

Abstract
Alzheimer's disease is thought to be caused by β-amyloid peptide (Aβ)-dependent synaptic dysfunction. However, the signaling pathways connecting Aβ and synaptic dysfunction remain elusive. Here we report that Aβ transiently increases the expression level of centaurin-α1 (CentA1) in neurons, which induces a Ras-dependent association of Elk-1 with mitochondria, leading to mitochondrial and synaptic dysfunction in organotypic hippocampal slices of rats. Downregulation of the CentA1–Ras-Elk-1 pathway restored normal mitochondrial activity, spine structural plasticity, spine density, and the amplitude and frequency of miniature EPSCs in Aβ-treated neurons, whereas upregulation of the pathway was sufficient to decrease spine density. Elevations of CentA1 and association of Elk-1 with mitochondria were also observed in transgenic mice overexpressing a human mutant form of amyloid precursor protein. Therefore, the CentA1–Ras-Elk-1 signaling pathway acts on mitochondria to regulate dendritic spine density and synaptic plasticity in response to Aβ in hippocampal neurons, providing new pharmacological targets for Alzheimer's disease.
Introduction
β-Amyloid (Aβ) peptide plays a major role in the pathogenesis of Alzheimer's disease (AD; Hardy and Allsop, 1991; Hardy and Selkoe, 2002). Application of Aβ to neurons causes reduced spine density, aberrant spine shape, decreased levels of molecules involved in synaptic signaling, and impaired long-term potentiation (LTP; Moolman et al., 2004; Almeida et al., 2005; Snyder et al., 2005; Hsieh et al., 2006; Jacobsen et al., 2006; Calabrese et al., 2007; Lacor et al., 2007; Shankar et al., 2007; Origlia et al., 2008; Tackenberg and Brandt, 2009; Wei et al., 2010; Sheng et al., 2012). Several signaling proteins, including calcineurin and glycogen synthase kinase-3β (GSK3β), are abnormally regulated by Aβ, leading to synaptic depression and decreased spine density (Pei et al., 1997; Peineau et al., 2007; Serenó et al., 2009; Tackenberg and Brandt, 2009; Wu et al., 2010). Moreover, Aβ-induced mitochondrial dysfunction has been reported to be required for impaired neuronal function (Du et al., 2008; Eckert et al., 2008; Hansson Petersen et al., 2008; Mattson et al., 2008; Wang et al., 2009; Rui et al., 2010). However, the signaling mechanisms by which Aβ induces mitochondrial and synaptic dysfunction are not fully understood.
Centaurin-α1 (CentA1) is upregulated in AD brain and accumulates in neuritic plaques (Reiser and Bernstein, 2002, 2004). However, whether CentA1 contributes to Aβ-dependent synaptic impairment has not been studied. CentA1 is a brain-specific ADP ribosylation factor (Arf) GTPase-activating protein localized to axons, dendrites, dendritic spines, and postsynaptic density (Hammonds-Odie et al., 1996; Kreutz et al., 1997; Aggensteiner and Reiser, 2003; Moore et al., 2007). During neuronal development, CentA1 is required for dendritic branching and spinogenesis (Kreutz et al., 1997; Moore et al., 2007). Moreover, CentA1 interacts with the mitochondrial permeability transition pore complex (mPTP) and regulates its function (Galvita et al., 2009). mPTP dysregulation is one of the Aβ-dependent cellular phenotypes that contribute to Aβ-induced neuronal dysfunction (Du et al., 2008).
CentA1 interacts with Ras and activates the Ras-E26-like-kinase 1 (Elk-1) pathway, increasing Elk-1-dependent transcription (Hayashi et al., 2006) induced by synaptic activity and neurotrophins, including BDNF (Sgambato et al., 1998; Vanhoutte et al., 1999; Kalita et al., 2006). Elk-1 is also present in extranuclear compartments including dendrites and axons (Sgambato et al., 1998). Extranuclear Elk-1 associates with mPTP in apoptotic neurons (Barrett et al., 2006) and has been implicated in neurodegenerative diseases including AD (Sharma et al., 2010). Because both CentA1 and Elk-1 can associate with mPTP and regulate its function (Barrett et al., 2006; Galvita et al., 2009), and because mitochondrial malfunction occurs in AD (Du et al., 2008; Hansson Petersen et al., 2008; Mattson et al., 2008; Wang et al., 2009), the CentA1-Ras-ERK-Elk-1 pathway at mPTP may play an essential role in AD.
In this study, we show that the CentA1–Ras-Elk-1 pathway links Aβ and synaptic dysfunction. We found that Aβ upregulates CentA1 and activates the Ras-Elk-1 pathway at mitochondria, which impairs mitochondrial activity. Downregulation of CentA1–Ras-Elk-1 signaling restores normal mitochondrial activity, synaptic function, and spine density in Aβ-treated neurons.
Materials and Methods
Animals.
Mouse studies were approved by the institutional animal care and use committee of Duke University in accordance with the National Institutes of Health guidelines for animal care. As a model for AD, we used male transgenic mice overexpressing a mutant human form (Swedish mutation) of amyloid precursor protein (J20 line; Mucke et al., 2000). Non-transgenic male littermates were used for controls.
DNA and reagents.
CentA1 and APP cDNAs were from OriGene Technologies, sh-CentA1 against rat centaurin-α1 was from SuperArray Bioscience, pFR-Luc transcription reporter and pFA2-Elk-1 (PathDetect Elk-1 trans-Reporting System) were from Stratagene, and pcDNA3-myc-Elk-1 and shElk-1 were kindly provided by Dr. T. Yoshida (University of Michigan) and Dr. M. Hetman (Kentucky Spinal Cord Injury Research Center), respectively. Aβ (1–42) was from rPeptide. For ballistic gene transfer, gold particles (10 mg) were coated with plasmids (50 μg total) and shot into organotypic hippocampal slices using the Helios gene gun system (Bio-Rad). For cotransfection, the gold particles were coated with multiple plasmids.
The following antibodies were used for Western blot analysis: goat anti-centaurin-ɑ1 (Abcam; 1:500); goat anti-Elk-1 (Santa Cruz Biotechnology; 1:500); rabbit anti-phospho-S383-Elk-1, rabbit anti-VDAC, and rabbit anti-COX IV (Cell Signaling Technology; 1:1000); mouse anti-GAPDH (Sigma; 1:1000); mouse anti-NeuN (Millipore; 1:1000); mouse anti-β-actin (Sigma, 1:2000); and HRP-labeled anti-mouse, anti-goat, or anti-rabbit IgG antibodies (BioRad; 1:5000).
Preparations.
Dissociated hippocampal cultures were prepared from newborn Sprague Dawley rats at postnatal day 0–1, as described previously (Szatmari et al., 2007). Neurons were cultured in basal medium Eagle (BME) supplemented with 10% heat-inactivated fetal bovine serum (HyClone), 35 mm glucose, 1 mm l-glutamine, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. Cytosine arabinoside (2.5 μm) was added to the cultures on day(s) in vitro (DIV) 2 to inhibit the proliferation of non-neuronal cells. Neurons were transfected by electroporation using the Amaxa Nucleofector System (Lonza Bioscience Cell Discovery) or by lipofection. Organotypic hippocampal slice cultures (300 μm thick) were prepared from Sprague Dawley rats at postnatal day 6 or 7, as described previously (Stoppini et al., 1991). After 1 week in culture, slices were transfected using biolistic gene transfer (McAllister, 2000). Aβ stock solution (100 μm) was prepared in 1% NH4OH as a vehicle and added directly to the culture medium at a final concentration of 1 μm. FTI-277 was dissolved in DMSO and added directly to the culture medium at a final concentration of 10 μm. Bongkrekic acid (Calbiochem) stock solution (0.4 mm) was prepared in DMSO as vehicle and added directly to the culture medium at a final concentration of 1 μm. Cyclosporin A (Tocris Bioscience) stock solution (0.4 mm) was prepared in DMSO as vehicle and added directly to the culture medium at a final concentration of 1 μm.
Image acquisition and glutamate uncaging.
A custom-built two-photon microscope with two Ti:sapphire lasers (Spectra-physics) was used. One laser was tuned to 920 nm to excite EGFP, and the second laser was tuned to 720 nm for glutamate uncaging. The beams were combined and passed through the same set of scan mirrors and objective (60×, 0.9 numerical aperture; Olympus). Fluorescence signal from a cooled PMT (Hamamatsu) was acquired using a data acquisition board controlled by ScanImage software (PCI-6110; National Instruments; Pologruto et al., 2003). Two-photon glutamate uncaging was performed in ACSF lacking Mg2+ in the presence of MNI-caged l-glutamate (2.5 mm) and TTX (1 μm). Brief pulses (5 mW, 6 ms, 30 pulses) of the uncaging beam were applied to the tip of the spine head.
Electrophysiology.
AMPAR-dependent mESPCs were recorded as described previously (Shankar et al., 2007). Briefly, whole-cell voltage–clamp recordings were obtained at a holding potential of −70 mV from visually identified pyramidal neurons in ACSF including 2 mm CaCl2, 1 mm MgCl2, 20 μm bicuculline, 1 μm TTX, 20 μm mibefradil, 100 μm picrotoxin, 20 μm nimodipine, and 50 μm APV. Mibefradil and nimodipine were used to inhibit voltage gated calcium channels and thereby to increase input resistance (Shankar et al., 2007). Currents were filtered at 2 kHz, digitized at 10 kHz, and acquired for 60 s periods. The intracellular solution contained 100 mm Cs-methanesulfonate, 20 mm CsCl, 10 mm HEPES, 10 mm EGTA, 4 mm MgCl2, 0.4 mm NaGTP, 4 mm MgATP, and 10 mm phosphocreatine. Series/input resistance (MΩ) was 18 ± 1/184 ± 5 for neurons treated with vehicle (final 0.01% NH4OH) + DMSO (final 0.25%), 17 ± 1/236 ± 18 M for vehicle + 10 μm FTI-277 (FTI), 21 ± 1/175 ± 10 for Aβ + DMSO, and 18 ± 1/231 ± 15 for Aβ + FTI. mEPSCs were analyzed using template matching (Clements and Bekkers, 1997).
Isolation of mitochondrial proteins.
Isolation of the mitochondrial fraction from mouse hippocampus and rat organotypic hippocampal slices was performed using mitochondrial isolation kit for tissue (Thermo Scientific). The mitochondrial isolation kit for cultured cells (Thermo Scientific) was used for dissociated neuron cultures.
Nuclear protein extraction.
Extraction of nuclear proteins was performed using a compartmental protein extraction kit (Millipore) according to the manufacturer's instructions.
Luciferase reporter gene assay.
Elk-1 activity was measured using the luciferase reporter gene assay by transfecting neurons with plasmids containing Gal4-UAS-luciferase (pFR-Luc) and a fusion of activation domain of Elk-1 and Gal4 DNA-binding domain (DBD) (pFA2-Elk-1). Elk-1 activation in the nucleus causes binding of Gal4-DBD to Gal4-UAS, leading to luciferase transcription. The transfection efficiency was determined with β-galactosidase (pEF1α-LacZ). Hippocampal neurons cultured on 24-well plates (plated at 5 × 105/well density) were cotransfected at DIV3 with pFR-Luc (0.3 μg/well), pFA2-Elk-1 (0.3 μg/well), and pEF1α-LacZ DNA (0.2 μg/well) with or without CentA1 (0.5 μg/well) expression plasmid using Lipofectamine 2000. Three days after transfection, neurons were treated with Aβ for 20 h. Luciferase and β-gal activities were measured using commercial kits from Promega. Transcriptional activity was determined as luciferase activity normalized to β-galactosidase activity and compared with unstimulated controls.
Mitochondrial activity assay.
Mitochondrial activity was measured with tetramethylrhodamine-methylester (TMRM) fluorescence in neurons treated with DMSO or FTI-277. Dissociated hippocampal neurons (DIV6) were treated as indicated for 24 h, followed by treatment with 100 nm TMRM (Invitrogen) for 30 min. The overall cellular fluorescence levels were visualized by fluorescence microscopy. The brightness of TMRM in each neuron was analyzed using ImageJ.
Results
Centaurin-α1 is upregulated by Aβ and is required for Aβ-induced loss of dendritic spines
Treatment of neurons with soluble Aβ serves as a cellular model of AD and causes synaptic dysfunction, including reduced spine density, aberrant spine shape, and reduced synaptic plasticity (Moolman et al., 2004; Almeida et al., 2005; Snyder et al., 2005; Hsieh et al., 2006; Jacobsen et al., 2006; Calabrese et al., 2007; Lacor et al., 2007; Shankar et al., 2007; Origlia et al., 2008; Tackenberg and Brandt, 2009; Wei et al., 2010). We first investigated whether Aβ treatment upregulates the expression level of CentA1 in hippocampal neurons. In both dissociated neurons and cultured organotypic hippocampal slices, the expression level of CentA1 increased by ∼2.5-fold (Fig. 1A,B). However, the level decayed to ∼1.3-fold by day 7 (Fig. 1B). To examine the effect of CentA1 upregulation after Aβ treatment, the CentA1 expression level was downregulated by short-hairpin RNA (shRNA) against CentA1 (sh-CentA1). sh-CentA1 decreased the basal level of CentA1 significantly and suppressed the Aβ-induced CentA1 upregulation (Fig. 1A). Cultured organotypic hippocampal slices were then transfected with sh-CentA1 or a scrambled shRNA (sh-SC) together with EGFP, and the dendritic spines of transfected CA1 pyramidal neurons were imaged using 2-photon laser-scanning microscopy. Consistent with previous studies (Snyder et al., 2005; Hsieh et al., 2006; Shankar et al., 2007; Tackenberg and Brandt, 2009), Aβ treatment for 7 d induced an ∼40% decrease of spine density in neurons expressing sh-SC (Fig. 1C, D). Downregulation of CentA1 with sh-CentA1 prevented Aβ from inducing spine loss (Fig. 1C, D). We also found that sh-CentA1 rescued the spine loss caused by overexpression of APP (Fig. 1E). Furthermore, when wild-type CentA1 (without Aβ treatment) was overexpressed, spine density was decreased to a degree similar to the spine density decrease seen with Aβ treatment (Fig. 1F,H). Aβ treatment of CentA1-overexpressing neurons did not show any additive effect (Fig. 1F,H). These results suggest that Aβ-induced upregulation of CentA1 is required and sufficient for decreasing spine density.
Figure 1.

CentA1-Ras signaling mediates Aβ-induced loss of dendritic spines. A, Expression level of CentA1 in dissociated hippocampal neurons treated with Aβ(1–42). Dissociated hippocampal neurons from P0 rats were transfected with sh-SC or sh-CentA1 using electroporation, cultured for 3 d and treated with vehicle (Veh; final 0.01% NH4OH) or 1 μm Aβ for 48 h. The level of CentA1 was analyzed by Western blotting and β-actin was used as a loading control. Numbers under blots represent normalized CentA1 expression in four independent experiments. B, Immunoblot analysis of the expression level of CentA1 in organotypic hippocampal slices treated with Aβ(1–42). Slices were prepared from postnatal day 6 rats and cultured for 1–2 weeks, then treated with Veh or 1 μm Aβ for 48 h and 7 d, respectively. The level of CentA1 was analyzed by Western blotting and β-actin was used as a loading control. Numbers under blots represent normalized CentA1 expression in 15 (48 h) and 22 (7 d) independent experiments. C, Representative images of apical dendrites from CA1 pyramidal neurons in organotypic slices. Slices were prepared from postnatal day 6 rats and cultured for 1–2 weeks. Neurons were biolistically transfected with sh-SC or sh-CentA1 together with EGFP, treated with either vehicle or 1 μm Aβ (1–42) for 7 d, and imaged with two-photon laser scanning microscopy. Scale bar, 5 μm. D, Quantification of spine density in neurons treated as in C. Error bars indicate SEM. *p < 0.01, ANOVA followed by post hoc test using least significant difference. The number of neurons was 8, 11, 8, and 8, respectively. E, Spine density in apical dendrites of CA1 pyramidal neurons transfected with sh-SC or sh-CentA1 together with an empty vector (Vec) or human amyloid precursor protein (APP). The number of neurons was 5, 6, and 5, respectively. Error bars indicate SEM. F, Images of apical dendrites of CA1 pyramidal neurons expressing EGFP and Vec or WT-CentA1. Scale bar, 5 μm. G, Images of apical dendrites of CA1 pyramidal neurons expressing EGFP and Vec or dominant-negative Ras (HRasS17N, DN-Ras). Scale bar, 5 μm. H, Quantification of spine density in neurons expressing EGFP and Vec, WT-CentA1, or DN-Ras and treated with Veh or 1 μm Aβ for 1 week. *p < 0.01. The number of neurons was 18, 9, 8, 6, and 6, respectively. I, Spine density in apical dendrites of CA1 pyramidal neurons treated with Veh or 1 μm Aβ together with DMSO or 10 μm FTI-277 for 1 week. *p < 0.01. Number of neurons was 8, 5, 6, and 11, respectively. Error bars indicate SEM.
CentA1 is involved in Aβ-induced impairment of spine structural plasticity
To elucidate the role of Aβ-induced upregulation of CentA1 in synaptic plasticity, we monitored the structural plasticity of dendritic spines induced by 2-photon glutamate uncaging in CA1 pyramidal neurons in cultured organotypic hippocampal slices (Fig. 2; Matsuzaki et al., 2004; Harvey et al., 2008; Lee et al., 2009; Murakoshi et al., 2011). Spine structural plasticity is associated with LTP and caused by rapid actin polymerization regulated by Ca2+/calmodulin-dependent kinase and small GTPase proteins (Matsuzaki et al., 2004; Lee et al., 2009; Murakoshi et al., 2011). We used slices slightly sooner after Aβ treatment (4–6 d) for uncaging than we did for spine density measurement (7 d) so that we could easily find spines for uncaging (Fig. 2). Under control conditions (neurons expressing control shRNA treated with vehicle for Aβ), the spine volume increased rapidly (∼1 min) after glutamate uncaging (transient phase) by 290 ± 67% and relaxed to an elevated level at 105 ± 32% increase lasting >30 min (sustained phase; Fig. 2), which is consistent with previous reports (Matsuzaki et al., 2004; Lee et al., 2009; Murakoshi et al., 2011). When neurons were treated with Aβ, the structural plasticity of spines was impaired significantly: the transient phase was decreased to 72 ± 32% and the sustained phase was decreased to 5.3 ± 11.7% (Fig. 2). Downregulation of CentA1 with sh-CentA1 restored normal structural plasticity to Aβ-treated neurons: the transient phase was 189 ± 56% and the sustained phase was 74 ± 25% (Fig. 2). sh-CentA1 without Aβ treatment slightly reduced the transient (116 ± 36%; range, −6% to 365%) and sustained phases (59 ± 17%; range, −11% to 153%) of spine enlargement (Fig. 2), suggesting that CentA1 is required for spine structural plasticity under normal conditions. These results suggest that Aβ-induced upregulation of CentA1 expression causes impaired structural plasticity of dendritic spines and that downregulation of CentA1 can restore normal plasticity. Therefore, CentA1 upregulation is required for Aβ -induced impairment of spine structural plasticity.
Figure 2.

CentA1 is required for Aβ-induced impairment of spine structural plasticity. A, Time-lapsed images of spine structural plasticity induced by 2-photon glutamate uncaging in neurons transfected with sh-SC or sh-CentA1 and treated with vehicle (Veh) or Aβ for 4–6 d. The arrows indicate the stimulated spines. Structural plasticity was induced by applying a low-frequency train of two-photon uncaging pulses (6 ms, 30 pulses, 0.5 Hz) to a single dendritic spine in zero extracellular Mg2+ and 2 mm MNI-caged glutamate. B, Time course of spine volume change in stimulated spines or adjacent spines (Adj) in neurons treated as in A. The number of samples (spine/neuron) was 10/8, 9/6, 10/7, and 10/8, respectively. C, Transient spine volume change (volume change averaged over 25–30 min subtracted by volume change at 2 min (Matsuzaki et al., 2004). *p < 0.05. Error bars indicate SEM. D, Sustained spine volume change (volume change averaged over 25–30 min (Matsuzaki et al., 2004). *p < 0.05. Error bars indicate SEM.
Ras-Elk-1 signaling mediates Aβ-induced spine loss and synaptic dysfunction
Binding of CentA1 to Ras activates Ras-extracellular signal-regulated protein kinase (ERK) signaling and subsequently induces Elk-1 transcription (Hayashi et al., 2006). Therefore, we investigated whether Ras-Elk-1 signaling is required for Aβ-induced synaptic dysfunction by imaging dendritic spines. When Ras signaling was inhibited by expression of a dominant-negative Ras mutant (HRasS17N; DN-Ras; Fig. 1G, H) or by a Ras inhibitor (FTI-277; Fig. 1I), Aβ failed to decrease spine density. Furthermore, downregulation of Elk-1 with shRNA against Elk-1 (sh-Elk-1) restored normal spine density in Aβ-treated neurons (Fig. 3A, B). In addition, overexpression of Elk-1 was sufficient to cause spine loss (Fig. 3D). These results suggest that Ras-Elk-1 signaling is involved in Aβ-dependent spine loss. sh-Elk-1 also rescued the spine loss caused by overexpression of CentA1 (Fig. 3C), suggesting that Elk-1 is downstream of CentA1.
Figure 3.

Elk-1 is involved in Aβ-mediated dendritic spine loss. A, Representative images of apical dendrites of CA1 pyramidal neurons in organotypic slices transfected with EGFP and pSuper (cloning vector for shRNA) or Elk-1-specific shRNA (sh-Elk-1) and treated with vehicle (Veh) or 1 μm Aβ for 7 d. Scale bar, 5 μm. B, Spine density in apical dendrites of CA1 pyramidal neurons treated as in A. *p < 0.01. The number of neurons was 9, 5, 6, and 8, respectively. Error bars indicate SEM. C, Spine density in apical dendrites of CA1 pyramidal neurons transfected with EGFP and one of the following combinations of constructs for 7 d: pSuper and empty cloning vector (Vec), pSuper and wild-type CentA1 (pSuper + WT-CentA1) or sh-Elk-1 and wild-type CentA1 (sh-Elk-1 + WT-CentA1). *p < 0.05. The number of neurons was 12, 8, and 8, respectively. Error bars indicate SEM. D, Spine density in apical dendrites of CA1 pyramidal neurons transfected with EGFP and cloning vector (pcDNA3) or wild-type Elk-1 (WT-Elk-1). *p < 0.01. The number of neurons was 10 and 16. Error bars indicate SEM.
To determine whether synaptic function, and not just spine density, is restored in neurons in which CentA1-Ras signaling is inhibited, we examined the effect of Aβ on AMPA receptor-mediated miniature EPSCs (mEPSCs) in the presence or absence of the Ras inhibitor FTI-277 (Fig. 4). Incubation of organotypic hippocampal slice cultures with Aβ for 6–8 d resulted in a significant reduction in the mean frequency (by 59%) and amplitude (by 14%) of AMPAR-mediated mEPSCs, which is consistent with previous studies (Hsieh et al., 2006; Shankar et al., 2007). FTI-277 prevented Aβ from reducing mEPSC frequency (Fig. 4A,B) and amplitude (Fig. 4A,C). Therefore, our results indicate that Ras-Elk-1 signaling mediates Aβ-induced reduction of spine density and impairment of excitatory synaptic function.
Figure 4.
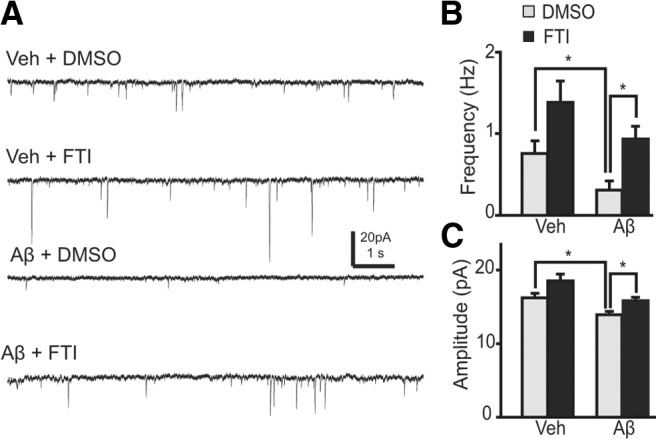
Ras is involved in Aβ-dependent reduction of mEPSC amplitude and frequency. A, Representative traces of mEPSCs in CA1 pyramidal neurons in organotypic slices, filtered with a 1 ms window. Slices were treated with vehicle (Veh) or 1 μm Aβ together with 0.25% DMSO or 10 μm FTI-277 (FTI; DMSO as vehicle) for 7 d. B, mEPSC frequency. *p < 0.05. The number of neurons was 38, 18, 14, and 20, respectively. Mean frequency of mEPSCs was calculated over each neuron and the mean and SEM of the averaged mEPSCs were plotted. C, mEPSC amplitude. Same data as in B.
Elk-1 association with mitochondria is involved in Aβ-mediated dendritic spine loss
Because we identified the location of Elk-1 as being downstream of CentA1 signaling in neurons, we investigated whether the Aβ-induced CentA1 upregulation induces ERK1/2-dependent phosphorylation of nuclear Elk-1 (S383) and subsequently activates the Elk-1-driven transcription. Immunoblot of nuclear extracts isolated from cultured organotypic hippocampal slices showed no increase in nuclear Elk-1 phosphorylation after Aβ treatment, suggesting that Elk-1 in the nucleus is not activated by Aβ (Fig. 5A). Supporting this result, CentA1-overexpression-induced Elk-1 transcription was suppressed by Aβ in dissociated hippocampal neurons (Fig. 5B). Therefore, these results suggest that CentA1-dependent Elk-1 transcription is not downstream of Aβ (Fig. 5A,B).
Figure 5.

Aβ-induced mitochondrial association of Elk-1 requires the CentA1-Ras-ERK pathway A, Nuclear Elk-1 phosphorylation/activation in organotypic slice cultures treated with vehicle (Veh) or 1 μm Aβ for 48 h followed by subcellular protein extraction and Western blotting of nuclear and cytosolic extracts for phospho-Elk-1 (Ser383). Blots were reprobed for total Elk-1 to ensure equal loading. GAPDH was used as a cytosolic marker and NeuN was used as a neuronal nuclear marker. Numbers under the blots represent relative Elk-1 phosphorylation/activation from four independent experiments. B, Elk-1 activity in dissociated hippocampal neurons quantified with the luciferase gene reporter assay. Neurons were transfected with either wild-type CentA1 (WT-CentA1) or empty vector (Vec) together with plasmids required for the assay (pFR-Luc, pFA2-Elk-1 and EF1ɑ-LacA), and treated with Veh (final 0.01% NH4OH) or 1 μm Aβ for 20 h. Error bars indicate SEM. *p < 0.05. C, Amount of Elk-1 in isolated mitochondria from hippocampal neuron cultures. Dissociated hippocampal neurons were electroporated with sh-SC or sh-CentA1, cultured for 3 d, treated with Veh or 1 μm Aβ for 48 h, followed by mitochondrial isolation. Elk-1 levels in mitochondrial fractions were determined by Western blotting. COX IV and VDAC were used as loading controls and mitochondrial markers. Numbers under blots represent COX-normalized changes in mitochondrial association of Elk-1 from seven independent experiments. D, Immunoblots of Elk-1 in whole lysates (total Elk-1; Top) and in mitochondria (Bottom) prepared from organotypic hippocampal slices treated with vehicle or 1 μm Aβ for 7 d. Numbers under the blots represent changes in expression of total Elk-1 normalized to actin (Top, n = 3) or mitochondrial Elk-1 normalized to VDAC (Bottom, n = 11). E, Ras signaling mediates mitochondrial association of Elk-1. Organotypic hippocampal slices were treated for 7 d with Veh or 1 μm Aβ together with 0.25% DMSO or 10 μm FTI-277 (DMSO as vehicle). The amount of total Elk-1 and Elk-1 from isolated mitochondria was analyzed by Western blotting. VDAC was used as the loading control and mitochondrial marker. NeuN was used to test the purity of the mitochondrial fractions. Numbers under blots represent normalized changes in Elk-1 mitochondrial association from five independent experiments. F, Organotypic hippocampal slices were treated for 7 d as in E in the presence or absence of FTI-277. Mitochondrial association and phosphorylation level of Elk-1 at Ser383 were determined by Western blotting, followed by VDAC reprobing as a loading control and mitochondrial marker. Numbers under the blots represent VDAC-normalized changes in mitochondrial Elk-1 phosphorylation from four independent experiments. G, Mitochondrial activity measured with TMRM fluorescence in neurons treated with DMSO or FTI-277. Dissociated hippocampal neurons (DIV6) were treated as in D for 24 h, followed by treatment with 100 nm TMRM (Invitrogen) for 30 min. The overall cellular fluorescence levels were visualized by fluorescence microscopy. The brightness of TMRM in each neuron was analyzed using ImageJ. Data are normalized to control condition (Veh + DMSO). Data represent average of four independent experiments. *p < 0.05. Error bars indicate SEM. H, Representative images of apical dendrites from CA1 pyramidal neurons in organotypic slices transfected with EGFP and treated with either vehicle or 1 μm Aβ (1–42) for 7 d, followed by cotreatment for additional 3 d with vehicle or 1 μm bongkrekic acid (BKA; DMSO as vehicle) or 1 μm cyclosporin A (CsA; DMSO as vehicle) and imaged with two-photon laser scanning microscopy. I, Quantification of spine density in neurons treated as in H. *p < 0.01. The number of neurons was 18, 16, 10, and 8, respectively. Error bars indicate SEM.
The reported association of Elk-1 with mPTP and its ability to open the mPTP in neurons exposed to DNA damage (Barrett et al., 2006) led us to investigate whether Aβ induces Elk-1 association with mitochondria and causes mitochondrial dysfunction. We found that Aβ increases the level of Elk-1 at mitochondria isolated from dissociated hippocampal neurons (Fig. 5C) and cultured organotypic hippocampal slices (Fig. 5D–F), whereas total Elk-1 level was not affected by Aβ treatment (Fig. 5D, top). Elk-1 association with mitochondria was abolished by sh-CentA1 in dissociated hippocampal neurons (Fig. 5C). Furthermore, the Ras inhibitor FTI-277 reduced the association of Elk-1 to the mitochondria (Fig. 5E,F). The phosphorylation level of Elk-1 at S383, the direct target of ERK1/2 (Whitmarsh et al., 1995), increased in the mitochondria after Aβ treatment and the increase was abolished by FTI-277 (Fig. 5F). These results suggest that Aβ causes the CentA1–Ras-ERK-dependent translocation of Elk-1 to mitochondria.
Measurement of mitochondrial activity using TMRM (Du et al., 2008) in dissociated hippocampal neurons demonstrated that Aβ decreases mitochondrial activity (Du et al., 2008), and inhibiting Ras with FTI-277 restored normal mitochondrial activity (Fig. 5G). Finally, we investigated whether mPTP opening by Elk-1 association is involved in Aβ-induced dendritic spine loss (Barrett et al., 2006). Inhibiting mPTP opening by bongkrekic acid or cyclosporin A rescued neurons from Aβ-induced reduction in spine density in cultured organotypic hippocampal slices (Fig. 5H,I), suggesting that opening of mPTP underlies Aβ-induced spine loss.
Finally, we investigated whether CentA1 upregulation and Elk-1 association with mitochondria also occur in a mouse model of AD, in which a mutant human form (Swedish mutation) of APP is overexpressed (APP mice; Fig. 6). Animals at 6 months of age are considered to correspond to the early stage of AD disease progression (Mucke et al., 2000; Jacobsen et al., 2006) and show a mild spine loss in the hippocampus (Jacobsen et al., 2006). Spine loss and learning deficits become progressively severe over the next several months (Jacobsen et al., 2006). At 3 months, before the onset of spine loss and behavioral phenotypes (Jacobsen et al., 2006), the level of CentA1 and mitochondrial Elk-1 in APP mice were similar to that in wild-type (non-transgenic littermates; Fig. 6A,B). At 6 months, the level of CentA1 and mitochondrial Elk-1 in APP mice increased by ∼2- and ∼5-fold compared with wild-type mice, respectively (Fig. 6A,B). Increase in CentA1 and mitochondrial Elk-1 in APP mice decayed at 8 months and even reversed at 12 months to ∼0.5-fold compared with wild-type animals (Fig. 6A–C). These results demonstrate that CentA1 upregulation and association of Elk-1 with mitochondria occur in APP mice in the early stage of disease progression.
Figure 6.

Increase in CentA1 and mitochondrial Elk-1 in a mouse model of AD. A, Expression level of CentA1 in hippocampi of APP mice overexpressing a mutant human form of amyloid precursor protein (APP) and in their non-transgenic littermates (WT). The level of CentA1 in hippocampi from 3-month-old (n = 5 WT, n = 6 APP), 6-month-old (n = 6 WT, n = 6 APP), 8-month-old (n = 5 WT, n = 7 APP), and 12-month-old (n = 4 WT, n = 5 APP) male mice was analyzed by Western blotting, followed by reprobing of the membranes for β-actin. B, The amount of Elk-1 in mitochondria isolated from hippocampi of male WT and APP mice. Mitochondrial lysates from hippocampi of WT and APP 3-months-old mice (n = 5 WT, n = 6 APP), 6-month-old mice (n = 11 WT, n = 11 APP), 8-month-old mice (n = 5 WT, n = 7 APP), and 12-month-old mice (n = 5 WT, n = 6 APP) were immunoblotted for Elk-1, followed by VDAC reprobing. C, Time dependence of CentA1 upregulation and Elk-1 mitochondrial association in APP mice compared with non-transgenic littermates. D, Hypothetical signaling scheme linking Aβ and neuronal dysfunctions.
Discussion
In this study, we show that Aβ increases the expression level of CentA1 in dissociated neurons (Fig. 1A) and organotypic hippocampal slices (Fig. 1B) within ∼48 h. Transient CentA1 increase also occurs in the hippocampus of APP mice (Fig. 6). We have presented evidence that this upregulation of CentA1 mediates Aβ-induced decreases in spine density and impaired synaptic plasticity and identified Ras-Elk-1 signaling at mitochondria to be downstream of CentA1, leading to neuronal dysfunction (Fig. 6D). Our findings suggest that the upregulation of CentA1 observed in the human AD brain is caused by Aβ, and that this Aβ-induced upregulation of CentA1 contributes to neuronal dysfunction in AD. Our results demonstrate that downregulation of the CentA1–Ras-Elk-1 pathway rescues Aβ-induced phenotypes in spine density (Fig. 1, Fig. 3), spine structural plasticity (Fig. 2), and mEPSC amplitude and frequency (Fig. 4) in organotypic hippocampal slice cultures.
Although CentA1 overexpression can activate Elk-1-dependent transcription (Fig. 5B; Hayashi et al., 2006), Aβ-induced CentA1 upregulation does not increase Elk-1 activity in the nucleus or Elk-1-dependent transcription (Fig. 5A,B). This finding is consistent with a previous study showing Aβ-mediated inhibition of BDNF-dependent Elk-1 transcription (Tong et al., 2004). These data suggest that the lack of upregulation of Elk-1-dependent transcription in Aβ-treated neurons is likely due to other Aβ-dependent actions that inhibit Elk-1-dependent transcription (Tong et al., 2004; Fig. 6D).
We found that Aβ causes mitochondrial association of Elk-1 in a CentA1-dependent (Fig. 5C) and Ras-dependent manner (Fig. 5E,F). It has been reported that Elk-1 association with mitochondria opens the mPTP, causing mitochondrial dysfunction (Barrett et al., 2006). Accordingly, inhibiting the opening of the mPTP using bongkrekic acid or cyclosporin A prevented spine loss in the presence of Aβ (Fig. 5H,I). Further, the Ras inhibitor FTI-277 inhibited both the association of Elk-1 with mitochondria (Fig. 5D,F) and Aβ-induced mitochondrial inactivation in dissociated neurons (Fig. 5G). These results support a model in which CentA1–Ras-dependent Elk-1 association with mitochondria causes mitochondrial dysfunction, leading to synaptic dysfunction (Fig. 6D). Consistent with our study, Ras signaling at the mitochondria has been implicated in cell death caused by mitochondrial dysfunction in tumor cell lines (Yagoda et al., 2007). Furthermore, cyclophilin D, an integral part of mPTP, has been implicated in Aβ-induced impairment of mitochondrial Ca2+ handling and synaptic dysfunction (Du et al., 2008).
In a mouse model of AD (APP mice), the onset of CentA1 elevation and Elk-1 association with mitochondria occurs around the age of 6 months (Fig. 6). This timing is consistent with the onset of many corresponding phenotypes, including spine loss, LTP impairment, and memory deficits (Mucke et al., 2000; Jacobsen et al., 2006). However, both CentA1 and mitochondrial Elk-1 decay when mice reach 8–12 months of age, whereas the phenotypes continue to progress. Therefore, the CentA1–Ras-Elk-1 signaling pathway at mitochondria is likely to underlie neuronal dysfunction in a relatively early stage of AD.
Footnotes
This work was supported by the Howard Hughes Medical Institute, National Institute of Mental Health, National Institute of Neurological Disorders and Stroke, Alzheimer's Association of America, Ruth K. Broad Biomedical Fellowship (to E.M.S.), Portuguese Foundation for Science and Technology (to A.F.O.), and a Young Investigator Award from Children's Tumor Foundation (to A.F.O.). We thank M. Ehlers, G. Feng, M. Hetman, W. Liedtke, and A. West for reagents, A. Wan, D. Kloetzer, and A. Sueki for technical assistance, and N. Hedrick, L. Colgan, and other members of the Yasuda laboratory for discussions.
The authors declare no competing financial interests.
References
- Aggensteiner M, Reiser G. Expression of the brain-specific membrane adapter protein p42IP4/centaurin alpha, a Ins(1,3,4,5)P4/PtdIns(3,4,5)P3 binding protein, in developing rat brain. Brain Res Dev Brain Res. 2003;142:77–87. doi: 10.1016/s0165-3806(03)00033-6. [DOI] [PubMed] [Google Scholar]
- Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, Snyder EM, Gouras GK. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis. 2005;20:187–198. doi: 10.1016/j.nbd.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Barrett LE, Van Bockstaele EJ, Sul JY, Takano H, Haydon PG, Eberwine JH. Elk-1 associates with the mitochondrial permeability transition pore complex in neurons. Proc Natl Acad Sci U S A. 2006;103:5155–5160. doi: 10.1073/pnas.0510477103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese B, Shaked GM, Tabarean IV, Braga J, Koo EH, Halpain S. Rapid, concurrent alterations in pre- and postsynaptic structure induced by naturally-secreted amyloid-beta protein. Mol Cell Neurosci. 2007;35:183–193. doi: 10.1016/j.mcn.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, Bekkers JM. Detection of spontaneous synaptic events with an optimally scaled template. Biophys J. 1997;73:220–229. doi: 10.1016/S0006-3495(97)78062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert A, Hauptmann S, Scherping I, Meinhardt J, Rhein V, Dröse S, Brandt U, Fändrich M, Müller WE, Götz J. Oligomeric and fibrillar species of beta-amyloid (A beta 42) both impair mitochondrial function in P301L tau transgenic mice. J Mol Med. 2008;86:1255–1267. doi: 10.1007/s00109-008-0391-6. [DOI] [PubMed] [Google Scholar]
- Galvita A, Grachev D, Azarashvili T, Baburina Y, Krestinina O, Stricker R, Reiser G. The brain-specific protein, p42(IP4) (ADAP 1) is localized in mitochondria and involved in regulation of mitochondrial Ca2+ J Neurochem. 2009;109:1701–1713. doi: 10.1111/j.1471-4159.2009.06089.x. [DOI] [PubMed] [Google Scholar]
- Hammonds-Odie LP, Jackson TR, Profit AA, Blader IJ, Turck CW, Prestwich GD, Theibert AB. Identification and cloning of centaurin-alpha. A novel phosphatidylinositol 3,4,5-trisphosphate-binding protein from rat brain. J Biol Chem. 1996;271:18859–18868. doi: 10.1074/jbc.271.31.18859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson Petersen CA, Alikhani N, Behbahani H, Wiehager B, Pavlov PF, Alafuzoff I, Leinonen V, Ito A, Winblad B, Glaser E, Ankarcrona M. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A. 2008;105:13145–13150. doi: 10.1073/pnas.0806192105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Harvey CD, Yasuda R, Zhong H, Svoboda K. The spread of Ras activity triggered by activation of a single dendritic spine. Science. 2008;321:136–140. doi: 10.1126/science.1159675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H, Matsuzaki O, Muramatsu S, Tsuchiya Y, Harada T, Suzuki Y, Sugano S, Matsuda A, Nishida E. Centaurin-alpha1 is a phosphatidylinositol 3-kinase-dependent activator of ERK1/2 mitogen-activated protein kinases. J Biol Chem. 2006;281:1332–1337. doi: 10.1074/jbc.M505905200. [DOI] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, Martone R, Morrison JH, Pangalos MN, Reinhart PH, Bloom FE. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:5161–5166. doi: 10.1073/pnas.0600948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalita K, Kharebava G, Zheng JJ, Hetman M. Role of megakaryoblastic acute leukemia-1 in ERK1/2-dependent stimulation of serum response factor-driven transcription by BDNF or increased synaptic activity. J Neurosci. 2006;26:10020–10032. doi: 10.1523/JNEUROSCI.2644-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutz MR, Böckers TM, Sabel BA, Hülser E, Stricker R, Reiser G. Expression and subcellular localization of p42IP4/centaurin-alpha, a brain-specific, high-affinity receptor for inositol 1,3,4,5-tetrakisphosphate and phosphatidylinositol 3,4,5-trisphosphate in rat brain. Eur J Neurosci. 1997;9:2110–2124. doi: 10.1111/j.1460-9568.1997.tb01378.x. [DOI] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Escobedo-Lozoya Y, Szatmari EM, Yasuda R. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature. 2009;458:299–304. doi: 10.1038/nature07842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron. 2008;60:748–766. doi: 10.1016/j.neuron.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK. Biolistic transfection of neurons. Sci STKE. 2000;2000:PL1. doi: 10.1126/stke.2000.51.pl1. [DOI] [PubMed] [Google Scholar]
- Moolman DL, Vitolo OV, Vonsattel JP, Shelanski ML. Dendrite and dendritic spine alterations in Alzheimer models. J Neurocytol. 2004;33:377–387. doi: 10.1023/B:NEUR.0000044197.83514.64. [DOI] [PubMed] [Google Scholar]
- Moore CD, Thacker EE, Larimore J, Gaston D, Underwood A, Kearns B, Patterson SI, Jackson T, Chapleau C, Pozzo-Miller L, Theibert A. The neuronal Arf GAP centaurin alpha1 modulates dendritic differentiation. J Cell Sci. 2007;120:2683–2693. doi: 10.1242/jcs.006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakoshi H, Wang H, Yasuda R. Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature. 2011;472:100–104. doi: 10.1038/nature09823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Origlia N, Righi M, Capsoni S, Cattaneo A, Fang F, Stern DM, Chen JX, Schmidt AM, Arancio O, Yan SD, Domenici L. Receptor for advanced glycation end product-dependent activation of p38 mitogen-activated protein kinase contributes to amyloid-beta-mediated cortical synaptic dysfunction. J Neurosci. 2008;28:3521–3530. doi: 10.1523/JNEUROSCI.0204-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei JJ, Tanaka T, Tung YC, Braak E, Iqbal K, Grundke-Iqbal I. Distribution, levels, and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J Neuropathol Exp Neurol. 1997;56:70–78. doi: 10.1097/00005072-199701000-00007. [DOI] [PubMed] [Google Scholar]
- Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, Lo E, Wu D, Saule E, Bouschet T, Matthews P, Isaac JT, Bortolotto ZA, Wang YT, Collingridge GL. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron. 2007;53:703–717. doi: 10.1016/j.neuron.2007.01.029. [DOI] [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL, Svoboda K. ScanImage: Flexible software for operating laser-scanning microscopes. Biomed Eng Online. 2003;2:13. doi: 10.1186/1475-925X-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiser G, Bernstein HG. Neurons and plaques of Alzheimer's disease patients highly express the neuronal membrane docking protein p42IP4/centaurin alpha. Neuroreport. 2002;13:2417–2419. doi: 10.1097/00001756-200212200-00008. [DOI] [PubMed] [Google Scholar]
- Reiser G, Bernstein HG. Altered expression of protein p42IP4/centaurin-alpha 1 in Alzheimer's disease brains and possible interaction of p42IP4 with nucleolin. Neuroreport. 2004;15:147–148. doi: 10.1097/00001756-200401190-00028. [DOI] [PubMed] [Google Scholar]
- Rui Y, Gu J, Yu K, Hartzell HC, Zheng JQ. Inhibition of AMPA receptor trafficking at hippocampal synapses by beta-amyloid oligomers: the mitochondrial contribution. Mol Brain. 2010;3:10. doi: 10.1186/1756-6606-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serenó L, Coma M, Rodríguez M, Sánchez-Ferrer P, Sánchez MB, Gich I, Agulló JM, Pérez M, Avila J, Guardia-Laguarta C, Clarimón J, Lleó A, Gómez-Isla T. A novel GSK-3beta inhibitor reduces Alzheimer's pathology and rescues neuronal loss in vivo. Neurobiol Dis. 2009;35:359–367. doi: 10.1016/j.nbd.2009.05.025. [DOI] [PubMed] [Google Scholar]
- Sgambato V, Vanhoutte P, Pagès C, Rogard M, Hipskind R, Besson MJ, Caboche J. In vivo expression and regulation of Elk-1, a target of the extracellular-regulated kinase signaling pathway, in the adult rat brain. J Neurosci. 1998;18:214–226. doi: 10.1523/JNEUROSCI.18-01-00214.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Callahan LM, Sul JY, Kim TK, Barrett L, Kim M, Powers JM, Federoff H, Eberwine J. A neurotoxic phosphoform of Elk-1 associates with inclusions from multiple neurodegenerative diseases. PLoS One. 2010;5:e9002. doi: 10.1371/journal.pone.0009002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Sabatini BL, Sudhof TC. Synapses and Alzheimer's disease. Cold Spring Harb Perspect Biol. 2012:4. doi: 10.1101/cshperspect.a005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Szatmari E, Kalita KB, Kharebava G, Hetman M. Role of kinase suppressor of Ras-1 in neuronal survival signaling by extracellular signal-regulated kinase 1/2. J Neurosci. 2007;27:11389–11400. doi: 10.1523/JNEUROSCI.3473-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tackenberg C, Brandt R. Divergent pathways mediate spine alterations and cell death induced by amyloid-{beta}, wild-type tau, and R406W tau. J Neurosci. 2009;29:14439–14450. doi: 10.1523/JNEUROSCI.3590-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong L, Balazs R, Thornton PL, Cotman CW. Beta-amyloid peptide at sublethal concentrations downregulates brain-derived neurotrophic factor functions in cultured cortical neurons. J Neurosci. 2004;24:6799–6809. doi: 10.1523/JNEUROSCI.5463-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhoutte P, Barnier JV, Guibert B, Pagès C, Besson MJ, Hipskind RA, Caboche J. Glutamate induces phosphorylation of Elk-1 and CREB, along with c-fos activation, via an extracellular signal-regulated kinase-dependent pathway in brain slices. Mol Cell Biol. 1999;19:136–146. doi: 10.1128/mcb.19.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Nguyen LN, Kessels HW, Hagiwara H, Sisodia S, Malinow R. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat Neurosci. 2010;13:190–196. doi: 10.1038/nn.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmarsh AJ, Shore P, Sharrocks AD, Davis RJ. Integration of MAP kinase signal transduction pathways at the serum response element. Science. 1995;269:403–407. doi: 10.1126/science.7618106. [DOI] [PubMed] [Google Scholar]
- Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, Spires-Jones T, Xie H, Arbel-Ornath M, Grosskreutz CL, Bacskai BJ, Hyman BT. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci. 2010;30:2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, Smith R, Lessnick SL, Sahasrabudhe S, Stockwell BR. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447:864–868. doi: 10.1038/nature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]