

Abstract
Familial Parkinson disease is associated with mutations in α-synuclein (α-syn), a presynaptic protein that has been localized not only to the cytosol, but also to mitochondria. We report here that wild-type α-syn from cell lines, and brain tissue from humans and mice, is present not in mitochondria but rather in mitochondria-associated endoplasmic reticulum (ER) membranes (MAM), a structurally and functionally distinct subdomain of the ER. Remarkably, we found that pathogenic point mutations in human α-syn result in its reduced association with MAM, coincident with a lower degree of apposition of ER with mitochondria, a decrease in MAM function, and an increase in mitochondrial fragmentation compared with wild-type. Although overexpression of wild-type α-syn in mutant α-syn-expressing cells reverted the fragmentation phenotype, neither overexpression of the mitochondrial fusion/MAM-tethering protein MFN2 nor inhibition/ablation of the mitochondrial fission protein DRP1 was able to do so, implying that α-syn operates downstream of the mitochondrial fusion/fission machinery. These novel results indicate that wild-type α-syn localizes to the MAM and modulates mitochondrial morphology, and that these behaviors are impaired by pathogenic mutations in α-syn. We believe that our results have far-reaching implications for both our understanding of α-syn biology and the treatment of synucleinopathies.
Introduction
Parkinson disease (PD) is characterized morphologically by the presence of intraneuronal inclusions, called Lewy bodies, consisting mainly of aggregates of α-synuclein (α-syn; Klein and Westenberger, 2012). Most cases of PD are sporadic, but ∼10% are familial, including dominant mutations in SNCA, which encodes α-syn (A53T, A30P, and E46K, and SNCA gene duplications and triplications).
In addition to its localization in the cytosol (Auluck et al., 2010), both the wild-type (WT) and mutated forms of α-syn interact with lipid membranes (Auluck et al., 2010). This binding can be detected only at high lipid–protein ratios, suggesting that α-syn interacts more efficiently with lipid raft-like domains (Fortin et al., 2004). These are specialized membrane subregions that are enriched in cholesterol and sphingolipids, conferring upon them the characteristic of being detergent-resistant membranes (DRMs). Although traditionally considered to be located only at the plasma membrane, recent work has shown the existence of intracellular lipid rafts, with a protein composition different from those located at the plasma membrane (Hayashi and Fujimoto, 2010). It has been suggested that the binding of α-syn to these lipid-rich domains determines its subcellular localization (Fortin et al., 2004).
Consistent with this view, α-syn has been reported to localize at or in mitochondria (Li et al., 2007; Cole et al., 2008, Devi et al., 2008; Parihar et al., 2008; Shavali et al., 2008). Indeed, the binding of α-syn to artificial membranes requires acidic phospholipids and cardiolipin, a mitochondrion-specific lipid. The localization of α-syn to mitochondria is also consistent with data showing altered mitochondrial function and dynamics both in cultured cells and in transgenic mice overexpressing WT and mutant forms of α-syn, similar to what has been seen in both sporadic and familial PD patients (Hsu et al., 2000). These alterations include complex I deficiency, increased oxidative stress, lipid abnormalities, and increased mitochondrial fragmentation (Schon and Przedborski, 2011). The regulation of mitochondrial dynamics (e.g., fission, fusion) is essential for maintaining cellular homeostasis (Schon and Przedborski, 2011).
Mitochondria are connected to the endoplasmic reticulum (ER) via mitochondria-associated ER membranes (MAM; Hayashi et al., 2009). MAM is a subregion of the ER with a unique lipid composition, enriched in cholesterol and anionic phospholipids, with the characteristics of a lipid raft (Hayashi and Fujimoto, 2010). MAM is involved in a number of key metabolic functions, including phospholipid and cholesterol metabolism (Hayashi et al., 2009). MAM is also enriched in proteins related to the control of mitochondrial division (Friedman et al., 2011) and dynamics (e.g., DRP1 and MFN2; Schon and Area-Gomez, 2013). Defects in MAM-localized proteins and/or disturbances in MAM function play a role in neurodegenerative diseases, including Alzheimer disease (Area-Gomez et al., 2012), and perhaps PD as well (Schon and Przedborski, 2011; Ottolini et al., 2013). Notably, α-syn influences the transfer of calcium between ER and mitochondria (Calì et al., 2012), a key MAM function (Hayashi et al., 2009).
We show here that α-syn, apart from its cytosolic localization, is present in MAM. We also show that cells containing pathogenic point mutations in α-syn have an altered distribution of this protein between the cytosol and MAM, which is associated with a decrease in MAM activity and ER–mitochondria apposition, along with an increase in mitochondrial fragmentation. The localization of α-syn at the ER–mitochondrial interface likely explains previous reports showing that α-syn is associated with mitochondria, and could also help explain the mitochondrial abnormalities seen in this form of PD. We believe that the presence of α-syn in MAM and its role in this compartment will help increase our understanding of α-syn function and its toxic effects in PD.
Materials and Methods
Cell lines, reagents, and tissues.
HeLa cells were obtained from ATCC. The BE(2)-M17 neuroblastoma cell line was a gift from Erwan Bezard (Université de Bordeaux). Drp1- and Mfn2-knock-out mouse embryonic fibroblasts (MEFs), as well as the relevant control MEFs, were kind gifts from David Chan (California Institute of Technology). For Western blotting and immunocytochemistry, we used antibodies recognizing α-syn (Cell Signaling Technology; no. 2628), DRP1 (BD Transduction Laboratories; no. 611113), ERLIN-2 (Cell Signaling Technology; no. 2959), HA tag (Sigma-Aldrich; no. H3663), protein kinase C (Sigma-Aldrich; no. P5704), OPA1 (BD Biosciences; no. 612606), and the α-subunit of mitochondrial ATP synthase (complex V, Invitrogen; no. 459240).
Human brain samples (3 males, 5 females) were obtained from the National Disease Research Interchange (NDRI). All of the samples had a postmortem interval of <8 h. Transgenic mice, expressing human WT (stock no. 010710; FVB), A53T (stock no. 004479; B6/C3), A30P α-syn (stock no. 008134; B6/SJL), and nontransgenic control mice, matched to their respective strain and gender, were obtained from The Jackson Laboratory; all mice (n = 4 per group) were males of age ∼13 weeks at the time of analysis.
DNA constructs.
α-Syn with an HA tag at the C-terminus was generated by PCR amplification of the human WT α-syn sequence (in uppercase letters; lowercase indicate noncoding region and restriction enzymes sites) using the forward primer: 5′-cgcgcggtcgacATGGATGTATTCATGAAAGGAC-3′ and the reverse primer: 5′-cgcgcgctcgagAGGCTTCAGGTTCGTAGTCTT-3′, and subcloned into the pCMV-HA-C (Clontech; no. 635690). cDNA constructs encoding mutant α-syn A53T-HA and α-syn A30P-HA were introduced in the previously cloned α-syn WT-HA construct by site-directed mutagenesis (Stratagene). Constructs were confirmed by DNA sequencing.
Other expression plasmids used: pCAS-α-syn-GFP (a kind gift from Dr Oren Levy, Columbia University), pDsRed2-Mito (Clontech; no. 632421), and GFP-Sec61-β (Addgene; no. 15108). All constructs were transfected at a 1:1 ratio using Lipofectamine 2000 (Invitrogen; no. 11668-027) in serum-free DMEM, and cell analyzed 24 h later.
Analysis of mitochondrial morphology.
For visualization of mitochondria, cells were transfected with pDsRed2-Mito, fixed 24 h later for 20 min in 4% paraformaldehyde and permeabilized in methanol at 4°C for 10 min. Coverslips were mounted with Fluoromount G (Southern Biotech; no. 0100-01) for analysis. Single-plane images were taken in a Leica SP5 confocal inverted microscope with a 63×/1.4 oil objective. Cells that presented a filamentous mitochondrial network were classified as normal and cells with a disrupted mitochondrial network were classified as fragmented. Data are mean values of five independent experiments and ∼150 cells/genotype. The same cells were used to measure mitochondrial length; analyses were performed as described previously (Magrané et al., 2009).
To assess the DRP1 by immunocytochemistry, M17 cells were transfected with pDsRed2-Mito to visualize mitochondria and with DRP1 antibody stained with Alexa 488 (Invitrogen). The same cells were treated with 50 μM mdivi-1 (Sigma #M0199) for 14 hours. The experiment was repeated three times. Representative images of each genotype were taken. To investigate the effects of DRP1 on mitochondrial morphology, we cotransfected Drp1-WT and -KO MEFs with pDsRed2-Mito and α-syn-HA constructs. Cells were classified as normal or fragmented, as above. Data are reported as mean values from ∼100 cells/genotype, from three independent experiments.
To analyze the effect of the expression of WT α-syn in M17 cells stably expressing α-syn mutations, we cotransfected the cells with a plasmid expressing WT α-syn-GFP and with pDsRed2-Mito. Mitochondrial fragmentation was assessed and quantified 24 h post-transfection as described above.
Mitochondrial importation of α-synuclein.
Mitochondria were isolated from cultured HeLa cells essentially as described previously (Becker et al., 2012). In brief, harvested cells were washed twice in PBS and resuspended in HMS-A buffer (220 mm mannitol, 70 mm sucrose, 20 mm HEPES, pH 7.6, 1 mm EDTA, 0.2% BSA, 1 mm PMSF). Cells were homogenized using a glass/Teflon homogenizer and homogenates were subjected to differential centrifugation. The mitochondrial fraction was washed in HMS-B buffer (HMS-A lacking BSA) and resuspended in HS buffer (20 mm HEPES, pH 7.6, 250 mm sucrose, 5 mm Mg acetate, 80 mm K acetate, 7.5 mm glutamate, 5 mm malate, 1 mm DTT, 0.2% BSA).
In vitro import into HeLa cell mitochondria was performed essentially as described previously (Becker et al., 2012). Radiolabeled proteins were synthesized by in vitro transcription and translation in the presence of [35S]-methionine/cysteine using the TNT-coupled reticulocyte lysate system (Promega). WT and mutant α-syn transcription template DNA was amplified from the HA-tagged plasmids by PCR, introducing the SP6 promoter and Kozak sequences via the forward (5′-gaattcatttaggtgacactatagaatacgccgccaccATGGATGTATTCATGAAAGGAC-3′) and reverse (5′-tcatcatcattaGGCTTCAGGTTCGTAGT-3′) primers. For an import reaction, 40 μg of mitochondria and radiolabeled proteins were diluted in 100 μl HS buffer containing 2 mm ATP. In control reactions, the membrane potential (Δψ) was dissipated by addition of 8 μm antimycin A, 0.5 μm valinomycin, and 2 μm oligomycin before import. Mock reactions contained radiolabeled proteins diluted in buffer but no mitochondria. Nonimported/protease-accessible proteins were digested by incubation with 100 μg/ml trypsin for 30 min on ice. Mitochondria were pelleted (12 min, 12,000 × g, 4°C), washed in HS buffer, and samples were analyzed by Tricine-SDS-PAGE and digital autoradiography.
Subcellular fractionation and Western blotting.
Purification of ER, MAM, and mitochondria, and of DRMs present in the crude mitochondrial fraction from HeLa cells transfected with α-syn-HA constructs and M17 cells stably expressing α-syn were performed as described previously (Area-Gomez et al., 2009, 2012). The samples (30 μg loaded in each lane) were electrophoresed through 4–20% Tris-glycine gels, transferred to 0.45 μm nitrocellulose membranes, and detected by immunoblotting with appropriate antibodies.
We evaluated OPA1 levels and cleavage in crude mitochondria isolated from the various M17 cell lines. Thirty micrograms of each sample were electrophoresed through 6% Tris-acetate gels, transferred to 0.45 μm nitrocellulose membranes, and detected by immunoblotting with the indicated antibodies.
Analysis of ER–mitochondrial apposition.
HeLa cells and Mfn2-WT and -KO MEFs transfected with α-syn-HA constructs, and M17 cells stably expressing α-syn, were cotransfected with GFP-Sec61-β (Addgene; plasmid no. 15108) and DsRed-Mito (Clontech; no. 632421) and fixed 24 h later. Single-plane images were acquired and interactions between mitochondria and ER were calculated using MetaMorph Imaging Series 7.1 image analysis software (Molecular Devices) determining the colocalization index using Mander's coefficient (de Brito and Scorrano, 2008).
Analysis of phospholipid synthesis in cultured cells.
Cells were incubated for 2 h in serum-free medium to ensure removal of exogenous lipids. The medium was then replaced with MEM containing 2.5 μCi/ ml of 3H-serine (3H-Ser) for the indicated periods of time. The cells were washed and collected in PBS, pelleted at 2500 × g for 5 min at 4°C, and resuspended in 0.5 ml water, removing a small aliquot for protein quantification. Lipid extraction was done by the Folch method. Briefly, three volumes of chloroform–methanol 2:1 were added to the samples and vortexed. After centrifugation at 8000 × g for 5 min, the organic phase was washed twice with two volumes of methanol/water 1:1, and the organic phase was blown to dryness under nitrogen. Dried lipids were resuspended in 60 μl of chloroform–methanol 2:1 and applied to a TLC plate. Phospholipids were separated using two solvents composed of petroleum ether/diethyl ether/acetic acid 84/15/1 v/v and chloroform/methanol/acetic acid/water 60/50/1/4 v/v. Development was performed by exposure of the plate to iodine vapor. The spots corresponding to the relevant phospholipids (identified using comigrating standards) were scraped and counted in a scintillation counter (Packard Tri-Carb 2900TR).
Statistical analyses.
All values are expressed as the mean ± SEM. Differences among means were analyzed using one- or two-way ANOVA, and when it showed significant differences, pair-wise comparisons between means were tested by Newman–Keuls post hoc testing. In all analyses, the null hypothesis was rejected at the 0.05 level.
Results
α-Syn is localized in MAM
It is well known that α-syn is located predominantly in the cytosol (Fortin et al., 2004). However, several groups have reported the presence of α-syn at or in mitochondria (Li et al., 2007; Cole et al., 2008; Devi et al., 2008; Parihar et al., 2008, Shavali et al., 2008), without any agreement among them regarding its precise suborganellar localization. Furthermore, none of the prediction algorithms, such as MitoProt, recognizes α-syn as a mitochondrially targeted protein. For this reason, we revisited this issue, starting with a standard in vitro import assay of [35S-Met/Cys]-labeled WT and mutant (A30P and A53T) α-syn into HeLa cell mitochondria, compared with that of a control matrix protein, malate dehydrogenase 2 (Mdh2). As expected, Mdh2 behaved like a classic mitochondrial imported protein, in that its importation increased steadily over time, was dependent upon mitochondrial membrane potential (Δψm), and the mature polypeptide was resistant to trypsin treatment (Fig. 1A). In contrast, both WT and mutant α-syn behaved in an anomalous manner. First, their apparent association with mitochondria did not increase over time, even in the presence of trypsin, but was still dependent upon Δψm (Fig. 1A). Second, we did not observe any evidence of a mitochondrially generated cleaved product, as the same ∼17 kDa protein was detected under all experimental conditions, including the mock control (lacking mitochondria). Third, trypsin treatment reduced the mitochondrial signal of α-syn, but only partially (Fig. 1A). Together, these results imply that both WT and mutant α-syn do not behave as canonical mitochondrially targeted proteins.
Figure 1.

Subcellular localization of α-synuclein. A, In vitro mitochondrial import assays. Import of [35S]Met/Cys-labeled α-syn and a control protein (Mdh2) into HeLa cell mitochondria for the indicated times. p, Precursor protein; m, mature processed protein. Imported proteins were separated by SDS-PAGE. Note that depletion of Δψ affected only the import of Mdh2. B, Western blot to detect the indicated proteins in standard subcellular fractionation of M17 and HeLa cells expressing WT α-syn and in normal human and mouse brain. The CM fraction was purified further into MAM and mitochondrial fractions. ATPase-α (complex V) was used as a mitochondrial marker, protein kinase C as a cytoplasmic marker, and ERLIN-2 as a MAM marker.
In light of this unusual behavior, we examined further the question of α-syn localization by subcellular fractionation. As expected, we detected α-syn in both the cytosolic and crude mitochondrial (CM) fractions from human dopaminergic neuroblastoma-derived M17 cells, from HeLa cells transfected transiently with WT α-syn, and from normal human and mouse brain (Fig. 1B). We then subfractionated the CM further, because apart from “pure” mitochondria, it is well known that CM also contains MAM (Area-Gomez et al., 2009). Western blot analysis showed that α-syn was clearly present in the MAM fraction, but not in mitochondria, from all cells and tissues analyzed (Fig. 1B). Together, these results indicate that WT α-syn is targeted not to pure mitochondria, but to MAM. We believe that the use of CM preparations and the proximity of MAM to mitochondria in this fraction is the reason why α-syn has been considered erroneously to be a mitochondrial protein.
Mutations in α-syn affect its localization to MAM
Previous results have shown that pathogenic mutations of α-syn affect its binding to lipid membranes (Auluck et al., 2010). For this reason, we asked whether pathogenic mutations in α-syn affect their MAM localization, using Western blot analysis of subcellular fractions from M17 cells stably expressing WT or mutant (A53T and A30P) α-syn (Fig. 2A). First, we noted a decrease in the amount of α-syn in the CM fraction in both α-syn mutants compared with WT (Fig. 2A). In the case of A30P, α-syn was expressed at levels similar to that in WT, but its relative distribution was shifted toward the cytoplasm (Fig. 2A). With respect to A53T, its distribution between cytosol and CM was similar to that of WT, but there was >60% lower expression of A53T compared with WT α-syn (Fig. 3A). Notably, this reduction in α-syn in the CM also occurred when we overexpressed WT and mutant forms of the protein in HeLa cells (Fig. 3B).
Figure 2.
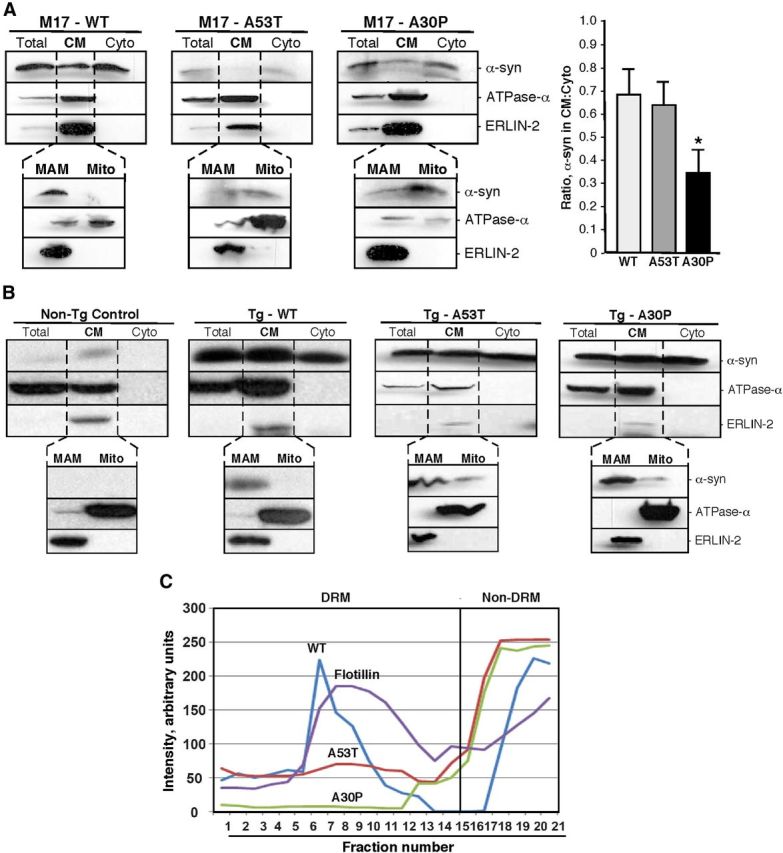
Localization of α-syn in M17 cells and transgenic mice. A, B, Western blots to detect the indicated proteins in standard subcellular fractionation of M17 cells stably expressing the indicated α-syn proteins (A; quantitation of the ratio of α-syn in the CM:Cyto fractions at right) and in transgenic mice expressing the indicated α-syn species (B). C, Localization of α-syn species in DRM and non-DRM fractions. CM (containing mitochondria and MAM) from transgenic mouse brain expressing WT and mutant α-syn were treated with Triton X-100 and then loaded onto a continuous 5–35% sucrose gradient, and the indicated fractions were collected and subjected to Western blotting to detect and quantitate α-syn (see Materials and Methods). Note that α-syn from WT and A53T, but not A30P, mice comigrate with flotillin, a marker of DRMs/lipid rafts.
Figure 3.

Expression of α-syn in M17 and HeLa cells. A, Left, Example of a Western blot of the expression of α-syn in EV and stably transfected WT, A53T, and A30P α-syn-expressing M17 cells relative to that of control β-actin. Right, Quantitation of the total α-syn in the blots exemplified at left, in arbitrary units; n = 3; asterisk denotes significance versus WT. B, Expression and quantitation of α-syn in HeLa cells, as in (A). In both cases, note reduced expression in A53T cells.
Upon further fractionation of CM from M17 cells into MAM and pure mitochondria, we found a lower degree of localization of both mutant α-syn species to MAM compared with WT, with a concomitantly higher degree of localization of α-syn to the pure mitochondrial fraction (Fig. 2A). This result agrees with previously published data showing that the A30P mutation reduces α-syn's affinity for membranes (Fortin et al., 2004). We also observed a similar decrease of α-syn in the CM and a concurrent increase of α-syn in the cytosol, as well as a shift of α-syn from the MAM to the pure mitochondria fractions, in brain tissues of transgenic mice expressing these mutations (Fig. 2B).
To explore these changes further, we used an alternative and complementary approach to analyze α-syn distribution in MAM. As noted above, MAM is an intracellular lipid raft with features of a DRM. We therefore extracted DRMs from the CM of transgenic mouse brains and analyzed them on sucrose gradients (Fig. 2C). WT α-syn was present in two peaks, one corresponding to non-DRMs (fractions 16–21) and the other to DRMs present in the CM, namely MAM, that colocalized with flotillin, a DRM/lipid raft marker (fractions 5–12). In contrast, both mutations showed lower amounts of α-syn in the DRMs, with a corresponding increase in the non-DRM fractions. The reduction of A30P in DRMs was far more pronounced than that of A53T, in general agreement with previous observations (Fortin et al., 2004) and with the other results described above.
The distance between ER and mitochondria tightly regulates the cross talk between both organelles, including calcium and phospholipid transfer (Hayashi et al., 2009). Mutation or ablation of some MAM-resident proteins can alter the distance between both compartments (de Brito and Scorrano, 2008). For this reason, and to determine the effect of α-syn on MAM behavior, we transfected cells with pDsRed2-Mito to detect mitochondria (in red) and with GFP-Sec61β to detect ER (in green), and analyzed and quantitated regions where the two signals were in close apposition. We found that the degree of ER–mitochondrial apposition was lower in M17 cells stably expressing the α-syn pathogenic mutations compared with the empty-vector (EV) control (Fig. 4). A similar experiment conducted in transiently transfected HeLa cells showed the same changes in ER–mitochondrial apposition in both the A53T and A30P overexpressing cells (Fig. 5).
Figure 4.
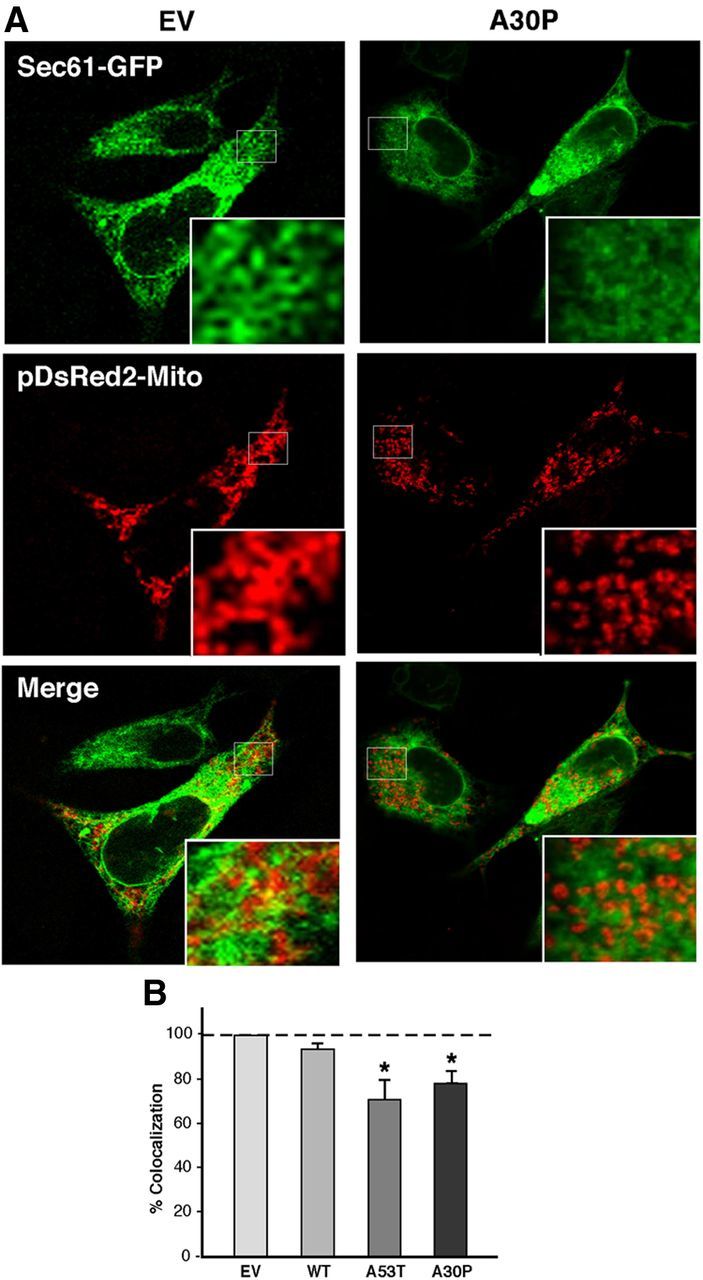
Apposition of ER and mitochondria in M17 cells. A, Examples of colocalization of ER labeled with GFP-Sec61-β (green), and mitochondria labeled with pDsRed2-mito (red), in WT- and A30P-expressing M17 cells. B, Quantitation of colocalization (as in A) by ImageJ analysis. Asterisk denotes significance versus EV.
Figure 5.

Apposition of ER and mitochondria in HeLa cells. A, Examples of colocalization of ER labeled with GFP-Sec61-β (green), and mitochondria labeled with pDsRed2-mito (red), in HeLa cells transiently expressing WT- and A30P-α-syn. B, Quantitation of colocalization (as in A) by ImageJ analysis, normalized to the baseline level of apposition value in EV cells (baseline = 100; average of 4 experiments ±SD). Asterisk denotes significance versus EV.
Finally, we analyzed MAM function in control and mutant cells by measuring the incorporation of radiolabeled serine into phosphatidylserine (PtdSer) and its conversion into phosphatidylethanolamine (PtdEtn), which is a recognized measurement of MAM activity and ER–mitochondrial communication (Voelker, 2005; Area-Gomez et al., 2012). As shown in Figure 6, both the A53T and A30P mutations caused a significant decrease in PtdSer and PtdEtn synthesis, indicating that these pathogenic mutations in α-syn induce a decrease in MAM activity that is associated with reduced ER–mitochondria connectivity.
Figure 6.

Phospholipid synthesis in M17 cells. Synthesis of 3H-PtdSer and 3H-PtdEtn after labeling the indicated M17 cells with 3H-Ser for the indicated times; n = 3 independent experiments. *Significant difference versus EV (horizontal lines; p < 0.05). Note the imputed reduction in MAM function in the two point mutations.
Together, these data indicate that α-syn is a MAM-resident protein that influences ER–mitochondrial apposition and MAM function, and that pathogenic mutations in α-syn affect its propensity to accumulate in this subcellular compartment, because it localizes either more to the cytosol or more to the mitochondria, or both (i.e., away from the MAM).
Mutations in α-syn cause mitochondrial fragmentation
The contacts between ER and mitochondria have been shown to play a fundamental role in the balance between fusion and fission of mitochondria (Friedman et al., 2011). Moreover, alterations in MAM proteins, such as mitofusin-2 (MFN2) and PACS-2, cause a lower degree of apposition between ER and mitochondria, and have been linked to an increase in mitochondrial fragmentation (de Brito and Scorrano, 2008). For these reasons, and in light of our data on MAM, we revisited the relationship between α-syn and mitochondrial fragmentation. Consistent with the results of others (Kamp et al., 2010; Nakamura et al., 2011), confocal analysis of M17 (Fig. 7) and HeLa (Fig. 8) cells revealed a higher degree of mitochondrial fragmentation in cells expressing the α-syn point mutations compared with EV controls.
Figure 7.
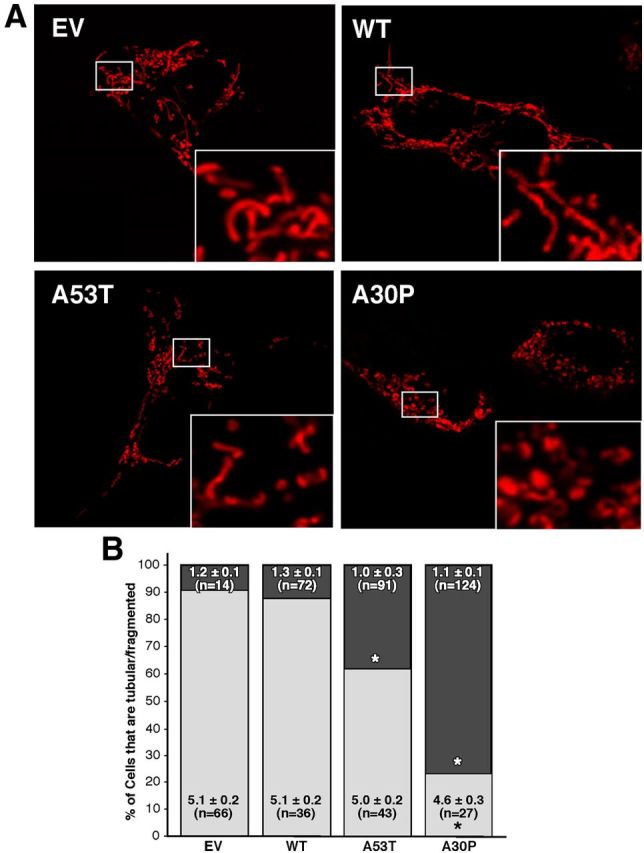
Mitochondrial fragmentation in M17 cells. A, Representative images of mitochondria in α-syn-expressing cells labeled with pDsRed2-Mito. Insets, Enlargements of the indicated regions (small boxes). B, Quantitation of fragmentation of the cells in A, measured as the percentage of all cells that contained predominantly fragmented (darker shading) versus tubular (lighter shading) mitochondria. Numbers within the boxes denote the average length (±SE) of the mitochondria in micrometers; n = number of mitochondria measured. *Significant difference versus EV (p < 0.05).
Figure 8.
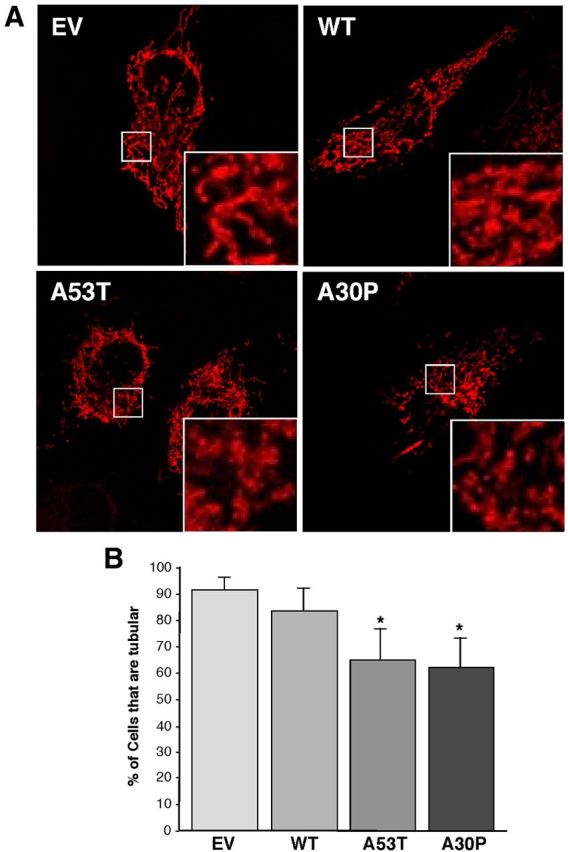
Mitochondrial fragmentation in HeLa cells. A, Representative images of mitochondria in α-syn-expressing cells labeled with pDsRed2-Mito. Insets, Enlargements of the indicated regions (small boxes). B, Quantitation of fragmentation of the cells in A, measured as the percentage of all cells that contained predominantly fragmented tubular mitochondria. *Significant difference versus EV (p < 0.05); n = 5 independent experiments.
We therefore assessed whether inhibiting DRP1 could rescue the α-syn-related fragmentation phenotype. DRP1 is recruited from the cytosol to the MOM, where it oligomerizes and severs the organelle (Westermann, 2008). We found that α-syn-related mitochondrial fragmentation was not altered by inhibition of the fission protein DRP1, as this phenotype was unchanged even in the presence of mdivi-1, a specific inhibitor of DRP1 (Fig. 9B; Cassidy-Stone et al., 2008). In agreement with the findings of others (Nakamura et al., 2011), we were able to recapitulate this same phenotype in cells where Drp1 had been ablated (Fig. 10). These results indicate that mutations in α-syn cause mitochondrial fragmentation through a DRP1-independent mechanism. Importantly, DRP1 function itself was essentially normal in the α-syn-mutant cells, as we found no apparent difference in the recruitment of DRP1 to the mitochondria between EV and α-syn-transfected M17 cells (i.e., the degree of DRP1 recruitment to mitochondria essentially paralleled the degree of fragmentation; Fig. 9A).
Figure 9.

Mitochondrial dynamics in M17 cells. A, DRP1 recruitment to mitochondria. Representative images of fluorescently labeled mitochondria (red) and anti-DRP1 (green) in M17 cells either untreated or treated with the DRP1 inhibitor mdivi-1. Arrowheads indicate examples of DRP1-positive foci. Other notation as in Figure 7. B, Inhibition of DRP1 with mdivi-1. Representative images of fluorescently labeled mitochondria (red) in M17 cells either untreated or treated with the DRP1 inhibitor mdivi-1. Note the persistence of fragmented mitochondria in the A53T and A30P cells even after mdivi-1 treatment.
Figure 10.

Mitochondrial dynamics in Drp1-null MEFs. A, Representative images of mitochondria in Drp1-WT and -KO MEFs labeled with MitoTracker Red (red) transiently expressing the indicated plasmid constructs. B, Quantitation of fragmented mitochondria from the experiments shown in A (n = 3). Note the persistence of fragmented mitochondria in the A53T and A30P cells even in the Drp1-KO cells. Asterisk denotes significance versus EV.
Conversely, the analysis of OPA1 expression by Western blot revealed a significant increase in the shorter forms and a concomitant decrease in the longer forms of this protein (generated by alternative splicing and processing; Duvezin-Caubet et al., 2006; Song et al., 2007) in crude mitochondrial fractions from M17 cells expressing A53T or A30P α-syn mutations (Fig. 11). This result suggests that the α-syn-mediated mitochondrial fragmentation phenotype may be due to an increase OPA1 cleavage, via a currently unknown mechanism.
Figure 11.

Western blot to detect OPA1 in CM fractions from M17 cells stably expressing the indicated forms of α-syn. Mitochondrial ATPase-α was used as a loading control.
On the other hand, and contrary to previous results (Kamp et al., 2010; Nakamura et al., 2011), we did not observe a significant increase in mitochondrial fragmentation in cells overexpressing WT α-syn. To explore this discrepancy further, we coexpressed pDsRed2-mito and WT α-syn-GFP in M17 cells already expressing either constitutively low levels of α-syn (i.e., in the EV cells) or expressing stably integrated α-syn (i.e., in the WT cells). Interestingly, we found that expression of α-syn-GFP caused a significant increase in mitochondrial fragmentation only in those cells already stably expressing WT α-syn (Fig. 12). Because the WT line expresses more α-syn (derived from both the integrated α-syn gene and the transiently expressed α-syn-GFP gene) than does the EV line (expressing only α-syn-GFP), these results suggest that there may be a minimum threshold level of α-syn expression above which mitochondrial fragmentation ensues. Surprisingly, the overexpression of WT α-syn-GFP had the opposite effect in the M17 cells already expressing A53T and A30P, i.e., there was a reduction in the proportion of cells with fragmented mitochondria, implying that WT α-syn can rescue the fragmented phenotype in α-syn mutant cells (Fig. 12). These results suggest that mutant α-syn promotes mitochondrial fragmentation via a fission/fusion-independent mechanism, either by its lower binding to MAM or by accumulating at the mitochondria. Furthermore, our data imply that mutations in α-syn cause a “gain-of-negative-function” effect that can be rescued by the WT form of the protein.
Figure 12.
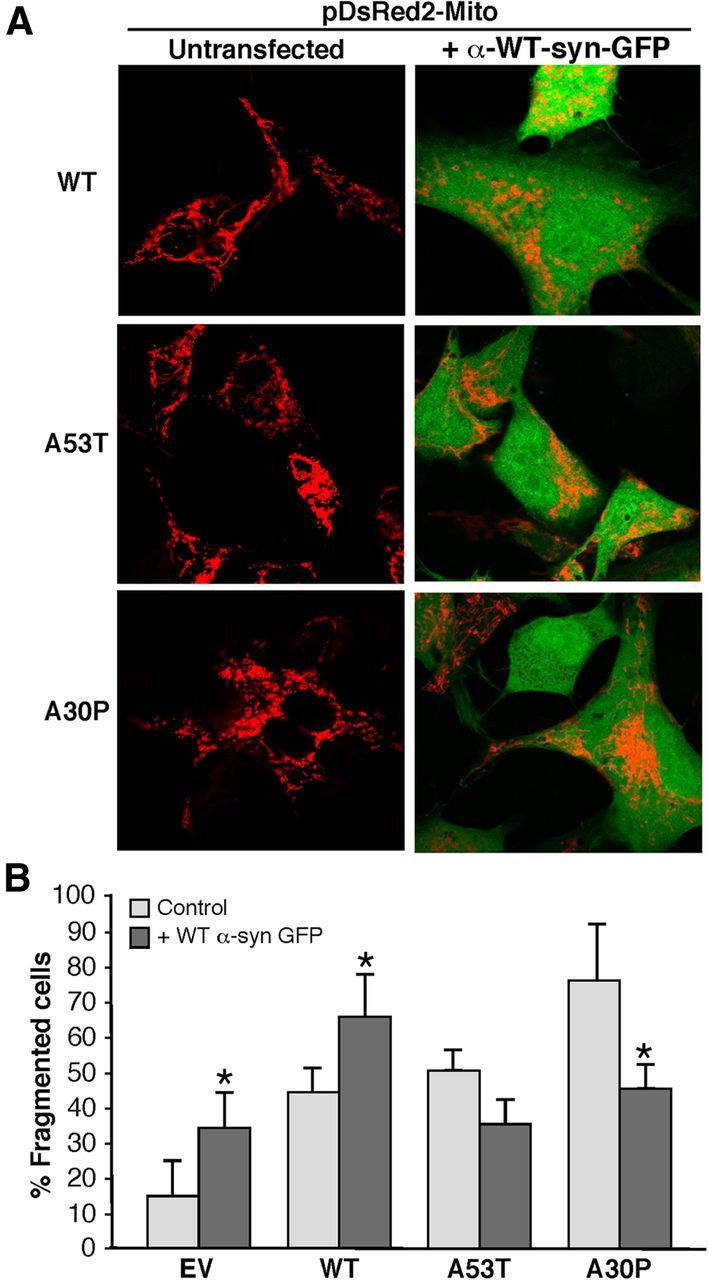
Relationship of WT α-syn expression to mitochondrial fragmentation. A, M17 cells stably transfected with an EV plasmid or with the indicated plasmids expressing α-syn species were transiently cotransfected with plasmids expressing pDsRed2-Mito (red) and WT α-syn-GFP (green) and visualized by confocal microscopy. Cells were inspected visually and scored for whether they contained predominantly tubular or predominantly fragmented mitochondria (red). Note that, compared with Figure 7, the fragmented phenotype in the α-syn mutant cells is ameliorated by the expression of WT α-syn-GFP. B, Quantitation of percentage fragmented cells shown in A. *Significant difference versus corresponding untransfected cells (p < 0.05).
Discussion
We show here that a portion of α-syn is located in MAM and not, as previously reported, in mitochondria (Li et al., 2007; Cole et al., 2008; Devi et al., 2008; Parihar et al., 2008; Shavali et al., 2008). Moreover, the association of α-syn with MAM can be altered by pathogenic α-syn mutations found in familial PD, which in turn are linked with reduced ER–mitochondrial connectivity and are correlated with an increased degree of mitochondrial fragmentation.
The localization of α-syn to MAM was supported by four lines of evidence. First, the in vitro import assays showed that the association of α-syn with mitochondria does not increase with time, although it decreases significantly when mitochondrial membrane potential is disrupted, indicating that α-syn is associated with a mitochondria-associated fraction, but in an anomalous way (i.e., MAM). Second, subcellular fractionation showed that α-syn was present in the MAM fraction. Third, isolation of lipid raft-like domains from the CM fraction as DRMs showed the presence of α-syn in this subcellular compartment. Finally, as with other known MAM proteins, mutations in α-syn affected its localization to this domain that was associated with a decrease in MAM function and in the degree of ER–mitochondrial apposition.
The presence of α-syn in synaptic terminals and vesicles could also be explained by its association with MAM, given that (1) synaptic vesicles are lipid rafts (Lv et al., 2008) and (2) ER–mitochondrial connections (McNulty, 1980) and known MAM markers (Mavlyutov et al., 2012) have been found closely juxtaposed to synaptic membranes. Similar connections were also observed in ganglion cell membranes adjacent to nerve endings, in so-called junctional subsurface organs, which are flat cisternae of ER (Taxi and Eugene, 1995) apposed to mitochondria (Watanabe and Burnstock, 1976).
The presence of α-syn in the MAM is also consistent with other observations. For example, it is well known that α-syn binds preferentially to lipid rafts (Fortin et al., 2004) and to membrane domains rich in acidic phospholipids (Davidson et al., 1998); notably, MAM is an intracellular lipid raft rich in cholesterol and acidic phospholipids (Hayashi and Fujimoto, 2010). Moreover, it was recently shown that α-syn affects calcium transport from the ER to the mitochondria (Calì et al., 2012). This trafficking was almost certainly via the MAM, as the major components of the calcium-transport machinery (e.g., IP3 receptors and the σ-1 receptor) between these two organelles are MAM-localized (Hayashi et al., 2009). Finally, α-syn was detected in a proteomic analysis of MAM isolated from mouse brain (Poston et al., 2013).
The relationship of α-syn to MAM is perturbed in the pathogenic situation. We found that both A30P and A53T showed a decrease in the amount of α-syn present in MAM, but for two different reasons. In the case of A30P, the lower localization in MAM is in accordance with previous data showing that the mutation disrupts the interaction of α-syn with lipid rafts (Fortin et al., 2004). With respect to A53T, however, the relative distribution of α-syn in M17 cells between the cytosol and the CM (containing MAM) was similar to that of WT α-syn (Fig. 2A), in agreement with previous reports showing that A53T α-syn has no deficiency in its ability to bind to lipid membranes (Fortin et al., 2004). What did change was the total amount of A53T α-syn, which was clearly reduced (by ∼50%) compared with WT, resulting in less protein in the CM and MAM (Fig. 2A), and less in DRMs (Fig. 2C). The reason for this reduction is unknown, but we note that it was observed in all tissues and cells analyzed (Figures 2, 3); perhaps this reduction was due to the tendency of A53T to aggregate and turn over extremely rapidly (Narhi et al., 1999). Thus, the net effect of both mutations was to reduce the absolute amount of α-syn in the MAM. The quantitative reduction in both A30P and A53T α-syn at the MAM was accompanied by a concomitant increase in these proteins in the pure mitochondrial fraction, but the reason for this behavior is currently unknown.
There is emerging evidence that mutations in α-syn affect mitochondrial morphology by fostering organellar fragmentation, but that modulating the mitodynamics machinery has no effect on this fragmentation phenotype (Kamp et al., 2010; Nakamura et al., 2011). Consistent with these observations, we recapitulated this phenotype in M17 and HeLa cells expressing A30P and A53T; likewise, fragmentation persisted even after ablation of DRP1 or inhibition of DRP1 with mdivi-1 (Cassidy-Stone et al., 2008). The DRP1-independence of mitochondrial fission/fusion due to mutations in α-syn is the first example of which we are aware where inhibition of mitochondrial fission is not accompanied by a corresponding increase in fusion (Knott and Bossy-Wetzel, 2008).
Conversely, and in contrast to previously published results (Kamp et al., 2010), we observed a clear increase in the cleavage of OPA1 in M17 cells overexpressing A30P and A53T α-syn mutations, a hitherto-undescribed aspect of α-syn behavior. Although it is well known that the cleavage of OPA1 into its shorter forms correlates with an increase in the degree of mitochondrial fragmentation (Duvezin-Caubet et al., 2006), its potential relationship to α-syn, either direct or indirect, is unclear at present. Perhaps the lack of binding of mutant α-syn to MAM and its concomitant accumulation on mitochondrial membranes (described above) caused a decrease in the mitochondrial membrane potential (Tanaka et al., 2001), which in turn provoked an increase in the cleavage of OPA1 and mitochondrial fragmentation (Duvezin-Caubet et al., 2006). In support of this speculation, overexpression of mutant forms of α-syn caused mitochondrial depolarization (Tanaka et al., 2001).
Because it is well known that mitochondrial distribution and dynamics are influenced by physical connections between the MOM and the ER via MAM (de Brito and Scorrano, 2008; Ciarlo et al., 2010), it may well be that MAM-localized α-syn mediates events that are downstream of the mitochondrial fission and fusion machinery. For example, since mitochondria-associated lipid rafts (i.e., MAM) have been shown to regulate mitochondrial dynamics (Ciarlo et al., 2010), alterations in MAM structure due to mutations in α-syn could affect the fusion/fission balance in an indirect way. In agreement with this speculation, we found that point mutations in α-syn reduced the degree of apposition between ER and mitochondria (Fig. 4).
The exact nature of this downstream effect is presently unknown. However, it has been shown that α-syn, when bound to lipid raft-like domains, changes its own conformation and that of the membranes to which it binds (Mizuno et al., 2012). Specifically, the binding of α-syn causes lipid reorganization in the target membrane that results in an increase in its curvature (Mizuno et al., 2012). It is possible that α-syn modifies MAM structure by inducing membrane curvature and contributing to the concentration of fusogenic lipids (i.e., phosphatidylethanolamine) in target membranes (Auluck et al., 2010), and forming a platform that helps in the assembly of the fusion “stalk” (Furt and Moreau, 2009), composed of various proteins, including MFN2 (Papanicolaou et al., 2012). Thus, the reduced binding of mutant α-syn to MAM may impair the formation of the fusion stalk and hence mitochondrial fusion/fission regardless of the behavior of specific mitodynamic proteins present therein.
If reduced association of mutant α-syn with MAM is pathogenic in PD, how do we explain the pathogenicity of duplications and triplications of WT SNCA? We believe that the stable overexpression of WT α-syn did not trigger mitochondrial fragmentation in our hands because its level was below a putative threshold to initiate such behavior. In agreement with this speculation, when we overexpressed WT α-syn in M17 cells already expressing stably integrated α-syn (i.e., in the “α-syn WT” line; Fig. 9), the degree of fragmentation increased to the level found in A53T and A30P cells (Fig. 6). On the other hand, the expression of a putatively similar amount of WT α-syn in A53T and A30P cells “rescued” the fragmentation phenotype due to these mutations (Fig. 9). This surprising result suggests that the dominant nature of familial PD associated with these two point mutations is due not to a gain of function, but rather to either a gain-of-negative-function or to haploinsufficiency (Kobayashi et al., 2003).
The localization of α-syn in MAM may help explain the mitochondrial abnormalities associated with pathogenic mutations in this protein, and the paradoxical nonresponsiveness of the mitochondrial fission/fusion machinery in cells expressing these mutants. Thus, aside from synaptic vesicles, MAM may represent another subcellular domain relevant to the function of this protein. Furthermore, an increasing number of PD-causing proteins have been linked to mitochondria, raising the possibility that MAM, either in addition to or instead of mitochondria, is the intracellular locus that underlies the intersection of many, if not all, of these apparently dissimilar PD-associated proteins.
Footnotes
This work was supported by a Lucien Côté Early Investigator Award in Clinical Genetics from the Parkinson's Disease Foundation (PDF-CEI-1364 and PDF-CEI-1240 to C.G.-L., PDF-RCE-1401 to S.P.), the NIH (Grants NS38370, ES017384, and NS070276 to S.P.), the U.S. Department of Defense (W911NF-12-1-0159 to E.A.S. and no. W81WXWH-08-1-0465 to S.P.), and the Ellison Medical Foundation, the Alzheimer's Drug Discovery Foundation, and the Marriott Mitochondrial Disorder Clinical Research Fund (E.A.S.).
The authors declare no competing financial interests.
References
- Area-Gomez E, de Groof AJ, Boldogh I, Bird TD, Gibson GE, Koehler CM, Yu WH, Duff KE, Yaffe MP, Pon LA, Schon EA. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am J Pathol. 2009;175:1810–1816. doi: 10.2353/ajpath.2009.090219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Area-Gomez E, Del Carmen Lara Castillo M, Tambini MD, Guardia-Laguarta C, de Groof AJ, Madra M, Ikenouchi J, Umeda M, Bird TD, Sturley SL, Schon EA. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012;31:4106–4123. doi: 10.1038/emboj.2012.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auluck PK, Caraveo G, Lindquist S. α-Synuclein: membrane interactions and toxicity in Parkinson's disease. Annu Rev Cell Dev Biol. 2010;26:211–233. doi: 10.1146/annurev.cellbio.042308.113313. [DOI] [PubMed] [Google Scholar]
- Becker D, Richter J, Tocilescu MA, Przedborski S, Voos W. Pink1 kinase and its membrane potential (Δψ)-dependent cleavage product both localize to outer mitochondrial membrane by unique targeting mode. J Biol Chem. 2012;287:22969–22987. doi: 10.1074/jbc.M112.365700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calì T, Ottolini D, Negro A, Brini M. α-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J Biol Chem. 2012;287:17914–17929. doi: 10.1074/jbc.M111.302794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciarlo L, Manganelli V, Garofalo T, Matarrese P, Tinari A, Misasi R, Malorni W, Sorice M. Association of fission proteins with mitochondrial raft-like domains. Cell Death Differ. 2010;17:1047–1058. doi: 10.1038/cdd.2009.208. [DOI] [PubMed] [Google Scholar]
- Cole NB, Dieuliis D, Leo P, Mitchell DC, Nussbaum RL. Mitochondrial translocation of α-synuclein is promoted by intracellular acidification. Exp Cell Res. 2008;314:2076–2089. doi: 10.1016/j.yexcr.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem. 2008;283:9089–9100. doi: 10.1074/jbc.M710012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvezin-Caubet S, Jagasia R, Wagener J, Hofmann S, Trifunovic A, Hansson A, Chomyn A, Bauer MF, Attardi G, Larsson NG, Neupert W, Reichert AS. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J Biol Chem. 2006;281:37972–37979. doi: 10.1074/jbc.M606059200. [DOI] [PubMed] [Google Scholar]
- Fortin DL, Troyer MD, Nakamura K, Kubo S, Anthony MD, Edwards RH. Lipid rafts mediate the synaptic localization of α-synuclein. J Neurosci. 2004;24:6715–6723. doi: 10.1523/JNEUROSCI.1594-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furt F, Moreau P. Importance of lipid metabolism for intracellular and mitochondrial membrane fusion/fission processes. Int J Biochem Cell Biol. 2009;41:1828–1836. doi: 10.1016/j.biocel.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Fujimoto M. Detergent-resistant microdomains determine the localization of sigma-1 receptors to the endoplasmic reticulum-mitochondria junction. Mol Pharmacol. 2010;77:517–528. doi: 10.1124/mol.109.062539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Rizzuto R, Hajnoczky G, Su TP. MAM: more than just a housekeeper. Trends Cell Biol. 2009;19:81–88. doi: 10.1016/j.tcb.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, Wong J, Takenouchi T, Hashimoto M, Masliah E. α-Synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol. 2000;157:401–410. doi: 10.1016/S0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamp F, Exner N, Lutz AK, Wender N, Hegermann J, Brunner B, Nuscher B, Bartels T, Giese A, Beyer K, Eimer S, Winklhofer KF, Haass C. Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 2010;29:3571–3589. doi: 10.1038/emboj.2010.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C, Westenberger A. Genetics of Parkinson's disease. Cold Spring Harb Perspect Med. 2012;2:a008888. doi: 10.1101/cshperspect.a008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott AB, Bossy-Wetzel E. Impairing the mitochondrial fission and fusion balance: a new mechanism of neurodegeneration. Ann N Y Acad Sci. 2008;1147:283–292. doi: 10.1196/annals.1427.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Krüger R, Markopoulou K, Wszolek Z, Chase B, Taka H, Mineki R, Murayama K, Riess O, Mizuno Y, Hattori N. Haploinsufficiency at the α-synuclein gene underlies phenotypic severity in familial Parkinson's disease. Brain. 2003;126:32–42. doi: 10.1093/brain/awg010. [DOI] [PubMed] [Google Scholar]
- Li WW, Yang R, Guo JC, Ren HM, Zha XL, Cheng JS, Cai DF. Localization of α-synuclein to mitochondria within midbrain of mice. Neuroreport. 2007;18:1543–1546. doi: 10.1097/WNR.0b013e3282f03db4. [DOI] [PubMed] [Google Scholar]
- Lv JH, He L, Sui SF. Lipid rafts association of synaptotagmin I on synaptic vesicles. Biochemistry (Mosc) 2008;73:283–288. doi: 10.1134/S0006297908030073. [DOI] [PubMed] [Google Scholar]
- Magrané J, Hervias I, Henning MS, Damiano M, Kawamata H, Manfredi G. Mutant SOD1 in neuronal mitochondria causes toxicity and mitochondrial dynamics abnormalities. Hum Mol Genet. 2009;18:4552–4564. doi: 10.1093/hmg/ddp421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavlyutov TA, Epstein ML, Liu P, Verbny YI, Ziskind-Conhaim L, Ruoho AE. Development of the sigma-1 receptor in C-terminals of motoneurons and colocalization with the N, N′-dimethyltryptamine forming enzyme, indole-N-methyl transferase. Neuroscience. 2012;206:60–68. doi: 10.1016/j.neuroscience.2011.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNulty JA. Ultrastructural observations on synaptic ribbons in the pineal organ of the goldfish. Cell Tissue Res. 1980;210:249–256. doi: 10.1007/BF00237613. [DOI] [PubMed] [Google Scholar]
- Mizuno N, Varkey J, Kegulian NC, Hegde BG, Cheng N, Langen R, Steven AC. Remodeling of lipid vesicles into cylindrical micelles by α-synuclein in an extended α-helical conformation. J Biol Chem. 2012;287:29301–29311. doi: 10.1074/jbc.M112.365817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Nemani VM, Azarbal F, Skibinski G, Levy JM, Egami K, Munishkina L, Zhang J, Gardner B, Wakabayashi J, Sesaki H, Cheng Y, Finkbeiner S, Nussbaum RL, Masliah E, Edwards RH. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein α-synuclein. J Biol Chem. 2011;286:20710–20726. doi: 10.1074/jbc.M110.213538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narhi L, Wood SJ, Steavenson S, Jiang Y, Wu GM, Anafi D, Kaufman SA, Martin F, Sitney K, Denis P, Louis JC, Wypych J, Biere AL, Citron M. Both familial Parkinson's disease mutations accelerate α-synuclein aggregation. J Biol Chem. 1999;274:9843–9846. doi: 10.1074/jbc.274.14.9843. [DOI] [PubMed] [Google Scholar]
- Ottolini D, Calì T, Negro A, Brini M. The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum-mitochondria tethering. Hum Mol Genet. 2013;22:2152–2168. doi: 10.1093/hmg/ddt068. [DOI] [PubMed] [Google Scholar]
- Papanicolaou KN, Phillippo MM, Walsh K. Mitofusins and the mitochondrial permeability transition: the potential downside of mitochondrial fusion. Am J Physiol Heart Circ Physiol. 2012;303:H243–H255. doi: 10.1152/ajpheart.00185.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parihar MS, Parihar A, Fujita M, Hashimoto M, Ghafourifar P. Mitochondrial association of alpha-synuclein causes oxidative stress. Cell Mol Life Sci. 2008;65:1272–1284. doi: 10.1007/s00018-008-7589-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poston CN, Krishnan SC, Bazemore-Walker CR. In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM) J Proteomics. 2013;79:219–230. doi: 10.1016/j.jprot.2012.12.018. [DOI] [PubMed] [Google Scholar]
- Schon EA, Area-Gomez E. Mitochondria-associated ER membranes in Alzheimer disease. Mol Cell Neurosci. 2013;55:26–36. doi: 10.1016/j.mcn.2012.07.011. [DOI] [PubMed] [Google Scholar]
- Schon EA, Przedborski S. Mitochondria: the next (neurode)generation. Neuron. 2011;70:1033–1053. doi: 10.1016/j.neuron.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shavali S, Brown-Borg HM, Ebadi M, Porter J. Mitochondrial localization of alpha-synuclein protein in alpha-synuclein overexpressing cells. Neurosci Lett. 2008;439:125–128. doi: 10.1016/j.neulet.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. 2007;178:749–755. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Engelender S, Igarashi S, Rao RK, Wanner T, Tanzi RE, Sawa A, L Dawson V, Dawson TM, Ross CA. Inducible expression of mutant α-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet. 2001;10:919–926. doi: 10.1093/hmg/10.9.919. [DOI] [PubMed] [Google Scholar]
- Taxi J, Eugene D. Effects of axotomy, deaffentiation, and reinnervation on sympathetic ganglionic synapses: a comparative study. In: Jeon K, Jarvik J, editors. International review of cytology, Vol 159, A survey of cell biology. London: Academic; 1995. pp. 195–209. [DOI] [PubMed] [Google Scholar]
- Voelker DR. Bridging gaps in phospholipid transport. Trends Biochem Sci. 2005;30:396–404. doi: 10.1016/j.tibs.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Burnstock G. Junctional subsurface organs in frog sympathetic ganglion cells. J Neurocytol. 1976;5:125–136. doi: 10.1007/BF01176186. [DOI] [PubMed] [Google Scholar]
- Westermann B. Molecular machinery of mitochondrial fusion and fission. J Biol Chem. 2008;283:13501–13505. doi: 10.1074/jbc.R800011200. [DOI] [PubMed] [Google Scholar]