

Alternative splicing has been increasingly recognized as an essential means to increase proteomic diversity from a limited number of genes. Alternative splicing can produce premature stop codons that result in RNA decay and disrupt protein domains and motifs that alter a protein’s regulatory and signaling properties.1 In addition to alternative splicing, alternative transcription, by regulating an alternative promoter or alternative 3′ end processing via selection of a more proximal polyadenlation site, contribute to transcriptome diversity and expression without regulating gene transcription.2,3 For example, both alternative splicing and the alternative 3′ end processing regulate inclusion of functional microRNA binding sites, altering the ability of these mRNAs to undergo translational repression.4
Although molecular approaches, such as DNA-array and RNA-seq (large-scale sequencing of RNA transcripts), are available to examine the transcriptome in great detail, a major roadblock for most scientists is the difficulty in analyzing and interpreting these data in a coherent and meaningful way. Such methods are particularly needed to understand how alternative isoforms regulate basic cell fate decisions in the early embryo and throughout development.
To examine the extent of alternative modes of transcript regulation in the control of cell-fate decisions and pluripotency, several laboratories have analyzed embryonic stem (ES) cells and derived differentiated cells by transcript-sensitive tiling arrays and RNA-seq. From these studies, several global observations emerged, including a greater diversity in isoforms expressed in ES cells,5 an enrichment in regulatory splicing motifs,6 loss of microRNA binding sites in ES cell transcripts7 and a tendency for alternative exons to occur in pathways of cell-cycle and pluripotency.7-9
While these global observations are useful in understanding these cellular transitions, they often highlight a large number of predictions that are difficult to test in a rigorous manner. For this reason, new tools are required that provide functional predictions that can be readily tested in living cells in addition to high-throughput experimental methods to test these predictions in a physiologically relevant manner. We recently developed an open-source application named AltAnalyze, with the aim of providing functional predictions that lead to testable hypotheses.7,8,10 Such hypotheses include how alternative exon expression affects the composition of protein domains, motifs or microRNA bindings (Fig. 1). These predictions can be further visualized in programs such as DomainGraph10 as a means to see which transcripts are specifically affected by alternative exon expression and how these changes affect protein-interaction networks and biological pathways.
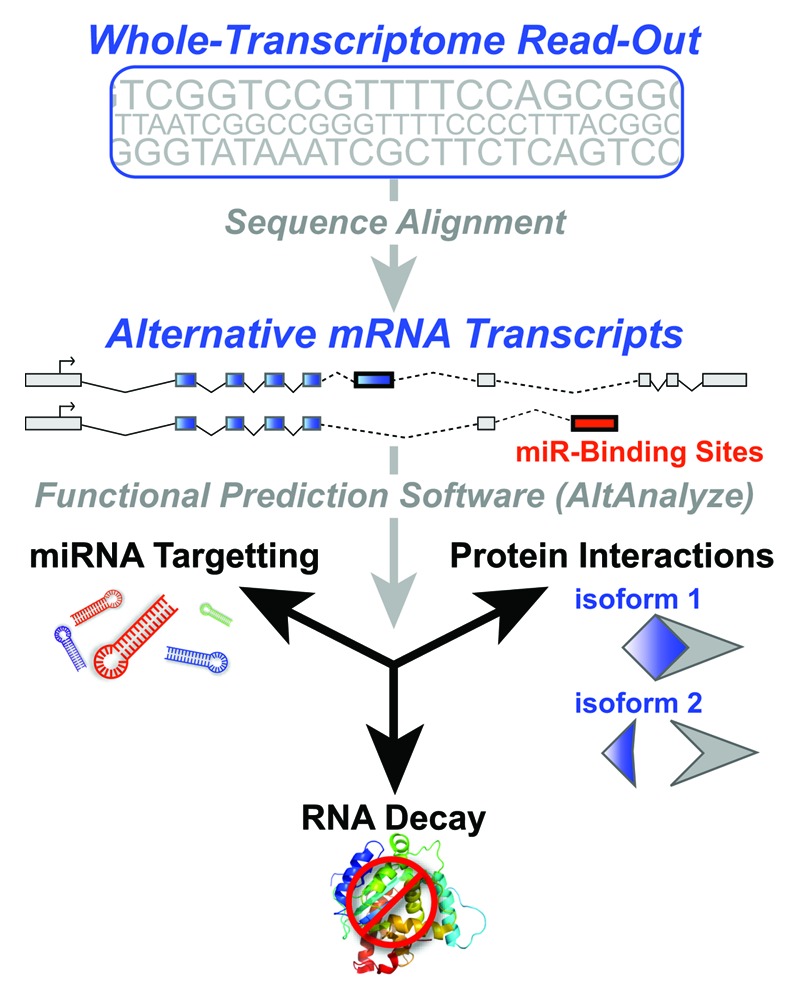
Figure 1. Functional implications of alternative modes of transcript regulation. Illustration depicting the analysis of whole-genome transcript expression data, in the context of alternative mRNA transcripts and functional outcomes with the application AltAnalyze. In the first step, Affymetrix exon or junction probeset sequences are aligned to genes, transcripts and exons. Next, alternative exons (thick black exon outline) that effect the composition of protein domains (exons aligning to a theoretical protein domain are shown in blue) and the presence of microRNA binding sites (red) are identified. Dashed lines indicate alternative exon-junctions. Predicted disruption of a protein-domain interaction is illustrated for the affected domain (blue) and an unaffected domain (gray).
To test the functional significance of alternative exons identified during ES cell differentiation, we exploited two cellular assays to compare isoform activity.8 To examine the loss of isoform expression for splice variants of the gene Tcf3, we selectively inhibited the expression of each isoform by mRNA knockdown. Alternative splicing of Tcf3 was predicted to alter the composition of a protein domain, critical for the regulation of pluripotency transcriptional targets.11 From this analysis, we identified differential expression of known lineage and tissue markers between isoform knockdowns and changes in the expression of transcriptional targets of Tcf3. MicroRNAs repress target protein expression by binding to complementary sequences in the 3′ untranslated region of mRNAs. Using a new screen, aimed at estimating the expression of a target protein by co-transcribing a florescent reporter, we could readily verify targeted repression of a specific splice-isoform of the gene ATP2A2 by several microRNAs computationally predicted to align to a single isoform of this gene. When analyzed in the context of differentially expressed microRNAs, such assays provide insight into more complex expression regulatory networks, overlooked by conventional screens. These assays can be reasonably scaled to accommodate a large number of targets (dozens to hundreds), highlighted by whole-genome transcript analyses and functional bioinformatics predictions. Such functional predictions can also provide useful hypotheses for downstream analyses, such as differences in signaling properties, co-factor binding, metabolic function and regulation by small RNAs.
The collection of whole-genome transcriptome data now presents the intriguing possibility of inferring complex signaling cascades from these data that extend beyond transcription to alternative modes of transcript regulation and the differential analysis of protein function. While such analyses will not become commonplace overnight, they offer the possibility of exploiting these data sets to a level that not imagined just a few years ago.
Salomonis N, et al. Proc Natl Acad Sci U S A. 2010;107:10514–9. doi: 10.1073/pnas.0912260107.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/12888
References
- 1.Cooper TA. Cell. 2005;120:1–2. doi: 10.1016/j.cell.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 2.Millevoi S, et al. Nucleic Acids Res. 2009;37:4672–83. doi: 10.1093/nar/gkp470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mayr C, et al. Cell. 2009;138:673–84. doi: 10.1016/j.cell.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sandberg R, et al. Science. 2008;320:1643–7. doi: 10.1126/science.1155390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu JQ, et al. Proc Natl Acad Sci U S A. 2010;107:5254–9. doi: 10.1073/pnas.0914114107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yeo GW, et al. PLoS Comput Biol. 2007;3:1951–67. doi: 10.1371/journal.pcbi.0030196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salomonis N, et al. PLoS Comput Biol. 2009;5:e1000553. doi: 10.1371/journal.pcbi.1000553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salomonis N, et al. Proc Natl Acad Sci U S A. 2010;107:10514–9. doi: 10.1073/pnas.0912260107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pritsker M, et al. Proc Natl Acad Sci U S A. 2005;102:14290–5. doi: 10.1073/pnas.0502132102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emig D, et al. Nucleic Acids Res. 2010;38(Suppl):W755-62. doi: 10.1093/nar/gkq405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pereira L, et al. Mol Cell Biol. 2006;26:7479–91. doi: 10.1128/MCB.00368-06. [DOI] [PMC free article] [PubMed] [Google Scholar]