

Abstract
Gi/o-protein-coupled receptors (GPCRs) ubiquitously inhibit neurotransmission, principally via Gβγ, which acts via a number of possible effectors. GPCR effector specificity has traditionally been attributed to Gα, based on Gα's preferential effector targeting in vitro compared with Gβγ's promiscuous targeting of various effectors. In synapses, however, Gβγ clearly targets unique effectors in a receptor-dependent way to modulate synaptic transmission. It remains unknown whether Gβγ specificity in vivo is due to specific Gβγ isoform-receptor associations or to spatial separation of distinct Gβγ pathways through macromolecular interactions. We thus sought to determine how Gβγ signaling pathways within axons remain distinct from one another. In rat hippocampal CA1 axons, GABAB receptors (GABABRs) inhibit presynaptic Ca2+ entry, and we have now demonstrated that 5-HT1B receptors (5-HT1BRs) liberate Gβγ to interact with SNARE complex C terminals with no effect on Ca2+ entry. Both GABABRs and 5-HT1BRs inhibit Ca2+-evoked neurotransmitter release, but 5-HT1BRs have no effect on Sr2+-evoked release. Sr2+, unlike Ca2+, does not cause synaptotagmin to compete with Gβγ binding to SNARE complexes. 5-HT1BRs also fail to inhibit release following cleavage of the C terminus of the SNARE complex protein SNAP-25 with botulinum A toxin. Thus, GABABRs and 5-HT1BRs both localize to presynaptic terminals, but target distinct effectors. We demonstrate that disruption of SNARE complexes and vesicle priming with botulinum C toxin eliminates this selectivity, allowing 5-HT1BR inhibition of Ca2+ entry. We conclude that receptor-effector specificity requires a microarchitecture provided by the SNARE complex during vesicle priming.
Introduction
Gi/o-protein-coupled receptor (GPCR)-dependent presynaptic inhibition occurs at all synapses, but is mediated by multiple mechanisms directly involved in evoked exocytosis. The most investigated mechanism is a membrane-delimited Gβγ-mediated inhibition of presynaptic Ca2+ entry (Herlitze et al., 1996; Ford et al., 1998; Brown and Sihra, 2008). Gβγ might also activate presynaptic G-protein inwardly rectifying channels (Michaeli and Yaka, 2010) and can modulate neurotransmission by directly interacting with the SNARE complex without altering Ca2+ entry (Blackmer et al., 2001; Takahashi et al., 2001; Gerachshenko et al., 2005). A region of the SNARE complex C terminus provides a target for the Ca2+ sensor synaptotagmin during the induction of exocytosis (Zhang et al., 2002). Gβγ competition with Ca2+-synaptotagmin at this region mediates presynaptic inhibition (Gerachshenko et al., 2005, 2009; Yoon et al., 2007; Wells et al., 2012). This region of the SNARE complex is also important for vesicle fusion. Cleavage of the C terminus of SNAP-25 with botulinum toxin A (BoNT/A) modifies fusion pore formation (Fang et al., 2008), and prevents GPCR-mediated inhibition (Gerachshenko et al., 2005; Delaney et al., 2007). Indeed, Ca2+-synaptotagmin interaction with the SNARE complex may mediate fusion pore expansion (Lai et al., 2013).
Mechanisms that target presynaptic Ca2+ channels and those that interact with the SNARE complex signal with Gβγ, but it is unknown whether, or how, Gβγ-selective targeting of closely localized effectors can occur. In the case of 5-HT1B receptors (5-HT1BRs), it has been shown that 5-HT1BRs target Ca2+ channels in the calyceal synapse of Held (Mizutani et al., 2006), but the C terminus of the SNARE complex in lamprey synapses (Gerachshenko et al., 2005) and chromaffin cells (Blackmer et al., 2005). GABABRs inhibit Ca2+ entry in the Calyx synapse of Held (Takahashi et al., 1998, 2005) and in hippocampal neurons (Wu and Saggau, 1995). Other GPCRs also target either SNARE complexes or Ca2+ channels, for example, noradrenergic-a2A receptors target the SNARE complex in the amygdala (Delaney et al., 2007), but may modulate Ca2+ channels in sensory neurons (Dunlap and Fischbach, 1978). Finally, GABABRs also inhibit spontaneous neurotransmitter release events independently of Ca2+ (Scanziani et al., 1992; Kabashima et al., 1997), an effect that was first shown for adenosine at the neuromuscular junction (Silinsky, 1984) and later in cortex (Cox et al., 2000). However, effects on spontaneous release may not directly involve evoked release (Wang and Armstrong, 2012), because spontaneous release can be mediated through independent pathways (Kavalali et al., 2011).
If two mechanisms, for example, Gβγ/SNARE complex interactions and Gβγ/Ca2+ channel interactions, coexist in one terminal, it raises fundamental questions about targeting of modulatory systems. Are Gβγ effectors selectively targeted and, if so, are effector identities determined by Gβγ properties or ultrastructurally with similar receptors and effectors as components of molecular machines? We demonstrate that two GPCRs, both Gαi/o coupled, using membrane-delimited mechanisms and which both inhibit CA1 to subicular excitation modulate neurotransmitter release through different effectors whose targets are governed by the microarchitecture of SNARE complexes during vesicle priming.
Materials and Methods
Hippocampal slice preparation.
Hippocampal slices (300 μm thickness) were prepared from 17- to 23-d-old male Sprague Dawley rats anesthetized with isoflurane and decapitated in accordance with institutional guidelines. All recordings were in the following solution (mm): 124 NaCl, 26 NaHCO3, 1.25 NaH2PO4, 3 KCl, 2 CaCl2, 1 MgCl2, and 10 d-glucose, bubbled with 95% O2-5% CO2. Recordings were at 28°C maintained by a flow heating system of the superfusate, which was constantly superfused over the preparation at 1–2 ml/min for all recordings. All pharmacological agents applied extracellularly were added to the superfusate.
Electrophysiology.
In hippocampal slices, CA1 and subicular pyramidal neurons were whole-cell clamped following microscopic visual identification under an upright microscope with the same lens used for imaging experiments. For postsynaptic subicular pyramidal cell recordings, patch pipettes (∼4 MΩ) contained solution of the following composition (in mm): 146 CsSO3, 2 MgCl2, 5 EGTA, and 9.1 HEPES, adjusted to pH 7.2 with CsOH. For imaging experiments recording from CA1 pyramidal neurons, the pipette composition was as follows (in mm): 146 KSO3, 2 MgCl2, 0.025 EGTA, and 9.1 HEPES, adjusted to pH 7.2 with KOH. Dyes were included in the patch solution (Fluo-5F, 200 μm; Alexa 594 hydrazide, 250 μm). Series resistance was monitored throughout the experiment by application of a 5 mV voltage step and recordings were discarded if the series resistance changed by >10% during the course of the experiment. All holding potentials for whole-cell voltage-clamp recordings were at −70 mV. For current-clamp recordings the membrane potential was maintained between −70 and −80 mV. Liquid junction potentials were not corrected. EPSCs in subicular neurons were evoked with focal stimuli (0.2 ms, <20 μA) to CA1 axons using glass-insulated tungsten microelectrodes.
Detection of spontaneous miniature EPSCs.
Spontaneous miniature EPSCs (mEPSCs) and asynchronous events in Sr2+ were low-pass filtered and digitized (5 kHz). Analysis was performed in Igor Pro (WaveMetrics). Detection and extraction of mEPSCs was semi-automated. Filtered data were differentiated to correct for baseline shifts. Thresholds were determined for differentiated control data and maintained for that cell following agonist application. Detected event amplitudes were mapped back onto raw data for visual comparison of amplitudes with mEPSCs to ensure that the detection algorithm functioned adequately. The algorithm is available from our website (http://alford.bios.uic.edu/Research/software.html). This algorithm was applied for asynchronous events in Sr2+. These events were detected over a time window (20–300 ms poststimulus). Two-population Kolmogorov–Smirnov goodness-of-fit tests determined statistical significance in cumulative histograms. Data are expressed as means ± SEM. Student paired two-tailed t tests were used to calculate significance, unless otherwise noted.
Imaging.
Line-scan confocal imaging (500 Hz, modified Bio-Rad MRC 600 confocal scan head attached to a custom microscope, http://alford.bios.uic.edu/Research/software.html) was used for Ca2+ transient recording in presynaptic varicosities following single action potentials stimulated with somatic whole-cell electrodes. Alexa 594 hydrazide was excited at 568 nm. Fluo-5F was separately excited (488 nm) and imaged in bandpass (510–560 nm). Images were taken separately to ensure no cross-channel bleed-through. This was confirmed with neurons filled with only one dye. No bleed-through image was discernible for either dye to the incorrect channel. Varicosities were identified 20–35 min after whole-cell access by imaging the Alexa 594 hydrazide dye and tracking the axon from the filled soma to its projection into the subiculum. Ca2+ transients at these varicosities were imaged in line scanning mode (500 Hz) for up to 1 s during stimuli to the soma to evoke action potentials. Image analysis was performed within ImageJ except anatomical reconstructions (VoxBlast; VayTek). Line scan images are represented as linear with the applied LUT mapping.
Gβγ competition binding assay.
The open reading frames for the SNARE component proteins were subcloned into the pGEX6p1 vector (GE Healthcare) for expression in bacteria as previously described (Yoon et al., 2007). To purify SNARE proteins, recombinant bacterially expressed glutathione S-transferase fusion proteins were expressed in Escherichia coli strain BL21(DE3). Protein expression was induced with 0.1 mm isopropyl β-d-thiogalactoside for 16 h at room temperature. Bacterial cultures were pelleted, washed with 1× PBS, and then resuspended in lysis buffer [25 mm HEPES-KOH, pH 8.0, 150 mm KCl, 1 mm EDTA, 0.2 mm PMSF, 10.7 μm leupeptin, 1.5 μm aprotinin, 1 μm pepstatin and 5 mm β-mercaptoethanol (BME)]. Cells were lysed with a sonic dismembrator at 4°C, 50% duty cycle, 20 s on, 40 s off, for 5 min total. GST-SNAP-25 and GST-H3 (the H3 domain of syntaxin1A) were purified from cleared lysates by affinity chromatography on glutathione Sepharose beads (GE Healthcare), following the manufacturer's instructions. While the proteins were bound to the beads, the beads were batch washed with two bed volumes of a buffer containing 25 mm HEPES-KOH, pH 8.0, 150 mm KCl, 1 mm EDTA, 1% Triton X-100, and 5 mm BME for 5 min at 4°C. This wash buffer was then exchanged with two bed volumes of a protease buffer containing 25 mm HEPES, pH 8.0, 150 mm KCl, 0.5% n-octyl β-d-glucopyranoside (OG), 10% glycerol, 1 mm EDTA, and 5 mm BME in a fresh tube for 5 min at 4°C. For GST-SNAP-25 on beads, the SNAP-25 was eluted by cleaving from GST with PreScission protease (GE Healthcare) overnight at 4°C in one bed volume of the protease buffer. Protein concentrations were determined with a Bradford assay kit (Pierce), and purity was verified by SDS/PAGE analysis.
For binary t-SNARE complex assembly a molar excess of 4 μm SNAP-25 was mixed with 3 μm GST-H3 on glutathione Sepharose beads in a buffer containing 25 mm HEPES, pH 8.0, 150 mm KCl, 0.25% OG, 10% glycerol, 5 mm BME, and 1 mm EDTA overnight at 4°C with gentle mixing. The binary t-SNARE complex was washed three times with protease buffer and eluted from the column by GST proteolytic cleavage with PreScission protease (GE Healthcare) for 4 h at 4°C as described above. Equimolar protein–protein interaction was confirmed by SDS-PAGE/Coomassie staining analysis.
Gβ1γ1 was purified from bovine retina as previously described (Mazzoni et al., 1991). Fluorescence labeling of Gβ1γ1 and binding assays were conducted as described previously (Phillips and Cerione, 1991). Briefly, purified Gβ1γ1 was dialyzed into labeling buffer (20 mm HEPES, pH 7.4, 5 mm MgCl2, 150 mm NaCl, and 10% Glycerol), then mixed with 2-(4′-Maleimidylanilino) naphthalene-6-sulfonic acid (MIANS) in a fivefold molar excess. The reaction proceeded for 3 h at 4°C before quenching with 5 mm 2-mercaptoethanol. The MIANS-Gβ1γ1 complex was separated from unreacted MIANS using a PD-10 desalting column (GE Healthcare). MIANS-Gβ1γ1 was stored in aliquots at −80°C.
Fluorescence measurements were performed in a fluorescence spectrophotometer (Cary Eclipse) at room temperature. Generally, MIANS-Gβ1γ1 was diluted into 0.1 ml of assay buffer [20 mm HEPES, pH 7.5, 5 mm MgCl2, 1 mm dithiothreitol, 0.1 m NaCl, 1 mm EDTA] to a final concentration of 20 nm. The MIANS fluorescence was monitored at an excitation wavelength of 322 nm and emission of 417 nm. Fluorescence was normalized by expressing all data as (F1–F0)/F0 (where baseline Gβγ-MIANS fluorescence mixed with t-SNARE = F0, fluorescence after addition of synaptotagmin I = F1) All proteins purified as GST fusion proteins were cleaved from GST with PreScission protease (GE Healthcare) before analysis. The fluorescent changes caused by the addition of SNARE complexes were monitored continuously. Note that the amplitude of the fluorescence increase is not a measure of the affinity of the complex, but rather reflects the specific site on fluorescently labeled Gβγ of interaction with each protein. There was no nonspecific binding of the free probe to the SNARE proteins and MIANS-Gβγ was resistant to photobleaching under experimental conditions (data not shown). For Gβγ/synaptotagmin competition assays, various concentrations of synaptotagmin with SNARE proteins were added to labeled Gβ1γ1 with the noted Ca2+or Sr2+ concentrations, and fluorescence changes were monitored. EC50 concentrations were determined by sigmoidal curve fitting with GraphPad Prism.
Results
Presynaptic GABAB and 5-HT1B receptors inhibit CA1-subicular synaptic transmission
To demonstrate that GABABRs and 5-HT1BRs inhibit neurotransmitter release from the same synapses, we stimulated CA1 pyramidal neuron axons in slices (<20 μA, 200 μs) and recorded postsynaptic responses in whole-cell voltage-clamped subicular pyramidal neurons (Fig. 1a). The GABAAR and NMDA receptor antagonists (bicuculline, 5 μm; d-2 amino-5-phosphonopentanoate, d-AP5, 50 μm respectively) were applied to isolate AMPA receptor (AMPAR)-mediated EPSCs. We applied the selective 5-HT1BR agonist CP93129 during whole-cell recordings, and the EPSCs were inhibited in a dose-dependent manner (Fig. 1b, CP93129; 50 nm inhibited to 55 ± 4% n = 13; 400 nm to 37 ± 3% p < 0.01, n = 14; all significant p < 0.01). The GABABR agonist, baclofen, similarly inhibited EPSCs profoundly (Fig. 1c; 1 μm to 48 ± 9% n = 7; 10 μm to 9 ± 2% n = 5; 100 μm to 7 ± 3%; all results significant, p < 0.01). Baclofen and CP93129 each inhibited synaptic transmission in every recording.
Figure 1.
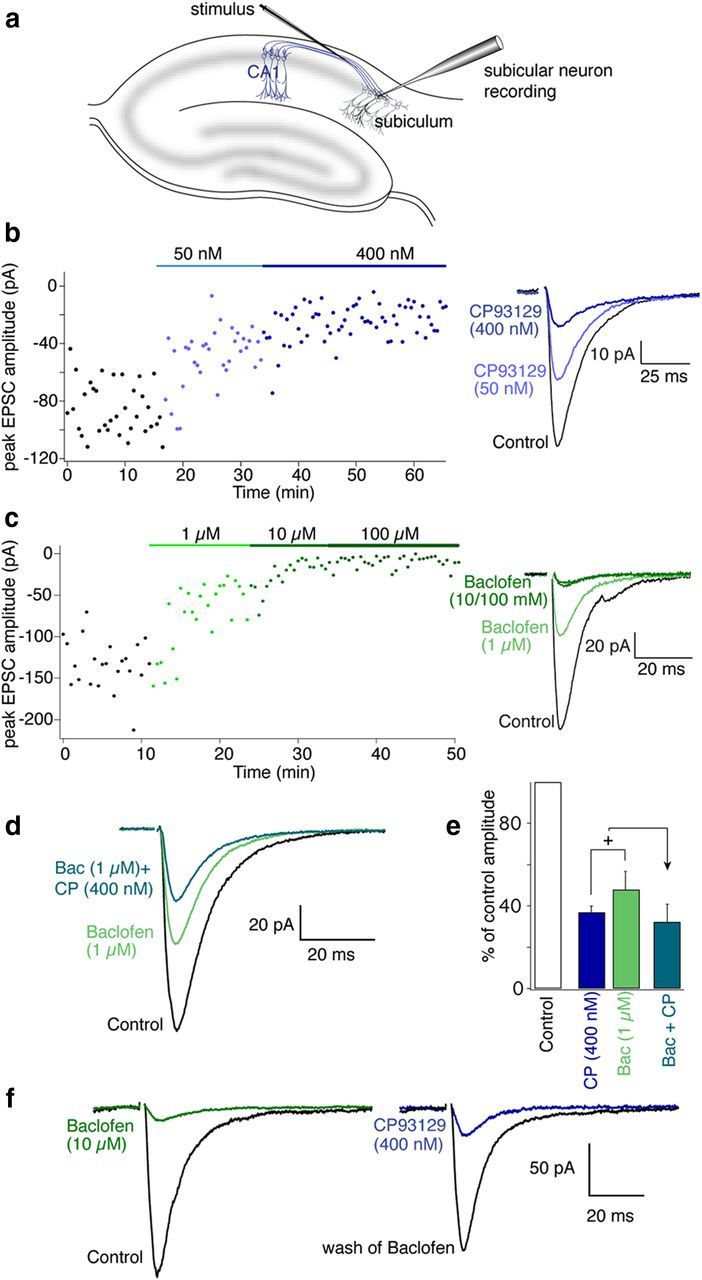
5-HT1B and GABAB receptors inhibit synaptic transmission at CA1-subicular synapses. a, CA1 pyramidal axons were stimulated close to the CA1- subiculum boundary with a glass-coated tungsten microelectrode. Subicular pyramidal neurons were recorded in whole-cell voltage-clamp mode. b, Plot of peak EPSC amplitude with time in Control (black), 50 nm CP93129 (light blue), and 400 nm CP93129 dark blue (stimuli at 30 s intervals). Right inset, Means of last 10 EPSCs in each condition from graph. c, Plot of peak EPSC amplitude in Control (black) baclofen (1, 10, and 100 μm; green). Right inset, Means of last 10 EPSCs in each condition. d, Means of 10 EPSCs in Control (black), baclofen (1 μm; green), and baclofen (1 μm) plus CP93129 (400 nm, blue/green). e, Histogram comparing mean inhibition by CP93129 and baclofen against the same doses combined indicating convergence of the receptor targets on the same synapses. f, Means of 10 EPSCs (left traces) in Control (black), baclofen (10 μm; green), after wash from baclofen (gray), and CP93129 (1 μm; blue).
Further supporting a conclusion that 5-HT1BRs and GABABRs are functionally colocalized to the same terminals, the combination of intermediate concentrations of baclofen and addition of CP93129 did not cause a linearly additive inhibition of responses. Instead, responses remaining in baclofen (1 μm) were inhibited by CP93129 (400 nm) significantly less effectively than in control (in 1 μm baclofen, 400 nm CP93129 reduced the response to 67 ± 9% of the response in baclofen; significantly less than the effect of CP93129 against control, p < 0.05; Fig. 1d,e; n = 7) This limited capacity to sum inhibitory effects indicates a convergence of targets; specifically that both receptors exist on the same presynaptic terminals. To confirm this, a further three experiments were performed in which a saturating dose of baclofen (10 μm) was applied to inhibit an evoked EPSC, baclofen was washed from the superfusate, and a saturating dose of CP93129 (1 μm) was applied (Fig. 1f). Baclofen inhibited the response to 11 ± 5% of control; CP93129 subsequently inhibited the response to 24 ± 8% of amplitude after wash of baclofen effects of both CP93129 and baclofen were significant (p < 0.01). Because both agonists can inhibit the response by >50% these receptors must be present on the same presynaptic terminals.
GPCR activation and presynaptic Ca2+ entry
Modulators of presynaptic Ca2+ channels are key regulators of neurotransmission and GPCRs modulate presynaptic Ca2+ entry (Miller, 1998; Brown and Sihra, 2008). Therefore, we measured presynaptic Ca2+ transients in single presynaptic varicosities of CA1 neurons projecting to neurons in the subiculum, the main hippocampal output. To image single presynaptic varicosities, CA1 neurons were recorded in current clamp with patch electrodes containing Alexa 594 hydrazide (Alexa, 250 μm) and a low affinity Ca2+ indicator, Fluo-5F (200 μm). After ∼30 min of dye diffusion, cells were imaged and axons tracked to the subiculum using the red fluorescent dye Alexa 594 (Fig. 2a). Single action potentials evoked by 2 ms depolarizing current pulses through the recording patch pipette caused presynaptic Ca2+ transients in axon varicosities, which were line scanned while confocally imaging Fluo-5F (Fig. 2b–e). To ensure that Ca2+ transients were unaffected by increasing dye/buffer concentration as it diffused into the axon, we recorded transients as the dye concentration increased. Values of baseline and peak increased (Fig. 2b–d,f) as the concentration of dye at the synapse increased (Fig. 2f,j), but signals expressed as ΔF/F remained constant with no change in peak amplitude of the response (Fig. 2e,i) or of decay rate (Fig. 2e,h), which is very sensitive to dye buffering capacity (Neher and Augustine, 1992; Jackson and Redman, 2003). Thus, at 200 μm, Fluo-5F buffering capacity is minimal compared with endogenous buffering and stably records action potential-induced presynaptic Ca2+ transients (n = 6 preparations).
Figure 2.
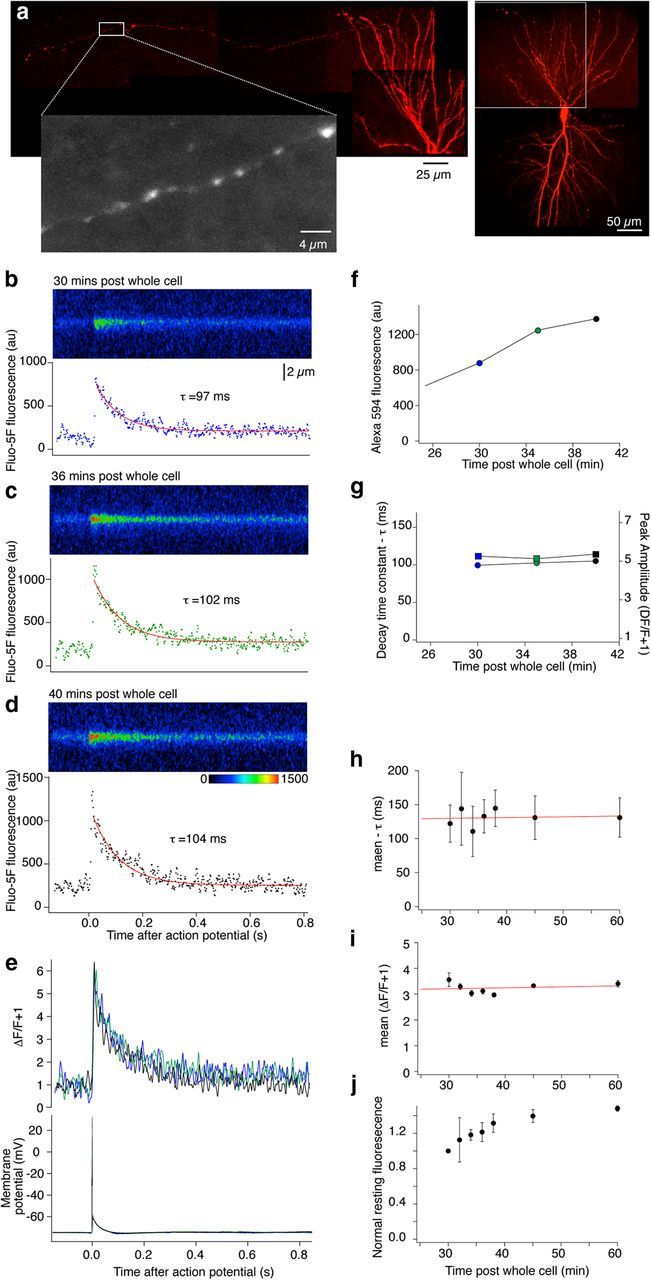
Ca2+ transients are reliably recorded in CA1 pyramidal neuron presynaptic terminals within the subiculum following labeling from a whole-cell electrode on the soma. a, Composite image of CA1 neuron reconstructed from 3D stacks obtained by scanning confocally. The neuron was filled with dyes from the electrode: Alexa (250 μm) to label the neuron (shown in red) and to trace axons to subiculum to identify presynaptic varicosities (expanded, grayscale) and Fluo-5F (200 μm) for Ca2+ measurements. b, After identifying a presynaptic varicosity the neuron was stimulated with single action potentials and individual Ca2+ transient responses were obtained by line scanning one terminal varicosity. No responses could be recorded from axons between the varicosities. Responses were recorded over time as the dye concentration rose by diffusion into the presynaptic varicosities (c, d). The integrated signals were calculated from these regions (graphs below line scan images). Peak values of signal intensity and rate of response decay were obtained from single exponential fits to these data. e, The data in b–d were expressed as (ΔF/F + 1) and overlaid. Colors from b–d. f, Graph demonstrating rise in intensity of Alexa 594 in this neuron's terminal over the time course of the experiment as the dye diffused into the terminal varicosities. g, Graph of decay rate (τ, left axis) and peak value of (ΔF/F + 1) for this presynaptic varicosity during the experiment. h, Mean decay rate (τ) and (i) the mean peak (ΔF/F + 1) during the course of the experiments (n = 6 preparations). j, Graph showing rise in intensity of Alexa 594 signal normalized to the first time point of recording in all terminals (same 6 preparations as h and i).
After recording control Ca2+ transients, we applied a saturating dose of CP93129 (1 μm, which reduces synaptic responses to <25% of control amplitudes), and imaged single action potential-evoked presynaptic Ca2+ transients as above. CP93129 altered neither Ca2+ entry (mean transient in CP93129 was 96 ± 7% of control; Fig. 3a–d; n = 6) nor electrophysiological properties of the neuron. Indeed, because we resolved individual Ca2+ responses to single action potentials in individual presynaptic terminals, it is clear that no modulation of presynaptic Ca2+ responses occurs and that no loss of action potential propagation was recorded. Sequences of individual response amplitudes from six neurons are shown (Fig. 3d) with no change in variance after CP93129 application (mean variances before and after CP93129 were 0.037 ± 0.012 and 0.038 ± 0.013 (ΔF/F)2).
Figure 3.
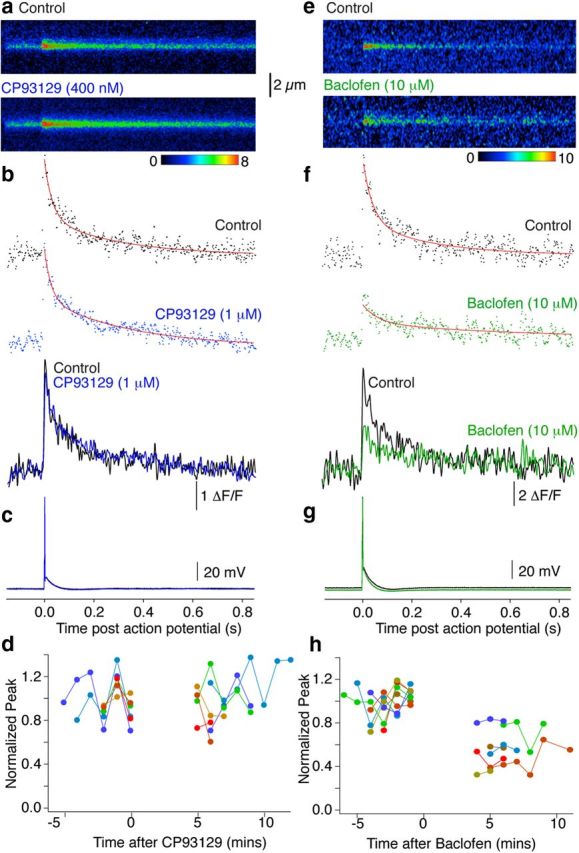
Differential inhibition of Ca2+ transients by 5-HT1BRs and GABABRs. a, Line scans (as in Fig. 2) from individual varicosities in the subiculum to resolve action potential-evoked (c) presynaptic Ca2+ transients (mean 3 responses in Control and 3 in CP93129; 1 μm). b, Signals were integrated for the sweeps over the terminal varicosities and amplitudes expressed as ΔF/F (Control, black; in CP93129, blue). Peak amplitudes were calculated from double exponential fits (b, red). Traces overlaid (below) show no effect of CP93129 application (time base identical, a–c). d, Peak amplitudes normalized to the mean Control amplitude before drug application were plotted against time for all cells. Each data point is the response to a single action potential in a single terminal varicosity. Colors represent results from individual terminals (n = 6). e–g, In another neuron baclofen (10 μm) imaged and stimulated under the same conditions as above significantly reduced Ca2+ transient amplitudes. h, Amplitudes of individual action potential-evoked Ca2+ transients demonstrate that in all seven neurons, although individual responses were reduced in amplitude in baclofen, failure of responses never occurred.
We performed similar experiments with the GABABR agonist baclofen (Fig. 3e–h). Baclofen slightly hyperpolarized the neuron (Fig. 3g), however, stimuli always resulted in action potentials. Baclofen inhibited presynaptic Ca2+ transients (to 56 ± 7% of control, p < 0.05; Fig. 3e,f; n = 7). Each of the Ca2+ transient events were recorded from one action potential in one presynaptic terminal, thus we can be certain that the Ca2+ inhibition recorded was not due to action potential failure in a subset of axons, because each action potential evoked a corresponding presynaptic Ca2+ transient. Indeed, in baclofen, no failure of presynaptic Ca2+ transients was recorded; rather we recorded a reduction in amplitude of each evoked response (Fig. 3h). While both baclofen and CP93129 inhibit exocytosis at this synapse, baclofen inhibits presynaptic Ca2+ entry, whereas CP93129 does not.
The presynaptic target of 5-HT1B receptors: spontaneous release
Inhibition of presynaptic Ca2+ entry by baclofen adequately explains its presynaptic inhibition should a fourth power relationship couple Ca2+ entry to neurotransmitter release (Dodge and Rahamimoff, 1967). Thus, if we take the fourth power of each of the Ca2+ signals after baclofen (10 μm) these would predict a reduction of the EPSC to 15 ± 7% of control. This is not significantly different from the observed reduction of the EPSC amplitude recorded in baclofen (9 ± 2% of control; 10 μm; Fig. 1).
In contrast, 5-HT1BRs must inhibit neurotransmitter release by another mechanism. The equivalent fourth power of the Ca2+ signal caused by CP93129 was to just 93 ± 20% of control (significantly different from the effect on synaptic transmission; p < 0.05). Thus, 5-HT1BRs must inhibit release by a mechanism that does not cause a reduction in presynaptic Ca2+ entry. One such mechanism was first identified at the neuromuscular junction, where adenosine inhibits ACh release by a Ca2+-independent mechanism that leads also to significant reductions in spontaneous neurotransmitter release (Silinsky, 1984, 2008). Thus, we tested whether 5-HT1BRs inhibit spontaneous release. mEPSCs were recorded in subicular neurons in tetrodotoxin (TTX; 1 μm) to prevent evoked release, and d-AP5 (50 μm) and bicuculline (5 μm) to isolate AMPAR-mediated EPSCs before and during application of CP93129 (400 nm; n = 6; Fig. 4a–d). Events were detected as indicated (see Materials and Methods) and showed a very similar kinetic profile to evoked events (Fig. 4e). The mean amplitudes and kinetic profiles of these events were compared before and during application of CP93129 (400 nm). CP93129 had no effect on amplitude (mean mEPSC amplitude in CP93129 was 99 ± 6% of control) or kinetic profile of the events (Fig. 4f), demonstrating that the effect of agonist was not postsynaptic. Furthermore, the amplitude distribution and cumulative amplitude distributions (Fig. 4g) before and in CP93129 were identical. Similarly the agonist had no effect on the frequency distribution or cumulative frequency distribution of the recorded events (frequency of events in CP93129 was 116 ± 21% of control; Fig. 4h, significance of distributions was tested with two-sample Kolmogorov–Smirnov tests). Thus, in contrast to neuromuscular effects of adenosine (Silinsky, 1984), 5-HT1BRs do not affect mEPSCs.
Figure 4.

Neither 5-HT1BRs nor GABABRs alter spontaneous release in the subiculum. a, Spontaneous events recorded in whole-cell voltage-clamped subicular pyramidal neurons. Recordings were in TTX, bicuculline and AP-5 to isolate AMPAR-mediated mEPSCs. b, The 5-HT1BR agonist, CP93129 (400 nm), was applied. c, d, Examples of detected events in a and b. e, The kinetic profile of averaged detected mEPSCs (gray) was compared with evoked responses obtained before addition of TTX in the same neurons (black). Events were scaled the same arbitrary amplitudes. f, Means of mEPSCs detected in Control (black) and in CP93129 (gray). The agonist altered neither amplitude nor kinetic profile. g, Mean event amplitude histograms were plotted for all cells examined before (gray) and after CP93129 (white). Bars indicate SEMs obtained comparing across preparations (n = 5). Cumulative histograms are plotted below (Control, black; CP93129, gray). SEMs represented as Control (black dashed line above curves) and in CP93129 (gray dashed line below curve). h, Event frequency distribution curves for the same datasets as g. Grayscale and SEMs marked in the same way. i, Spontaneous events were recorded as in a. j, The GABABR agonist, baclofen (1 μm), was applied. k, l, Examples of detected events in i and j. m, Means of mEPSCs detected in Control (black) and in baclofen (gray). The agonist altered neither amplitude nor kinetic profile. n, Mean event amplitude histograms were plotted in Control and baclofen (10 μm) as for g. o, Event frequency distribution histograms for the same datasets as in n.
The lack of effect of 5-HT1BR activation on spontaneous release is consistent with an inhibitory effect targeting an aspect of Ca2+-evoked exocytosis. As a comparison similar experiments were performed on spontaneous release during the application of baclofen. Spontaneous mEPSCs were again recorded in subicular neurons in TTX (1 μm), d-AP5 (50 μm), and bicuculline (5 μm) to isolate AMPAR-mediated EPSCs before and during application of baclofen (10 μm; n = 5; Fig. 4i–l). The mean amplitudes and kinetic profiles of these events were compared before and during application of baclofen, which had no effect on amplitude (mean mEPSC amplitude in baclofen was 106 ± 17% of control) or kinetic profile of the events (Fig. 4m) or on the amplitude distribution and cumulative amplitude distributions (Fig. 4n). Similarly baclofen had no effect on the frequency distribution or cumulative frequency distribution of the recorded events (frequency of events in baclofen was 94 ± 22% of control; Fig. 4o; significance of distributions were tested with two-sample Kolmogorov–Smirnov tests). Thus, neither GABABRs nor 5-HT1BRs modify spontaneous release, which occurs independently of Ca2+-entry through Ca2+ channels and is likely activated by Doc2 rather than synaptotagmin I (Groffen et al., 2010; Pang et al., 2011).
The presynaptic target of 5-HT1B receptors: asynchronous release
Baclofen may clearly inhibit neurotransmitter release by reducing presynaptic Ca2+ entry. However, CP93129 and 5-HT1BRs do not modify presynaptic Ca2+ signals and yet only act on Ca2+-evoked release. A second such mechanism by which GPCRs inhibit neurotransmitter release with no inhibitory effect on presynaptic Ca2+ entry has been identified in lamprey synapses (Blackmer et al., 2001; Gerachshenko et al., 2005), in amygdala (Delaney et al., 2007), and in chromaffin cells (Blackmer et al., 2005). Gβγ inhibits release by a competitive interaction with synaptotagmin I at the C-terminal region of the SNARE complex (Yoon et al., 2007). Synaptotagmin binds Ca2+ to evoke vesicle fusion, and Ca2+-synaptotagmin I competes with Gβγ at the C-terminal region of the SNARE complex in vitro, and in lamprey synapses (Blackmer et al., 2005; Gerachshenko et al., 2005; Yoon et al., 2007). Substitution of Sr2+ for Ca2+ in the superfusate provides an approach to identifying whether Gβγ interacts with this C-terminal region of the SNARE complex.
Sr2+ evokes asynchronous vesicle fusion following evoked entry through Ca2+ channels (Mellow et al., 1978) but does not cause synaptotagmin to interact with SNARE complexes (Shin et al., 2003; Bhalla et al., 2005). Given that Sr2+ enters through Ca2+ channels and yet does not cause synaptotagmin SNARE interactions, it provides an independent approach to determine whether GPCRs act at Ca2+ channels or via synaptotagmin I/SNARE complex interactions. Thus, we first determined whether Sr2+-synaptotagmin I competes with Gβγ at SNARE complexes using a sensitive quantitative in vitro fluorescence assay (Phillips and Cerione, 1991; Yoon et al., 2007). Gβ1γ1 (20 nm assay concentration) was labeled with an environmentally sensitive probe, MIANS, which shows enhanced fluorescence in more hydrophobic local conditions, indicating interaction with a binding partner. The assay was used to quantify synaptotagmin I C2AB fragment competition with MIANS-labeled Gβγ for binding to t-SNARE complexes comprising the syntaxin1A H3 domain and SNAP-25 in increasing concentrations of either Sr2+ or Ca2+. We used a synaptotagmin I mutant (K326A, K327A) that exhibits wild-type SNARE binding affinity but which has a reduced propensity to oligomerize (Bai et al., 2004). MIANS-Gβγ fluorescence increases following interaction with t-SNARE complexes, but this increase is significantly reduced by 20 nm synaptotagmin I when applied in the presence of Ca2+ (Yoon et al., 2007). As a control, MIANS-Gβγ does not show significant interactions with C2AB alone. Thus, we tested competition between Gβγ and C2AB at t-SNARE complexes substituting Sr2+ for Ca2+. The interaction between 20 nm MIANS-Gβγ and 1 μm t-SNARE complex was only slightly reduced by 20 nm C2AB in increasing Sr2+ concentrations up to 250 μm (Fig. 5a; normalized fluorescence reduced to 69.8 ± 4.0% of the response in 0 Sr2+). To confirm earlier results, the experiment was repeated with Ca2+ and the effect with Sr2+ was substantially and significantly less than that seen for 250 μm Ca2+ (Fig. 5b; normalized fluorescence reduced to 5.8 ± 8.9% of response in 0 Ca2+, p < 0.01 compared with effect of Sr2+).
Figure 5.
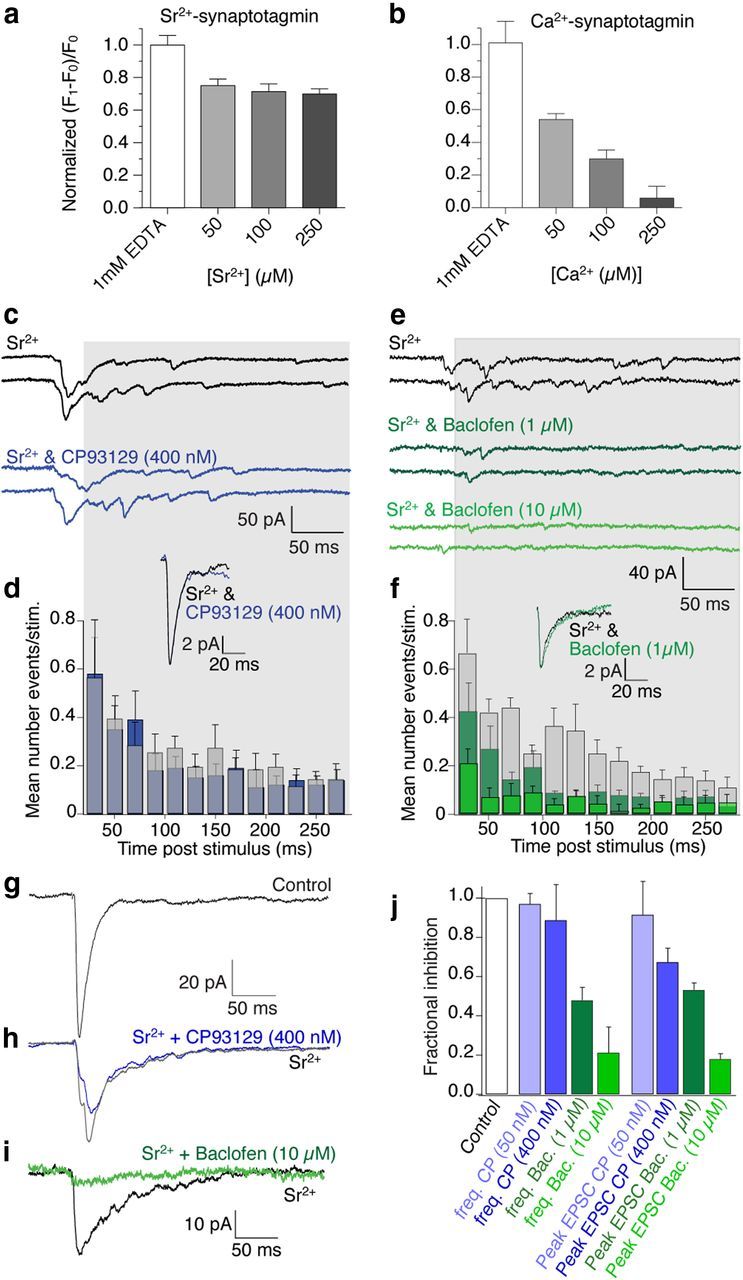
GABABRs but not 5-HT1BRs inhibit Sr2+-evoked neurotransmission. a, Graphs of normalized fluorescence of MIANS-labeled Gβγ bound to t-SNARE complexes in vitro. Synaptotagmin I in increasing [Sr2+] slightly reduces Gβγ binding to t-SNAREs. Binding of 1 μm t-SNARE to 20 nm MAINS-Gβγ was measured by fluorescence enhancement. b, In contrast, synaptotagmin I in Ca2+ substantially reduced fluorescence [Ca2+] dependently. c, Examples of Sr2+-evoked EPSCs in subicular pyramidal neurons and in CP93129 (400 nm; blue) following stimulation of CA1 pyramidal neuron axons. d, Frequency distribution from gray region across traces in c; mean number of events/stimulus from all neurons of 40 stimuli in each neuron in Sr2+ and in Sr2+ plus CP93129 (blue). Inset shows mean of detected asynchronous events in Control and in CP93129 showing no effect on detected event amplitudes or kinetic profiles. e, Sr2+-evoked EPSCs in another cell (Control, black) and in baclofen (1 μm; dark green and 10 μm; green). f, Frequency distribution from gray region across traces in e plotted in Control (black) and baclofen (1 μm; dark green and 10 μm; green). Data are from all examined neurons. Inset shows mean of detected asynchronous events in Control and in baclofen showing no effect on detected event amplitudes or kinetic profiles even as the frequency of events was reduced. g, Mean of EPSCs in Ca2+ (Control). h, Sr2+ (gray), Sr2+ + CP93129 (blue; 400 nm). i, Mean of EPSCs in Sr2+ (black) and Sr2+ + baclofen (10 μm; green). j, Summary of CP93129 effects (50 nm; n = 3 and 400 nm; n = 4) and baclofen (1 μm; n = 5 and 10 μm; n = 4) against frequencies and peak amplitudes in Sr2+.
Thus, unlike Ca2+-synaptotagmin, Sr2+-synaptotagmin does not compete with Gβγ to inhibit fusion, and while Sr2+ entry to the neuron will be modified by GPCRs acting at Ca2+ channels, Gβγ targeting the SNARE complex cannot modify the effects of Sr2+ because there is no subsequent competition between Gβγ and Sr2+-synaptotagmin at the SNARE complex Figure 5a). During recordings from subicular neurons in hippocampal slices, we replaced extracellular Ca2+ with Sr2+. Synchronous EPSCs (stimulated as Fig. 1) were reduced in amplitude while asynchronous events typical of Sr2+ (Mellow et al., 1982) were recorded (Fig. 5c,d). Asynchronous events were detected from 20 ms poststimulus to the end of the sweep (Fig. 5c,d, shading; 40 control sweeps, 40 CP93129 sweeps). Time/frequency histograms of mean frequencies of events for all neurons revealed the event frequency distribution. CP93129 had no effect on this distribution nor on overall event frequency (frequency in CP93129, was 97 ± 5% at 50 nm and 88 ± 12% at 400 nm events in Sr2+ alone; Fig. 5d; n = 7). Averaging detected events revealed that neither amplitudes nor kinetics of Sr2+-evoked asynchronous events were affected by CP93129 (Fig. 5d, inset).
Baclofen substantially reduces presynaptic Ca2+ entry to CA1 neuron terminals in the subiculum. Consequently, it may be used as a positive control to demonstrate the effect of inhibiting Ca2+ channels on Sr2+-evoked asynchronous events, because if Ca2+ channels are inhibited their permeation by Sr2+ will be correspondingly reduced. In contrast to CP93129, baclofen (1 μm) substantially reduced Sr2+-evoked event frequencies (to 47 ± 6%, n = 5) and nearly eliminated them at a higher concentration (10 μm; Fig. 5e,f; to 21 ± 12%, n = 4), but had no effect on kinetics (Fig. 5f, inset) or the mean amplitude of asynchronous events that were detected (Fig. 5f, inset).
Contrasting inhibitions by baclofen and CP93129 on Sr2+-evoked release are most notable on asynchronous events. Even so, CP93129-mediated inhibition of the early EPSC in Sr2+ was substantially less than in Ca2+ (EPSC was 92 ± 2% of peak in Sr2+ at 50 nm CP93129 and 67 ± 7% at 400 nm; Fig. 5 h; significantly less inhibition than in Ca2+, p < 0.01; reductions to 55 and 37%). In contrast to CP93129, inhibition by baclofen in Sr2+ was similar to that in Ca2+ (1 μm baclofen peak EPSC reduced to 53 ± 3% of amplitude in Sr2+, in 10 μm baclofen reduced to 18 ± 3%, n = 6; Fig. 5i). Even the small inhibition by CP93129 may be explained by residual Ca2+ that cannot be removed (Xu-Friedman and Regehr, 1999).
Similarly to effects on event frequencies, neither amplitudes nor the distribution of Sr2+-evoked asynchronous events were affected by CP93129. This was shown to help clarify that no postsynaptic effects of these receptors account for the observed inhibition. As for event frequency data (Fig. 5), event amplitudes were plotted against time in a window from 20 to 300 ms after the stimulus (Fig. 6a–d; analysis of same data as Fig. 5; in all seven neurons mean event amplitude in 400 nm CP93129 was 108 ± 5% of event amplitude in Sr2+ alone). Time/amplitude histograms and cumulative plots of mean event amplitudes for all neurons were plotted to show no effect on the amplitude distribution of events by CP93129.
Figure 6.
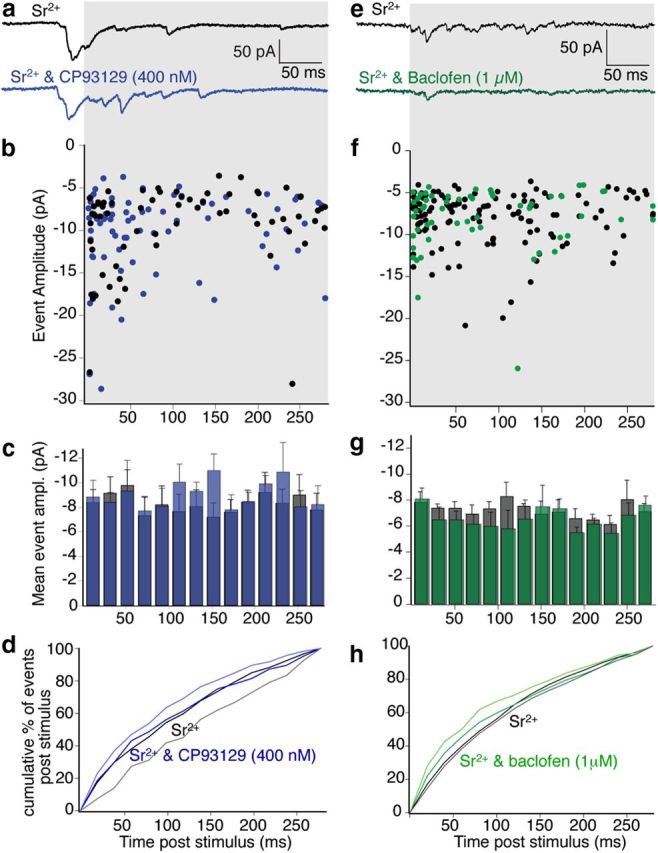
Asynchronous event amplitude distributions for evoked events in Sr2+ were not affected either by 5-HT1B or GABABR activation. a, Examples of Sr2+-evoked EPSCs in subicular pyramidal neurons and in CP93129 (400 nm; blue) following stimulation of CA1 pyramidal neuron axons (as for Fig. 5; to indicate relative timing of events in the sweep). b, Event amplitudes from the same 40 sweeps used for frequency distribution (Fig. 5) plotted against time of detection poststimulus in Control (black) and CP93129 (blue) during the region of the traces shown in gray background. c, Amplitude distribution from gray region across traces in a; mean amplitude of events from 40 stimuli in Sr2+ (gray) and in Sr2+ plus CP93129 (blue). d, Cumulative amplitude distribution of data plotted in histogram. Sr2+ (black) and Sr2+ and CP93129 (blue) counting events from the first to last bin in the sweep. Cumulative SEs are plotted for Sr2+ in (gray lower line) and Sr2+ and CP93129 (light blue line). (Data in c and d are from all recorded neurons.) e–h, Data obtained in Sr2+ and Sr2+ plus baclofen (1 μm) analyzed as for the data using CP93129. (Data in g and h are from all recorded neurons.)
Similar analysis was performed for neurons recorded in an intermediate baclofen dose (1 μm), at which sufficient numbers of events were detected. Event amplitudes were plotted across a similar time window in Sr2+ and after addition of baclofen (1 μm; Fig. 6e,f). Time/amplitude histograms and cumulative plots of mean event amplitudes for all neurons showed no effect on the amplitude distribution of events by baclofen (Fig. 6 g,h). The mean event amplitudes in baclofen (1 and 10 μm) were 93 ± 3 and 89 ± 5%, respectively, of the amplitudes in Sr2+ alone (n = 5 and 4, respectively). These data also indicate that neither agonist modified the postsynaptic response to detected events.
Spontaneous mEPSCs in Ca2+-containing Ringer (Fig. 4) were unaffected by CP93129 as were events in Sr2+. Furthermore, CP93129 was equally effective in inhibiting evoked EPSCs in Ca2+ when the postsynaptic patch pipette contained GDPβS to inactivate effects of postsynaptic GPCRs (data not shown). These results confirm that 5-HT1BRs act presynaptically. Critically, the Sr2+ data confirms the Ca2+ imaging data demonstrating that 5-HT1BRs do not modify presynaptic Ca2+ entry, but are instead consistent with 5-HT modifying synaptotagmin/SNARE complex interactions (Blackmer et al., 2001, 2005; Takahashi et al., 2001; Gerachshenko et al., 2005; Yoon et al., 2007) because Sr2+-synaptotagmin I does not interact with the SNARE complex (Fig. 5a) to cause exocytosis (Shin et al., 2003; Bhalla et al., 2005).
The presynaptic target of 5-HT1B receptors: BoNT/A cleavage of C-terminal SNAP-25
Because Sr2+ does not recruit synaptotagmin I to evoke release and Sr2+-synaptotagmin does not compete effectively with Gβγ at SNARE complexes, it is likely that 5-HT1BRs modify fusion by targeting the SNARE complex C-terminal region. Consequently, we investigated how 5-HT1BR signal transduction interacts with this region of the SNARE complex. BoNT/A reduces presynaptic inhibition mediated by 5-HT via Gβγ in lamprey (Gerachshenko et al., 2005), noradrenaline in amygdala (Delaney et al., 2007), and Gβγ in PC12 cells (Blackmer et al., 2005). BoNT/A cleavage of the C terminus of SNAP-25 favors synaptotagmin I/SNARE interactions over those with Gβγ (Yoon et al., 2007) without preventing SNARE complex formation. Thus, we determined the effects of BoNT/A on CP93129-mediated presynaptic inhibition.
Hippocampal slices were incubated (2–4 h) in light/heavy chain BoNT/A (1–2 nm) to cleave the C-terminal 9 residues from SNAP-25. It remains unclear whether SNARE complex formation during priming might protect SNAP-25 from BoNT/A (Xu et al., 1998), thus, we eliminated any residual primed vesicles unaffected by BoNT/A during the incubation period. Subicular pyramidal neurons were recorded as in Figure 1 and stimuli were applied to CA1 axons. Residual primed vesicles were eliminated by stimulation (400 stimuli, 1 Hz; Fig. 7a). EPSCs were markedly depressed during the 1 Hz stimulation period, but partially recovered, albeit to a still significantly inhibited response (66.7 ± 1% of control, p < 0.05, n = 5; Fig. 7a) when stimulation was slowed to 1/15 Hz. CP93129 applied to these reduced responses caused much less inhibition, indicating that 5-HT1BR effectors target the SNARE complex (EPSC amplitudes were 77 ± 5% of post 1 Hz amplitude; n = 5, 50 nm, and 67 ± 1%, 400 nm n = 5; Fig. 7a,b). This inhibition was significantly less than that seen in control recordings (Fig. 7e).
Figure 7.

BoNT/A treatment substantially reduces 5-HT1B receptor-mediated inhibition. a, Graph of EPSC amplitudes recorded in subicular pyramidal neurons and stimulation of CA1 pyramidal neuron axons after BoNT/A pretreatment. Four hundred stimuli (1 Hz; open circles) eliminated pre-BoNT/A primed vesicles. Short-term depression from initial amplitude partially recovered at 1/15 Hz stimulation (filled circles). CP93129 (400 nm) was then applied (gray filled circles). b, Mean of EPSCs before (black, thin line) and post 1 Hz stimulation (black) in CP93129 (gray) after BoNT/A treatment. c, EPSCs were recorded and evoked as in a and slices treated identically except BoNT/A toxin was not added to the media. Evoked EPSC amplitudes are graphed during 1 Hz stimulation for 400 stimuli (○). After a period of recovery at low frequency (1/15 Hz, black) CP93129 (gray) caused inhibition as in Control slices with no period of 1 Hz stimulation. d, Means of EPSCs before 1 Hz stimulation (Control, thin black), after recovery from this stimulation (post 1 Hz, black), and during CP93129 application (gray). e, Summary of CP93129 effects on EPSC amplitudes for all cells in Control, (50, 400 nm; n = 13, 14), following BoNT/A, 1 Hz stimulation (50, 400 nm, n = 5), and following 1 Hz stimulation with no prior BoNT/A treatment (50, 400 nm, n = 3).
To ensure that the 1 Hz prestimulation did not compromise CP93129-mediated inhibition, slices were treated as controls without BoNT/A. Four hundred stimuli (1 Hz, applied as before) caused less short-term depression (Fig. 7c). EPSCs recovered to 82 ± 1% of control at 1/15 Hz stimulation (Fig. 7c,d). Subsequent application of CP9319 inhibited EPSCs to the same extent as it did in applications with no 1 Hz stimulation (for 50 and 400 nm to 46 ± 4 and 34 ± 6% of post 1 Hz amplitudes; Fig. 7c–e; n = 3). Thus, 5-HT1BR-mediated inhibition is curtailed following SNAP-25 cleavage with BoNT/A.
5-HT1BRs inhibit release to a much reduced extent after cleavage of the C-terminal 9 residues of SNAP-25 and the subsequent elimination of previously primed vesicles and their associated SNARE complexes. However, while most BoNTs cannot access their SNARE complex target proteins after the formation of ternary SNARE complexes during vesicle priming (Hayashi et al., 1994; Pellegrini et al., 1994), it is not clear whether this is true of BoNT/A. Indeed, following BoNT/A treatment, SNARE complexes can be formed, although with reduced efficacy at promoting vesicle fusion (Otto et al., 1995). It was possible to determine whether BoNT/A, like other BoNTs, fails to access primed vesicle SNAREs or whether BoNT/A treatment alone caused nonspecific damage to the synapses leading to a loss of 5-HT1BR-mediated effects. Slices were treated with BoNT/A as for Figure 7. However, after BoNT/A treatment, no prestimulus train was given, thus leaving the primed vesicle pool intact before application of agonist. After obtaining control-evoked EPSCs at 15 s intervals, CP93129 reduced responses as for controls (to 29 ± 3% at 400 nm; Fig. 8a,b). Thus, consistent with a final target of the 5-HT1BR on the primed vesicle (Gerachshenko et al., 2005), the readily releasable pool must be depleted after BoNT/A treatment in order for BoNT/A to prevent 5-HT1BR-mediated inhibition.
Figure 8.

BoNT/A does not prevent effects of 5-HT1B receptors on the previously primed vesicle pool. a, In slices pretreated with BoNT/A toxin as for Figure 7, EPSCs were evoked as above but at 1/15 Hz for 35 stimuli with no period of 1 Hz stimulation. In these cells with no loss of preprimed vesicle pool, CP93129 inhibited EPSCs by similar amounts to Control responses. b, Means of EPSCs after BoNT/A treatment (from a) but before drug application (BoNT/A treated, black), and during CP93129 application (gray).
From results using Ca2+ imaging, Sr2+-evoked asynchronous release, and BoNT/A it is clear that 5-HT1BRs target the SNARE complex to modify Ca2+-synaptotagmin binding, while GABABRs inhibit presynaptic Ca2+ entry.
Selective targeting of Gβγ effector targets and the SNARE complex
GABABRs and 5-HT1BRs are colocalized in the same synaptic pathway between CA1 and subicular pyramidal neurons but inhibit exocytosis by different mechanisms. These mechanisms nevertheless both use Gβγ subunits (Blackmer et al., 2001; Catterall and Few, 2008). Ca2+ channels may localize to the SNARE complex N terminus (Sheng et al., 1994, 1996), while 5-HT1BR-liberated Gβγ interacts with the SNARE complex C terminus (Blackmer et al., 2001, 2005; Yoon et al., 2007). It is interesting that 5-HT1BRs do not, under normal circumstances, modulate presynaptic Ca2+ while GABABRs do. We hypothesized that the SNARE complex separates receptor/effector complexes. To test this, we prevented SNARE complex formation by cleavage of syntaxin by applying BoNT/C (Hayashi et al., 1994).
Recordings were made from CA1 pyramidal neurons in current-clamp mode. Similarly to Figures 2 and 3, the electrodes were filled with Alexa 594 hydrazide (250 μm) and the Ca2+ indicator Fluo-5F (200 μm). In addition, the electrodes contained light chain BoNT/C (5 nm), which was infused into CA1 neurons with the dyes. After 35 min of whole-cell recording to allow diffusion of the toxin and the dyes throughout the neuron and its axon, 200 action potentials (1 Hz) were evoked by short depolarizing current pulses (2 ms) through the patch electrode. This was done to eliminate any residual primed vesicles whose SNARE complexes are known to prevent BoNT/C access to its target, syntaxin (Hayashi et al., 1994). Presynaptic varicosities were identified as in Figures 2 and 3 and subsequently line scanned with 488 nm excitation during single action potentials. The 5-HT1BR agonist CP93129 (1 μm; Fig. 9a,d) now significantly inhibited Ca2+ transients in all of the terminals recorded (to 52 ± 7% of control amplitude n = 8; not significantly different from the effect of baclofen under control conditions). Like baclofen, this inhibition was not due to a failure of action potentials to evoke presynaptic responses, because in all cases an identifiable Ca2+ transient was evoked by stimulation in single varicosities before and during application of CP93129. Thus, 5-HT1BRs inhibit Ca2+ entry after syntaxin cleavage.
Figure 9.
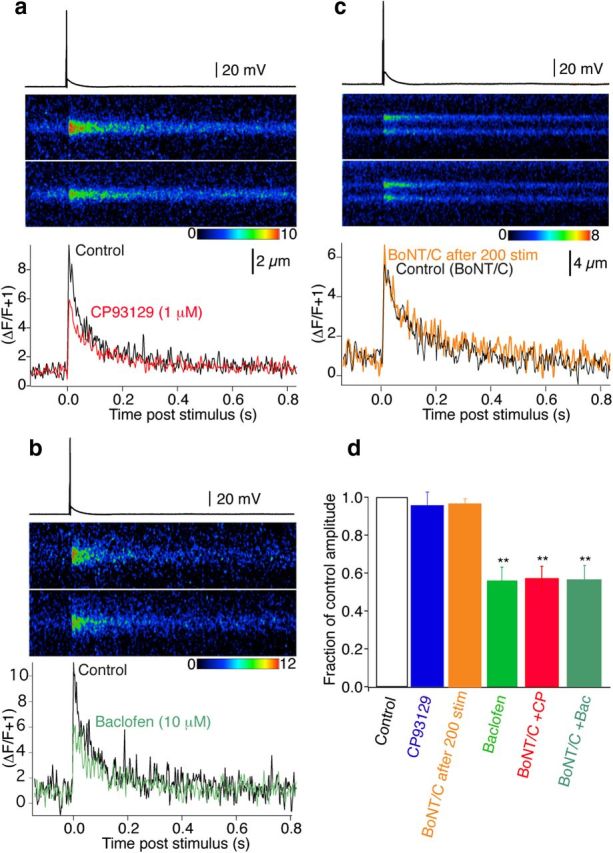
BoNT/C reveals 5-HT1B receptor-mediated inhibition of presynaptic Ca2+ transients. CA1 pyramidal neurons were recorded with electrodes containing BoNT/C, Alexa, and Fluo-5F. Two hundred action potentials (1 Hz, 35 min post whole cell) eliminated primed vesicles before recording. a, Action potential-evoked Ca2+ transients recorded by line scanning over CA1 pyramidal neuron presynaptic varicosities in the subiculum (top trace Control, bottom in CP93129; 1 μm). Graph (bottom) shows the integrated signals of three averaged responses in Control (black) and CP93129 (1 μm; red). b, Similar CA1 presynaptic varicosity recording before (Control, black) and in baclofen (10 μm; green) also after perfusion with BoNT/C. c, Similar CA1 presynaptic Ca2+ transient before (Control BoNT/C, black) following infusion of dyes and BoNT/C, and after 200 stimuli (BoNT/ after 200 stim, orange) Stimuli to exhaust the primed vesicle pool did not significantly alter the time course or amplitude of the transient. d, Summary of BoNT/C and GPCR effects on Ca2+ transients (orange, 200 stimuli after BoNT/C application, n = 3; blue, CP93129, n = 8; green, baclofen, n = 7) both in Control conditions. Then after priming eliminated with BoNT/C (red, CP93129, n = 5; dark green, baclofen, n = 4). BoNT/C revealed reduction of Ca2+ transients in CP93129. **p < 0.01.
It has been demonstrated that pre-existing Gβγ modulation of Ca2+ channels in expression systems is reduced by BoNT/C (Zamponi et al., 1997; Jarvis et al., 2000). It is important to consider that in the latter experiments only the SNARE complex proteins, plus Ca2+ channels and Gβγ, were present. Intact synapses, in contrast, represent a substantially more complex situation, in which BoNT/C in synapses cleaves syntaxin, but consequently prevents SNARE formation and further vesicle priming. Therefore, we determined whether pretreatment with BoNT/C modifies baclofen inhibition of Ca2+ transients. In cells infused with BoNT/C and stimulated (200 action potentials) identically to experiments with CP93129, baclofen (10 μm) inhibited presynaptic Ca2+ transients similarly to controls without BoNT/C (to 55 ± 6%, n = 5, compared with 56 ± 7% with no BoNT/C treatment; Fig. 9b,d).
To ensure that BoNT/C treatment and subsequent action potential-evoked depriming did not alter the Ca2+ transient amplitude, in a further three neurons recorded as above, Ca2+ transients were evoked after loading of the terminal but before application of 200 action potential at 1 Hz. This response was compared with that obtained after the 1 Hz period of stimulation to remove any primed vesicles. No significant alteration in the Ca2+ transient was seen (Fig. 9c,d; response amplitude after 1 Hz stimulation was 96 ± 3% of control. Decay rate of the transient was also similar: before 200 stimuli τ = 154 ± 29 ms, after 200 stimuli τ = 147 ± 30 ms).
In these presynaptic terminals, it is likely that GABABRs and 5-HT1BRs functionally colocalize within the same terminals. For imaging experiments, all recorded terminals (n = 12 in total) in which baclofen was applied alone showed inhibition of Ca2+ entry, while after BoNT/C treatment all eight tested terminals showed inhibition of Ca2+ entry by CP93129. The probability of randomly sampling only single receptor-expressing terminals would be extremely low (p = 0.0007 and p = 0.004, for GABABRs and 5-HT1BRs, respectively) indicating coexpression of GABAB and 5-HT1B receptors. Nevertheless, to confirm that both receptors are functional on the same presynaptic terminals four further Ca2+ imaging experiments were performed in which BoNT/C was applied to the recorded neurons to test efficacy of both GABABRs and 5-HT1BRs on the same terminal. Neurons were again recorded with electrodes containing BoNT/C, Fluo-5F, and Alexa 594 hydrazide as above. In two of these neurons after identifying presynaptic varicosities, control Ca2+ transients were recorded. Baclofen (1 μm) was applied, transients recorded, and baclofen washed from the superfusate (Baclofen reduced responses to 68 and 86% of control). Following wash 200 stimuli were applied to exhaust the primed vesicle pool. CP93129 (1 μm) was applied to test whether responses could be inhibited (responses were inhibited to 36 and 55% of the pre-CP93129 control response). In a further two experiments similar recordings were made. However, after identification of the presynaptic varicosity, 200 stimuli were immediately applied to exhaust the synaptic vesicle pool (Fig. 10a–c). Control Ca2+ transients were recorded and after CP93129 (1 μm) was applied the responses were reduced to 58 and 56% of control. CP93129 has proved very difficult to wash from the superfusate, thus a high dose of baclofen (100 μm) was applied to test whether a further reduction in the Ca2+ transient could be recorded. Baclofen reduced the remaining responses by a further 58 and 75% of the responses in CP93129 (Fig. 10a,b). Baclofen caused a slight hyperpolarization of the recorded neuron; however, this did not prevent the firing of a somatic action potential (Fig. 10c), and in each case this action potential evoked a measurable Ca2+ transient at the presynaptic varicosity, demonstrating no spike failure (Fig. 3h). The CP93129 and baclofen-dependent inhibition of Ca2+ transients at the same presynaptic varicosities confirms that GABABRs and 5-HT1BRs are found at the same synapse (Fig. 10d).
Figure 10.

5-HT1BRs and GABABRs are located on the same presynaptic terminals. CA1 pyramidal neurons were recorded with electrodes containing BoNT/C, Alexa, and Fluo-5F. Two hundred action potentials (1 Hz, 35 min post whole cell) eliminated primed vesicles before recording. a, Action potential-evoked Ca2+ transients recorded by line scanning over CA1 pyramidal neuron presynaptic varicosities in the subiculum. (top trace, Control; CP93129, bottom; 1 μm). To make clear the sequential reduction of Ca2+ transient amplitude the image data from over the varicosity is shown as ΔF/F in pseudocolor after background subtraction from the images. b, Graph shows the integrated signals of three averaged responses in Control (black), CP93129 (1 μm; red), and in CP93129 (1 μm) plus baclofen (100 μm; purple). c, Action potentials that evoked Ca2+ transients in a and b. d, Schematic of presynaptic GPCR targets. At primed vesicles 5-HT1BRs and their associated G-proteins interact with SNARE C terminals. GABABRs interact with Ca2+ channels. e, BoNT/C treatment prevents priming, freeing 5-HT1BRs to modulate Ca2+ channels similarly to GABABRs.
Discussion
GPCR-mediated inhibition of neurotransmitter release at presynaptic terminals (Starke, 1972) is considered ubiquitous (de Jong and Verhage, 2009). However, GPCRs may inhibit release by a number of mechanisms. In this study we have demonstrated two such mechanisms mediated by distinct Gαi/o-coupled receptors that coexist on CA1 pyramidal neuron presynaptic terminals: GABABR-mediated inhibition of Ca2+ channels and 5-HT1BR-mediated Gβγ interaction with the SNARE complex. We propose that the SNARE complex itself segregates these signaling pathways (Fig. 10d).
GPCRs can regulate neurotransmitter release by modifying presynaptic Ca2+ entry (Wu and Saggau, 1995) by a membrane delimited action of Gβγ on Ca2+ channels (Herlitze et al., 1996; Tedford and Zamponi, 2006) or alternatively by pathways that neither alter, nor require Ca2+ entry. These latter effects were first revealed by receptor-mediated inhibition of spontaneous release events (mEPSCs) independently of evoked Ca2+ entry and have been identified in the peripheral and central nervous systems (Silinsky, 1984; Stephens, 2009). Similar receptors may mediate both these forms of presynaptic inhibition. For example, GABABRs are typically thought to inhibit Ca2+ entry (Wu and Saggau, 1995; Takahashi et al., 1998) but can also modify spontaneous event frequencies independently of Ca2+ entry (Kabashima et al., 1997). In hippocampus, GABABRs have been implicated in both effects within the CA3 region (Lei and McBain, 2003). A third mechanism by which Gβγ inhibits neurotransmitter release by directly interacting with the SNARE complex has also been identified. 5-HT1BRs (Blackmer et al., 2001; Takahashi et al., 2001), noradrenergic α2ARs (Delaney et al., 2007), and mGluRs (Zhang et al., 2011; Upreti et al., 2013) may all act via this mechanism, which does not involve a change in presynaptic Ca2+ entry. Instead Gβγ acts directly in the pathway of Ca2+-evoked exocytosis by interfering with Ca2+-dependent synaptotagmin binding to SNARE complexes (Blackmer et al., 2001; Gerachshenko et al., 2005; Delaney et al., 2007; Yoon et al., 2007).
Gβγ interactions in the presynaptic terminal are modified by the release machinery itself. Membrane delimited actions of Gβγ on Ca2+ channels may be augmented by a direct interaction of Gβγ with various release machinery proteins (Dunlap and Fischbach, 1978; Holz et al., 1989; Herlitze et al., 1996; Ikeda, 1996). For example, SNARE complexes, Ca2+ channels, and Gβγ (Jarvis et al., 2002) may interact to augment Gβγ-mediated inhibition of Ca2+ channels, while GPCR-mediated inhibition of Ca2+ entry can be prevented by BoNT/C cleavage of syntaxin 1A in ciliary ganglion neurons (Stanley and Mirotznik, 1997). However, GPCR-mediated Ca2+ channel inhibition is also seen in heterologous expression systems such as COS-7 cells with no coexpression of SNARE complex proteins and in isolated membrane patches with no presynaptic structures (Meir et al., 2000), indicating that formation of a complex with SNARE proteins is not required for modulation. Another presynaptic protein, cysteine string protein, can also augment Gβγ-mediated voltage-dependent inhibition of Ca2+ channels (Magga et al., 2000) while the ancillary Ca2+ channel subunits (Cavβ) promote voltage-dependent G-protein modulation of Ca2+ channels (Dresviannikov et al., 2009). Perhaps any one of these or other protein–protein interactions may predispose Ca2+ channels to be modulated by Gβγ. Of these, interaction with intact syntaxin 1A is one, but clearly structural relationships with other proteins are essential for GPCR modulation of Ca2+ channels. In this study we show that GABABRs substantially modulate Ca2+ transients in hippocampal nerve terminals, even after cleavage of syntaxin 1A by treatment with BoNT/C.
It is less clear whether interacting proteins are necessary for Gβγ to inhibit neurotransmitter release by its direct, but Ca2+-synaptotagmin-dependent, action on the SNARE complex (Blackmer et al., 2001; Takahashi et al., 2001). This inhibition involves competition between Gβγ and synaptotagmin for binding to the SNARE complex, and has been characterized by sensitivity to BoNT/A toxin (Blackmer et al., 2005; Gerachshenko et al., 2005; Delaney et al., 2007; Zhang et al., 2011), which alters the relative affinities of Ca2+-synaptotagmin and Gβγ to the SNARE complex (Yoon et al., 2007).
5-HT1BRs clearly inhibit release from CA1 neurons by using this direct interaction with the SNARE complex (Fig. 10d). The evidence for this comes from a number of results that we have presented in this study. 5-HT1BRs strongly inhibit neurotransmitter release with no effect on presynaptic Ca2+ entry measured directly from individual presynaptic terminals. 5-HT1BR-mediated inhibition is prevented by pretreatment with BoNT/A, which modifies competition between Gβγ and Ca2+-synaptotagmin at the C-terminal region of SNARE complexes. We also show that, in contrast to GABABRs, these receptors do not modify Sr2+-evoked release. This is significant because, unlike Ca2+ as shown in this study and in earlier work (Bhalla et al., 2005), Sr2+ does not cause synaptotagmin binding to SNARE complexes leaving no direct target for Gβγ to interfere with exocytosis. Together with earlier work indicating that Gβγ can compete with Ca2+-dependent synaptotagmin binding to the C-terminal region of the SNARE complex (Blackmer et al., 2005; Gerachshenko et al., 2005; Yoon et al., 2007; Wells et al., 2012), these data imply that 5-HT1BRs cause a direct interaction of Gβγ on SNARE complexes. Finally, 5-HT1BRs do not modify spontaneous event frequencies. However, the receptor clearly interferes with Ca2+-dependent release–albeit not by altering Ca2+ entry but instead by altering the interaction of Ca2+-synaptotagmin with the SNARE complex. These data also indicate that Gβγ and Doc2, a proposed mediator of spontaneous release (Groffen et al., 2010; Pang et al., 2011), do not share a binding site on SNARE complexes. Gβγ interacts with the very C-terminal region of SNAP-25, overlapping the C-terminal region of the synaptotagmin I interaction site (Blackmer et al., 2005; Yoon et al., 2007; Wells et al., 2012). Perhaps Doc2 interacts at a less C-terminal region of the SNARE complex (Groffen et al., 2010). Since Sr2+-evoked asynchronous release is also likely mediated by Doc2 (albeit different isoforms; Pang et al., 2011), and not synaptotagmin I, this could also explain why 5-HT1BR has no effect on asynchronous release.
The effect of GABABRs is much simpler. These receptors substantially inhibit Ca2+ (and Sr2+) entry to the presynaptic terminal. GABABRs inhibit Ca2+ entry sufficiently to account for baclofen's effect on EPSCs if a fourth power relationship couples Ca2+ to exocytosis (Dodge and Rahamimoff, 1967).
We propose that functional separation of these pathways is provided by a microarchitecture, by which we mean Gβγ prefers its nearest effector, as determined by the location of the associated GPCRs within the active zone. In synapses, Ca2+ channels may localize to SNARE complex N terminus (Jarvis et al., 2000, 2002). GPCRs, particularly GABABRs (Padgett and Slesinger, 2010), form molecular machines by complexing with effectors and downstream targets, including Ca2+ channels (Park et al., 2010). Indeed, the GB1 GABABR subunit C-terminal region is required for the receptor to associate with Ca2+ channels and to fully inhibit neurotransmitter release (Laviv et al., 2011). Similarly, in the case of 5-HT1BRs (Svenningsson et al., 2006), association of the receptor to molecular targets is required for functional inhibition of neurotransmitter release. Gβγ, liberated by 5-HT1BRs, targets SNARE complex C-terminals to inhibit release (Blackmer et al., 2001, 2005; Gerachshenko et al., 2005). This result places effectors of 5-HT1BRs and GABABRs at opposite ends of primed SNARE complexes, and this separation appears to be sufficient to confer Gβγ specificity.
Perhaps SNARE complexes functionally separate targets of Gβγ. G-protein or GPCR interactions with the SNARE complex may prevent 5-HT1BR-mediated Gβγ release from interacting with Ca2+ channels but not GABABR released Gβγ. Physical disruption of this microarchitecture by cleavage of syntaxin and subsequent disruption of vesicle priming with BoNT/C reveals equally effective targeting of Ca2+ channels by both receptors (Fig. 10e). Thus, receptor selectivity to their respective targets is not necessarily a property of a particular Gβγ isoform, but may be a function of the structure in which the signal transduction cascade is embedded. BoNT/C treatment has also been previously shown to modify Ca2+ entry to presynaptic terminals (Bergsman and Tsien, 2000; Degtiar et al., 2000), albeit at a much slower time course than recorded in this study. It is conceivable that an alteration of the nanodomains of Ca2+ channels may also alter their availability to Gβγ in the terminal. However, the loss of primed vesicles by stimulation after BoNT/C application did not alter Ca2+ entry, and the effect of baclofen on this Ca2+ entry was also not altered by BoNT/C treatment. When 5-HT1BRs and their effector Gβγ are no longer anchored to their target at the C-terminal region of the SNARE complex Gβγ may inhibit presynaptic Ca2+ channels. Similarly, presynaptic Ca2+ channels will also no longer be associated with SNARE complexes (Fig. 10d,e). Of course, ultimately, properties of this signal transduction system must govern its target during vesicle priming. These results do not exclude the possibility that Gβγ identity plays a role in effector targeting. Indeed, there is precedent for such specificity (Macrez-Leprêtre et al., 1997; Wu et al., 1998; Mahon et al., 2006). If formation of primed SNARE complexes ensures G-protein target specificity, then priming must take just milliseconds to recruit modulatory proteins to the correct region of the SNARE complex as it is formed.
Footnotes
This work is supported by National Institute of Neurological Disorders and Stroke, RO1NS52699 and MH84874 to S.A., F31NS063662 to E.H., and R01EY010291 to H.E.H. We thank J. Richmond, T. Searl, M. Alpert, E. Church, and S. Ramachandran for critical reading of this manuscript and invaluable discussions.
The authors declare no competing financial interests.
References
- Bai J, Wang CT, Richards DA, Jackson MB, Chapman ER. Fusion pore dynamics are regulated by synaptotagmin*t-SNARE interactions. Neuron. 2004;41:929–942. doi: 10.1016/S0896-6273(04)00117-5. [DOI] [PubMed] [Google Scholar]
- Bergsman JB, Tsien RW. Syntaxin modulation of calcium channels in cortical synaptosomes as revealed by botulinum toxin C1. J Neurosci. 2000;20:4368–4378. doi: 10.1523/JNEUROSCI.20-12-04368.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla A, Tucker WC, Chapman ER. Synaptotagmin isoforms couple distinct ranges of Ca2+, Ba2+, and Sr2+ concentration to SNARE-mediated membrane fusion. Mol Biol Cell. 2005;16:4755–4764. doi: 10.1091/mbc.E05-04-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmer T, Larsen EC, Takahashi M, Martin TF, Alford S, Hamm HE. G protein betagamma subunit-mediated presynaptic inhibition: regulation of exocytotic fusion downstream of Ca2+ entry. Science. 2001;292:293–297. doi: 10.1126/science.1058803. [DOI] [PubMed] [Google Scholar]
- Blackmer T, Larsen EC, Bartleson C, Kowalchyk JA, Yoon EJ, Preininger AM, Alford S, Hamm HE, Martin TF. G protein betagamma directly regulates SNARE protein fusion machinery for secretory granule exocytosis. Nat Neurosci. 2005;8:421–425. doi: 10.1038/nn1423. [DOI] [PubMed] [Google Scholar]
- Brown DA, Sihra TS. Presynaptic signaling by heterotrimeric G-proteins. Handb Exp Pharmacol. 2008:207–260. doi: 10.1007/978-3-540-74805-2_8. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Few AP. Calcium channel regulation and presynaptic plasticity. Neuron. 2008;59:882–901. doi: 10.1016/j.neuron.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Cox CL, Denk W, Tank DW, Svoboda K. Action potentials reliably invade axonal arbors of rat neocortical neurons. Proc Natl Acad Sci U S A. 2000;97:9724–9728. doi: 10.1073/pnas.170278697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degtiar VE, Scheller RH, Tsien RW. Syntaxin modulation of slow inactivation of N-type calcium channels. J Neurosci. 2000;20:4355–4367. doi: 10.1523/JNEUROSCI.20-12-04355.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong AP, Verhage M. Presynaptic signal transduction pathways that modulate synaptic transmission. Curr Opin Neurobiol. 2009;19:245–253. doi: 10.1016/j.conb.2009.06.005. [DOI] [PubMed] [Google Scholar]
- Delaney AJ, Crane JW, Sah P. Noradrenaline modulates transmission at a central synapse by a presynaptic mechanism. Neuron. 2007;56:880–892. doi: 10.1016/j.neuron.2007.10.022. [DOI] [PubMed] [Google Scholar]
- Dodge FA, Jr, Rahamimoff R. Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J Physiol. 1967;193:419–432. doi: 10.1113/jphysiol.1967.sp008367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dresviannikov AV, Page KM, Leroy J, Pratt WS, Dolphin AC. Determinants of the voltage dependence of G protein modulation within calcium channel beta subunits. Pflugers Arch. 2009;457:743–756. doi: 10.1007/s00424-008-0549-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlap K, Fischbach GD. Neurotransmitters decrease the calcium component of sensory neurone action potentials. Nature. 1978;276:837–839. doi: 10.1038/276837a0. [DOI] [PubMed] [Google Scholar]
- Fang Q, Berberian K, Gong LW, Hafez I, Sørensen JB, Lindau M. The role of the C terminus of the SNARE protein SNAP-25 in fusion pore opening and a model for fusion pore mechanics. Proc Natl Acad Sci U S A. 2008;105:15388–15392. doi: 10.1073/pnas.0805377105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CE, Skiba NP, Bae H, Daaka Y, Reuveny E, Shekter LR, Rosal R, Weng G, Yang CS, Iyengar R, Miller RJ, Jan LY, Lefkowitz RJ, Hamm HE. Molecular basis for interactions of G protein betagamma subunits with effectors. Science. 1998;280:1271–1274. doi: 10.1126/science.280.5367.1271. [DOI] [PubMed] [Google Scholar]
- Gerachshenko T, Blackmer T, Yoon EJ, Bartleson C, Hamm HE, Alford S. Gbetagamma acts at the C terminus of SNAP-25 to mediate presynaptic inhibition. Nat Neurosci. 2005;8:597–605. doi: 10.1038/nn1439. [DOI] [PubMed] [Google Scholar]
- Gerachshenko T, Schwartz E, Bleckert A, Photowala H, Seymour A, Alford S. Presynaptic G-protein-coupled receptors dynamically modify vesicle fusion, synaptic cleft glutamate concentrations, and motor behavior. J Neurosci. 2009;29:10221–10233. doi: 10.1523/JNEUROSCI.1404-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groffen AJ, Martens S, Díez Arazola R, Cornelisse LN, Lozovaya N, de Jong AP, Goriounova NA, Habets RL, Takai Y, Borst JG, Brose N, McMahon HT, Verhage M. Doc2b is a high-affinity Ca2+ sensor for spontaneous neurotransmitter release. Science. 2010;327:1614–1618. doi: 10.1126/science.1183765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, McMahon H, Yamasaki S, Binz T, Hata Y, Südhof TC, Niemann H. Synaptic vesicle membrane fusion complex: action of clostridial neurotoxins on assembly. EMBO J. 1994;13:5051–5061. doi: 10.1002/j.1460-2075.1994.tb06834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- Holz GG, 4th, Kream RM, Spiegel A, Dunlap K. G proteins couple alpha-adrenergic and GABAb receptors to inhibition of peptide secretion from peripheral sensory neurons. J Neurosci. 1989;9:657–666. doi: 10.1523/JNEUROSCI.09-02-00657.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein beta gamma subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- Jackson MB, Redman SJ. Calcium dynamics, buffering, and buffer saturation in the boutons of dentate granule-cell axons in the hilus. J Neurosci. 2003;23:1612–1621. doi: 10.1523/JNEUROSCI.23-05-01612.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis SE, Magga JM, Beedle AM, Braun JE, Zamponi GW. G protein modulation of N-type calcium channels is facilitated by physical interactions between syntaxin 1A and Gbetagamma. J Biol Chem. 2000;275:6388–6394. doi: 10.1074/jbc.275.9.6388. [DOI] [PubMed] [Google Scholar]
- Jarvis SE, Barr W, Feng ZP, Hamid J, Zamponi GW. Molecular determinants of syntaxin 1 modulation of N-type calcium channels. J Biol Chem. 2002;277:44399–44407. doi: 10.1074/jbc.M206902200. [DOI] [PubMed] [Google Scholar]
- Kabashima N, Shibuya I, Ibrahim N, Ueta Y, Yamashita H. Inhibition of spontaneous EPSCs and IPSCs by presynaptic GABAB receptors on rat supraoptic magnocellular neurons. J Physiol. 1997;504:113–126. doi: 10.1111/j.1469-7793.1997.113bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET, Chung C, Khvotchev M, Leitz J, Nosyreva E, Raingo J, Ramirez DM. Spontaneous neurotransmission: an independent pathway for neuronal signaling? Physiology. 2011;26:45–53. doi: 10.1152/physiol.00040.2010. [DOI] [PubMed] [Google Scholar]
- Lai Y, Diao J, Liu Y, Ishitsuka Y, Su Z, Schulten K, Ha T, Shin YK. Fusion pore formation and expansion induced by Ca2+ and synaptotagmin 1. Proc Natl Acad Sci U S A. 2013;110:1333–1338. doi: 10.1073/pnas.1218818110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviv T, Vertkin I, Berdichevsky Y, Fogel H, Riven I, Bettler B, Slesinger PA, Slutsky I. Compartmentalization of the GABAB receptor signaling complex is required for presynaptic inhibition at hippocampal synapses. J Neurosci. 2011;31:12523–12532. doi: 10.1523/JNEUROSCI.1527-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei S, McBain CJ. GABA B receptor modulation of excitatory and inhibitory synaptic transmission onto rat CA3 hippocampal interneurons. J Physiol. 2003;546:439–453. doi: 10.1113/jphysiol.2002.034017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrez-Leprêtre N, Kalkbrenner F, Morel JL, Schultz G, Mironneau J. G protein heterotrimer Galpha13beta1gamma3 couples the angiotensin AT1A receptor to increases in cytoplasmic Ca2+ in rat portal vein myocytes. J Biol Chem. 1997;272:10095–10102. doi: 10.1074/jbc.272.15.10095. [DOI] [PubMed] [Google Scholar]
- Magga JM, Jarvis SE, Arnot MI, Zamponi GW, Braun JE. Cysteine string protein regulates G protein modulation of N-type calcium channels. Neuron. 2000;28:195–204. doi: 10.1016/S0896-6273(00)00096-9. [DOI] [PubMed] [Google Scholar]
- Mahon MJ, Bonacci TM, Divieti P, Smrcka AV. A docking site for G protein betagamma subunits on the parathyroid hormone 1 receptor supports signaling through multiple pathways. Mol Endocrinol. 2006;20:136–146. doi: 10.1210/me.2005-0169. [DOI] [PubMed] [Google Scholar]
- Mazzoni MR, Malinski JA, Hamm HE. Structural analysis of rod GTP-binding protein, Gt. Limited proteolytic digestion pattern of Gt with four proteases defines monoclonal antibody epitope. J Biol Chem. 1991;266:14072–14081. [PubMed] [Google Scholar]
- Meir A, Bell DC, Stephens GJ, Page KM, Dolphin AC. Calcium channel beta subunit promotes voltage-dependent modulation of alpha 1 B by G beta gamma. Biophys J. 2000;79:731–746. doi: 10.1016/S0006-3495(00)76331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellow AM, Phillips TE, Silinsky EM. On the conductance pathway traversed by strontium in mediating the asynchronous release of acetylcholine by motor nerve impulses. Br J Pharmacol. 1978;63:229–232. doi: 10.1111/j.1476-5381.1978.tb09750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellow AM, Perry BD, Silinsky EM. Effects of calcium and strontium in the process of acetylcholine release from motor nerve endings. J Physiol. 1982;328:547–562. doi: 10.1113/jphysiol.1982.sp014283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaeli A, Yaka R. Dopamine inhibits GABA(A) currents in ventral tegmental area dopamine neurons via activation of presynaptic G-protein coupled inwardly-rectifying potassium channels. Neuroscience. 2010;165:1159–1169. doi: 10.1016/j.neuroscience.2009.11.045. [DOI] [PubMed] [Google Scholar]
- Miller RJ. Presynaptic receptors. Annu Rev Pharmacol Toxicol. 1998;38:201–227. doi: 10.1146/annurev.pharmtox.38.1.201. [DOI] [PubMed] [Google Scholar]
- Mizutani H, Hori T, Takahashi T. 5-HT1B receptor-mediated presynaptic inhibition at the calyx of Held of immature rats. Eur J Neurosci. 2006;24:1946–1954. doi: 10.1111/j.1460-9568.2006.05063.x. [DOI] [PubMed] [Google Scholar]
- Neher E, Augustine GJ. Calcium gradients and buffers in bovine chromaffin cells. J Physiol. 1992;450:273–301. doi: 10.1113/jphysiol.1992.sp019127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto H, Hanson PI, Chapman ER, Blasi J, Jahn R. Poisoning by botulinum neurotoxin A does not inhibit formation or disassembly of the synaptosomal fusion complex. Biochem Biophys Res Commun. 1995;212:945–952. doi: 10.1006/bbrc.1995.2061. [DOI] [PubMed] [Google Scholar]
- Padgett CL, Slesinger PA. GABAB receptor coupling to G-proteins and ion channels. Adv Pharmacol. 2010;58:123–147. doi: 10.1016/S1054-3589(10)58006-2. [DOI] [PubMed] [Google Scholar]
- Pang ZP, Bacaj T, Yang X, Zhou P, Xu W, Südhof TC. Doc2 supports spontaneous synaptic transmission by a Ca(2+)-independent mechanism. Neuron. 2011;70:244–251. doi: 10.1016/j.neuron.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HW, Jung H, Choi KH, Baik JH, Rhim H. Direct interaction and functional coupling between voltage-gated CaV1.3 Ca2+ channel and GABAB receptor subunit 2. FEBS Lett. 2010;584:3317–3322. doi: 10.1016/j.febslet.2010.07.014. [DOI] [PubMed] [Google Scholar]
- Pellegrini LL, O'Connor V, Betz H. Fusion complex formation protects synaptobrevin against proteolysis by tetanus toxin light chain. FEBS Lett. 1994;353:319–323. doi: 10.1016/0014-5793(94)01070-6. [DOI] [PubMed] [Google Scholar]
- Phillips WJ, Cerione RA. Labeling of the beta gamma subunit complex of transducin with an environmentally sensitive cysteine reagent. Use of fluorescence spectroscopy to monitor transducin subunit interactions. J Biol Chem. 1991;266:11017–11024. [PubMed] [Google Scholar]
- Scanziani M, Capogna M, Gähwiler BH, Thompson SM. Presynaptic inhibition of miniature excitatory synaptic currents by baclofen and adenosine in the hippocampus. Neuron. 1992;9:919–927. doi: 10.1016/0896-6273(92)90244-8. [DOI] [PubMed] [Google Scholar]
- Sheng ZH, Rettig J, Takahashi M, Catterall WA. Identification of a syntaxin-binding site on N-type calcium channels. Neuron. 1994;13:1303–1313. doi: 10.1016/0896-6273(94)90417-0. [DOI] [PubMed] [Google Scholar]
- Sheng ZH, Rettig J, Cook T, Catterall WA. Calcium-dependent interaction of N-type calcium channels with the synaptic core complex. Nature. 1996;379:451–454. doi: 10.1038/379451a0. [DOI] [PubMed] [Google Scholar]
- Shin OH, Rhee JS, Tang J, Sugita S, Rosenmund C, Südhof TC. Sr2+ binding to the Ca2+ binding site of the synaptotagmin 1 C2B domain triggers fast exocytosis without stimulating SNARE interactions. Neuron. 2003;37:99–108. doi: 10.1016/S0896-6273(02)01145-5. [DOI] [PubMed] [Google Scholar]
- Silinsky EM. On the mechanism by which adenosine receptor activation inhibits the release of acetylcholine from motor nerve endings. J Physiol. 1984;346:243–256. doi: 10.1113/jphysiol.1984.sp015019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silinsky EM. Selective disruption of the mammalian secretory apparatus enhances or eliminates calcium current modulation in nerve endings. Proc Natl Acad Sci U S A. 2008;105:6427–6432. doi: 10.1073/pnas.0708814105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley EF, Mirotznik RR. Cleavage of syntaxin prevents G-protein regulation of presynaptic calcium channels. Nature. 1997;385:340–343. doi: 10.1038/385340a0. [DOI] [PubMed] [Google Scholar]
- Starke K. Influence of extracellular noradrenaline on the stimulation-evoked secretion of noradrenaline from sympathetic nerves: evidence for an receptor-mediated feed-back inhibition of noradrenaline release. Naunyn Schmiedebergs Arch Pharmacol. 1972;275:11–23. doi: 10.1007/BF00505064. [DOI] [PubMed] [Google Scholar]
- Stephens GJ. G-protein-coupled-receptor-mediated presynaptic inhibition in the cerebellum. Trends Pharmacol Sci. 2009;30:421–430. doi: 10.1016/j.tips.2009.05.008. [DOI] [PubMed] [Google Scholar]
- Svenningsson P, Chergui K, Rachleff I, Flajolet M, Zhang X, El Yacoubi M, Vaugeois JM, Nomikos GG, Greengard P. Alterations in 5-HT1B receptor function by p11 in depression-like states. Science. 2006;311:77–80. doi: 10.1126/science.1117571. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Freed R, Blackmer T, Alford S. Calcium influx-independent depression of transmitter release by 5-HT at lamprey spinal cord synapses. J Physiol. 2001;532:323–336. doi: 10.1111/j.1469-7793.2001.0323f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T. Dynamic aspects of presynaptic calcium currents mediating synaptic transmission. Cell Calcium. 2005;37:507–511. doi: 10.1016/j.ceca.2005.01.018. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Kajikawa Y, Tsujimoto T. G-Protein-coupled modulation of presynaptic calcium currents and transmitter release by a GABAB receptor. J Neurosci. 1998;18:3138–3146. doi: 10.1523/JNEUROSCI.18-09-03138.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedford HW, Zamponi GW. Direct G protein modulation of Cav2 calcium channels. Pharmacol Rev. 2006;58:837–862. doi: 10.1124/pr.58.4.11. [DOI] [PubMed] [Google Scholar]
- Upreti C, Zhang XL, Alford S, Stanton PK. Role of presynaptic metabotropic glutamate receptors in the induction of long-term synaptic plasticity of vesicular release. Neuropharmacology. 2013;66:31–39. doi: 10.1016/j.neuropharm.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Armstrong WE. Tonic regulation of GABAergic synaptic activity on vasopressin neurones by cannabinoids. J Neuroendocrinol. 2012;24:664–673. doi: 10.1111/j.1365-2826.2011.02239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells CA, Zurawski Z, Betke KM, Yim YY, Hyde K, Rodriguez S, Alford S, Hamm HE. Gβγ inhibits exocytosis via interaction with critical residues on soluble N-ethylmaleimide-sensitive factor attachment protein-25. Mol Pharmacol. 2012;82:1136–1149. doi: 10.1124/mol.112.080507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Benovic JL, Hildebrandt JD, Lanier SM. Receptor docking sites for G-protein betagamma subunits. Implications for signal regulation. J Biol Chem. 1998;273:7197–7200. doi: 10.1074/jbc.273.13.7197. [DOI] [PubMed] [Google Scholar]
- Wu LG, Saggau P. GABAB receptor-mediated presynaptic inhibition in guinea-pig hippocampus is caused by reduction of presynaptic Ca2+ influx. J Physiol. 1995;485:649–657. doi: 10.1113/jphysiol.1995.sp020759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Binz T, Niemann H, Neher E. Multiple kinetic components of exocytosis distinguished by neurotoxin sensitivity. Nat Neurosci. 1998;1:192–200. doi: 10.1038/642. [DOI] [PubMed] [Google Scholar]
- Xu-Friedman MA, Regehr WG. Presynaptic strontium dynamics and synaptic transmission. Biophys J. 1999;76:2029–2042. doi: 10.1016/S0006-3495(99)77360-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon EJ, Gerachshenko T, Spiegelberg BD, Alford S, Hamm HE. Gbetagamma interferes with Ca2+-dependent binding of synaptotagmin to the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex. Mol Pharmacol. 2007;72:1210–1219. doi: 10.1124/mol.107.039446. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G proteins and protein kinase C mediated by the calcium channel alpha1 subunit. Nature. 1997;385:442–446. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]
- Zhang XL, Upreti C, Stanton PK. Gβγ and the C terminus of SNAP-25 are necessary for long-term depression of transmitter release. PLoS One. 2011;6:e20500. doi: 10.1371/journal.pone.0020500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Kim-Miller MJ, Fukuda M, Kowalchyk JA, Martin TF. Ca2+-dependent synaptotagmin binding to SNAP-25 is essential for Ca2+-triggered exocytosis. Neuron. 2002;34:599–611. doi: 10.1016/S0896-6273(02)00671-2. [DOI] [PubMed] [Google Scholar]