E415A. Importantly, if σ32 is membrane associated via transient protein–protein and/or protein–lipid interactions, some σ32 may dissociate from the membrane during cell lysis, as was demonstrated for FtsY, another peripheral membrane protein [69],[70]. Therefore, although we report that ∼50% of σ32 is membrane-associated, the fraction of σ32 that is actually IM-localized may be significantly higher.
IM-associated σ32 may provide regulatory flexibility not possible for IM-tethered σ32. For example, during times of high stress, σ32 may be able to dissociate from the membrane to escape homeostatic control. These excursions could be transient if SRP were able to transport σ32 posttranslationally, a possibility suggested by the fact that full-length, fully folded σ32 binds to SRP (Figures 2 and 3 and Figure S1). Additionally, IM-tethered σ32 is more rapidly degraded than IM-associated σ32, suggesting that tethering makes σ32 a better FtsH substrate. This could diminish the ability of the cell to regulate the rate at which FtsH degrades σ32, which is of physiological significance during temperature upshift [8]. The transient reduction in σ32 degradation following increased temperature contributes significantly to the rapid build-up of σ32 during heat shock [8].
Membrane localization is widely used to control σ factors [71],[72]. The inactive B. subtilis SigK pro-protein is membrane inserted; cleavage of its N-terminal pro-sequence releases SigK [73],[74]. Cleavage is coordinated with passage of a checkpoint in spore development to provide just-in-time SigK activity [75]. Additionally, many σ factors are held in an inactive state at the membrane by cognate membrane-spanning anti-σ factors and released as transcriptionally active proteins when stress signals lead to degradation of their anti-σ [71],[76]. IM-localization of σ32 serves a conceptually distinct role as σ32 is equally active in the cytoplasm or at the IM. Instead, the localization process itself is the key regulatory step in two ways: localization is both regulated by protein folding status and is prerequisite for proper function of the homeostatic control circuit.
The SRP-SR co-translational targeting system has an important role in maintaining proteostasis. SRP-SR minimizes aggregation and misfolding of the approximately 20%–30% of proteins destined for the IM, by making their translation coincident with membrane insertion. Our finding, that SRP/SR-mediated transit of σ32 to the IM is also critical for proper control of the HSR, points to a significant new regulatory role for the co-translational targeting apparatus in protein-folding homeostasis. This finding also raises important mechanistic questions. Our in vitro interaction results suggest a direct, but weak, interaction between full-length σ32 and the M-domain of SRP. The prevailing paradigm suggests that the M-domain interacts only with nascent polypeptides with particularly hydrophobic signal sequences. It is possible that σ32 is detected co-translationally, as the Region 2.1 N-terminal α-helical structure, which resembles a hydrophobic signal sequence, may be recognized by the SRP. Alternatively, we note that the SRP chloroplast homolog (cpSRP54) has a dedicated posttranslational targeting mechanism for several fully translated membrane proteins [77], and E. coli SRP, alone or in combination with additional accessory factors (e.g., other σ32 interactors, such as chaperones or SecA), may target mature σ32 to the membrane in vivo. It remains to be determined whether an interaction between full-length σ32 and SRP, or a novel co-translational targeting interaction by the SRP-SR system, mediates transit of σ32 to the membrane.
Materials and Methods
Strains, Plasmids, and Growth Conditions
All strains used were derivatives of the E. coli K-12 strain MG1655, CAG48238 [25],[39]. For chaperone overexpression experiments, mutations were transduced with phage P1 into strains carrying chromosomal Para-groEL/S [78] or PA1/lacO-1-dnaK/J-lacIq [14]. Mutant alleles in secY [51] and secA [39] were transferred to various strain backgrounds through P1 transduction. The SecAL43P mutant used here is a SecA(ts) allele, with general defects in protein export [40],[41]. For propagation and transfer of the R6K pir plasmid, pKNG101, strains DH5σ λpir and SM10 λpir were used, respectively. Plasmids pET21a and pTrc99A were used as expression plasmids. For construction of pRM5 (6×HIS-rpoH), the rpoH gene was PCR-amplified from the chromosomal DNA of W3110 and cloned into the EcoRI-SalI sites of pTTQ18 [79]. Then, the T52amber mutation was introduced into pRM5 by site-directed mutagenesis, yielding pRM17 (6×HIS-σ32T52amber). pEVOL-pBpF (Addgene) carried evolved Methanocaldococcus jannaschii aminoacyl-tRNA synthetase/suppressor tRNA for incorporation of a photoreactive amino acid analog, p-benzoylphenylalanine (pBPA), into the amber codon site. All strains were grown in LB medium. When required, antibiotics were added to the medium as follows: 100 µg/ml ampicillin, 30 µg/ml kanamycin, 20 µg/ml chloramphenicol, and 25 µg/mL streptomycin.
Isolation of pftsY::Tn5 Mutant
Strain CAG48275 [25], which is ΔlacX74, contains the prophage JW2 (PhtpG-lacZ), and a chromosomal dnaK/J locus driven from PA1/lacO-1 under control of lacIq [14] was grown in LB, induced with 1 mM IPTG to overexpress DnaK/J chaperones, treated with Tn5, and plated at 30°C on X-gal indicator plates containing kanamycin to select for strains containing Tn5. Blue colonies were picked and tested for higher σ32 activity and for feedback resistance to excess DnaK/J [25]. Tn5 insertion sites were determined by DNA sequencing.
β-Galactosidase Assay
Overnight cultures (LB medium) were diluted 250-fold and grown to exponential phase (OD600 = 0.05–0.5). Samples were taken at intervals starting at OD600 = 0.05, and σ32 activity was monitored by measuring β-galactosidase activity expressed from the σ32-dependent htpG promoter, as done previously [25].
Protein Purification
The following proteins were purified essentially as described: 6×H-tagged, Strep-6×H-tagged, and untagged WTσ32 or I54Nσ32 [80], FtsY, Ffh, 4.5S RNA [81], and SecA [82]. Chaperones were removed from σ32 with an additional wash containing 10 mM ATP, 10 mM MgCl2, and 25 uM of both peptides, CALLLSAARR and MQERITLKDYAM, synthesized by Elim Biopharmaceuticals, Inc (Hayward, CA).
In Vivo Co-Immunoprecipitations
Cells were grown to OD600∼0.35 in LB medium at 30°C, harvested, washed two times with 1× PBS, resuspended in Lysis Buffer (20 mM Hepes-KOH, 150 mM NaCl, 10 mM EDTA, 10% glycerol, pH 7.5), and lysed by passing 4× through an Avestin EmulsiFlex-C5 cell homogenizer at 15,000 psi. Cellular debris was spun out and the supernatants were incubated with anti-Ffh or anti-FtsY antibodies at 4°C for 14 h by rotation. TrueBlot anti-Rabbit Ig IP Beads (eBioscience) were added and the supernatants rotated for an additional 2 h at 4°C. Immunocomplexes were isolated by centrifugation and washed 5× in Lysis Buffer without EDTA, and eluted in TCA Resuspension Buffer (100 mM Tris (pH 11.0), 3% SDS) containing LDS Sample Buffer (Invitrogen). Proteins were separated by 10% SDS-PAGE, analyzed by immunoblotting using anti-σ70 and anti-σ32 antibodies, and imaged using fluorescent secondary antibodies (as described below).
Identification of Direct Protein–Protein/Domain Interactions
Detection of a direct protein–protein/domain interaction was carried out exactly as previously described [83]. Proteins were separated on 10% SDS-PAGE. Partially proteolyzed Ffh was obtained by incubating 400 µg of purified Ffh with 4 µg of Glu-C endopeptidase (New England Biolabs) at 25°C in 10 mM Na-HEPES (pH 7.5), 150 mM NaCl, 1 mM DTT, 10 mM MgCl2, and 10% glycerol. An aliquot of the reaction was taken out at various times (0, 5, 10, 15, 30, 45, 60, 120, 180, and 330 min) and stopped by addition of 5× volume of 5× SDS-sample loading buffer. The samples were then analyzed by blot overlay with σ32 as the probe.
In Vivo Crosslinking, 6×HIS-tag Affinity Isolation and Co-Immunoprecipitation
In vivo crosslinking experiments were carried out essentially as described previously [84]. Strains of CAG48238 carrying pEVOL-pBpF were further transformed with pRM5 or pRM17. Cells were grown at 30°C in L medium containing 0.02% arabinose and 1 mM pBPA, induced with 1 mM IPTG for 1 h, and UV-irradiated for 0 or 10 min at 4°C. For analysis of whole cell samples, total cellular proteins were precipitated with 5% trichloroacetic acid, solublized in SDS sample buffer, and analyzed by 7.5% SDS-PAGE and immunoblotting.
Co-immunoprecipitations were carried out as follows: UV-irradiated cells were suspended in 10 mM Tris-HCl (pH 8.1) and disrupted by sonication at 0°C. After removal of total membranes by ultracentrifugation, proteins were precipitated with 5% trichloroacetic acid, washed with acetone, and solubilized in buffer containing 50 mM TrisHCl (pH 8.1), 1% SDS, 1 mM EDTA. The samples were then diluted 33-fold with NP40 buffer (50 mM TrisHCl (pH 8.1), 150 mM NaCl, 1% NP40). After clarification, supernatants were incubated with anti-Ffh antibodies and TrueBlot anti-Rabbit Ig IP Beads (eBioscience) at 4°C for 13 h with rotation. Immunocomplexes were isolated by centrifugation, washed 2 times with NP40 buffer and then once with 10 mM TrisHCl (pH 8.1), and dissolved in SDS sample buffer. Proteins were separated by 7.5% SDS-PAGE and analyzed by immunoblotting using anti-Ffh and anti-σ32 antibodies, TrueBlot anti-Rabbit IgG (eBioscience), and Can Get Signal immunoreaction enhancer solution (TOYOBO Life Science, Japan).
For 6×HIS-tag affinity isolation, UV-irradiated cells were suspended in 10 mM Tris-HCl (pH 8.1) containing 6 M urea and disrupted by sonication at 0°C. After clarification by ultracentrifugation, the soluble fraction was loaded onto the TALON resin (TAKARA BIO, Inc., Japan). After washing the resin with wash buffer (50 mM TrisHCl (pH 7.0), 300 mM KCl, 6 M urea, 20 mM imidazole), bound proteins were eluted with wash buffer containing 300 mM imidazole. Proteins were precipitated with 5% trichloroacetic acid, solublized in SDS sample buffer, and analyzed by 7.5% SDS-PAGE and immunoblotting.
Gel Filtration
Purified proteins were run on a Superdex 200 PC 3.2/30 column, pre-equilibrated with Buffer A (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 10 mM MgCl2, 2 mM DTT). Purified proteins or protein complexes were run with Buffer A at a flow rate of 40 µL/min, and collected fractions were analyzed by SDS-PAGE and immunoblotting for σ32. SRP was formed by incubating purified Ffh with 1.5× molar excess of purified 4.5S RNA on ice for 10 min. To form SRP-σ32 complexes, 3 µM of purified WTσ32 or I54Nσ32 was mixed with 10× molar excess of SRP; proteins were incubated on ice for 30 min before analysis by gel filtration.
Construction of σ32-PhoA Fusion
A 52-σ32-Tn5PhoA fusion was initially isolated by random screening for PhoA+ clones on PhoA indicator plates—using a strain carrying a TnphoA transposon probe [85] on a low-copy plasmid and Plac-rpoH (encoding σ32) on a multicopy plasmid. The fusion used in this article (N52-σ32-PhoA lacking the transposon but containing the first 52 amino acids of WTσ32) was subsequently constructed by standard recombinant DNA techniques. Direct construction of fusions past amino acid 52 of σ32 was very unstable, precluding their analysis.
Cell Fractionation
Cells were grown to OD600 = 0.3–0.4, harvested, and resuspended in ice-cold Buffer B (10 mM Tris-Acetate (pH 7.4), 10 mM Mg(OAc)2, 60 mM NH4Cl, 1 mM EDTA, supplemented with 1 mM PMSF) to an OD600 of 15. Cells were immediately lysed by passaging the extracts through an Avestin EmulsiFlex-C5 cell homogenizer at 15,000 psi, and subjected to low-speed centrifugation to remove cell debris and un-lysed cells. Membranes were collected by ultracentrifugation in an Optima benchtop centrifuge (Beckman–Spinco) with a TLA 100.3 rotor (60 min; 52,000 rpm; 4°C). The supernatant was saved as the soluble fraction, while the pellet was washed 3× with Buffer B and then resuspended in Buffer C (50 mM HEPES-KOH pH 7.6, 50 mM KCl, 1 mM EDTA, 1 mM EGTA, 0.5% n-Dodecyl β-D-maltoside, and 5% glycerol). Both the soluble and membrane fractions were precipitated in trichloroacetic acid (13% vol/vol), incubated on ice for 30 min, and then overnight at 4°C. Precipitated proteins were then washed with ice-cold acetone and analyzed by SDS-PAGE and immunoblotted for σ32 (Neoclone), β′ (Neoclone), σ70 (Neoclone), RseA [86], and RuvB (Abcam) with fluorescent secondary antibodies (LI-COR Biosciences) used for detection. The percentage of σ32 in each fraction was determined by direct scanning and analyzing bands with ImageJ software (National Institutes of Health).
RNA Polymerase Pull-Downs
Cells were grown to OD600 = 0.35–0.45, harvested, and resuspended in ice-cold Buffer D (50 Tris-HCl, pH 8.0, 0.1 mM EDTA, 150 mM NaCl, and 5% glycerol) to an OD600 of 20. Lysozyme was added to 0.75 mg/mL and cells were incubated on ice for 30 min, followed by sonication, then subjected to low-speed centrifugation to remove cell debris and unlysed cells. Lysates were then incubated with pre-equilibrated, pre-blocked (Buffer D containing 5% Bovine Serum Albumin, 0.1 mg/mL dextran) Softag 4 Resin (Neoclone) overnight at 4°C. Bound proteins were washed 3× with Buffer D and eluted with 4× LDS NuPAGE Buffer (Life Technologies). To collect lysates and eluted proteins, 0.05 µM of Strep-6×H-tagged σ32 was added as a loading and blotting control during analysis by SDS-PAGE and Western blotting against σ32.
Construction of 3L-Pf3 Fusion Proteins
The 3L-Pf3 genetic sequence was created by carrying out standard polymerase chain reaction using the following overlapping oligos: 5′-atgcaatccgtgattactgatgtgacaggccaactgacagcggtgcaagc-3′, 5′-taccattggtggtgctattcttctcctgattgttctggccgctgttgtgctggg-3′, 5′-aaagaattgcgctttgatccagcgaatacccagcacaacagcggccagaa-3′, and 5′-aagaatagcaccaccaatggtagtgatatcagcttgcaccgctgtcagtt-3′. The stitched oligos were then cloned using TOPO TA cloning (Invitrogen) and sequenced. To construct chromosomal 3L-Pf3-σ32, PCR was carried out to stitch the 3L-Pf3 gene sequence flanked by the first 500 base pairs of the σ32 open reading frame and 500 base pairs upstream of the start codon, and subsequently cloned into the pKNG101 suicide vector. The 3L-Pf3 sequence was then integrated 5′ and in-frame with the chromosomal rpoH gene by double homologous recombination. Counterselection of sacB on pKNG101 was carried out on 10% sucrose media (5 g/L Yeast Extract, 10 g/L Tryptone, 15 g/L Bacto Agar, 10% sucrose) [25],[87]. Clones were sequenced to verify chromosomal integration of the 3L-Pf3 sequence in the correct reading frame.
To construct pTrc99A expressing 3L-Pf3-FliA, flgM and fliA (in that order) were cloned as an operon, with the sequence 5′-ccgtctagaattaaagAGGAGaaaggtacc-3′ added between the two genes in the vector; the Shine-Dalgarno site is designated in uppercase. Two plasmids were created—one with just flgM and fliA, unmodified, and one where the 3L-Pf3 sequence was cloned 5′ to and in-frame with fliA. Clones were sequenced to verify correct sequences and proper reading frame. Expression was from the leaky pTrc promoter, and experiments were only carried out after fresh transformation into the parental CAG48238 strain. Levels of FliA were analyzed by SDS-PAGE and immunoblotting with antibodies against FliA (Abcam).
Immunoblotting
Cells were re-suspended in equal volumes of Buffer C, with the addition of trichloroacetic acid (final 13% vol/vol), kept on ice overnight, and the precipitate collected by centrifugation. Pellets were washed with acetone and resuspended in 1× LDS NuPAGE Buffer (Life Technologies). Serial dilutions of WT and mutant samples were loaded onto a polyacrylamide gel, and proteins transferred to nitrocellulose membranes. The blots were first probed with primary antibodies and then with anti-primary fluorescence-conjugated secondary antibody (Licor). Immunoblots were scanned at the appropriate wavelengths for detection. Fold increase (protein level experiments) was estimated by comparison with a dilution series of samples from the WT strain. Fold decrease after addition of chloramphenicol (protein stability experiments) was determined by direct scanning and analyzing bands with ImageJ software (National Institutes of Health).
Supporting Information
σ32Δ21aa, a C-terminal truncation of σ32, is defective in binding to RNA polymerase in vivo . (A) Immunoprecipitation of RNA polymerase-bound native σ32 and σ32Δ21aa. σ32Δ21aa was expressed from pTrc99A in ΔftsH cells, induced to levels comparable to endogenous σ32, grown to mid-exponential at 30°C in LB medium and the amount of σ32 bound to the anti-β′ resin (Softag4; Neoclone) and remaining σ32 in the supernatant was quantified by immunoblotting using a polyclonal antibody against σ32. Comparable amounts of total cellular lysates (TCL; left lane) and corresponding RNA-polymerase immunoprecipitations (RNAP IP; right lane) are shown. Purified σ32, tagged at the N-terminus with a Strep and 6×Histidine (Strep-6×H) tag, was used as a loading and blotting control. Results of a representative experiment are shown. (B) Quantification of RNA polymerase-bound native σ32 and σ32Δ21aa expressed in the same strain background (ΔftsH). Averages of four independent experiments are shown.
(TIF)
Membrane fractionation of σ32 is independent of RNA polymerase binding. ftsH E415A cells expressing either WTσ32Δ21aa or I54Nσ32Δ21aa were subjected to cellular fractionation (see Materials and Methods), and soluble and membrane fractions were resolved by SDS-PAGE and analyzed by immunoblotting for σ32, σ70, and the β′ subunit of RNA polymerase. Ectopically expressed σ32Δ21aa or I54Nσ32Δ21aa were present at levels comparable to native σ32 and were distinguished from endogenous σ32 on a 10% SDS-PAGE gel. All fractionation experiments were performed ≥8 times, and % fractionation was calculated from experiments where probed cytoplasmic (RuvB) and membrane (RseA) proteins separated properly.
(TIF)
σ32 interacts with SecA through protein–protein interaction analysis. Purified SecA was run on a 10% SDS-PAGE gel (along with FtsY and Ffh), transferred to nitrocellulose, re-natured, and incubated with purified WTσ32. The Coomassie-stained gel of the prey proteins (FtsY, Ffh, and SecA; left) and the nitrocellulose membrane containing the transferred prey proteins, probed with polyclonal anti-σ32 antibodies (right), are shown. The Coomassie-stained gel section of FtsY and Ffh and the corresponding σ32-incubated nitrocellulose membrane probed with anti-σ32 antibodies are also shown in Figure 2C.
(TIF)
Fusing the 3L-Pf3 peptide to the N-terminus of WTσ32 coding sequence significantly increases its membrane localization. (A) Schematic representation of membrane-tethered 3L-Pf3-WTσ32 (IM-WTσ32). The amino acids corresponding to the 3L-Pf3 and σ32 are shown as open or enclosed dark circles, respectively. (B) Soluble (lanes 1 and 3) and membrane (lanes 2 and 4) fractions from cellular fractionations (described in Materials and Methods) were separated by SDS-PAGE and immunoblotted for the indicated proteins shown on the right.
(TIF)
Levels of σ32 and σ32 variants in varying strain backgrounds. Strains were grown to OD600∼0.35, precipitated by addition of TCA to 13% final (vol/vol). Levels of σ32 and σ32 variants were determined by quantitative immunoblotting (see Materials and Methods). The experiment was carried out ≥5 times, with an example blot shown. These are the raw data used to obtain level values for σ32 and its variants shown in Table 3. Averaged quantification of the amount β′ served as a loading control, and levels of FtsH and FtsY are additionally shown. The genetic backgrounds of the mutant strains are shown below the blots. The specific protein probed on each blot is shown to the right. Note that IM-σ32 and IM-I54Nσ32 run as a smear, most likely because the membrane localization signal adopts multiple conformations during SDS-PAGE electrophoresis. To minimize this problem, gels were run very slowly (60–80 volts). Amount of IM-σ32 variants was calculated over the entire smear. Additionally, there is a contaminating band in all samples marked with an asterisk (*) that runs approximately at the same molecular weight as IM-σ32. This contaminating band prevents accurate quantification of samples with low amounts of IM-σ32 (lanes 2, 7, and 9).
(TIF)
The 3L-Pf3 peptide does not alter the stability of the FliA σ. (A) Addition of the 3L-Pf3 peptide to the N-terminus of FliA σ does not affect its cellular levels. Total cellular lysates were separated on SDS-PAGE and immunoblotted for FliA. WT fliA or 3L-Pf3-fliA was expressed from uninduced pTrc99A in the MG1655 background, and the fliA variants expressed are shown (at top). MG1655 carrying only pTrc99A (Vector) shows the endogenous levels of FliA. The lower band present in the 3L-Pf3-FliA lysate is endogenous FliA. Experiments were performed at least three times. The representative experiment shown demonstrates that addition of the 3L-PF3 peptide does not alter the amount of the FliA present in the lysate. As both FliA and 3L-Pf3-FliA are expressed from the same transcriptional and translational start points, we conclude that the 3L-Pf3 tag does not destabilize FliA. Thus, even though targeted to the membrane, 3L-Pf3FliA is not degraded by the membrane localized FtsH protein, which preferentially degrades membrane proteins. (B) Addition of the 3L-Pf3 peptide to the N-terminus of FliA increases its membrane localization. Soluble and membrane fractions from cellular fractionations of MG1655 carrying fliA or 3L-Pf3-fliA expressed on pTrc99A were separated on SDS-PAGE and immunoblotted for FliA. Percentage of membrane-localized FliA is plotted. Averages of four independent experiments are shown.
(TIF)
Growth defects in I54Nσ32 and p ftsY ::Tn 5 are relieved when the endogenous σ32 is membrane-tethered. (A) Early exponential growth comparison of WT, IM-WTσ32, I54Nσ32, and IM-I54Nσ32. (B) Early exponential growth comparison of WT, IM-WTσ32, pftsY::Tn5 mutant, and the double mutant pftsY::Tn5, IM-WTσ32. Cellular density (OD600) was plotted over time in (A) and (B). Experiments for both (A) and (B) were carried out three times, and an example growth curve obtained is shown. (C) IM-tethering of σ32 in mutant strains restores growth rates to that of WT. Doubling times were calculated as the inverse of the slope of the cultures growing in early exponential phase in LB at 30°C. Strain mutations are shown on the left. The exact values of the doubling times for each strain are shown on the right and are an average of three experiments. (D) Membrane-tethering of σ32 in the pftsY::Tn5 mutant restores transition into stationary phase growth to that of WT. The pftsY::Tn5 mutant transitions into stationary phase growth significantly earlier and at a lower OD600 than both WT and the double mutant pftsY::Tn5, IM-WTσ32. Cellular density (OD600) was plotted over time. Growth curves are an average of three biological replicates.
(TIF)
Acknowledgments
We thank Byuong-Mo Koo for experimental expertise and Hana El-Samad, Jonathan Weissman, Jeffery Cox, Tania Baker (Massachusetts Institute of Technology), Christophe Herman (Baylor College of Medicine), Hiroyuki Mori (Institute for Virus Research Kyoto University), and members of the Gross Lab for discussion and input.
Abbreviations
- HSPs
heat shock proteins
- HSR
heat shock response
- IM
inner membrane
- pBPA
p-benzoylphenylalanine
- PhoA
alkaline phosphatase
- S.A.
specific activity
- SR
signal receptor
- SRP
signal recognition particle
Funding Statement
This work was supported by National Institutes of Health Grant GM-36278 (to C.A.G.), a National Science Foundation Graduate Research Fellowship (to B.L.), and the Jane Coffin Child Memorial Fund (to S.N). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Anckar J, Sistonen L (2011) Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem 80: 1089–1115 doi:10.1146/annurev-biochem-060809-095203 [DOI] [PubMed] [Google Scholar]
- 2. Morimoto RI (2008) Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes & Development 22: 1427–1438 doi:10.1101/gad.1657108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Morimoto RI (2012) The heat shock response: systems biology of proteotoxic stress in aging and disease. Cold Spring Harbor Symposia on Quantitative Biology 76: 91–99 doi:10.1101/sqb.2012.76.010637 [DOI] [PubMed] [Google Scholar]
- 4. Yamamori T, Yura T (1980) Temperature-induced synthesis of specific proteins in Escherichia coli: evidence for transcriptional control. J Bacteriol 142: 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grossman AD, Erickson JW, Gross CA (1984) The htpR gene product of E. coli is a sigma factor for heat-shock promoters. Cell 38: 383–390. [DOI] [PubMed] [Google Scholar]
- 6. Taylor WE, Straus DB, Grossman AD, Burton ZF, Gross CA, et al. (1984) Transcription from a heat-inducible promoter causes heat shock regulation of the sigma subunit of E. coli RNA polymerase. Cell 38: 371–381. [DOI] [PubMed] [Google Scholar]
- 7. Straus DB, Walter WA, Gross CA (1989) The activity of sigma32 is reduced under conditions of excess heat shock protein production in Escherichia coli . Genes & Development 3: 2003–2010 doi:10.1101/gad.3.12a.2003 [DOI] [PubMed] [Google Scholar]
- 8. Straus DB, Walter WA, Gross CA (1987) The heat shock response of E. coli is regulated by changes in the concentration of σ32 . Nature 329: 348–351. [DOI] [PubMed] [Google Scholar]
- 9. Taura T, Kusukawa N, Yura T, Ito K (1989) Transient shut off of Escherichia coli heat shock protein synthesis upon temperature shift down. Biochem Biophys Res Commun 163: 438–443. [DOI] [PubMed] [Google Scholar]
- 10. Morita M, Kanemori M, Yanagi H, Yura T (1999) Heat-induced synthesis of sigma32 in Escherichia coli: structural and functional dissection of rpoH mRNA secondary structure. J Bacteriol 181: 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morita MT, Tanaka Y, Kodama TS, Kyogoku Y, Yanagi H, et al. (1999) Translational induction of heat shock transcription factor σ32: evidence for a built-in RNA thermosensor. Genes & Development 13: 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guisbert E, Yura T, Rhodius VA, Gross CA (2008) Convergence of molecular, modeling, and systems approaches for an understanding of the Escherichia coli heat shock response. Microbiology and Molecular Biology Reviews 72: 545–554 doi:10.1128/MMBR.00007-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guisbert E, Herman C, Lu CZ, Gross CA (2004) A chaperone network controls the heat shock response in E. coli . Genes & Development 18: 2812–2821 doi:10.1101/gad.1219204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tomoyasu T, Ogura T, Tatsuta T, Bukau B (1998) Levels of DnaK and DnaJ provide tight control of heat shock gene expression and protein repair in Escherichia coli . Mol Microbiol 30: 567–581. [DOI] [PubMed] [Google Scholar]
- 15. Gamer J, Multhaup G, Tomoyasu T, McCarty JS, Rüdiger S, et al. (1996) A cycle of binding and release of the DnaK, DnaJ and GrpE chaperones regulates activity of the Escherichia coli heat shock transcription factor sigma32. EMBO J 15: 607–617. [PMC free article] [PubMed] [Google Scholar]
- 16. Gamer J, Bujard H, Bukau B (1992) Physical interaction between heat shock proteins DnaK, DnaJ, and GrpE and the bacterial heat shock transcription factor sigma32. Cell 69: 833–842. [DOI] [PubMed] [Google Scholar]
- 17. Liberek K, Galitski TP, Zylicz M, Georgopoulos C (1992) The DnaK chaperone modulates the heat shock response of Escherichia coli by binding to the sigma32 transcription factor. Proc Natl Acad Sci USA 89: 3516–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Herman C, Thévenet D, D'Ari R, Bouloc P (1995) Degradation of sigma32, the heat shock regulator in Escherichia coli, is governed by HflB. Proc Natl Acad Sci USA 92: 3516–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kanemori M, Nishihara K, Yanagi H, Yura T (1997) Synergistic roles of HslVU and other ATP-dependent proteases in controlling in vivo turnover of sigma32 and abnormal proteins in Escherichia coli . J Bacteriol 179: 7219–7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tomoyasu T, Gamer J, Bukau B, Kanemori M, Mori H, et al. (1995) Escherichia coli FtsH is a membrane-bound, ATP-dependent protease which degrades the heat-shock transcription factor sigma32. EMBO J 14: 2551–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Straus D, Walter W, Gross CA (1990) DnaK, DnaJ, and GrpE heat shock proteins negatively regulate heat shock gene expression by controlling the synthesis and stability of sigma32. Genes & Development 4: 2202–2209 doi:10.1101/gad.4.12a.2202 [DOI] [PubMed] [Google Scholar]
- 22. Blaszczak A, Georgopoulos C, Liberek K (1999) On the mechanism of FtsH-dependent degradation of the sigma32 transcriptional regulator of Escherichia coli and the role of the Dnak chaperone machine. Mol Microbiol 31: 157–166. [DOI] [PubMed] [Google Scholar]
- 23. Horikoshi M, Yura T, Tsuchimoto S, Fukumori Y, Kanemori M (2004) Conserved region 2.1 of Escherichia coli heat shock transcription factor σ32 is required for modulating both metabolic stability and transcriptional activity. J Bacteriol 186: 7474–7480 doi:10.1128/JB.186.22.7474-7480.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Obrist M, Narberhaus F (2005) Identification of a turnover element in region 2.1 of Escherichia coli sigma32 by a bacterial one-hybrid approach. J Bacteriol 187: 3807–3813 doi:10.1128/JB.187.11.3807-3813.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yura T, Guisbert E, Poritz M, Lu CZ, Campbell E, et al. (2007) Analysis of sigma32 mutants defective in chaperone-mediated feedback control reveals unexpected complexity of the heat shock response. Proc Natl Acad Sci USA 104: 17638–17643 doi:10.1073/pnas.0708819104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Suzuki HH, Ikeda AA, Tsuchimoto SS, Adachi K-IK, Noguchi AA, et al. (2012) Synergistic binding of DnaJ and DnaK chaperones to heat shock transcription factor σ32 ensures its characteristic high metabolic instability: implications for heat shock protein 70 (Hsp70)-Hsp40 mode of function. J Biol Chem 287: 19275–19283 doi:10.1074/jbc.M111.331470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bernstein HD, Hyndman JB (2001) Physiological basis for conservation of the signal recognition particle targeting pathway in Escherichia coli . J Bacteriol 183: 2187–2197 doi:10.1128/JB.183.7.2187-2197.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bourgaize DB, Phillips TA, VanBogelen RA, Jones PG, Neidhardt FC, et al. (1990) Loss of 4.5S RNA induces the heat shock response and lambda prophage in Escherichia coli . J Bacteriol 172: 1151–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Poritz MA, Bernstein HD, Strub K, Zopf D, Wilhelm H, et al. (1990) An E. coli ribonucleoprotein containing 4.5S RNA resembles mammalian signal recognition particle. Science 250: 1111–1117. [DOI] [PubMed] [Google Scholar]
- 30. Nonaka G, Blankschien M, Herman C, Gross CA, Rhodius VA (2006) Regulon and promoter analysis of the E. coli heat-shock factor, sigma32, reveals a multifaceted cellular response to heat stress. Genes & Development 20: 1776–1789 doi:10.1101/gad.1428206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wild J, Kamath-Loeb A, Ziegelhoffer E, Lonetto M, Kawasaki Y, et al. (1992) Partial loss of function mutations in DnaK, the Escherichia coli homologue of the 70-kDa heat shock proteins, affect highly conserved amino acids implicated in ATP binding and hydrolysis. Proc Natl Acad Sci USA 89: 7139–7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tomoyasu T, Arsène F, Ogura T, Bukau B (2001) The C terminus of σ32 is not essential for degradation by FtsH. J Bacteriol 183: 5911–5917 doi:10.1128/JB.183.20.5911-5917.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Qi HYH, Bernstein HDH (1999) SecA is required for the insertion of inner membrane proteins targeted by the Escherichia coli signal recognition particle. J Biol Chem 274: 8993–8997. [DOI] [PubMed] [Google Scholar]
- 34. Schiebel E, Driessen AJ, Hartl FU, Wickner W (1991) Delta mu H+ and ATP function at different steps of the catalytic cycle of preprotein translocase. Cell 64: 927–939. [DOI] [PubMed] [Google Scholar]
- 35. van der Wolk JP, de Wit JG, Driessen AJ (1997) The catalytic cycle of the Escherichia coli SecA ATPase comprises two distinct preprotein translocation events. EMBO J 16: 7297–7304 doi:10.1093/emboj/16.24.7297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hartl FU, Lecker S, Schiebel E, Hendrick JP, Wickner W (1990) The binding cascade of SecB to SecA to SecY/E mediates preprotein targeting to the E. coli plasma membrane. Cell 63: 269–279. [DOI] [PubMed] [Google Scholar]
- 37. Economou A, Wickner W (1994) SecA promotes preprotein translocation by undergoing ATP-driven cycles of membrane insertion and deinsertion. Cell 78: 835–843. [DOI] [PubMed] [Google Scholar]
- 38. Neumann-Haefelin C, Schäfer U, Müller M, Koch HG (2000) SRP-dependent co-translational targeting and SecA-dependent translocation analyzed as individual steps in the export of a bacterial protein. EMBO J 19: 6419–6426 doi:10.1093/emboj/19.23.6419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wild J, Walter WA, Gross CA, Altman E (1993) Accumulation of secretory protein precursors in Escherichia coli induces the heat shock response. J Bacteriol 175: 3992–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Oliver DB, Beckwith J (1981) E. coli mutant pleiotropically defective in the export of secreted proteins. Cell 25: 765–772. [DOI] [PubMed] [Google Scholar]
- 41. Schmidt MG, Rollo EE, Grodberg J, Oliver DB (1988) Nucleotide sequence of the secA gene and secA(Ts) mutations preventing protein export in Escherichia coli . J Bacteriol 170: 3404–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Angelini S, Deitermann S, Koch H-G (2005) FtsY, the bacterial signal-recognition particle receptor, interacts functionally and physically with the SecYEG translocon. EMBO Rep 6: 476–481 doi:10.1038/sj.embor.7400385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kuhn P, Weiche B, Sturm L, Sommer E, Drepper F, et al. (2011) The bacterial SRP receptor, SecA and the ribosome use overlapping binding sites on the SecY translocon. Traffic 12: 563–578 doi:10.1111/j.1600-0854.2011.01167.x [DOI] [PubMed] [Google Scholar]
- 44. Jiang Y, Cheng Z, Mandon EC, Gilmore R (2008) An interaction between the SRP receptor and the translocon is critical during cotranslational protein translocation. J Cell Biol 180 6: 1149–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mori H, Ito K (2006) Different modes of SecY-SecA interactions revealed by site-directed in vivo photo-cross-linking. Proc Natl Acad Sci USA 103: 16159–16164 doi:10.1073/pnas.0606390103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zimmer J, Nam Y, Rapoport TA (2008) Structure of a complex of the ATPase SecA and the protein-translocation channel. Nature 455: 936–943 doi:10.1038/nature07335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. van der Sluis EO, Nouwen N, Koch J, de Keyzer J, van der Does C, et al. (2006) Identification of two interaction sites in SecY that are important for the functional interaction with SecA. J Mol Biol 361: 839–849 doi:10.1016/j.jmb.2006.07.017 [DOI] [PubMed] [Google Scholar]
- 48. Cheng Z (2004) Identification of cytoplasmic residues of Sec61p involved in ribosome binding and cotranslational translocation. J Cell Biol 168: 67–77 doi:10.1083/jcb.200408188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Becker T, Bhushan S, Jarasch A, Armache JP, Funes S, et al. (2009) Structure of monomeric yeast and mammalian Sec61 complexes interacting with the translating ribosome. Science 326: 1369–1373 doi:10.1126/science.1178535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Prinz A, Behrens C, Rapoport TA, Hartmann E, Kalies KU (2000) Evolutionarily conserved binding of ribosomes to the translocation channel via the large ribosomal RNA. EMBO J 19: 1900–1906 doi:10.1093/emboj/19.8.1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shimohata N, Nagamori S, Akiyama Y, Kaback HR, Ito K (2007) SecY alterations that impair membrane protein folding and generate a membrane stress. J Cell Biol 176: 307–317 doi:10.1083/jcb.200611121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ito K, Akiyama Y, Yura T, Shiba K (1986) Diverse effects of the MalE-LacZ hybrid protein on Escherichia coli cell physiology. J Bacteriol 167: 201–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Manoil C, Beckwith J (1986) A genetic approach to analyzing membrane protein topology. Science 233: 1403–1408. [DOI] [PubMed] [Google Scholar]
- 54. Prinz WA, Spiess C, Ehrmann M, Schierle C, Beckwith J (1996) Targeting of signal sequenceless proteins for export in Escherichia coli with altered protein translocase. EMBO J 15: 5209–5217. [PMC free article] [PubMed] [Google Scholar]
- 55. Serek J, Bauer-Manz G, Struhalla G, van den Berg L, Kiefer D, et al. (2004) Escherichia coli YidC is a membrane insertase for Sec-independent proteins. EMBO J 23: 294–301 doi:10.1038/sj.emboj.7600063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tatsuta T, Tomoyasu T, Bukau B, Kitagawa M, Mori H, et al. (1998) Heat shock regulation in the ftsH null mutant of Escherichia coli: dissection of stability and activity control mechanisms of sigma32 in vivo. Mol Microbiol 30: 583–593. [DOI] [PubMed] [Google Scholar]
- 57. Zhang X, Rashid R, Wang K, Shan S-O (2010) Sequential checkpoints govern substrate selection during cotranslational protein targeting. Science 328: 757–760 doi:10.1126/science.1186743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jensen CG, Pedersen S (1994) Concentrations of 4.5S RNA and Ffh protein in Escherichia coli: the stability of Ffh protein is dependent on the concentration of 4.5S RNA. J Bacteriol 176: 7148–7154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bukau B, Walker GC (1990) Mutations altering heat shock specific subunit of RNA polymerase suppress major cellular defects of E. coli mutants lacking the DnaK chaperone. EMBO J 9: 4027–4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bahl H, Echols H, Straus DB, Court D, Crowl R, et al. (1987) Induction of the heat shock response of E. coli through stabilization of sigma32 by the phage lambda cIII protein. Genes & Development 1: 57–64 doi:10.1101/gad.1.1.57 [DOI] [PubMed] [Google Scholar]
- 61. Petersen TN, Brunak S, Heijne von G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nature Publishing Group 8: 785–786 doi:10.1038/nmeth.1701 [DOI] [PubMed] [Google Scholar]
- 62. Krogh A, Larsson B, Heijne von G, Sonnhammer ELL (2001) Predicting transmembrane protein topology with a hidden markov model: application to complete genomes. J Mol Biol 305: 567–580 doi:10.1006/jmbi.2000.4315 [DOI] [PubMed] [Google Scholar]
- 63. Zylicz M, Nieradko J, Taylor K (1983) Escherichia coli dnaJ- and dnaK- gene products: synthesis in minicells and membrane-affinity. Biochem Biophys Res Commun 110: 176–180. [DOI] [PubMed] [Google Scholar]
- 64. Bardwell JCJ, Tilly KK, Craig EE, King JJ, Zylicz MM, et al. (1986) The nucleotide sequence of the Escherichia coli K12 dnaJ+ gene. A gene that encodes a heat shock protein. J Biol Chem 261: 1782–1785. [PubMed] [Google Scholar]
- 65. Kostyal DA, Farrell M, McCabe A, Mei Z, Firshein W (1989) Replication of an RK2 miniplasmid derivative in vitro by a DNA/membrane complex extracted from Escherichia coli: involvement of the dnaA but not dnaK host proteins and association of these and plasmid-encoded proteins with the inner membrane. Plasmid 21: 226–237. [DOI] [PubMed] [Google Scholar]
- 66. Wickström D, Wagner S, Simonsson P, Pop O, Baars L, et al. (2011) Characterization of the consequences of YidC depletion on the inner membrane proteome of E. coli using 2D blue native/SDS-PAGE. J Mol Biol 409: 124–135 doi:10.1016/j.jmb.2011.03.068 [DOI] [PubMed] [Google Scholar]
- 67. Wickstrom (2011) Consequences of depletion of the signal recognition particle in Escherichia coli . J Biol Chem 286: 4598–4609 doi:10.1074/jbc.M109.081935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lasserre J-P, Beyne E, Pyndiah S, Lapaillerie D, Claverol S, et al. (2006) A complexomic study of Escherichia coli using two-dimensional blue native/SDS polyacrylamide gel electrophoresis. Electrophoresis 27: 3306–3321 doi:10.1002/elps.200500912 [DOI] [PubMed] [Google Scholar]
- 69. Mircheva M, Boy D, Weiche B, Hucke F, Graumann P, et al. (2009) Predominant membrane localization is an essential feature of the bacterial signal recognition particle receptor. BMC Biol 7: 76 doi:10.1186/1741-7007-7-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Luirink J, Hagen-Jongman ten CM, van der Weijden CC, Oudega B, High S, et al. (1994) An alternative protein targeting pathway in Escherichia coli: studies on the role of FtsY. EMBO J 13: 2289–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Heinrich J, Wiegert T (2009) Regulated intramembrane proteolysis in the control of extracytoplasmic function sigma factors. Res Microbiol 160: 696–703 doi:10.1016/j.resmic.2009.08.019 [DOI] [PubMed] [Google Scholar]
- 72. Österberg S, Peso-Santos TD, Shingler V (2011) Regulation of alternative sigma factor use. Annu Rev Microbiol 65: 37–55 doi:10.1146/annurev.micro.112408.134219 [DOI] [PubMed] [Google Scholar]
- 73. Kroos L, Kunkel B, Losick R (1989) Switch protein alters specificity of RNA polymerase containing a compartment-specific sigma factor. Science 243: 526–529. [DOI] [PubMed] [Google Scholar]
- 74. Lu S, Halberg R, Kroos L (1990) Processing of the mother-cell sigma factor, sigma K, may depend on events occurring in the forespore during Bacillus subtilis development. Proc Natl Acad Sci USA 87: 9722–9726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cutting S, Oke V, Driks A, Losick R, Lu S, et al. (1990) A forespore checkpoint for mother cell gene expression during development in B. subtilis . Cell 62: 239–250. [DOI] [PubMed] [Google Scholar]
- 76. Hughes KT, Mathee K (1998) The anti-sigma factors. Annu Rev Microbiol 52: 231–286. [DOI] [PubMed] [Google Scholar]
- 77. Li X, Henry R, Yuan J, Cline K, Hoffman NE (1995) A chloroplast homologue of the signal recognition particle subunit SRP54 is involved in the posttranslational integration of a protein into thylakoid membranes. Proc Natl Acad Sci USA 92: 3789–3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. McLennan N, Masters M (1998) GroE is vital for cell-wall synthesis. Nature 392: 139 doi:10.1038/32317 [DOI] [PubMed] [Google Scholar]
- 79. Stark MJM (1987) Multicopy expression vectors carrying the lac repressor gene for regulated high-level expression of genes in Escherichia coli . Gene 51: 255–267 doi:10.1016/0378-1119(87)90314-3 [DOI] [PubMed] [Google Scholar]
- 80. Koo B-M, Rhodius VA, Campbell EA, Gross CA (2009) Dissection of recognition determinants of Escherichia coli σ32 suggests a composite −10 region with an “extended −10” motif and a core −10 element. Mol Microbiol 72: 815–829 doi:10.1111/j.1365-2958.2009.06690.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Peluso P, Shan S-O, Nock S, Herschlag D, Walter P (2001) Role of SRP RNA in the GTPase Cycles of Ffh and FtsY. Biochemistry 40: 15224–15233 doi:10.1021/bi011639y [DOI] [PubMed] [Google Scholar]
- 82. Kusters I, van den Bogaart G, Kedrov A, Krasnikov V, Fulyani F, et al. (2011) Quaternary structure of SecA in solution and bound to SecYEG probed at the single molecule level. Structure/Folding and Design 19: 430–439 doi:10.1016/j.str.2010.12.016 [DOI] [PubMed] [Google Scholar]
- 83. Wu Y, Li Q, Chen X-Z (2007) Detecting protein–protein interactions by far western blotting. Nat Protoc 2: 3278–3284 doi:10.1038/nprot.2007.459 [DOI] [PubMed] [Google Scholar]
- 84. Narita S-I, Masui C, Suzuki T, Dohmae N, Akiyama Y (2013) Protease homolog BepA (YfgC) promotes assembly and degradation of β-barrel membrane proteins in Escherichia coli . Proc Natl Acad Sci doi:10.1073/pnas.1312012110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Manoil C, Beckwith J (1985) TnphoA: a transposon probe for protein export signals. Proc Natl Acad Sci USA 82: 8129–8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Alba BM (2002) DegS and YaeL participate sequentially in the cleavage of RseA to activate the sigma E-dependent extracytoplasmic stress response. Genes & Development 16: 2156–2168 doi:10.1101/gad.1008902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Blomfield IC, Vaughn V, Rest RF, Eisenstein BI (1991) Allelic exchange in Escherichia coli using the Bacillus subtilis sacB gene and a temperature-sensitive pSC101 replicon. Mol Microbiol 5: 1447–1457. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
σ32Δ21aa, a C-terminal truncation of σ32, is defective in binding to RNA polymerase in vivo . (A) Immunoprecipitation of RNA polymerase-bound native σ32 and σ32Δ21aa. σ32Δ21aa was expressed from pTrc99A in ΔftsH cells, induced to levels comparable to endogenous σ32, grown to mid-exponential at 30°C in LB medium and the amount of σ32 bound to the anti-β′ resin (Softag4; Neoclone) and remaining σ32 in the supernatant was quantified by immunoblotting using a polyclonal antibody against σ32. Comparable amounts of total cellular lysates (TCL; left lane) and corresponding RNA-polymerase immunoprecipitations (RNAP IP; right lane) are shown. Purified σ32, tagged at the N-terminus with a Strep and 6×Histidine (Strep-6×H) tag, was used as a loading and blotting control. Results of a representative experiment are shown. (B) Quantification of RNA polymerase-bound native σ32 and σ32Δ21aa expressed in the same strain background (ΔftsH). Averages of four independent experiments are shown.
(TIF)
Membrane fractionation of σ32 is independent of RNA polymerase binding. ftsH E415A cells expressing either WTσ32Δ21aa or I54Nσ32Δ21aa were subjected to cellular fractionation (see Materials and Methods), and soluble and membrane fractions were resolved by SDS-PAGE and analyzed by immunoblotting for σ32, σ70, and the β′ subunit of RNA polymerase. Ectopically expressed σ32Δ21aa or I54Nσ32Δ21aa were present at levels comparable to native σ32 and were distinguished from endogenous σ32 on a 10% SDS-PAGE gel. All fractionation experiments were performed ≥8 times, and % fractionation was calculated from experiments where probed cytoplasmic (RuvB) and membrane (RseA) proteins separated properly.
(TIF)
σ32 interacts with SecA through protein–protein interaction analysis. Purified SecA was run on a 10% SDS-PAGE gel (along with FtsY and Ffh), transferred to nitrocellulose, re-natured, and incubated with purified WTσ32. The Coomassie-stained gel of the prey proteins (FtsY, Ffh, and SecA; left) and the nitrocellulose membrane containing the transferred prey proteins, probed with polyclonal anti-σ32 antibodies (right), are shown. The Coomassie-stained gel section of FtsY and Ffh and the corresponding σ32-incubated nitrocellulose membrane probed with anti-σ32 antibodies are also shown in Figure 2C.
(TIF)
Fusing the 3L-Pf3 peptide to the N-terminus of WTσ32 coding sequence significantly increases its membrane localization. (A) Schematic representation of membrane-tethered 3L-Pf3-WTσ32 (IM-WTσ32). The amino acids corresponding to the 3L-Pf3 and σ32 are shown as open or enclosed dark circles, respectively. (B) Soluble (lanes 1 and 3) and membrane (lanes 2 and 4) fractions from cellular fractionations (described in Materials and Methods) were separated by SDS-PAGE and immunoblotted for the indicated proteins shown on the right.
(TIF)
Levels of σ32 and σ32 variants in varying strain backgrounds. Strains were grown to OD600∼0.35, precipitated by addition of TCA to 13% final (vol/vol). Levels of σ32 and σ32 variants were determined by quantitative immunoblotting (see Materials and Methods). The experiment was carried out ≥5 times, with an example blot shown. These are the raw data used to obtain level values for σ32 and its variants shown in Table 3. Averaged quantification of the amount β′ served as a loading control, and levels of FtsH and FtsY are additionally shown. The genetic backgrounds of the mutant strains are shown below the blots. The specific protein probed on each blot is shown to the right. Note that IM-σ32 and IM-I54Nσ32 run as a smear, most likely because the membrane localization signal adopts multiple conformations during SDS-PAGE electrophoresis. To minimize this problem, gels were run very slowly (60–80 volts). Amount of IM-σ32 variants was calculated over the entire smear. Additionally, there is a contaminating band in all samples marked with an asterisk (*) that runs approximately at the same molecular weight as IM-σ32. This contaminating band prevents accurate quantification of samples with low amounts of IM-σ32 (lanes 2, 7, and 9).
(TIF)
The 3L-Pf3 peptide does not alter the stability of the FliA σ. (A) Addition of the 3L-Pf3 peptide to the N-terminus of FliA σ does not affect its cellular levels. Total cellular lysates were separated on SDS-PAGE and immunoblotted for FliA. WT fliA or 3L-Pf3-fliA was expressed from uninduced pTrc99A in the MG1655 background, and the fliA variants expressed are shown (at top). MG1655 carrying only pTrc99A (Vector) shows the endogenous levels of FliA. The lower band present in the 3L-Pf3-FliA lysate is endogenous FliA. Experiments were performed at least three times. The representative experiment shown demonstrates that addition of the 3L-PF3 peptide does not alter the amount of the FliA present in the lysate. As both FliA and 3L-Pf3-FliA are expressed from the same transcriptional and translational start points, we conclude that the 3L-Pf3 tag does not destabilize FliA. Thus, even though targeted to the membrane, 3L-Pf3FliA is not degraded by the membrane localized FtsH protein, which preferentially degrades membrane proteins. (B) Addition of the 3L-Pf3 peptide to the N-terminus of FliA increases its membrane localization. Soluble and membrane fractions from cellular fractionations of MG1655 carrying fliA or 3L-Pf3-fliA expressed on pTrc99A were separated on SDS-PAGE and immunoblotted for FliA. Percentage of membrane-localized FliA is plotted. Averages of four independent experiments are shown.
(TIF)
Growth defects in I54Nσ32 and p ftsY ::Tn 5 are relieved when the endogenous σ32 is membrane-tethered. (A) Early exponential growth comparison of WT, IM-WTσ32, I54Nσ32, and IM-I54Nσ32. (B) Early exponential growth comparison of WT, IM-WTσ32, pftsY::Tn5 mutant, and the double mutant pftsY::Tn5, IM-WTσ32. Cellular density (OD600) was plotted over time in (A) and (B). Experiments for both (A) and (B) were carried out three times, and an example growth curve obtained is shown. (C) IM-tethering of σ32 in mutant strains restores growth rates to that of WT. Doubling times were calculated as the inverse of the slope of the cultures growing in early exponential phase in LB at 30°C. Strain mutations are shown on the left. The exact values of the doubling times for each strain are shown on the right and are an average of three experiments. (D) Membrane-tethering of σ32 in the pftsY::Tn5 mutant restores transition into stationary phase growth to that of WT. The pftsY::Tn5 mutant transitions into stationary phase growth significantly earlier and at a lower OD600 than both WT and the double mutant pftsY::Tn5, IM-WTσ32. Cellular density (OD600) was plotted over time. Growth curves are an average of three biological replicates.
(TIF)