

Abstract
CSF from the subarachnoid space moves rapidly into the brain along paravascular routes surrounding penetrating cerebral arteries, exchanging with brain interstitial fluid (ISF) and facilitating the clearance of interstitial solutes, such as amyloid β, in a pathway that we have termed the “glymphatic” system. Prior reports have suggested that paravascular bulk flow of CSF or ISF may be driven by arterial pulsation. However, cerebral arterial pulsation could not be directly assessed. In the present study, we use in vivo two-photon microscopy in mice to visualize vascular wall pulsatility in penetrating intracortical arteries. We observed that unilateral ligation of the internal carotid artery significantly reduced arterial pulsatility by ∼50%, while systemic administration of the adrenergic agonist dobutamine increased pulsatility of penetrating arteries by ∼60%. When paravascular CSF–ISF exchange was evaluated in real time using in vivo two-photon and ex vivo fluorescence imaging, we observed that internal carotid artery ligation slowed the rate of paravascular CSF–ISF exchange, while dobutamine increased the rate of paravascular CSF–ISF exchange. These findings demonstrate that cerebral arterial pulsatility is a key driver of paravascular CSF influx into and through the brain parenchyma, and suggest that changes in arterial pulsatility may contribute to accumulation and deposition of toxic solutes, including amyloid β, in the aging brain.
Introduction
Anatomically, paravascular spaces (or Virchow-Robin spaces) are compartments containing interstitial fluid (ISF) or CSF that surround surface and penetrating blood vessels of the brain, and are bounded by one or more leptomeningeal layers. One key function of these paravascular spaces these paravascular spaces is to enable the exchange of ISF and CSF, which facilitates the clearance of interstitial solutes and wastes from the brain parenchyma (Cserr and Ostrach, 1974; Ichimura et al., 1991; Yamada et al., 1991; Abbott, 2004). Although this process is central to the proper maintenance of the brain's extracellular environment, the mode of this exchange is unclear.
In a series of studies, Grady and colleagues reported that subarachnoid CSF enters the brain along paravascular spaces surrounding cortical penetrating arteries, traveling along the outside of vessels to reach the basal lamina of the terminal cerebral capillary bed (Rennels et al., 1985, 1990). Ligation of the brachiocephalic artery reduced CSF tracer influx into the parenchyma (Rennels et al., 1985). Based upon these findings, we proposed that cerebral arteries provide an anatomical pathway to facilitate efficient CSF–ISF exchange in the brain, and that arterial pulsation provides the driving force for this process. In contrast, Weller and colleagues have asserted that interstitial solutes, including amyloid β (Aβ), exit the brain parenchyma along intramural basement membranes of cerebral arteries, but have likewise proposed that arterial pulsations provide the driving force for this process (Schley et al., 2006).
In four recent studies, we have confirmed and extended the findings of Grady and colleagues (Rennels et al., 1985; 1990), demonstrating by in vivo two-photon microscopy (Iliff et al., 2012; Xie et al., 2013), by ex vivo fluorescence imaging (Yang et al., 2013), and by dynamic contrast-enhanced magnetic resonance imaging (MRI; Iliff et al., 2013; Strittmatter, 2013) that a large proportion of subarachnoid CSF rapidly re-enters the brain along paravascular pathways surrounding penetrating arteries, reaching the terminal capillary beds, and exchanging with ISF throughout the brain. This paravascular CSF–ISF exchange was facilitated by astroglial water transport via the aquaporin-4 (AQP4) water channel and supports the clearance of interstitial solutes, such as soluble Aβ, from the brain parenchyma (Iliff et al., 2012). Based upon its similarity in function to the peripheral lymphatic system, and its dependence upon astroglial water flux, we termed this network the “glymphatic” system.
Prior studies evaluating the role of arterial pulsation in CSF–ISF exchange have been unable to directly observe cerebral vascular pulsatility (Rennels et al., 1985; Hadaczek et al., 2006). In the present study, we use in vivo two-photon microscopy to conduct high temporal resolution line scanning to directly measure the amount of vessel wall movement within the paravascular space with each cardiac cycle, integrating vessel wall deflection over 3 s to quantify vessel wall “pulsatility.” Using this approach, we directly evaluate paravascular CSF influx into the brain parenchyma and establish the contribution of arterial pulsatility to CSF–ISF exchange. Utilizing interventions that both increase and decrease cerebrovascular pulsatility, we provide evidence indicating that penetrating arterial pulsation is a major driver of paravascular CSF influx and subsequent CSF–ISF exchange.
Materials and Methods
Animals.
All experiments were approved by the University Committee on Animal Resources of the University of Rochester and Oregon Health & Science University and performed according to guidelines from the National Institutes of Health. Unless otherwise noted, 8–12-week-old male C57BL/6 mice (Charles River) were used in experiments. NG2-DsRed mice were used to identify arteries/arterioles versus veins/venules by endogenous fluorescence: arteries and arterioles express vascular smooth muscle DsRed; veins and venules lack vascular smooth muscle DsRed (Iliff et al., 2012).
In initial experiments, animals were anesthetized with a combination of ketamine (0.12 mg/g, i.p.) and xylazine (0.01 mg/g, i.p.). To ensure that observed effects were the result of experimental manipulations rather than cardiovascular depression resulting from ketamine/xylazine anesthesia, a second group of animals was anesthetized with isofluorane (4% induction, 1.5–3.0% maintenance) with room air supplemented with 17% O2. Arterial blood pressure, heart rate, and blood gasses were monitored via a femoral arterial catheter. Intracranial pressure (ICP) was monitored (Millar Instruments) via a small burr hole drilled over the right somatosensory cortex.
For unilateral internal carotid artery ligation, the right common, internal, and external carotid arteries were surgically isolated. The internal carotid was carefully ligated by a 6-O silk suture and the wound closed. Cerebral blood flow (CBF) was monitored by laser Doppler flowmetry (LDF), with the probe placed directly over the cranial window or the thin-skull site immediately before internal carotid artery ligation. CBF was normalized to average values acquired for 30 s before ligation.
Intracisternal tracer injection was conducted beginning 30 min after ligation, after CBF had normalized. For systemic dobutamine administration, a femoral venous catheter was inserted and dobutamine (40 μg/kg in saline) was infused over 10 min. Intracisternal tracer injection was conducted beginning 30 min after the start of dobutamine infusion. Anesthetized mice were fixed in a stereotaxic frame and a 30 ga needle was inserted into the cisterna magna. All fluorescent CSF tracers were initially constituted in artificial CSF (aCSF) at a concentration of 0.5%. This includes Alexa-647-conjugated ovalbumin [OA-647; molecular weight (MW), 45 kDa] and FITC-conjugated dextran-40 (FITC-d40, fixable; MW, 40 kDa; both from Invitrogen). In mice anesthetized with ketamine/xylazine, 10 μl of CSF tracer was injected at a rate of 2 μl/min over 5 min with a syringe pump (Harvard Apparatus). In a series of pilot studies (D.M. Zeppenfeld, J.J. Iliff, and H. Benveniste, unpublished results), we found that under isofluorane anesthesia, this injection rate resulted in the nonphysiological reflux of CSF tracer from the cisternal spaces into the fourth, third, and lateral ventricles. Thus in isofluorane-anesthetized mice, 10 μl CSF tracer injections were conducted at a rate of 1 μl/min for 10 min, which did not result in reflux into the ventricular space. In agreement with prior measurements of ICP in the mouse and rat during similar infusion protocols (Yang et al., 2013), the effect of this injection paradigm upon ICP was mild (∼2.5 mmHg) and transient, normalizing within 5 min of the cessation of tracer infusion (Fig. 1H). Given that rapid paravascular CSF influx continues long after the normalization of these shifts in ICP, the paravascular CSF fluxes observed in the present and prior studies (Iliff et al., 2012, 2013; Yang et al., 2013) appear to represent physiological fluxes and not artifacts of changes in ICP resulting from CSF tracer infusion.
Figure 1.
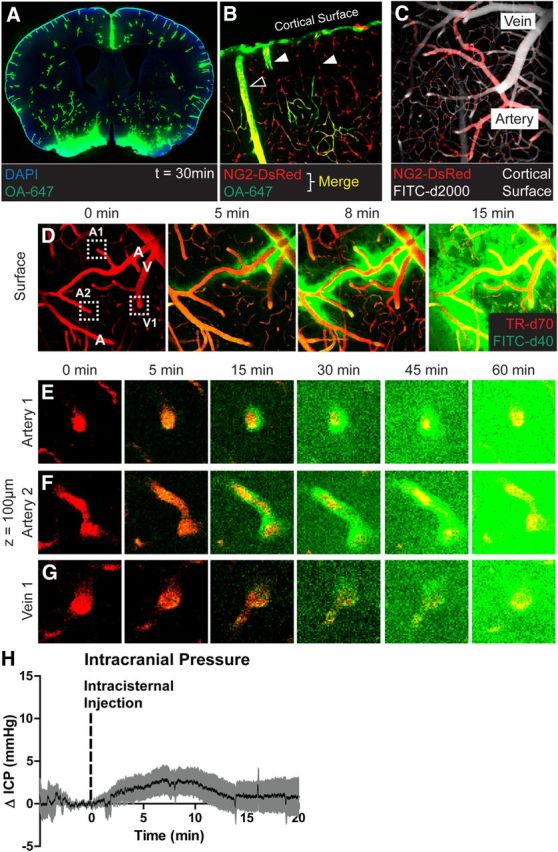
CSF enters the brain along para-arterial pathways. A, Fluorescent tracer (OA-647; MW, 45 kDa) injected intracisternally into the subarachnoid CSF enters the brain parenchyma along paravascular pathways. B, Ex vivo confocal imaging in NG2-DsRed mice showed that 15 min after injection, CSF tracer enters the brain along DsRed-positive penetrating arteries (hollow arrowheads), but not along DsRed-negative ascending veins (filled arrowheads). C, In vivo two-photon imaging in NG2-DsRed mice after fluorescence angiography (intravenous FITC-conjugated dextran-2000; MW, 2000 kDa) shows that cortical arteries and veins can be readily distinguished in these mice. D–G, Time-lapse in vivo two-photon imaging of florescent CSF tracer (FITC-d40; MW, 40 kDa) influx into the cortex after intracisternal injection. Cerebral vasculature is imaged by intra-arterial injection of Texas Red-conjugated dextran-70 (TR-d70; MW, 70 kDa). D, At the cortical surface, CSF tracer moves via para-arterial spaces. E, F, 100 μm below the cortical surface, CSF tracer enters the cortex along penetrating arteries, then exchanges with the surrounding interstitium. G, After para-arterial influx, CSF tracer is also evident along ascending veins. H, Measurement of ICP during intracisternal CSF tracer infusion (1 μl/min for 10 min) resulted in mild (∼2.5 mmHg) elevation of ICP that resolved rapidly upon cessation of infusion.
Ex vivo fluorescence imaging.
To visualize tracer movement from the subarachnoid space of the cisterna magna into the brain parenchyma, animals were perfusion fixed (4% paraformaldehyde in PBS) 30 min after intracisternal tracer injection. Vibratome slices measuring 100 μm were cut, mounted with Prolong Anti-Fade Gold with DAPI (Invitrogen), and imaged ex vivo by conventional fluorescence microscopy. Multichannel whole-slice montages were acquired with the Virtual Slice module of StereoInvestigator Software (Microbrightfield). This included separate DAPI and far-red emission channels. Exposure levels were determined based upon uninjected control slices, then maintained constant throughout the study. To quantify tracer movement into fixed slices, slice images were analyzed in ImageJ software (National Institutes of Health) as described in detail previously (Iliff et al., 2012; Yang et al., 2013). For each slice, color channels were split and whole-slice, cortical, white matter, hippocampal, and subcortical (including striatum, thalamus and hypothalamus) regions of interest (ROIs) were defined based upon the DAPI emission channel. The color channel corresponding to the tracer was background-subtracted based upon an ROI outside of the slice area and the mean ROI fluorescence intensity was calculated. Approximately 8–12 slices per animal were imaged in this manner, and mean fluorescence intensity was averaged among anterior [+0.5 − (−1.0) mm relative to bregma] and posterior [(−1.0) − (−2.5) mm relative to bregma] slices within each animal to generate a single biological replicate. The effect of internal carotid artery ligation or systemic dobutamine treatment upon tracer influx into the brain was evaluated by two-way ANOVA with Bonferroni's post hoc test to determine differences in individual regions.
In vivo two-photon laser scanning microscopy.
For initial in vivo imaging, ketamine/xylazine-anesthetized animals were intubated and artificially ventilated with room air using a small animal ventilator (CWE) at ∼100 breaths/min and tidal volume of 0.3–0.4 ml. Body temperature was kept at 37°C with a temperature-controlled warming pad. A craniotomy (3 mm in diameter) was made over the cortex 1 mm lateral and 0.5 mm posterior to bregma. The dura was left intact, covered with aCSF, and sealed with a glass coverslip. In a second series of studies, isofluorane-anesthetized animals were tracheotomized and ventilated (∼100 breaths/min; tidal volume, 0.2 ml) with room air supplemented with 22% O2. A thin-skull preparation was performed as described by Shih et al. (2012) over the same brain region to permit the visualization of the cerebral vasculature without opening the skull. The femoral artery was cannulated for mean arterial blood pressure monitoring and the measurement of arterial blood gas values. Blood gas values were maintained within physiological ranges [pH 7.35 ± 0.01 (range, 7.31–7.43); PaO2, 147.1 ± 11.2 mmHg (range, 105.8–208.0 mmHg); PaCO2, 31.2 ± 1.8 mmHg (range, 24.7–43.7 mmHg)]. To visualize the vasculature, 0.1 ml of blood–brain barrier-impermeable Texas Red-conjugated dextran-70 (MW, 70 kDa; 1% in saline; Invitrogen) was injected intra-arterially immediately before imaging. Two different systems were used for in vivo imaging: a Mai Tai laser (SpectraPhysics) attached to a confocal scanning system (Fluoview 300, Olympus) and an upright microscope (IX51W, Olympus), and a Chameleon laser (Coherent) attached to combined scan head/upright stand (LSM 7 MP, Zeiss). A 20× (0.9 numerical aperture) water-immersion lens was used to image the cortex. Excitation wavelength was 870 nm. The cerebral vasculature was first imaged with 512 × 512 pixel frames from the surface to a depth of 240 μm with 5 μm z-steps. After intracisternal injection of CSF tracer, tracer movement into the cortex was conducted with dual-channel (FITC and Texas Red) 512 × 512 pixel image acquisition (Fig. 2A). The cortex was repeatedly scanned from the surface to 240 μm below the surface with 20 μm z-steps at 1 min intervals for the duration of the experiment. Image analysis was conducted with ImageJ software (National Institutes of Health) with the UCSD (University of California, San Diego) plugin set. Following imaging, penetrating arterioles were distinguished from penetrating venules on the basis of morphology: surface arteries passing superficially to surface veins and exhibiting less branching at superficial cortical depths. Imaging planes 100 μm below the cortical surface were selected for the analysis of intracisternal tracer penetration. To define para-arterial tracer movement, a circular ROI, 25 pixels (29.75 μm) in diameter, was defined surrounding three penetrating arteries. To define tracer movement into brain tissue surrounding paravascular spaces, donut-shaped ROIs were defined that had an external diameter of 150 pixels (178.5 μm) and an internal diameter of 50 pixels (59.5 μm, thus excluding the paravascular ROI). These were centered upon the penetrating arteries. Mean pixel intensity within these ROIs was measured at each time point. Within each animal at each time point para-arterial and surrounding tissue ROIs were separately averaged to generate values for a single biological replicate. When tracer movement along penetrating arteries or into surrounding brain tissue was compared between sham and internal carotid artery ligation treatment groups, a two-way repeated-measures ANOVA was used followed by Bonferroni's post hoc test.
Figure 2.
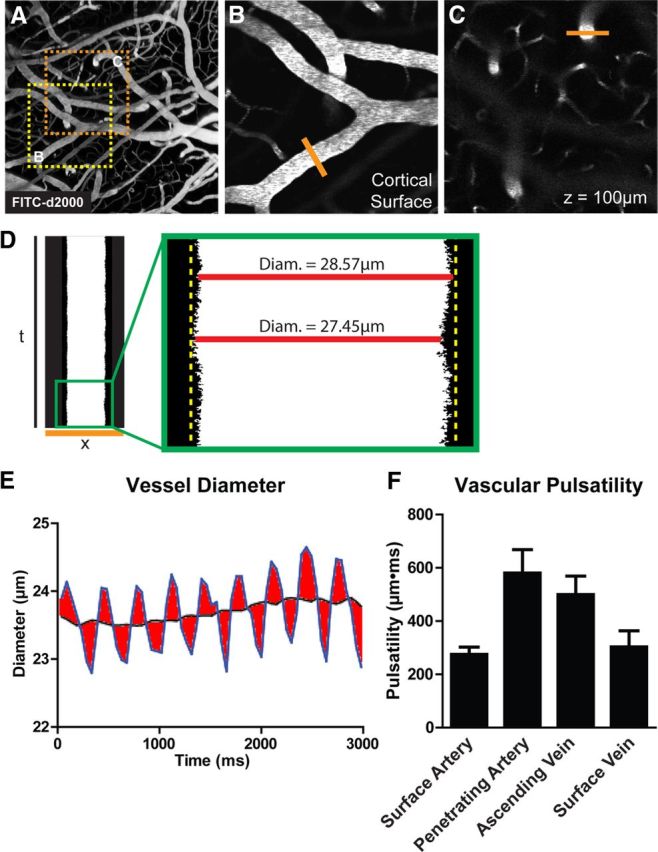
Measurement of vascular pulsatility by in vivo two-photon imaging. A, The cerebral vasculature was visualized by in vivo two-photon fluorescence angiography after intravenous injection of FITC-conjugated detran-2000 (FITC-d2000; MW, 2000 kDa). B, C, Cortical surface arteries and veins (B), and penetrating arteries and ascending veins (C) were selected and X–T line scans (orange lines) were generated orthogonal to the vessel axis. D, E, Vessel diameter was measured and plotted as a function of time. Vascular pulsatility was defined as the absolute value of the integral of the vascular diameter approximately a running average over a 3000 ms epoch. F, Vascular pulsatility was measured in cortical surface arteries (SA), penetrating arteries (PA), ascending veins (AV), and surface veins (SV). Pulsatility was greatest in penetrating arteries and veins compared with surface vessels.
To measure vessel diameters, 3000 ms X–T line scans were acquired orthogonal to the vessel axis in surface arteries, surface veins, penetrating arteries, and penetrating veins (Fig. 2B,C). Penetrating vessel line scans were acquired 50–150 μm below the cortical surface. Vessel diameter was extracted from X–T plots (Fig. 2D) and plotted versus time using custom Matlab and ImageJ software. Steady-state vessel diameters were calculated as the mean value over the 3000 ms epoch. Vessel wall pulsatility (derived units μm*ms) was calculated as the absolute value of area under the diameter–time plot, integrated about the running average over the 3000 ms epoch (Fig. 2E). During the course of developing this analysis, we normalized these values to the mean vessel diameter and calculated pulsatility upon a per-cardiac cycle basis. Neither treatment of these data substantially altered the results; so the simpler calculation of vessel wall pulsatility was used. Vessel pulsatility was calculated from 10 to 45 vessels per group taken from 6 to 8 animals per treatment (sham, internal carotid artery ligation, vehicle injection, and dobutamine injection). Differences in vessel pulsatility between different vessel types was evaluated by one-way ANOVA, with Tukey's post hoc test to evaluate differences between vessel segments. The effect of treatment on vascular pulsatility and diameter was evaluated by two-way ANOVA, with Bonferroni's post hoc test to evaluate differences among individual vascular segments. Differences in heart rate were evaluated by paired t test.
Statistics.
In all figures, data are presented as the mean ± SEM. All statistics were performed using Prism software (Graphpad). A p value <0.05 was considered significant. The statistical treatment of each dataset is detailed individually in the methodological description above.
Results
Following intracisternal injection, CSF tracers rapidly entered into the brain chiefly along paravascular pathways (Iliff et al., 2012, 2013; Xie et al., 2013). This is evident in Figure 1A, a whole-slice montage generated from a mouse fixed 30 min after intracisternal injection of OA-647. Tracer penetration was observed throughout all brain regions, but appeared greatest along the large vessels penetrating from the ventral brain surface. When CSF tracer was injected into reporter mice in which the cerebral arteries can be distinguish from veins by the expression of DsRed in smooth muscle cells (Fig. 1B,C; Iliff et al., 2012), fluorescent tracer entered the brain specifically along penetrating arteries to reach the terminal capillary beds (Fig. 1B). When intracisternal CSF tracer (FITC-d40) influx into the cortex was imaged in vivo by two-photon microscopy, tracer appeared initially along pial surface arteries (Fig. 1D). This was in agreement with recent MRI-based work that demonstrated that large surface arteries provide key routes for rapid CSF movement throughout the brain (Iliff et al., 2013). From these surface arteries, CSF tracer moved rapidly into the cortex along penetrating arteries, then exchanged with the surrounding ISF (Fig. 1E,F). Only at later time points did CSF tracer appear along ascending veins (Fig. 1G), suggesting that the primary route of CSF influx into the brain parenchyma was along paravascular spaces surrounding penetrating arteries. In prior studies, we found that rates of tracer infusion that had no effect upon ICP resulted in the same manner of paravascular CSF influx into the brain (Iliff et al., 2013; Yang et al., 2013), as observed in the present and prior studies (Iliff et al., 2012; Xie et al., 2013). In the present study, when ICP was monitored during CSF tracer infusion, changes in ICP were mild and transient (Fig. 1H), normalizing within 5 min of the cessation of infusion. Because paravascular CSF influx continues long after the normalization of ICP (Fig. 1D–F; Iliff et al., 2012, 2013; Xie et al., 2013; Yang et al., 2013), this paravascular influx of CSF tracer into the cortex appears to be physiological and does not represent an artifact of the effect of tracer infusion upon ICP.
Effect of unilateral internal carotid artery ligation on CSF–ISF exchange
Prior studies have suggested that paravascular movement of CSF (Rennels et al., 1985) and ISF (Hadaczek et al., 2006) may be driven in part by arterial pulsation. In each of these studies, the authors sought to alter cerebral arterial pulsation via surgical or pharmacological manipulation, yet the effect of these interventions upon cerebral arterial pulsation could not be directly assessed. We took advantage of the high temporal resolution of line scanning to visualize the rapid movement of the vascular wall within the paravascular space using in vivo two-photon microscopy (Fig. 2A–E) and could thereby directly assess changes in vascular pulsatility while evaluating the effect of these changes upon para-arterial CSF–ISF exchange. When vascular pulsatility was evaluated at different levels of the cerebrovascular tree, including surface arteries, penetrating arteries, ascending veins, and surface veins, the greatest vascular pulsatility was observed along penetrating arteries, followed by ascending veins (Fig. 2F). To our knowledge, this is the first time that this imaging approach has been used to directly measure differences in vascular pulsatility along the cerebrovascular tree. The fact that wall pulsatility was greater along the penetrating arteries than along surface arteries was surprising, but may reflect the respective influence of arterial blood pressure pulse wave amplitude, differences in vessel wall thickness (and thus elasticity), and investment with surrounding connective tissue along these vascular segments.
Following the approach of Rennels et al. (1985), we first attempted to reduce cerebral arterial pulsatility by unilateral ligation of the internal carotid artery. Carotid artery ligation did not affect systemic blood pressure (Fig. 3A). As expected, CBF (measured by LDF) was initially reduced following ligation, but gradually recovered to baseline values between 5 and 30 min postligation (Fig. 3B). In initial experiments, we evaluated the effect of carotid artery ligation upon cerebral vascular pulsatility via a conventional cranial window preparation, which includes a local craniotomy. Because of the risk of swelling through the craniotomy, ketamine/xylazine anesthesia was used, resulting in cardiovascular depression. In these experiments, internal carotid artery ligation significantly reduced vascular pulsatility along penetrating cortical arteries, but not along other segments of the vascular tree (Fig. 3C; **p < 0.01 sham vs ligation, two-way ANOVA). Carotid artery ligation resulted in an initial dilation of cerebral arteries that corresponded to the recovery CBF 5–10 min postligation (Fig. 3B). However, between 10 and 30 min after ligation, vessel diameter at all levels of the vascular tree normalized to baseline levels (Fig. 3D). Similarly, heart rate at 30 min post-ligation was not altered by carotid artery ligation (Fig. 3E).
Figure 3.
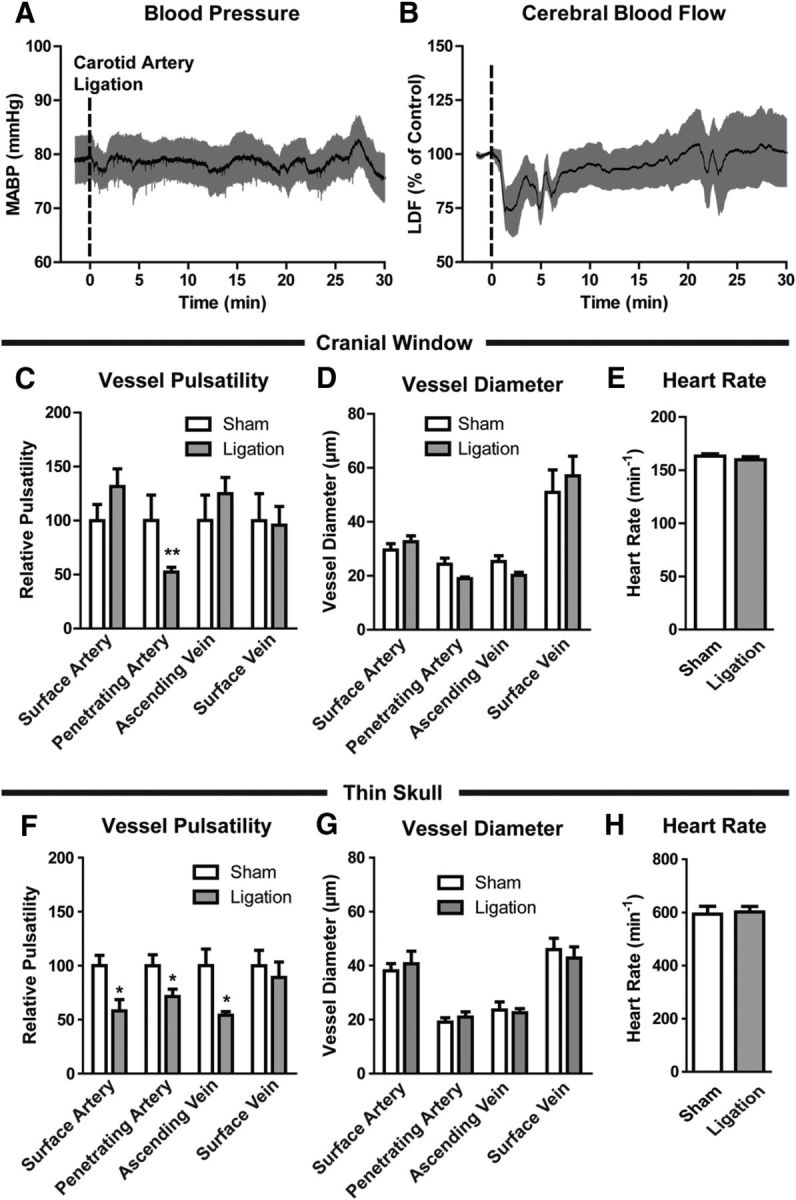
Reduced cortical arterial pulsatility after internal carotid artery ligation. A, Unilateral internal carotid artery ligation did not appreciably alter systemic blood pressure. B, Immediately following carotid artery ligation, CBF measured by LDF was reduced ∼25%, recovering to baseline values within 30 min. C, When imaged via an in vivo cranial window preparation, cerebral vessel wall pulsatility was significantly reduced in penetrating cortical arteries, but remained unchanged in surface arteries, ascending veins, and surface veins (**p < 0.01 sham vs ligation; 2-way ANOVA; n = 16–45 vessels per group). D, E, Unilateral internal carotid artery ligation did not significantly alter the diameter of cortical surface arteries, penetrating arteries, ascending veins, surface veins, or heart rate. F, When imaged by a thin-skull preparation that leaves the skull intact, internal carotid artery ligation significantly reduced cerebral arterial wall pulsatility in surface arteries, penetrating arteries, and ascending veins (*p < 0.05 sham vs ligation; 2-way ANOVA; n = 12–15 vessels per group). G, H, Carotid artery ligation did not alter cortical vascular diameter or heart rate when imaging was conducted through a thin-skull preparation.
To evaluate that potential role of craniotomy or cardiovascular depression upon these findings, we conducted a second set of experiments in which animals were anesthetized with isofluorane and cerebral arterial pulsatility was imaged through a thin-skull preparation (in which the skull is thinned to 10–20 μm, but not pierced; Shih et al., 2012). Using the thin-skull preparation, we observed that vascular pulsatility along pial surface arteries, penetrating arteries, and ascending veins were significantly reduced following internal carotid artery ligation (Fig. 3F; *p < 0.05 sham vs ligation, two-way ANOVA). As in the cranial window experiments, carotid artery ligation had no effect upon vessel diameter at any level of the vascular tree (Fig. 3G) or upon heart rate (Fig. 3H).
We next evaluated whether reducing cerebral arterial pulsatility by unilateral internal carotid artery ligation slowed paravascular CSF–ISF exchange. When CSF influx into the cortex was imaged by in vivo two-photon microscopy via a cranial window (Fig. 4A,B), CSF tracer (FITC-d40) accumulated steadily within the paravascular spaces surrounding penetrating arterioles (Fig. 4C), then moved into the surrounding parenchyma (Fig. 4D). Carotid artery ligation significantly slowed the movement of CSF tracer into and through the cortex (Fig. 4B–D; *p < 0.05 sham vs ligation, repeated measure two-way ANOVA). Interestingly, CSF tracer movement into the proximal paravascular space (Virchow-Robin space) was attenuated to a greater extent than tracer movement into the surrounding parenchyma (Fig. 4, compare C, D). This suggests that reducing arterial pulsation slowed the movement of tracer along the paravascular space to a greater degree than it slowed movement into and through the surrounding interstitial space. When CSF tracer influx was evaluated in ex vivo fixed brain slices, a similar effect was observed (Fig. 4E,F). Within both anterior (Fig. 4G) and posterior (Fig. 4H) slices, tracer intensity in ipsilateral brain structures was significantly reduced compared with sham levels (*p < 0.05, **p < 0.01, ***p < 0.001 sham vs ipsilateral, two-way ANOVA). CSF–ISF exchange within the anterior slices appeared to be less sensitive to carotid artery ligation than in the posterior slices. As both the anterior and posterior cortical regions fall largely within the blood supply of the middle cerebral artery (Dorr et al., 2007), one possible explanation for these differences is that, because posterior sections are located more distally along the middle cerebral artery, these experience greater changes in pulsatility than more proximal anterior slices. Tracer influx into the contralateral hemisphere was not significantly different than tracer influx into sham tissue (Fig. 4G,H). However, values in these regions tended to fall to a level intermediate between the sham levels and the ipsilateral values. When CSF tracer injections were conducted under isofluorane anesthesia, ex vivo tracer imaging in brain slices fixed 30 min postinjection produced similar results. Following internal carotid artery ligation, CSF influx into the ipsilateral cortex was reduced to 72.5 ± 8.1% of sham values, while tracer influx into the contralateral hemisphere was largely preserved at 97.2 ± 11.6% (*p < 0.05 ipsilateral vs contralateral ligation, two-way ANOVA). These results demonstrate that unilateral internal carotid artery ligation significantly reduces cerebral vascular pulsatility along penetrating cortical arteries concurrently with slowing paravascular CSF–ISF exchange throughout the brain.
Figure 4.
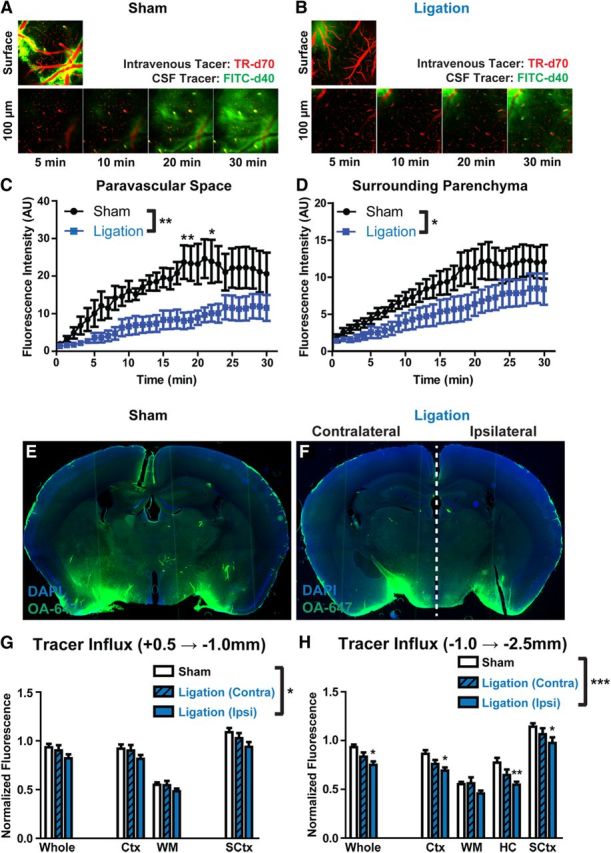
Internal carotid artery ligation reduces paravascular CSF influx. A, B, Paravascular influx of CSF tracer (FITC-d40; MW, 40 kDa) was first evaluated by in vivo two-photon microscopy at the cortical surface, 100 and 160 μm below the cortical surface in sham (A) animals and those undergoing unilateral carotid artery ligation (B). C, Quantification of paravascular CSF tracer 100 μm below the surface demonstrated that carotid artery ligation significantly slowed CSF tracer influx (*p < 0.05, **p < 0.01; 2-way repeated-measures ANOVA; n = 6 animals per group). D, A similar reduction in CSF tracer penetration into the surrounding parenchyma was observed 100 μm below the cortical surface (*p < 0.05; 2-way repeated-measures ANOVA; n = 6 animals per group). E, F, CSF tracer (OA-647; MW, 45 kDa) influx into deeper brain tissue was evaluated by ex vivo fluorescence imaging 30 min after intracisternal tracer injection. G, Quantification of fluorescence intensity in anterior [+0.5 mm − (−1.0) mm relative to bregma] brain regions showed that compared with sham-treated animals, CSF tracer influx was reduced ipsilateral to carotid artery ligation, but not contralateral (*p < 0.05, sham vs ipsilateral side; 2-way ANOVA; n = 4–6 animals per group). H, A greater reduction was evident in posterior [−1.0 mm − (−2.5) mm relative to bregma] brain regions (*p < 0.05, **p < 0.01, ***p < 0.001, sham vs ipsilateral side; 2-way ANOVA; n = 4–6 animals per group). Ctx, Cerebral cortex; WM, subcortical white matter; HC, hippocampus; SCtx, subcortical structures, including striatum, thalamus, and hypothalamus.
Effect of systemic dobutamine administration on CSF–ISF exchange
We next determined whether increasing arterial pulsatility by the systemic administration of the β1 adrenergic agonist dobutamine would correspondingly increase the rate of CSF–ISF exchange along paravascular pathways. When injected systemically (intraperitoneally), dobutamine acutely increased systolic, diastolic, and mean arterial blood pressure (Fig. 5A,B; *p < 0.05 vs baseline, two-way repeated-measures ANOVA). After peaking acutely, blood pressure normalized within 30 min of the dobutamine injection (Fig. 5A,B), the time at which CSF tracer was injected. Heart rate, however, was significantly increased 30 min after dobutamine injection (Fig. 5C; *p < 0.05 vs control, one-way repeated-measures ANOVA). When evaluated by in vivo two-photon angiography, a significant elevation in pulsatility was observed along penetrating arteries (Fig. 5D; *p < 0.05 control vs dobutamine, two-way ANOVA) 30 min after dobutamine treatment. These changes were not observed along surface arteries, ascending veins, or surface veins. At 30 min post-infusion, systemic dobutamine did not significantly alter vessel diameter anywhere along the cerebrovascular tree (Fig. 5E).
Figure 5.
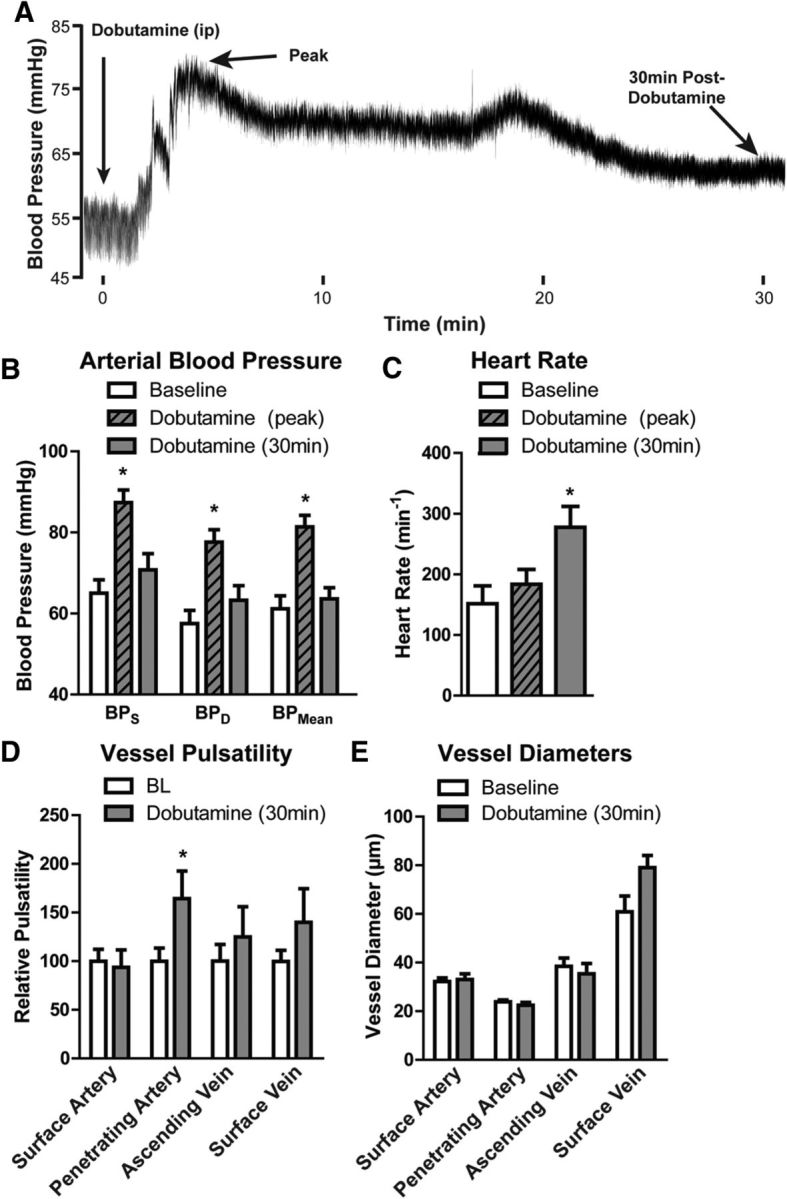
Increased arterial pulsation after systemic dobutamine administration. The effect of systemic dobutamine (40 μg/kg, i.v.) on cortical vessel pulsatility was evaluated by in vivo two-photon angiography 30 min after administration. A, Raw arterial blood pressure trace following systemic dobutamine administration shows an acute effect of the drug that peaks within 10 min of injection and largely resolves over the first 30 min postinjection (the time when intracisternal tracer injections took place). B, Systolic (BPS), diastolic (BPD), and mean (BPMean) arterial blood pressure were significantly increased immediately after systemic dobutamine injection (*p < 0.05 vs baseline values, repeated-measures 1-way ANOVA; n = 8 per group). However, this response resolved within the first 30 min postinjection. C, Heart rate was not altered during the peak blood pressure response to dobutamine. However, 30 min after dobutamine injection, heart rate was significantly elevated compared with baseline values (*p < 0.05, repeated-measures 1-way ANOVA; n = 8 per group). D, 30 min after systemic dobutamine injection, vessel pulsatility was significantly increased only within penetrating arteries but not within other vessel types (*p < 0.05, 2-way ANOVA; n = 10–42 vessels per group). E, Dobutamine did not significantly alter the diameter of surface arteries (SA), penetrating arteries (PA), ascending veins (AV), or surface veins (SV).
To test whether systemic dobutamine administration increased paravascular CSF–ISF exchange, we evaluated intracisternal fluorescent CSF tracer influx into the brain parenchyma beginning 30 min after dobutamine injection. Animals were fixed 30 min after tracer injection (1 h after the start of dobutamine injection), and fluorescent tracer influx into the brain was measured ex vivo in fixed slices. Compared with vehicle-injected controls, CSF tracer influx into the brain parenchyma was significantly increased following dobutamine injections (Fig. 6A–D; *p < 0.05 control vs dobutamine, two-way ANOVA). This effect was largely uniform throughout different brain regions, and did not appear to differ between anterior [+0.5 − (−1.0) mm relative to bregma; Fig. 6C] and posterior [(−1.0) − (−2.5) mm relative to bregma; Fig. 6D]. These results demonstrate that systemic dobutamine administration increases vascular pulsatility along penetrating cortical arteries and accelerates CSF–ISF exchange throughout the brain.
Figure 6.

Systemic dobutamine administration increases paravascular CSF tracer influx. The effect of systemic dobutamine administration upon paravascular influx of subarachnoid CSF tracer was evaluated by ex vivo fluorescence imaging 30 min after intracisternal tracer injection. A, B, Compared with sham animals (A), animals treated with dobutamine 30 min before the start of tracer infusion exhibited a marked increase in the influx of CSF tracer into brain tissue (B). C, D, Quantification of CSF tracer influx into anterior (C) and posterior (D) brain regions demonstrated that systemic dobutamine administration significantly increased CSF tracer influx in the parenchyma (*p < 0.05, 2-way ANOVA; n = 4–6 animals per group). Ctx, Cortex; WM, subcortical white matter; HC, hippocampus; SCtx, subcortical structures.
Discussion
In the present study, we take advantage of the high temporal resolution of in vivo two-photon line scanning to directly visualize cerebral arterial wall pulsatility within surrounding paravascular spaces, demonstrating that unilateral internal carotid artery ligation markedly reduces vascular wall pulsatility along cerebral arteries, whereas systemic administration of the adrenergic agonist dobutamine elevates vascular wall pulsatility along cerebral arteries. In vivo and ex vivo analysis of fluorescent CSF tracer influx into and through the brain parenchyma demonstrated that reducing cerebral arterial pulsatility slowed paravascular CSF–ISF exchange in the brain, while increasing pulsatility with dobutamine accelerated the rate of paravascular CSF influx into brain tissue. These findings demonstrate that cerebral arterial pulsation is a significant driver of paravascular CSF–ISF exchange in the brain.
It has been widely accepted that paravascular spaces provide a pathway for the clearance of interstitial solutes from the brain parenchyma (Cserr and Ostrach, 1974; Ichimura et al., 1991; Yamada et al., 1991; Abbott, 2004). Prior studies by Grady and colleagues (Rennels et al., 1985, 1990) reported that CSF enters the brain along paravascular spaces surrounding cerebral arteries and exchanges with surrounding ISF. More recent studies in fixed tissue slices from Weller, Carare, and colleagues have suggested that ISF solutes, such as Aβ, are cleared from the brain in the opposite direction along cerebral arteries, traveling outward along basement membranes between vascular smooth muscle cells within the tunica media, thus providing the basis for the deposition of Aβ along penetrating and leptomeningeal arteries in cerebral amyloid angiopathy (Weller et al., 2008; Hawkes et al., 2011). Whether these vascular and paravascular pathways represent independent routes of solute movement is unclear.
Our group has recently reported that these paravascular channels constitute a brain-wide system by which CSF exchanges with ISF to facilitate the rapid clearance of interstitial wastes, including soluble Aβ, from the brain (Iliff et al., 2012; Strittmatter, 2013). In vivo two-photon imaging demonstrated that intracisternal CSF tracer moves rapidly along paravascular spaces surrounding cortical vessels at the pial surface (Iliff et al., 2012; Xie et al., 2013). The rapid movement of CSF tracer along paravascular spaces at the brain surface compared with the rate of movement within the wider subarachnoid space (exhibited in the present study and shown by MRI in supplemental videos in Iliff et al., 2013), combined with the apparent restriction of small MW tracers (3 kDa) to these spaces (Iliff et al., 2012), suggests that these spaces represent distinct subcompartments through which CSF is moving rapidly to enter the brain parenchyma and exchange with the surrounding ISF. Whole-brain dynamic contrast-enhanced MRI imaging suggests that CSF first enters this pathway proximally at leptomeningeal paravascular spaces within the cisternal compartments (Iliff et al., 2013). ISF and interstitial solutes are cleared from the brain in large part along paravascular channels surrounding large-caliber draining veins (Iliff et al., 2012), presumably spilling solutes, such as Aβ, into the subarachnoid CSF for subsequent elimination via the choroid plexus (Ghersi-Egea et al., 1996; Fujiyoshi et al., 2011), or through bulk CSF reabsorption through arachnoid granulations or cervical lymphatics (Johanson et al., 2008).
CSF influx and interstitial solute clearance are governed in part by the properties of the neuropil interposed between paravascular influx and clearance pathways. Reducing parenchymal water conductance via genetic deletion of the perivascular astrocytic water channel AQP4 dramatically slowed paravascular CSF influx and the clearance of interstitial Aβ from the brain (Iliff et al., 2012). Sleep state-dependent changes in cortical extracellular volume similarly determined the rates of paravascular CSF influx and interstitial Aβ clearance (Xie et al., 2013).
While these factors appear to define the resistance of the parenchyma to ISF and solute flux, the mechanical drivers of paravascular CSF–ISF exchange remain unclear. Both ISF and CSF movement along vascular and paravascular pathways have been proposed to be driven by arterial pulsation. In their initial description of the para-arterial movement of CSF tracer into the brain, Rennels et al. (1985) reported that when cranial arterial pulse pressure was experimentally reduced in the dog by ligating the brachiocephalic artery, the paravascular movement of CSF tracer into the brain was slowed. In a more recent study focusing upon convective bulk flow-mediated movement of different sized particles through the brain interstitium, interventions that increased blood pressure and heart rate (intravenous epinephrine) increased the interstitial movement of albumin, liposomes, or virions, while those that reduced blood pressure and heart rate (hypovolemia) slowed the interstitial movement of albumin, liposomes, or virions (Hadaczek et al., 2006). Additionally, a recent modeling study defined a mechanism for pulsation-driven fluid transport along paravascular spaces (Wang and Olbricht, 2011). Weller and colleagues have similarly published a biophysical model proposing arterial wall pulsation as a key driving force for solute efflux along the arterial wall in the opposite direction of pulse wave propagation (Schley et al., 2006). Each of these studies, however, was limited by its inability to directly measure cerebral arterial wall pulsatility. In the present study, in vivo two-photon line scanning was used to directly measure for the first time vessel wall pulsatility at multiple levels of the cerebrovascular tree, enabling us to determine whether the same interventions that increase or decrease penetrating cortical arterial pulsatility have a corresponding effect on paravascular CSF influx into the brain parenchyma. This novel approach has potential application beyond the present study, such as in defining the contributions that changes in arterial wall pulsatility make to the failure of Aβ or the movement of cells through the paravascular spaces.
The present results demonstrate that cerebral arterial pulsatility is an important driver of paravascular CSF–ISF exchange. The observation that penetrating cerebral arteries exhibit the greatest pulsatility as well as the greatest sensitivity to internal carotid artery occlusion and systemic dobutamine treatment suggests that, at least among the local cerebral vessels surveyed (including surface arteries, surface veins, and ascending veins), pulsatile motion within this population makes the greatest contribution to paravascular CSF–ISF exchange. Because rapid paravascular CSF movement occurs along leptomeningeal paravascular spaces quite proximal to penetrating dorsal cortical arteries evaluated in the present study (Iliff et al., 2013), we cannot rule out the possibility that arterial pulsation within these proximal compartments, which are also presumably altered by internal carotid artery ligation and systemic dobutamine administration, provides the driving force to propel paravascular CSF flux along the entire cerebrovascular tree.
Internal or common carotid artery ligations or stenoses are commonly used experimental models of cerebral hypoperfusion and vascular dementia in rodents (Jiwa et al., 2010), resulting in accelerated parenchymal amyloid plaque deposition, cerebral amyloid angiopathy progression, and cognitive decline in mouse models of Alzheimer's disease (Kitaguchi et al., 2009; Okamoto et al., 2012; Pimentel-Coelho et al., 2013). Although traditionally considered clinically distinct from cerebral vascular cognitive impairment, Alzheimer's disease is increasingly regarded as a pathological process that is strongly influenced by cerebrovascular dysfunction, connected through cerebral hypoperfusion, blood–brain barrier compromise, inflammatory cell infiltration, and hemorrhagic susceptibility (Roher et al., 2003; Beach et al., 2007; Brundel et al., 2012; Yarchoan et al., 2012). The observation that cerebral arterial pulsatility contributes to paravascular CSF–ISF exchange suggests the possibility that age-related and cerebrovascular disease-related reductions in arterial pulsatility may additionally contribute to failure of the clearance of interstitial waste, including Aβ, from the aged brain (Weller et al., 2008).
Cerebral arteries from aged patients exhibit reduced mechanical compliance. In a study measuring elastin functionality and compliance of human posterior cerebral arteries, Fonck et al. (2009) reported that arteries from aged patients exhibited less elastin expression and function than arteries from middle-aged patients, in addition to a loss of distensibility in response to changes in pressure. In cerebral amyloid angiopathy, Aβ is deposited within the walls of leptomeningeal and cerebral arteries, leading to alterations in the composition of the basement membrane and the eventual destruction of vascular smooth muscles within the tunica media (Weller et al., 2009). In both cases, such alterations in the arterial wall may slow paravascular CSF–ISF exchange. The interplay between these two influences—loss of arterial pulsatility reducing solute clearance, and the potential effect of paravascular solute deposition (such as Aβ) upon cerebral arterial pulsatility—may constitute a feedforward pathogenic cycle driving neurodegeneration. This suggests that treatments that target cerebrovascular compliance may provide a therapeutic opportunity against neurodegenerative processes associated particularly with paravascular deposition of protein or peptide aggregates.
Footnotes
This work was supported by the National Institutes of Health (J.J.I., M.N.), the United States Department of Defense (M.N.), the Harold and Leila Y. Mathers Charitable Foundation (M.N.), and the American Heart Association (J.J.I.).
The authors declare no competing financial interests.
References
- Abbott NJ. Evidence for bulk flow of brain interstitial fluid: significance for physiology and pathology. Neurochem Int. 2004;45:545–552. doi: 10.1016/j.neuint.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Beach TG, Wilson JR, Sue LI, Newell A, Poston M, Cisneros R, Pandya Y, Esh C, Connor DJ, Sabbagh M, Walker DG, Roher AE. Circle of Willis atherosclerosis: association with Alzheimer's disease, neuritic plaques and neurofibrillary tangles. Acta Neuropathol. 2007;113:13–21. doi: 10.1007/s00401-006-0136-y. [DOI] [PubMed] [Google Scholar]
- Brundel M, de Bresser J, van Dillen JJ, Kappelle LJ, Biessels GJ. Cerebral microinfarcts: a systematic review of neuropathological studies. J Cereb Blood Flow Metab. 2012;32:425–436. doi: 10.1038/jcbfm.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cserr HF, Ostrach LH. Bulk flow of interstitial fluid after intracranial injection of blue dextran 2000. Exp Neurol. 1974;45:50–60. doi: 10.1016/0014-4886(74)90099-5. [DOI] [PubMed] [Google Scholar]
- Dorr A, Sled JG, Kabani N. Three-dimensional cerebral vasculature of the CBA mouse brain: a magnetic resonance imaging and micro computed tomography study. Neuroimage. 2007;35:1409–1423. doi: 10.1016/j.neuroimage.2006.12.040. [DOI] [PubMed] [Google Scholar]
- Fonck E, Feigl GG, Fasel J, Sage D, Unser M, Rüfenacht DA, Stergiopulos N. Effect of aging on elastin functionality in human cerebral arteries. Stroke. 2009;40:2552–2556. doi: 10.1161/STROKEAHA.108.528091. [DOI] [PubMed] [Google Scholar]
- Fujiyoshi M, Tachikawa M, Ohtsuki S, Ito S, Uchida Y, Akanuma S, Kamiie J, Hashimoto T, Hosoya K, Iwatsubo T, Terasaki T. Amyloid-beta peptide(1–40) elimination from cerebrospinal fluid involves low-density lipoprotein receptor-related protein 1 at the blood-cerebrospinal fluid barrier. J Neurochem. 2011;118:407–415. doi: 10.1111/j.1471-4159.2011.07311.x. [DOI] [PubMed] [Google Scholar]
- Ghersi-Egea JF, Gorevic PD, Ghiso J, Frangione B, Patlak CS, Fenstermacher JD. Fate of cerebrospinal fluid-borne amyloid beta-peptide: rapid clearance into blood and appreciable accumulation by cerebral arteries. J Neurochem. 1996;67:880–883. doi: 10.1046/j.1471-4159.1996.67020880.x. [DOI] [PubMed] [Google Scholar]
- Hadaczek P, Yamashita Y, Mirek H, Tamas L, Bohn MC, Noble C, Park JW, Bankiewicz K. The “perivascular pump” driven by arterial pulsation is a powerful mechanism for the distribution of therapeutic molecules within the brain. Mol Ther. 2006;14:69–78. doi: 10.1016/j.ymthe.2006.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes CA, Härtig W, Kacza J, Schliebs R, Weller RO, Nicoll JA, Carare RO. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol. 2011;121:431–443. doi: 10.1007/s00401-011-0801-7. [DOI] [PubMed] [Google Scholar]
- Ichimura T, Fraser PA, Cserr HF. Distribution of extracellular tracers in perivascular spaces of the rat brain. Brain Res. 1991;545:103–113. doi: 10.1016/0006-8993(91)91275-6. [DOI] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med. 2012;4:147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Lee H, Yu M, Feng T, Logan J, Nedergaard M, Benveniste H. Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J Clin Invest. 2013;123:1299–1309. doi: 10.1172/JCI67677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiwa NS, Garrard P, Hainsworth AH. Experimental models of vascular dementia and vascular cognitive impairment: a systematic review. J Neurochem. 2010;115:814–828. doi: 10.1111/j.1471-4159.2010.06958.x. [DOI] [PubMed] [Google Scholar]
- Johanson CE, Duncan JA, 3rd, Klinge PM, Brinker T, Stopa EG, Silverberg GD. Multiplicity of cerebrospinal fluid functions: new challenges in health and disease. Cerebrospinal Fluid Res. 2008;5:10. doi: 10.1186/1743-8454-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitaguchi H, Tomimoto H, Ihara M, Shibata M, Uemura K, Kalaria RN, Kihara T, Asada-Utsugi M, Kinoshita A, Takahashi R. Chronic cerebral hypoperfusion accelerates amyloid beta deposition in APPSwInd transgenic mice. Brain Res. 2009;1294:202–210. doi: 10.1016/j.brainres.2009.07.078. [DOI] [PubMed] [Google Scholar]
- Okamoto Y, Yamamoto T, Kalaria RN, Senzaki H, Maki T, Hase Y, Kitamura A, Washida K, Yamada M, Ito H, Tomimoto H, Takahashi R, Ihara M. Cerebral hypoperfusion accelerates cerebral amyloid angiopathy and promotes cortical microinfarcts. Acta Neuropathol. 2012;123:381–394. doi: 10.1007/s00401-011-0925-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimentel-Coelho PM, Michaud JP, Rivest S. Effects of mild chronic cerebral hypoperfusion and early amyloid pathology on spatial learning and the cellular innate immune response in mice. Neurobiol Aging. 2013;34:679–693. doi: 10.1016/j.neurobiolaging.2012.06.025. [DOI] [PubMed] [Google Scholar]
- Rennels ML, Gregory TF, Blaumanis OR, Fujimoto K, Grady PA. Evidence for a ‘paravascular’ fluid circulation in the mammalian central nervous system, provided by the rapid distribution of tracer protein throughout the brain from the subarachnoid space. Brain Res. 1985;326:47–63. doi: 10.1016/0006-8993(85)91383-6. [DOI] [PubMed] [Google Scholar]
- Rennels ML, Blaumanis OR, Grady PA. Rapid solute transport throughout the brain via paravascular fluid pathways. Adv Neurol. 1990;52:431–439. [PubMed] [Google Scholar]
- Roher AE, Esh C, Kokjohn TA, Kalback W, Luehrs DC, Seward JD, Sue LI, Beach TG. Circle of Willis atherosclerosis is a risk factor for sporadic Alzheimer's disease. Arterioscler Thromb Vasc Biol. 2003;23:2055–2062. doi: 10.1161/01.ATV.0000095973.42032.44. [DOI] [PubMed] [Google Scholar]
- Schley D, Carare-Nnadi R, Please CP, Perry VH, Weller RO. Mechanisms to explain the reverse perivascular transport of solutes out of the brain. J Theor Biol. 2006;238:962–974. doi: 10.1016/j.jtbi.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Shih AY, Mateo C, Drew PJ, Tsai PS, Kleinfeld D. A polished and reinforced thinned-skull window for long-term imaging of the mouse brain. J Vis Exp. 2012;pii:3742. doi: 10.3791/3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ. Bathing the brain. J Clin Invest. 2013;123:1013–1015. doi: 10.1172/JCI68241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Olbricht WL. Fluid mechanics in the perivascular space. J Theor Biol. 2011;274:52–57. doi: 10.1016/j.jtbi.2011.01.014. [DOI] [PubMed] [Google Scholar]
- Weller RO, Subash M, Preston SD, Mazanti I, Carare RO. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathol. 2008;18:253–266. doi: 10.1111/j.1750-3639.2008.00133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller RO, Preston SD, Subash M, Carare RO. Cerebral amyloid angiopathy in the aetiology and immunotherapy of Alzheimer disease. Alzheimers Res Ther. 2009;1:6. doi: 10.1186/alzrt6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Meenakshisundaram T, O'Donnell J, Christensen DJ, Nicholson C, Iliff JJ, Takano T, Deane R, Nedergaard M. Sleep drives interstitial metabolite clearance from the adult brain. Science. 2013;342:373–377. doi: 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada S, DePasquale M, Patlak CS, Cserr HF. Albumin outflow into deep cervical lymph from different regions of rabbit brain. Am J Physiol. 1991;261:H1197–H1204. doi: 10.1152/ajpheart.1991.261.4.H1197. [DOI] [PubMed] [Google Scholar]
- Yang L, Kress BT, Weber HJ, Thiyagarajan M, Wang B, Deane R, Benveniste H, Iliff JJ, Nedergaard M. Evaluating glymphatic pathway function utilizing clinically relevant intrathecal infusion of CSF tracer. J Transl Med. 2013;11:107. doi: 10.1186/1479-5876-11-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarchoan M, Xie SX, Kling MA, Toledo JB, Wolk DA, Lee EB, Van Deerlin V, Lee VM, Trojanowski JQ, Arnold SE. Cerebrovascular atherosclerosis correlates with Alzheimer pathology in neurodegenerative dementias. Brain. 2012;135:3749–3756. doi: 10.1093/brain/aws271. [DOI] [PMC free article] [PubMed] [Google Scholar]