

Abstract
Genetic studies of human diseases have identified multiple genetic risk loci for various fibrotic diseases. This has provided insights into the myriad of biological pathways potentially involved in disease pathogenesis. These discoveries suggest that alterations in immune responses, barrier function, metabolism and telomerase activity may be implicated in the genetic risks for fibrotic diseases. In addition to genetic disease-risks, the identification of genetic disease-modifiers associated with disease complications, severity or prognosis provides crucial insights into the biological processes implicated in disease progression. Understanding the biological processes driving disease progression may be critical to delineate more effective strategies for therapeutic interventions. This review provides an overview of current knowledge and gaps regarding genetic disease-risks and genetic disease-modifiers in human fibrotic diseases.
Keywords: fibrosis, auto-immunity, genetics, GWAS, disease progression
Introduction
Fibrosis arises as the result of excessive connective tissue and extracellular matrix deposition. It emerges from an aberrant or uncontrolled repair response often triggered by tissue damage that may be initiated by radiation, mechanical injury or infections and results in scar formation. In the context of auto-immunity, with sustained immune activation, the injury and repair phases persist and lead to scar tissue formation that disrupts organ architecture and function with a frequently fatal outcome (Figure 1).
Figure 1.
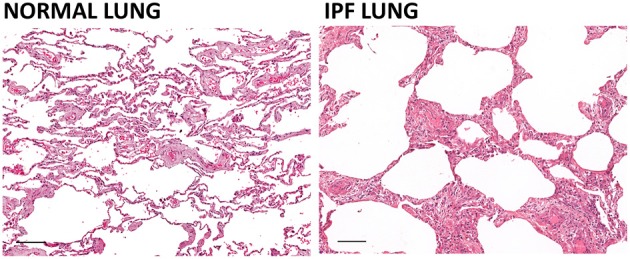
Effect of pulmonary fibrosis on lung architecture. The architecture of the lung in idiopathic pulmonary fibrosis (IPF) is characterized by a so-called “honeycomb” pattern with airways separated by bands of inflamed fibrous connective tissue and to a lesser extent, smooth muscle. The modifications of the lung architecture induced by the fibrosis lead to compromised diffusion of oxygen and carbon dioxide and impaired pulmonary function. Hematoxylin and eosin staining of human normal lung and IPF lung (scale bar = 100 um). Courtesy of Dr. Robert Dunstan.
In the last decade, the scientific community has successfully collaborated through consortia to unravel the genetic basis of susceptibility for many diseases. Genome-Wide Association Studies (GWAS) have identified numerous genetic polymorphisms that confer higher risk for diseases and have provided insights into the biological processes that contribute to disease susceptibility. One key finding is the substantial overlap of genetic loci associated with disease risk across a variety of complex immune diseases (Cotsapas and Hafler, 2013). This highlights the complexity of the etiology of clinical phenotypes that have an immune basis but are also largely influenced by environmental factors and can affect different target organs. Although the target organs may be different, a common complication of these diverse immune-mediated diseases is the abundance of fibrotic processes and scar tissue formation. This likely reflects that, when altered, many processes such as inflammation, barrier function and metabolism may result in sustained tissue injury, impaired repair processes and ultimately fibrosis (Figure 2).
Figure 2.
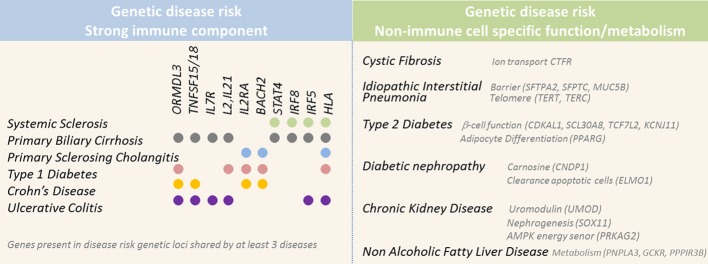
Selected genes located in genetic risk loci associated with higher susceptibility for diseases with fibrotic complications. Genetic studies have successfully identified numerous genetic risk loci associated with higher susceptibility for diseases associated with fibrosis. Left panel displays diseases associated with a strong immune component and the genetic risk loci implicated in at least three of these diseases. Right panel displays diseases associated with risk loci implicating genes involved in non-immune function, such as barrier and metabolic functions.
Earlier genetic studies focused on signals that distinguish between disease and healthy status using “case-control” studies. Recent efforts have sought to identify genetic factors influencing clinical outcomes with emerging “case-case” studies, looking at patient subgroups that follow different disease courses. The hope is this will provide insights into the pathogenic processes dictating disease progression and severity. Emerging results on genetic disease-modifiers show limited overlap with genetic loci involved in disease risk, highlighting the point that disease initiation and disease progression are not necessarily driven by the same mechanisms. Interestingly, these studies also allow us to determine how genetics might contribute to milder clinical outcomes, as illustrated by the recent discovery of a common polymorphism in FOXO3 locus affecting the TGF-β pathway which appears associated with improved prognosis in Crohn's disease and rheumatoid arthritis (Lee et al., 2013a). Further investigations will define whether, across diseases involving different organ systems, genetic variants affecting a set of common key biological pathways might favor more susceptibility to fibrotic complications.
Diseases associated with lung fibrosis
Idiopathic interstitial pneumonias and idiopathic pulmonary fibrosis
Familial forms of idiopathic pulmonary fibrosis (IPF) account for 2–20% of IPF patients, supporting a strong genetic component in the development of the disease [reviewed in Kropski et al. (2013)]. Investigations on families have identified genetic variants in SFTPC (encoding surfactant protein C), SFTPA2 (encoding surfactant protein A2), MUC5B (encoding a mucin constituent of the mucus), as well as TERT and TERC (encoding components of the telomerase complex) to be associated with pulmonary fibrosis (Nogee et al., 2001; Thomas et al., 2002; Armanios et al., 2007; Tsakiri et al., 2007; Wang et al., 2009; van Moorsel et al., 2010; Ono et al., 2011; Seibold et al., 2011). The genetic association of genetic polymorphisms in MUC5B-MUC2-TOLLIP as well as TERT and TERC loci with high risk for pulmonary fibrosis has been confirmed by recent results from GWAS comparing 4683 controls and 1616 cases of fibrotic idiopathic interstitial pneumonias (IIP) including 77% of IPF cases with independent replication cohort (Fingerlin et al., 2013). These observations provide justification for investigating familial diseases with linkage studies, as well as large GWAS approaches.
The implication of genetic polymorphisms in SFTPA2, SFTPC, MUC5B as well as DSP (encoding desmoplakin) as risk factors for pulmonary fibrosis suggest that the integrity of the barrier function is critically important in maintaining lung homeostasis (Fingerlin et al., 2013). Coding mutations in SFTPC associated with pulmonary fibrosis lead to aberrant pro-surfactant protein C intermediate products which cause alterations in protein maturation [reviewed in Tanjore et al. (2013), Thurm et al. (2013)]. These mutations are often, but not always, detected in conjunction with activation of the Unfolded Protein Response (UPR) pathway, which has also been reported in the alveolar epithelial cells of IPF patients carrying the SFTPC L188Q mutation (Tanjore et al., 2012; Thurm et al., 2013). SFTPA2 mutations induce retention of surfactant protein A in the endoplasmic reticulum and similarly lead to UPR activation (Wang et al., 2009; Maitra et al., 2010). Two proposed mechanisms link UPR activation to tissue injury and fibrosis: UPR activation likely increases the loss of epithelial cells after injury and may also be involved in promoting epithelial-to-mesenchymal (EMT) transition (Tanjore et al., 2011; Zhong et al., 2011). Both these mechanisms likely favor the cycle of aberrant injury and repair that is typical of fibrotic responses. Recent studies have, however, also revealed that SFTPA1 and SFTPC mutations induce excessive TGF-β secretion (Maitra et al., 2012, 2013). Depending on the mutations, this effect does not always depend on UPR activation. This brings into question the importance of the role of UPR activation in promoting profibrotic phenotype associated with the polymorphisms identified by genetic studies.
The rs35705950 risk Single Nucleotide Polymorphism (SNP) for pulmonary fibrosis in the MUC5B region has generated a lot of interest. MUC5B expression is reported to be higher in the lung of IPF vs. healthy subjects, and in subjects carrying rs35705950 risk allele (Seibold et al., 2011). MUC5B encodes a member of the mucin family, which contains highly glycosylated proteins that are component of mucus secretions that protect the epithelial layer. Perhaps unexpectedly, rs35705950 was recently associated with improved survival in an IPF (Peljto et al., 2013). This supports the concept that underlying mechanisms of disease initiation and disease progression may be quite distinct. Given these recent revelations, it has now been proposed that the increased expression of MUC5B predisposes to IPF, but probably also has a beneficial role in enhancing the mucosal host defense during tissue damage. Additional SNPs in the MUC5B region including in the TOLLIP and MUC2 loci are also associated with higher risk for IPF and the biological contribution of these SNPs in the increased risk for IPF remains to be elucidated (Fingerlin et al., 2013).
As MUC5B, SFTPC, and SFTPA1 are expressed by alveolar type II cells, this raises the possibility that injury of these cells is a critical pathogenic mechanism in pulmonary fibrosis (Seibold et al., 2013). This hypothesis is supported by the observation that lung fibrosis occurs following ablation of alveolar type II in genetically modified mice using diphtheria toxin (DT) receptor transgene under the control of Sftpc promoter (Sisson et al., 2010). However, lung fibrosis was not observed in a different genetic mouse model where the DT expression is controlled by an inducible Cre recombinase knocked into the Sftpc locus (Barkauskas et al., 2013). This discrepancy in phenotype could reflect different levels of cell ablation between the two systems or the differential contribution of additional lung cell populations that mediate the lung epithelium repair.
Different studies in human and mouse models have attempted to provide biological insights into the association of TERT and TERC polymorphisms with pulmonary fibrosis. Several reports showed shortened telomeres in IIP patients with or without mutation in genes encoding components of the telomerase complex, however, this was not replicated in a recent investigation of an IPF Mexican cohort (Alder et al., 2008; Cronkhite et al., 2008; Diaz de Leon et al., 2010; Liu et al., 2013a). In mouse models, two independent groups reported that Tert and Terc KO mice do not present spontaneous lung fibrosis and that Terc deficiency does not affect bleomycin-induced lung fibrosis (Liu et al., 2007; Degryse et al., 2012; Liu et al., 2013a). Degryse et al. did not observe any phenotype on bleomycin-induced lung fibrosis using Tert deficient mice, while Liu et al. reports a protective effect using similar disease model but a different Tert deficient strain. The reason of the discrepancy of these results is yet to be elucidated. However, the protection phenotype observed in Tert deficient mice in the bleomycin-induced lung fibrosis was similar with mice from 2nd and 4th generation despite shortening of the telomeres. This led the authors to speculate that this effect may not be dependent on telomere length (Liu et al., 2013a). Supporting the notion that TERT and TERC deficiency might contribute to pulmonary fibrosis by mechanisms dependent on telomerase activity but not necessarily telomere length, telomerase activity is induced in IPF and NSIP fibroblasts and systemic sclerosis lung compared to healthy donor samples (Fridlender et al., 2007; Liu et al., 2013a). Furthermore, telomerase activity was recently shown to regulate Wnt signaling, mitochondrial function and oxidative stress (Park et al., 2009; Sahin et al., 2011). These pathways are known to be activated in human and mouse disease tissues and the inhibition of Wnt/beta catenin pathways and oxidative stress decreases fibrosis in mouse models and are therefore currently considered as attractive therapeutic fibrosis targets (Lam and Gottardi, 2011; Hecker et al., 2012).
The examples described above show genetic studies have successfully unraveled key components of the genetic architecture of IIP and IPF by leveraging the strong genetic signals associated with disease in familial cases. Functional in-vivo characterizations of the genetic polymorphisms associated with pulmonary fibrosis are now starting to provide insights into potential mechanisms that remain to be further validated. Emerging efforts to evaluate the role of the susceptibility loci for pulmonary fibrosis have led to unexpected results, as demonstrated by the discovery of the association of MUC5B variant with disease risk, but improved disease prognosis. Additional genetic polymorphisms are proposed to affect IPF severity, such as TLR3 L412F, and FcγRIIa R131H variants, which further reflect the influence of immune mechanisms in IPF progression (Bournazos et al., 2010; O'Dwyer et al., 2013). Polymorphisms in the angiotensinogen promoter are also described to be associated with further decline of pulmonary function in IPF subjects perhaps consistent with results from mouse models in which the angiotensin pathway promotes fibrosis (Molina-Molina et al., 2008; Dang et al., 2013). However, these results are yet to be replicated in well-powered studies. These examples clearly demonstrate the need for genetic studies of disease progression to further understand pathogenesis, alongside development of mouse models and in-vitro/ex-vivo models of human, cells and tissues to fully validate the leads provided by genetic studies.
Interstitial lung disease in systemic sclerosis
Systemic Sclerosis (SSc) is thought to be a chronic systemic autoimmune disease with limited genetic component because of the rare familial cases and low concordance for disease in monozygotic twins (4.7%) (Feghali-Bostwick et al., 2003). However, the concordance in monozygotic twins for the presence of antinuclear antibodies in SSc is very high (90 vs. 40% for dizygotic twins), suggesting that the auto-immunity component of SSc is highly inheritable, but that the disease phenotype may be influenced by other factors that are largely not dependent on genetics. Despite limited disease heritability, several GWAS have detected genetic associations with risk for SSc and appear to have confirmed the role of the immune response in the disease risk. Many of the identified risk loci are shared with Rheumatoid Arthritis and Systemic Lupus Erythematous, including alleles located in MHC, STAT4, CD247, and IRF5 loci (Radstake et al., 2010; Allanore et al., 2011; Gorlova et al., 2011). These genes suggest that dysregulation of different components of the immune response influence auto-immunity. For example, STAT4 regulates signaling from IL-12 and IL-23 receptors in T-cells and from IFN receptor in monocytes and NK cells, while IRF5 is a transcription factor in the type 1 interferon pathway, and CD247 encodes for a subunit of the T-cell receptor and modulates T-cell activation [reviewed in Romano et al. (2011)]. However, precise functional consequences of the risk alleles discovered in these loci still remain to be elucidated. These studies come with great challenges for ex-vivo studies using samples from patients carrying risk and non-risk alleles, and with the development of mouse models with knock-in of risk alleles for in-vivo studies.
Interstitial lung disease (ILD) is one complication of SSc and is most often associated with diffuse cutaneous disease and the presence of anti-topoisomerase I antibodies (Steen et al., 1988; Assassi et al., 2010). Genetic candidate approach studies (albeit with limited sample size) have identified genetic polymorphisms associated with SSc-ILD in CTGF, HGF, MMP12, which encode known regulators of fibrotic responses, and in IRAK1 and NLRP1, which encode proteins involved innate immune responses (Fonseca et al., 2007; Manetti et al., 2010; Dieudé et al., 2011a,b; Hoshino et al., 2011; Sharif et al., 2012) (see Table 1). Similar to the above example where the MUC5B rs35705950 SNP is associated with IPF susceptibility but with improved prognosis, the IRF5 SNP rs4728142 confers higher risk for SSc, but also longer survival with milder ILD (Sharif et al., 2012). Combination of the risk alleles at STAT4 SNP rs7574865 and IRF5 SNP rs2004640 leads to increased risk for ILD, highlighting that studies of genetic interactions may be relevant for disease (Dieudé et al., 2009). Observations such as this reflect the complexity of these diseases.
Table 1.
Genetic polymorphisms proposed to be associated with SSc-ILD.
| Association with phenotypes | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Variants | Genes | Population | Discovery | Replication | Replication | Odd ratio | p = value | Anti-SLC70 | SSc | Expression | Ref |
| rs2276109 | MMP12 | Italian | 250/263 | 2.94 (95% CI 1.25–6.95) | p = 0.01 | Yes | ILD | higher level of MMP12 | Manetti et al., 2010 | ||
| CTGF -945GG | CTGF | UK | 200/188 | 300/312 | 3.1 (95% CI, 1.9–5.0) | p < 0.001 | Yes | ILD | higher level of CTGF | Fonseca et al., 2007 | |
| HGF -1652 TT | HGF | Japanese | 159/103 | 155/0 | 8.1 (95% CI 2.5–26.0) | p = 0.0004 | NA | ESLD | lower level of HGF | Hoshino et al., 2011 | |
| rs1059702 | IRAK1 | EU | 849/625 | 495/509 | 466/1083 | 2.09 (95% CI 1.35–3.24) | p = 0.0009 | Yes | ILD | Dieudé et al., 2011a, 2009 | |
| rs8182352 | NLRP1 | EU | 870/962 | 532/324 | 527/301 | 1.19 (95% CI 1.05–1.36) | p = 0.0065 | Yes | ILD | Dieudé et al., 2011b | |
| rs2004640 | IRF5 | French | ## 179/374 | ## 134/374 | 1.786 (95% Cl 1.25–2.58) | p = 0.002 | NA | ILD | Dieudé et al., 2009 | ||
| rs7574865 | STAT4 | ||||||||||
| VARIANTS ASSOCIATED WITH IMPROVED PROGNOSIS | |||||||||||
| rs4728142 | IRF5 | Caucasian | 914 cases | 529 cases | 0.75 (95% CI 0.62–0.90) | p = 0.002 | Longer survival | Lower level of IRF5 | Sharif et al., 2012 | ||
Candidate gene approach studies with limited power but increasing sample sizes have reported several candidate polymorphisms that may confer risk for SSc-ILD. Discovery and Replication stages show numbers of case and control patients. ## symbol indicate a case-case study.
While IPF and SSc-ILD present with distinct clinical features, they are both characterized by the presence of fibrotic lesions in the lung at end stage disease. Similar gene expression profiles are detected in lung explants from IPF and SSc patients, suggesting some overlap in pathogenic mechanisms (Hsu et al., 2011; Murray et al., 2012). This hypothesis was tested with three independent studies that investigated the MUC5B SNP rs35705950 risk allele for IPF in SSc-ILD. There was no association with SSc-ILD, while the association with IPF was confirmed by all groups (Peljto et al., 2012; Borie et al., 2013; Stock et al., 2013). This result further highlights the differences in pathogenic mechanisms associated with IPF and SSc-ILD, even when the disease tissue gene expression profiles may be similar.
Understanding the genetic architecture associated with SSc-ILD will be crucial to provide biological insights into the pathogenic mechanisms driving this debilitating disease. Breakthrough discoveries will require well-powered studies and comprehensive genetic analysis with meta-analysis of genome-wide data rather than candidate gene studies.
Diseases associated with renal fibrosis
Tubulointerstitial fibrosis is a feature of progression of chronic kidney diseases (CKD) and diabetic nephropathy (DN). The incidence of end stage renal disease in African Americans is known to be 3–4-fold higher compared to non-African Americans (Li et al., 2004). This excess risk is thought to be mainly due to genetic polymorphisms in the MY9H/APOL1 region with a non-synonymous SNP in APOL1 locus (Kao et al., 2008; Kopp et al., 2008). Interestingly, this polymorphism appears to result from a positive selection in population of African ancestry, due to a functional advantage over sleeping sickness (Genovese et al., 2010).
Chronic kidney disease
GWAS have identified genetic polymorphisms associated with renal function and susceptibility to CKD. Genetic polymorphisms in UMOD, SOX11, and PRKAG2 loci appear associated with CKD (Köttgen et al., 2009, 2010; Gudbjartsson et al., 2010). Mutations in UMOD are linked to familial kidney diseases, and common polymorphisms in the UMOD locus were shown to be associated with risk for CKD in two GWAS scans (Köttgen et al., 2009, 2010; Vyletal et al., 2010). UMOD encodes uromodulin, which is released in the urine and plays a protective role against urinary tract infections and ischemia-induced acute kidney injury, as shown in studies of Umod-deficient animals (Bates et al., 2004; Mo et al., 2004; El-Achkar et al., 2008). The underlying mechanisms are unclear as uromodulin appears to have cell-specific effects that could be both pro-inflammatory (on macrophages and neutrophils) or anti-inflammatory [reviewed in El-Achkar and Wu (2012)].
SOX11 appears essential for embryonic development as Sox11-deficient mice die at birth with many malformations (Hargrave et al., 1997; Sock et al., 2004). Sox11 was shown to control the expression of Wnt4 in Xenopus (Murugan et al., 2012). Wnt4 signaling is known to play a key role in nephrogenesis, as its activation promotes renal fibrosis in mouse models (Kispert et al., 1998; Surendran et al., 2002). In addition, missense mutations in WNT4 locus are associated with renal hypodysplasia in humans (Vivante et al., 2013). Thus, one may speculate that SOX11 genetic variants associated with CKD might affect renal function through dysregulation of Wnt4 pathway; this hypothesis remains to be tested.
PRKAG2 encodes a subunit of the energy sensor AMP-activated protein kinase (AMPK) whose role in renal homeostasis has been extensively studied (Hallows et al., 2010). In the context of tissue injury, activation of AMPK inhibits Epithelial-to-Mesenchymal Transition EMT and Reactive Oxygen Species (ROS) production induced by known pro-fibrotic factors in renal fibrosis, such as TGF-β, angiotensin II and high glucose (Lee et al., 2013b). It is also reported to promote Monocyte-to-Fibroblast transition (Yang et al., 2013). AMPK activity was shown to be protective in non-diabetic and in high fat diet-induced renal disease models (Declèves et al., 2011; Satriano et al., 2013). The beneficial effects of metformin, an AMPK activator, on renal function are recognized, but its use in CKD is currently at the center of controversial debates due to potential risk of lactic acidosis in the context of renal deficiency, (Ekström et al., 2012; Rocha et al., 2013).
Diabetic nephropathy
DN is a common complication of type 1 and type 2 diabetes, which have been associated with very distinct disease risk loci [Figure 2 and reviewed in Ntzani and Kavvoura (2012), Polychronakos and Li (2011)]. Results of genetics studies are extensively discussed in two recent reviews (Gu and Brismar, 2012; Palmer and Freedman, 2012), we therefore will focus only on the genetic association of ELMO1, CNDP1, and FRDM3 loci with DN risk, as they were detected in both GWAS and candidate gene approach studies.
ELMO1 encodes Engulfment and cell motility 1 and regulates Rac signaling and biological processes linked actin cytoskeleton remodeling. ELMO1 plays an established role in the clearance of apoptotic cells (Park et al., 2007; Elliott et al., 2010; van Ham et al., 2012), leading to the hypothesis that ELMO1 regulates homeostasis upon kidney injury by ensuring clearance of apoptotic cells and that impairment of this function might promote DN. ELMO1 is also known to contribute to the development of vasculature and to the production of extracellular matrix protein (ECM), which both may affect renal fibrosis (Shimazaki et al., 2006; Epting et al., 2010).
CNDP1 encodes carnosinase that hydrolyzes carnosine, an anti-oxidant molecule. Carnosine is a protective factor in several animal models of renal disease and was shown to inhibit TGF-β and ECM production by mesangial cells in hyperglycemic conditions (Köppel et al., 2011; Riedl et al., 2011; Menini et al., 2012). While CNDP1 polymorphisms are suspected to affect the level of its substrate carnosine, this remains to be demonstrated.
Little is known about the biological function of FRDM3, however, its locus is proposed to be associated with defective renal function in rats, based on Quantitative Trait Loci analysis (Garrett et al., 2010). It was recently suggested that FRDM3 risk SNP for DN may affect BMP signaling, a hypothesis that remains to be validated (Martini et al., 2013).
Genetic studies have identified many genetic polymorphisms that confer risk for CKD and DN using the gene candidate approach, but GWAS often have not confirmed these associations (Gu and Brismar, 2012; Palmer and Freedman, 2012). Current studies include only cross-sectional measurements of renal function, and genetic factors affecting disease progression of renal diseases are yet to be elucidated.
Diseases associated with liver fibrosis
Autoimmune liver diseases
The genetic architecture of autoimminue liver diseases such as Primary Sclerosing Cholangitis (PSC) and Primary Biliary Cirrhosis (PBC) was recently studied through a series of GWAS (Hirschfield et al., 2009; Liu et al., 2010; Mells et al., 2011; Melum et al., 2011; Liu et al., 2013a,b). Inflammation and tissue damage is thought to trigger sustained aberrant tissue repair responses that ultimately lead to the replacement of the organ by scar fibrotic tissue. Susceptibility loci largely overlap with the loci detected in other complex immune diseases affecting different organs: PSC and PBC shared common risk loci with multiple sclerosis, celiac disease, inflammatory bowel disease (IBD), rheumatoid arthritis and type 1 diabetes (Mells et al., 2013). Concurrent autoimmune disorders are commonly present in PSC and PBC patients (PSC is often seen in patients with IBD, type 1 diabetes and autoimmune thyroid disease and PBC is often seen in patients with Sjogren's syndrome, Raynaud Phenomenon, autoimmune thyroid disease and rheumatoid arthritis (Mells et al., 2013), which may explain the result of the genetic studies. Due to this co-occurrence of auto-immune diseases, case-case studies focusing on disease progression will be especially critical in PBC and PSC in order to identify pathogenic mechanisms that could be targeted by therapies.
Non-alcoholic fatty liver disease
Non-Alcoholic Fatty Liver Disease (NAFLD) is strongly associated with obesity, type 2 diabetes and dyslipidemia. The disease is characterized by steatosis with an increased hepatic Free Fatty Acid flux and cellular damage that trigger inflammatory and fibrotic responses. Genetic polymorphisms in the PNPLA3 locus that encodes for adiponutrin have been associated with NAFLD in many genetics studies using the candidate approach, and with well-powered GWAS (Daly et al., 2011). Adiponutrin is a triacylglycerol hydrolase, and the I148M variant associated with NAFLD induces accumulation of triacylglycerol and hepatic steatosis (He et al., 2010; Li et al., 2012). In independent studies, PNPLA3 locus has also been associated with NAFLD progression and fibrosis (Speliotes et al., 2011; Kitamoto et al., 2013). Additional potential genetic disease-modifiers associated with fibrosis reported in these studies are NCAN, GCKR, LYPLAL1, SAMM50, and PARVB loci. LYLPAL1 encodes a lysophospholipase and GCKR encodes glucokinase regulatory protein that regulates both glucose metabolism and lipogenesis. GCKR and NCAN variants affect circulating triglyceride levels (Gorden et al., 2013; Shen et al., 2013). Altogether, this suggests that risk for NAFLD and its progression could be largely influenced by genetic factors regulating lipid metabolism.
Genetic variants of angiotensin II receptor 1 have also been reported to be linked to fibrosis in NAFLD in two different studies, but with some inconsistencies in effects of AGTR1 variants/alleles perhaps due to cohort ethnicities (Yoneda et al., 2009; Zain et al., 2013). The use of blockers of angiotensin receptor in patients with liver fibrosis has yielded different results, therefore their beneficial effect in NAFLD remains controversial (Yokohama et al., 2004; Abu Dayyeh et al., 2011; Hirata et al., 2013). Thus, understanding the role of ATGR1 genetic polymorphisms in the progression of NAFLD liver fibrosis, and defining the relevant patient population, might be crucial to evaluate the potential beneficial role of angiotensin receptor blockers in NAFLD progression.
Diseases associated with intestinal fibrosis
Intestinal fibrosis is a common complication occurring with intestinal inflammation such as that seen with IBD, which comprises both Crohn's Disease (CD) and Ulcerative Colitis (UC) (Speca et al., 2012). The GWAS approach was very successful in identifying more than a hundred genetic risk factors for IBD (Franke et al., 2010; Anderson et al., 2011). These discoveries highlighted a major role for inflammatory pathways controlling innate and adaptive immune responses, mucosal barrier function, endoplasmic reticulum stress and oxidative stress in the disease pathogenesis (Khor et al., 2011). The chronic inflammatory injury in IBD triggers unrelenting mucosal injury/repair processes, and this ongoing damage/repair cycle is thought to underlie the intestinal fibrosis and strictures that are commonly seen in CD patients. In a subset of CD patients, the fibrosis and strictures can lead to intestinal obstruction and thus surgery. Immunosuppressive and anti-inflammatory treatments have little effects on intestinal fibrosis once the process has started, suggesting that non-immune pathways must be playing a role in the progression of fibrosis.
It is somehow surprising that there are only a few reports on genetic polymorphisms associated with higher risk for intestinal fibrosis, despite the strong success in recruiting a large number of patients for the IBD GWAS efforts. Candidate-gene approach studies with small size cohorts have implicated NOD2 and CX3CR1 polymorphisms as higher risk factors for stricturing CD and the subsequent need for surgery (Abreu et al., 2002; Lesage et al., 2002; Brand et al., 2006; Seiderer et al., 2006; Sabate et al., 2008). NOD2 is a known risk factor for CD and is presumed to primarily control innate immune response to bacterial products, while CX3CR1 is a chemokine receptor involved in leukocyte recruitment. In addition, GWAS to identify genetic polymorphisms associated with CD severity has recently implicated several loci including IL12B, RXRA/COL5A1, AHR, and FOXO3 loci in severe clinical phenotypes defined by need for surgery (Dubinsky et al., 2013; Lee et al., 2013b). However, their specific association to structuring CD is unclear. It is the hope that future studies will able to identify genetic risk factors associated with the structuring CD to better understand the pathogenesis of the development of intestinal fibrosis in IBD.
Diseases associated with skin fibrosis
Skin fibrosis represents a cardinal feature of several diseases with debilitating skin pathologies, including keloid disease/hypertrophic scars, systemic sclerosis and nephrogenic systemic fibrosis. While the etiology of skin fibrosis remains poorly understood, growing evidence supports the hypothesis that fibrosis arises from aberrant tissue injury (e.g., vasculopathy) and repair (e.g., wound healing) responses.
A number of studies in recent years have investigated the genetic basis of skin fibrosis, especially in keloid disease (KD) where higher prevalence in ethnicities with darker pigmentation. Linkage studies in familial keloids have been reported, with suggested candidate genes involved in fibroblast proliferation (EGFR), inflammation (TNFAIP6), and TGF-β signaling (SMADs) (Marneros et al., 2004; Yan et al., 2007). Supporting the potential role of inflammation and/or immune activation in the pathogenesis of KD, polymorphisms in the HLA region are associated with increased risk for KD in Caucasian, Chinese and Black populations (Brown et al., 2008, 2010; Lu et al., 2008). More recently, two independent GWAS of KD in Japanese and Chinese populations identified risk SNPs in chromosomal regions 1q41 and 15q21.3 (NEDD4 locus) (Nakashima et al., 2010; Zhu et al., 2013). It will be of great interest to understand how these loci may confer disease susceptibility for KD whose etiology is poorly understood.
Several candidate gene and GWAS have been carried out in systemic sclerosis (SSc), with most of the genetic variants identified being immune regulatory genes as mentioned earlier in this review. While these findings clearly support a major role of autoimmunity in SSc genetics, it is less clear whether these SSc susceptibility loci are directly involved in SSc skin fibrosis. SSc can be divided in two subtypes and the extent of the skin fibrosis is greater in diffuse SSc than in limited SSc. GWAS in SSc detected only one locus in ZC3H10/ESYT1 region conferring susceptibility preferentially for the diffuse clinical phenotype (Gorlova et al., 2011). It remains to be elucidated whether there is relationship between this risk locus and any pathogenic mechanisms linked to the skin fibrosis in diffuse SSc. Since 90% of the SSc patients have Raynaud's syndrome preceding their onset of skin hardening by several years, and vasculopathy is often viewed that may be a key disease-driving cause of SSc, it is somewhat surprising that no vasculature-related genes have been described from the SSc GWAS studies so far.
In addition to risk factors for SSc, an allele in CAV1 locus (encoding caveolin 1) was recently shown in a French cohort and replicated in an Italian cohort to confer protection against SSc and in particular limited SSc (Manetti et al., 2012). This protective allele was shown to be associated with an increased expression of caveolin 1 in skin from both healthy subjects and SSc patients. Caveolin 1 is a component of membrane caveolae that is proposed to regulate TGF-β receptor degradation (Del Galdo et al., 2008a). Confirming an anti-fibrotic role of caveolin 1, Cav1-deficient mice develop spontaneous lung and skin fibrosis (Drab et al., 2001; Del Galdo et al., 2008a,b). Caveolin 1 expression is decreased in many human fibrosis tissues including SSc skin and lung, IPF lung and keloid-derived fibroblasts, which suggest that the caveolin 1-mediated regulatory pathway may represent a new therapeutic opportunity in fibrotic diseases (Wang et al., 2006; Del Galdo et al., 2008a,b; Zhang et al., 2011).
Fibrosis progression: promising studies in cystic fibrosis and HCV-induced fibrosis
Cystic fibrosis arises as the result of an abnormal transport of salt due to mutations in CTFR. Although this is a Mendelian disorder, additional genetic factors are emerging as disease modifiers due to their influence on disease severity. Polymorphisms in MUC5AC may affect the severity of cystic fibrosis lung disease highlighting further the role of mucin in maintaining lung homeostasis (Guo et al., 2011). A recent GWAS meta-analysis including more than 3000 patients detected a SNP in a large intergenic region near EHF and APIP to be associated with disease severity (Wright et al., 2011). Additional suggestive (close to genome-wide significance) associations were reported in AGTR2 and in AHRR regions, indicating a role for angiotensin and xenobiotic sensing pathways in the severity of cystic fibrosis. As mentioned previously, the angiotensin pathway may be involved in the progression of NAFLD. Strikingly, angiotensin receptor blockade protects from experimental lung fibrosis and Ahr-deficient mice develop hepatic fibrosis (Fernandez-Salguero et al., 1995; Andreola et al., 2004; Waseda et al., 2008; Yaguchi et al., 2013). Further studies will be required to test the hypothesis that these pathways may be critical in influencing disease progression in cystic fibrosis and may lead to additional therapeutic approach for this Mendelian disorder.
Genetic disease-modifiers in HCV-induced fibrosis were recently identified in a GWAS meta-analysis including more than 2000 patients (Patin et al., 2012). Genetic polymorphisms in RNF7 and MERTK were associated with fibrosis progression and also point to the previously mentioned involvement of oxidative stress and the clearance of apoptotic cells in fibrotic diseases (Duan et al., 1999; Scott et al., 2001; Zizzo et al., 2012). Different candidate gene approach studies detected rs12785878 near DHCR7 to be associated with 25-hydroxyvitamin D [25(OH)D] serum levels, liver stiffness in chronic liver diseases, and progression of liver fibrosis in HCV patients (Grünhage et al., 2012; Petta et al., 2013). The same SNP was associated with development of hepatocarcinoma, but not with progression rate of liver fibrosis in HCV patients (Lange et al., 2013). Strikingly, vitamin D receptor was demonstrated to be key in the control of liver fibrosis by affecting SMAD3-mediated transcriptional response in mouse model, supporting the notion that this pathway might be essential in the control of liver fibrosis (Ding et al., 2013).
Together these studies confirm that genetics may play a critical role in influencing disease progression independently of the cause of the fibrosis (Mendelian or infectious disease). Understanding the underlying biological pathways associated with these disease modifiers, and how they influence fibrosis, may lead to new leads for therapeutic strategies.
Concluding remarks
Genetic studies have successfully identified polymorphisms associated with susceptibility for diseases with fibrotic complications. On-going functional studies attempt to elucidate the underlying pathogenic mechanisms. After a decade of human genetics studies focusing on disease risk, emerging results from genetic studies of disease progression suggest a multi-hit paradigm in which disease initiation and disease progression are not necessarily driven by the same mechanisms (Figure 3). Early discoveries on fibrosis progression point to pathways already shown in mouse models to control fibrotic responses, such as vitamin D and xenobiotic sensing pathways. Perhaps future genetic studies on disease progression will identify more genes and pathways identified in mouse models to control fibrotic responses.
Figure 3.

Overview of biological processes and pathways proposed to be associated with disease risk and disease progression. Left panel shows the biological processes associated with the genes located in the risk loci associated with higher susceptibility for disease. Right panel shows the biological pathways associated with genes located in the loci associated with disease progression. Further genetic studies of disease progression are needed to confirm initial results from limited sample sizes. SSc, systemic#sclerosis; PSC, primary sclerosing cholangitis; PBC, primary biliary cirrhosis; T1D, type 1 diabetes; T2D, type 2 diabetes; IBD, inflammatory bowel disease; IIP, idiopathic interstitial pneumonias; CKD, chronic kidney disease; NAFLD, non-alcoholic fatty liver disease; ILD, interstitial lung disease; IPF, idiopathic pulmonary fibrosis.
The identification of genetic disease modifiers comes with great challenges with a requirement for clinical annotations to inform on disease progression or severity with well-powered case-case studies rather than case-control studies to understand disease progression in human fibrotic diseases. However, elucidating the genetic basis of disease severity is crucial to understand pathogenic mechanisms and may be even more relevant to highlight biological pathways for therapeutic interventions.
Conflict of interest statement
Agnès Gardet, Timothy S. Zheng, and Joanne L. Viney are employees of Biogen Idec.
Acknowledgments
We are grateful to Dr. Robert W. Dunstan, DVM, MS, DACVP, Distinguished Investigator in the Translational Pathology Laboratory at Biogen Idec for kindly providing histology images to illustrate the effect of fibrotic tissues on lung architecture. We thank Dr. Christine Loh for carefully reviewing of this manuscript and her constructive comments.
References
- Abreu M. T., Taylor K. D., Lin Y.-C., Hang T., Gaiennie J., Landers C. J., et al. (2002). Mutations in NOD2 are associated with fibrostenosing disease in patients with Crohn's disease. Gastroenterology 123, 679–688 10.1053/gast.2002.35393 [DOI] [PubMed] [Google Scholar]
- Abu Dayyeh B. K., Yang M., Dienstag J. L., Chung R. T. (2011). The effects of angiotensin blocking agents on the progression of liver fibrosis in the HALT-C trial cohort. Dig. Dis. Sci. 56, 564–568 10.1007/s10620-010-1507-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alder J. K., Chen J. J.-L., Lancaster L., Danoff S., Su S. C., Cogan J. D., et al. (2008). Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. U.S.A. 105, 13051–13056 10.1073/pnas.0804280105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allanore Y., Saad M,., Dieudé P., Avouac J., Distler J. H., Amouyel P., et al. (2011). Genome-wide scan identifies TNIP1, PSORS1C1, and RHOB as novel risk loci for systemic sclerosis. PLoS Genet. 7:e1002091 10.1371/journal.pgen.1002091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson C. A., Boucher G., Lees C. W., Franke A., D'Amato M., Taylor K. D., et al. (2011). Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 43, 246–252 10.1038/ng.764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreola F., Calvisi D. F., Elizondo G., Jakowlew S. B., Mariano J., Gonzalez F. J., et al. (2004). Reversal of liver fibrosis in aryl hydrocarbon receptor null mice by dietary vitamin A depletion. Hepatology 39, 157–166 10.1002/hep.20004 [DOI] [PubMed] [Google Scholar]
- Armanios M. Y., Chen J. J.-L., Cogan J. D., Alder J. K., Ingersoll R. G., Markin C., et al. (2007). Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 356, 1317–1326 10.1056/NEJMoa066157 [DOI] [PubMed] [Google Scholar]
- Assassi S., Sharif R., Lasky R. E., McNearney T. A., Estrada-Y-Martin R. M., Draeger H., et al. (2010). Predictors of interstitial lung disease in early systemic sclerosis: a prospective longitudinal study of the GENISOS Cohort. Arthritis Res. Ther. 12:R166 10.1186/ar3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkauskas C. E., Cronce M. J., Rackley C. R., Bowie E. J., Keene D. R., Stripp B. R., et al. (2013). Type 2 alveolar cells are stem cells in adult lung. J. Clin. Invest. 123, 3025–3036 10.1172/JCI68782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates J. M., Raffi H. M., Prasadan K., Mascarenhas R., Laszik Z., Maeda N., et al. (2004). Tamm-horsfall protein knockout mice are more prone to urinary tract infection: rapid communication. Kidney Int. 65, 791–797 10.1111/j.1523-1755.2004.00452.x [DOI] [PubMed] [Google Scholar]
- Borie R., Crestani B., Dieude P., Nunes H., Allanore Y., Kannengiesser C., et al. (2013). The MUC5B variant is associated with idiopathic pulmonary fibrosis but not with systemic sclerosis interstitial lung disease in the european caucasian population. PLoS ONE 8:e70621 10.1371/journal.pone.0070621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bournazos S., Grinfeld J., Alexander K. M., Murchison J. T., Wallace W. A., McFarlane P., et al. (2010). Association of FcγRIIa R131H polymorphism with idiopathic pulmonary fibrosis severity and progression. BMC Pulm. Med. 10:51 10.1186/1471-2466-10-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand S., Hofbauer K., Dambacher J., Schnitzler F., Staudinger T., Pfennig S., et al. (2006). Increased expression of the chemokine fractalkine in crohn's disease and association of the fractalkine receptor T280M polymorphism with a fibrostenosing disease phenotype. Am. J. Gastroenterol. 101, 99–106 10.1111/j.1572-0241.2005.00361.x [DOI] [PubMed] [Google Scholar]
- Brown J. J., Ollier W. E. R., Arscott G., Bayat A. (2010). Association of HLA-DRB1* and keloid disease in an afro-caribbean population. Clin. Exp. Dermatol. 35, 305–310 10.1111/j.1365-2230.2009.03506.x [DOI] [PubMed] [Google Scholar]
- Brown J. J., Ollier W. E. R., Thomson W., Bayat A. (2008). Positive association of HLA-DRB1*15 with keloid disease in caucasians. Int. J. Immunogenet. 35, 303–307 10.1111/j.1744-313X.2008.00780.x [DOI] [PubMed] [Google Scholar]
- Cotsapas C., Hafler D. A. (2013). Immune-mediated disease genetics: the shared basis of pathogenesis. Trends Immunol. 34, 22–26 10.1016/j.it.2012.09.001 [DOI] [PubMed] [Google Scholar]
- Cronkhite J. T., Xing C., Raghu G., Chin K. M., Torres F., Rosenblatt R. L., et al. (2008). Telomere shortening in familial and sporadic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 178, 729–737 10.1164/rccm.200804-550OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly A. K., Ballestri S., Carulli L., Loria P., Day C. P. (2011). Genetic determinants of susceptibility and severity in nonalcoholic fatty liver disease. Expert Rev. Gastroenterol. Hepatol. 5, 253–263 10.1586/egh.11.18 [DOI] [PubMed] [Google Scholar]
- Dang M.-T. T., Gu C., Klavanian J. I., Jernigan K. A., Friderici K. H., Cui Y., et al. (2013). Angiotensinogen promoter polymorphisms predict low diffusing capacity in U.S. and spanish IPF cohorts. Lung 191, 353–360 10.1007/s00408-013-9476-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Declèves A.-E., Mathew A. V., Cunard R., Sharma K. (2011). AMPK mediates the initiation of kidney disease induced by a high-fat diet. J. Am. Soc. Nephrol. 22, 1846–1855 10.1681/ASN.2011010026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degryse A. L., Xu X. C., Newman J. L., Mitchell D. B., Tanjore H., Polosukhin V. V., et al. (2012). Telomerase deficiency does not alter bleomycin-induced fibrosis in mice. Exp. Lung Res. 38, 124–134 10.3109/01902148.2012.658148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Galdo F., Lisanti M. P., Jimenez S. A. (2008a). Caveolin-1, transforming growth factor-beta receptor internalization, and the pathogenesis of systemic sclerosis. Curr. Opin. Rheumatol. 20, 713–719 10.1097/BOR.0b013e3283103d27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Galdo F., Sotgia F., de Almeida C. J., Jasmin J.-F., Musick M., Lisanti M. P., et al. (2008b). Decreased expression of caveolin 1 in patients with systemic sclerosis: crucial role in the pathogenesis of tissue fibrosis. Arthritis Rheum. 58, 2854–2865 10.1002/art.23791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz de Leon A., Cronkhite J. T., Katzenstein A.-L. A., Godwin J. D., Raghu G., Glazer C. S., et al. (2010). Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS ONE 5:10680 10.1371/journal.pone.0010680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieudé P., Bouaziz M., Guedj M., Riemekasten G., Airò P., Müller M., et al. (2011a) Evidence of the contribution of the X chromosome to systemic sclerosis susceptibility: association with the functional IRAK1 196Phe/532Ser haplotype. Arthritis Rheum. 63, 3979–3987 10.1002/art.30640 [DOI] [PubMed] [Google Scholar]
- Dieudé P., Guedj M., Wipff J.,., Ruiz B., Riemekasten G., Airo P., et al. (2011b). NLRP1 Influences the systemic sclerosis phenotype: a new clue for the contribution of innate immunity in systemic sclerosis-related fibrosing alveolitis pathogenesis. Ann. Rheum. Dis. 70, 668–674 10.1136/ard.2010.131243 [DOI] [PubMed] [Google Scholar]
- Dieudé P., Guedj M., Wipff J., Ruiz B., Hachulla E., Diot E., et al. (2009). STAT4 Is a genetic risk factor for systemic sclerosis having additive effects with IRF5 on disease susceptibility and related pulmonary fibrosis. Arthritis Rheum. 60, 2472–2479 10.1002/art.24688 [DOI] [PubMed] [Google Scholar]
- Ding N., Yu R. T., Subramaniam N., Sherman M. H., Wilson C., Rao R., et al. (2013). A vitamin D receptor/SMAD genomic circuit gates hepatic fibrotic response. Cell 153, 601–613 10.1016/j.cell.2013.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drab M., Verkade P., Elger M., Kasper M., Lohn M., Lauterbach B., et al. (2001). Loss of caveolae, vascular dysfunction, and pulmonary defects in Caveolin-1 gene-disrupted mice. Science 293, 2449–2452 10.1126/science.1062688 [DOI] [PubMed] [Google Scholar]
- Duan H., Wang Y., Aviram M., Swaroop M., Loo J. A., Bian J., et al. (1999). SAG, a novel zinc RING finger protein that protects cells from apoptosis induced by redox agents. Mol. Cell. Biol. 19, 3145–3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubinsky M. C., Kugathasan S., Kwon S., Haritunians T., Wrobel I., Wahbeh G., et al. (2013). Multidimensional prognostic risk assessment identifies association between IL12B variation and surgery in Crohn's disease. Inflamm. Bowel Dis. 19, 1662–1670 10.1097/MIB.0b013e318281f275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekström N., Schiöler L., Svensson A.-M., Eeg-Olofsson K., Jonasson J. M., Zethelius B., et al. (2012). Effectiveness and safety of metformin in 51 675 patients with Type 2 diabetes and different levels of renal function: a cohort study from the swedish national diabetes register. BMJ Open 2, 1–10 10.1136/bmjopen-2012-001076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Achkar T. M., Wu X.-R. (2012). Uromodulin in kidney injury: an instigator, bystander, or protector? Am. J. Kidney Dis. 59, 452–461 10.1053/j.ajkd.2011.10.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Achkar T. M., Wu X.-R., Rauchman M., McCracken R., Kiefer S., Dagher P. C. (2008). Tamm-horsfall protein protects the kidney from ischemic injury by decreasing inflammation and altering TLR4 expression. Am. J. Physiol. Renal Physiol. 295, F534–F544 10.1152/ajprenal.00083.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott M. R., Zheng S., Park D., Woodson R. I., Reardon M. A., Juncadella I. J., et al. (2010). Unexpected requirement for ELMO1 in clearance of apoptotic germ cells in vivo. Nature 467, 333–337 10.1038/nature09356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epting D., Wendik B., Bennewitz K., Dietz C. T., Driever W., Kroll J. (2010). The Rac1 regulator ELMO1 controls vascular morphogenesis in Zebrafish. Circ. Res. 107, 45–55 10.1161/CIRCRESAHA.109.213983 [DOI] [PubMed] [Google Scholar]
- Feghali-Bostwick C., Medsger T. A., Jr., Wright T. M. (2003). Analysis of systemic sclerosis in twins reveals low concordance for disease and high concordance for the presence of antinuclear antibodies. Arthritis Rheum. 48, 1956–1963 10.1002/art.11173 [DOI] [PubMed] [Google Scholar]
- Fernandez-Salguero P., Pineau T., Hilbert D. M., McPhail T., Lee S. S., Kimura S., et al. (1995). Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science 268, 722–726 10.1126/science.7732381 [DOI] [PubMed] [Google Scholar]
- Fingerlin T. E., Murphy E., Zhang W., Peljto A. L., Brown K. K., Steele M. P., et al. (2013). Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 45, 613–620 10.1038/ng.2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca C., Lindahl G. E., Ponticos M., Sestini P., Renzoni E. A., Holmes A. M., et al. (2007). A polymorphism in the CTGF promoter region associated with systemic sclerosis. N. Engl. J. Med. 357, 1210–1220 10.1056/NEJMoa067655 [DOI] [PubMed] [Google Scholar]
- Franke A., McGovern D. P. B., Barrett J. C., Wang K., Radford-Smith G. L., Ahmad T., et al. (2010). Genome-wide meta-analysis Increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat. Genet. 42, 1118–1125 10.1038/ng.717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridlender Z. G., Cohen P. Y., Golan O., Arish N., Wallach-Dayan S., Breuer R. (2007). Telomerase activity in bleomycin-induced epithelial cell apoptosis and lung fibrosis. Eur. Respir. J. 30, 205–213 10.1183/09031936.00009407 [DOI] [PubMed] [Google Scholar]
- Garrett M. R., Pezzolesi M. G., Korstanje R. (2010). Integrating human and rodent data to identify the genetic factors involved in chronic kidney disease. J. Am. Soc. Nephrol. 21, 398–405 10.1681/ASN.2009080881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G., Friedman D. J., Ross M. D., Lecordier L., Uzureau P., Freedman B. I., et al. (2010). Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329, 841–845 10.1126/science.1193032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorden A., Yang R., Yerges-Armstrong L. M., Ryan K. A., Speliotes E., Borecki I. B., et al. (2013). Genetic variation at NCAN locus is associated with inflammation and fibrosis in non-alcoholic fatty liver disease in morbid obesity. Hum. Hered. 75, 34–43 10.1159/000346195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlova O., Martin J.-E., Rueda B., Koeleman B. P. C., Ying J., Teruel M., et al. (2011). Identification of novel genetic markers associated with clinical phenotypes of systemic sclerosis through a genome-wide association strategy. PLoS Genet. 7:e1002178 10.1371/journal.pgen.1002178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünhage F., Hochrath K., Krawczyk M., Höblinger A., Obermayer-Pietsch B., Geisel J., et al. (2012). Common genetic variation in vitamin D metabolism is associated with liver stiffness. Hepatology 56, 1883–1891 10.1002/hep.25830 [DOI] [PubMed] [Google Scholar]
- Gu H. F., Brismar K. (2012). Genetic association studies in diabetic nephropathy. Curr. Diabetes Rev. 8, 336–344 10.2174/157339912802083522 [DOI] [PubMed] [Google Scholar]
- Gudbjartsson D. F., Holm H., Indridason O. S., Thorleifsson G., Edvardsson V., Sulem P., et al. (2010). Association of variants at UMOD with chronic kidney disease and kidney stones-role of age and comorbid diseases. PLoS Genet. 6:e1001039 10.1371/journal.pgen.1001039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X., Pace R. G., Stonebraker J. R., Commander C. W., Dang A. T., Drumm M. L., et al. (2011). Mucin variable number tandem repeat polymorphisms and severity of cystic fibrosis lung disease: significant association with MUC5AC. PLoS ONE 6:e25452 10.1371/journal.pone.0025452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallows K. R., Mount P. F., Pastor-Soler N. M., Power D. A. (2010). Role of the energy sensor amp-activated protein kinase in renal physiology and disease. Am. J. Physiol. Renal Physiol. 298, F1067–F1077 10.1152/ajprenal.00005.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargrave M., Wright E., Kun J., Emery J., Cooper L., Koopman P. (1997). Expression of the Sox11 gene in mouse embryos suggests roles in neuronal maturation and epithelio-mesenchymal induction. Dev. Dyn. 210, 79–86 [DOI] [PubMed] [Google Scholar]
- He S., McPhaul C., Li J. Z., Garuti R., Kinch L., Grishin N. V., et al. (2010). A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J. Biol. Chem. 285, 6706–6715 10.1074/jbc.M109.064501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecker L., Cheng J., Thannickal V. J. (2012). Targeting NOX enzymes in pulmonary fibrosis. Cell. Mol. Life Sci. 69, 2365–2371 10.1007/s00018-012-1012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata T., Tomita K., Kawai T., Yokoyama H., Shimada A., Kikuchi M., et al. (2013). Effect of telmisartan or losartan for treatment of nonalcoholic fatty liver disease: fatty liver protection trial by telmisartan or losartan study (FANTASY). Int. J. Endocrinol. 2013:587140 10.1155/2013/587140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschfield G. M., Liu X., Xu C., Lu Y., Xie G., Lu Y., et al. (2009). Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. N. Engl. J. Med. 360, 2544–2555 10.1056/NEJMoa0810440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino K., Satoh T., Kawaguchi Y., Kuwana M. (2011). Association of hepatocyte growth factor promoter polymorphism with severity of interstitial lung disease in japanese patients with systemic sclerosis. Arthritis Rheum. 63, 2465–2472 10.1002/art.30415 [DOI] [PubMed] [Google Scholar]
- Hsu E., Shi H., Jordan R. M., Lyons-Weiler J., Pilewski J. M., Feghali-Bostwick C. A. (2011). Lung tissues in patients with systemic sclerosis have gene expression patterns unique to pulmonary fibrosis and pulmonary hypertension. Arthritis Rheum. 63, 783–794 10.1002/art.30159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao W. H. L., Klag M. J., Meoni L. A., Reich D., Berthier-Schaad Y., Li M., et al. (2008). MYH9 is associated with nondiabetic end-stage renal disease in African Americans. Nat. Genet. 40, 1185–1192 10.1038/ng.232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khor B., Gardet A., Xavier R. J. (2011). Genetics and pathogenesis of inflammatory bowel disease. Nature 474, 307–317 10.1038/nature10209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kispert A., Vainio S., McMahon A. P. (1998). Wnt-4 is a mesenchymal signal for epithelial transformation of metanephric mesenchyme in the developing kidney. Development 125, 4225–4234 [DOI] [PubMed] [Google Scholar]
- Kitamoto T., Kitamoto A., Yoneda M., Hyogo H., Ochi H., Nakamura T., et al. (2013). Genome-wide scan revealed that polymorphisms in the PNPLA3, SAMM50, and PARVB genes are associated with development and progression of nonalcoholic fatty liver disease in Japan. Hum. Genet. 132, 783–792 10.1007/s00439-013-1294-3 [DOI] [PubMed] [Google Scholar]
- Köppel H., Riedl E., Braunagel M., Sauerhoefer S., Ehnert S., Godoy P., et al. (2011). L-carnosine inhibits high-glucose-mediated matrix accumulation in human mesangial cells by interfering with TGF-β production and signalling. Nephrol., Dial. Transplant. 26, 3852–3858 10.1093/ndt/gfr324 [DOI] [PubMed] [Google Scholar]
- Kopp J. B., Smith M.W., Nelson G. W., Johnson R. C., Freedman B. I., Bowden D. W., et al. (2008). MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat. Genet. 40, 1175–1184 10.1038/ng.226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köttgen A., Glazer N. L., Dehghan A., Hwang S.-J., Katz R., Li M., et al. (2009). Multiple loci associated with indices of renal function and chronic kidney disease. Nat. Genet. 41, 712–717 10.1038/ng.377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köttgen A., Pattaro C., Böger C. A., Fuchsberger C., Olden M., Glazer N. L., et al. (2010). New loci associated with kidney function and chronic kidney disease. Nat. Genet. 42, 376–384 10.1038/ng.568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kropski J. A., Lawson W. E., Young L. R., Blackwell T. S. (2013). Genetic studies provide clues on the pathogenesis of idiopathic pulmonary fibrosis. Dis. Models Mech. 6, 9–17 10.1242/dmm.010736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam A. P., Gottardi C. J. (2011). B-catenin signaling: a novel mediator of fibrosis and potential therapeutic target. Curr. Opin. Rheumatol. 23, 562–567 10.1097/BOR.0b013e32834b3309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange C. M., Miki D., Ochi H., Nischalke H.-D., Bojunga J., Bibert S., et al. (2013). Genetic analyses reveal a role for vitamin D insufficiency in HCV-associated hepatocellular carcinoma development. PLoS ONE 8:e64053 10.1371/journal.pone.0064053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. C., Espéli M., Anderson C. A., Linterman M. A., Pocock J. M., Williams N. J., et al. (2013a). Human SNP links differential outcomes in inflammatory and infectious disease to a FOXO3-regulated pathway. Cell 155, 57–69 10.1016/j.cell.2013.08.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. H., Kim J. H., Kim J. S., Chang J. W., Kim S. B., Park J. S., et al. (2013b). AMP-activated protein kinase inhibits TGF-β-, angiotensin II-, aldosterone-, high glucose-, and albumin-induced epithelial-mesenchymal transition. Am. J. Physiol. Renal Physiol. 304, F686–F697 10.1152/ajprenal.00148.2012 [DOI] [PubMed] [Google Scholar]
- Lesage S., Zouali H., Cézard J.-P., Colombel J.-F., Belaiche J., Almer S., et al. (2002). CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 Patients with Inflammatory Bowel Disease. Am. J. Hum. Genet. 70, 845–857 10.1086/339432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. Z., Huang Y., Karaman R., Ivanova P. T., Brown H. A., Roddy T., et al. (2012). Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J. Clin. Invest. 122, 4130–4144 10.1172/JCI65179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., McAlpine D. D., Liu J., Li S., Collins A. J. (2004). Differences between blacks and whites in the incidence of end-stage renal disease and associated risk factors. Adv. Ren. Replace. Ther. 11, 5–13 10.1053/j.arrt.2003.10.005 [DOI] [PubMed] [Google Scholar]
- Liu T., Chung M. J., Ullenbruch M., Yu H., Jin H., Hu B., et al. (2007). Telomerase activity is required for bleomycin-induced pulmonary fibrosis in mice. J. Clin. Invest. 117, 3800–3809 10.1172/JCI32369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T., Ullenbruch M., Choi Y. Y., Yu H., Ding L., Xaubet A., et al. (2013a). Telomerase and telomere length in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 49, 260–268 10.1165/rcmb.2012-0514OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. Z., Hov J. R., Folseraas T., Ellinghaus E., Rushbrook S. M., Doncheva N. T., et al. (2013b). Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat. Genet. 45, 670–675 10.1038/ng.2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Invernizzi P., Lu Y., Kosoy R., Lu Y., Bianchi I., et al. (2010). Genome-wide meta-analyses identify three loci associated with primary biliary cirrhosis. Nat. Genet. 42, 658–660 10.1038/ng.627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W.-S., Wang J.-F., Yang S., Xiao F.-L., Quan C., Cheng H., et al. (2008). Association of HLA-DQA1 and DQB1 alleles with keloids in chinese hans. J. Dermatol. Sci. 52, 108–117 10.1016/j.jdermsci.2008.04.010 [DOI] [PubMed] [Google Scholar]
- Maitra M., Cano C. A., Garcia C. K. (2012). Mutant surfactant A2 proteins associated with familial pulmonary fibrosis and lung cancer induce TGF-β 1 secretion. Proc. Natl. Acad. Sci. U.S.A. 109, 21064–21069 10.1073/pnas.1217069110 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Maitra M., Dey M., Yuan W.-C., Nathanielsz P. W., Garcia C. K. (2013). Lung fibrosis associated surfactant protein A1 and C variants induce latent TGF-β 1 secretion in lung epithelial cells. J. Biol. Chem. 288, 27159–27171 10.1074/jbc.M113.475335 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Maitra M., Wang Y., Gerard R. D., Mendelson C. R., Garcia C. K. (2010). Surfactant protein A2 mutations associated with pulmonary fibrosis lead to protein instability and endoplasmic reticulum stress. J. Biol. Chem. 285, 22103–22113 10.1074/jbc.M110.121467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manetti M., Allanore Y., Saad M., Fatini C., Cohignac V., Guiducci S., et al. (2012). Evidence for caveolin-1 as a new susceptibility gene regulating tissue fibrosis in systemic sclerosis. Ann. Rheum. Dis. 71, 1034–1041 10.1136/annrheumdis-2011-200986 [DOI] [PubMed] [Google Scholar]
- Manetti M., Ibba-Manneschi L., Fatini C., Guiducci S., Cuomo G., Bonino C., et al. (2010). Association of a functional polymorphism in the matrix metalloproteinase-12 promoter region with systemic sclerosis in an italian population. J. Rheumatol. 37, 1852–1857 10.3899/jrheum.100237 [DOI] [PubMed] [Google Scholar]
- Marneros A. G., Norris J. E. C., Watanabe S., Reichenberger E., Olsen B. R. (2004). Genome scans provide evidence for keloid susceptibility loci on chromosomes 2q23 and 7p11. J. Invest. Dermatol. 122, 1126–1132 10.1111/j.0022-202X.2004.22327.x [DOI] [PubMed] [Google Scholar]
- Martini S., Nair V., Patel S. R., Eichinger F., Nelson R. G., Weil E. J., et al. (2013). From single nucleotide polymorphism to transcriptional mechanism: a model for FRMD3 in diabetic nephropathy. Diabetes 62, 2605–2612 10.2337/db12-1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mells G. F., Floyd J. A. B., Morley K. I., Cordell H. J., Franklin C. S., Shin S.-Y., et al. (2011). Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat. Genet. 43, 329–332 10.1038/ng.789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mells G. F., Kaser A., Karlsen T. H. (2013). Novel insights into autoimmune liver diseases provided by genome-wide association studies. J. Autoimm. 46, 41–54 10.1016/j.jaut.2013.07.004 [DOI] [PubMed] [Google Scholar]
- Melum E., Franke A., Schramm C., Weismüller T. J., Gotthardt D. N., Offner F. A., et al. (2011). Genome-wide association analysis in primary sclerosing cholangitis identifies two non-HLA susceptibility loci. Nat. Genet. 43, 17–19 10.1038/ng.728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menini S., Iacobini C., Ricci C., Scipioni A., Fantauzzi C. B., Giaccari A., et al. (2012). D-carnosine octylester attenuates atherosclerosis and renal disease in ApoE null mice fed a western diet through reduction of carbonyl stress and inflammation. Br. J. Pharmacol. 166, 1344–1356 10.1111/j.1476-5381.2012.01834.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo L., Zhu X.-H., Huang H.-Y., Shapiro E., Hasty D. L., Wu X.-R. (2004). Ablation of the tamm-horsfall protein gene increases susceptibility of mice to bladder colonization by Type 1-fimbriated escherichia coli. Am. J. Physiol. Renal Physiol. 286, F795–F802 10.1152/ajprenal.00357.2003 [DOI] [PubMed] [Google Scholar]
- Molina-Molina M., Xaubet A., Li X., Abdul-Hafez A., Friderici K., Jernigan K., et al. (2008). Angiotensinogen gene G-6A polymorphism influences idiopathic pulmonary fibrosis disease progression. Eur. Respir. J. 32, 1004–1008 10.1183/09031936.00015808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray L. A., Rubinowitz A., Herzog E. L. (2012). Interstitial lung disease: is interstitial lung disease the same as scleroderma lung disease? Curr. Opin. Rheumatol. 24, 656–662 10.1097/BOR.0b013e3283588de4 [DOI] [PubMed] [Google Scholar]
- Murugan S., Shan J., Kühl S. J., Tata A., Pietilä I., Kühl M., et al. (2012). WT1 and Sox11 regulate synergistically the promoter of the Wnt4 gene that encodes a critical signal for nephrogenesis. Exp. Cell Res. 318, 1134–1145 10.1016/j.yexcr.2012.03.008 [DOI] [PubMed] [Google Scholar]
- Nakashima M., Chung S., Takahashi A., Kamatani N., Kawaguchi T., Tsunoda T., et al. (2010). A genome-wide association study identifies four susceptibility loci for keloid in the japanese population. Nat. Genet. 42, 768–771 10.1038/ng.645 [DOI] [PubMed] [Google Scholar]
- Nogee L. M., Dunbar A. E., 3rd., Wert S. E., Askin F., Hamvas A., Whitsett J. A. (2001). A mutation in the surfactant Protein C gene associated with familial interstitial lung disease. N. Engl. J. Med. 344, 573–579 10.1056/NEJM200102223440805 [DOI] [PubMed] [Google Scholar]
- Ntzani E. E., Kavvoura F. K. (2012). Genetic risk factors for Type 2 diabetes: insights from the emerging genomic evidence. Curr. Vasc. Pharmacol. 10, 147–155 10.2174/157016112799305030 [DOI] [PubMed] [Google Scholar]
- O'Dwyer D. N., Armstrong M. E., Trujillo G., Cooke G., Keane M. P., Fallon P. G., et al. (2013). The toll-like receptor 3 L412F polymorphism and disease progression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. [Epub ahead of print]. 10.1164/rccm.201304-0760OC [DOI] [PubMed] [Google Scholar]
- Ono S., Tanaka T., Ishida M., Kinoshita A., Fukuoka J., Takaki M., et al. (2011). Surfactant protein C G100S mutation causes familial pulmonary fibrosis in Japanese Kindred. Eur. Respir. J. 38, 861–869 10.1183/09031936.00143610 [DOI] [PubMed] [Google Scholar]
- Palmer N. D., Freedman B. I. (2012). Insights into the genetic architecture of diabetic nephropathy. Curr. Diab. Rep. 12, 423–431 10.1007/s11892-012-0279-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D., Tosello-Trampont A.-C., Elliott M. R., Lu M., Haney L. B., Ma Z., et al. (2007). BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature 450, 430–434 10.1038/nature06329 [DOI] [PubMed] [Google Scholar]
- Park J.-I., Venteicher A. S., Hong J. Y., Choi J., Jun S., Shkreli M., et al. (2009). Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 460, 66–72 10.1038/nature08137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patin E., Kutalik Z., Guergnon J., Bibert S., Nalpas B., Jouanguy E., et al. (2012). Genome-wide association study identifies variants associated with progression of liver fibrosis from HCV infection. Gastroenterology 143, 1244–1252.e1–12 10.1053/j.gastro.2012.07.097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peljto A. L., Steele M. P., Fingerlin T. E., Hinchcliff M. E., Murphy E., Podlusky S., et al. (2012). The pulmonary fibrosis-associated MUC5B promoter polymorphism does not influence the development of interstitial pneumonia in systemic sclerosis. Chest 142, 1584–1588 10.1378/chest.12-0110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peljto A. L., Zhang Y., Fingerlin T. E., Ma S.-F., Garcia J. G. N., Richards T. J., et al. (2013). Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA 309, 2232–2239 10.1001/jama.2013.5827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petta S., Grimaudo S., Marco V. D., Scazzone C., Macaluso F. S., Cammà C., et al. (2013). Association of vitamin D serum levels and its common genetic determinants, with severity of liver fibrosis in Genotype 1 chronic hepatitis C patients. J. Viral Hepat. 20, 486–493 10.1111/jvh.12072 [DOI] [PubMed] [Google Scholar]
- Polychronakos C., Li Q. (2011). Understanding Type 1 diabetes through genetics: advances and prospects. Nat. Rev. Genet. 12, 781–792 10.1038/nrg3069 [DOI] [PubMed] [Google Scholar]
- Radstake T. R., Gorlova O., Rueda B., Martin J. E., Alizadeh B. Z., Palomino-Morales R., et al. (2010). Genome-wide association study of systemic sclerosis identifies CD247 as a new susceptibility locus. Nat Genet. 42, 426–429 10.1038/ng.565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl E., Pfister F., Braunagel M., Brinkkötter P., Sternik P., Deinzer M., et al. (2011). Carnosine prevents apoptosis of glomerular cells and podocyte loss in STZ diabetic rats. Cell. Physiol. Biochem. 28, 279–288 10.1159/000331740 [DOI] [PubMed] [Google Scholar]
- Rocha A., Almeida M., Santos J., Carvalho A. (2013). Metformin in patients with chronic kidney disease: strengths and weaknesses. J. Nephrol. 26, 55–60 10.5301/jn.5000166 [DOI] [PubMed] [Google Scholar]
- Romano E., Manetti M., Guiducci S., Ceccarelli C., Allanore Y., Matucci-Cerinic M. (2011). The genetics of systemic sclerosis: an update. Clin. Exp. Rheumatol. 29(2 Suppl 65): S75–S86 10.1007/s11926-011-0221-7 [DOI] [PubMed] [Google Scholar]
- Sabate J.-M., Ameziane N., Lamoril J., Jouet P., Farmachidi J.-P., Soulé J.-C., et al. (2008). The V249I polymorphism of the CX3CR1 gene is associated with fibrostenotic disease behavior in patients with Crohn's disease. Eur. J. Gastroenterol. Hepatol. 20, 748–755 10.1097/MEG.0b013e3282f824c9 [DOI] [PubMed] [Google Scholar]
- Sahin E., Colla S., Liesa M., Moslehi J., Müller F. L., Guo M., et al. (2011). Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 470, 359–365 10.1038/nature09787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satriano J., Sharma K., Blantz R. C., Deng A. (2013). Induction of AMPK activity corrects early pathophysiological alterations in the subtotal nephrectomy model of chronic kidney disease. Am. J. Physiol. Renal Physiol. 305, F727–F733 10.1152/ajprenal.00293.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott R. S., McMahon E. J., Pop S. M., Reap E. A., Caricchio R., Cohen P. L., et al. (2001). Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 411, 207–211 10.1038/35075603 [DOI] [PubMed] [Google Scholar]
- Seibold M. A., Smith R. W., Urbanek C., Groshong S. D., Cosgrove G. P., Brown K. K., et al. (2013). The idiopathic pulmonary fibrosis honeycomb cyst contains a mucocilary pseudostratified epithelium. PLoS ONE 8:e58658 10.1371/journal.pone.0058658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibold M. A., Wise A. L., Speer M. C., Steele M. P., Brown K. K., Loyd J. E., et al. (2011). A Common MUC5B promoter polymorphism and pulmonary fibrosis. N. Engl. J. Med. 364, 1503–1512 10.1056/NEJMoa1013660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiderer J., Brand S., Herrmann K. A., Schnitzler F., Hatz R., Crispin A., et al. (2006). Predictive value of the CARD15 variant 1007fs for the diagnosis of intestinal stenoses and the need for surgery in Crohn's disease in clinical practice: results of a prospective study. Inflamm. Bowel Dis. 12, 1114–1121 10.1097/01.mib.0000235836.32176.5e [DOI] [PubMed] [Google Scholar]
- Sharif R., Mayes M. D., Tan F. K., Gorlova O. Y., Hummers L. K., Shah A. A., et al. (2012). IRF5 polymorphism predicts prognosis in patients with systemic sclerosis. Ann. Rheum. Dis. 71, 1197–1202 10.1136/annrheumdis-2011-200901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y., Wu L., Xi B., Liu X., Zhao X., Cheng H., et al. (2013). GCKR variants increase triglycerides while protecting from insulin resistance in chinese children. PLoS ONE 8:e55350 10.1371/journal.pone.0055350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazaki A., Tanaka Y., Shinosaki T., Ikeda M., Watada H., Hirose T., et al. (2006). ELMO1 increases expression of extracellular matrix proteins and inhibits cell adhesion to ECMs. Kidney Int. 70, 1769–1776 10.1038/sj.ki.5001939 [DOI] [PubMed] [Google Scholar]
- Sisson T. H., Mendez M., Choi K., Subbotina N., Courey A., Cunningham A., et al. (2010). Targeted injury of type ii alveolar epithelial cells induces pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 181 254–263 10.1164/rccm.200810-1615OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sock E., Rettig S. D., Enderich J., Bösl M. R., Tamm E. R., Wegner M. (2004). Gene targeting reveals a widespread role for the high-mobility-group transcription factor Sox11 in tissue remodeling. Mol. Cell. Biol. 24, 6635–6644 10.1128/MCB.24.15.6635-6644.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speca S., Giusti I., Rieder F., Latella G. (2012). Cellular and molecular mechanisms of intestinal fibrosis. World J. Gastroenterol. 18, 3635–3661 10.3748/wjg.v18.i28.3635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speliotes E. K., Yerges-Armstrong L. M., Wu J., Hernaez R., Kim L. J., Palmer C. D., et al. (2011). Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 7:e1001324 10.1371/journal.pgen.1001324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen V. D., Powell D. L., Medsger T. A., Jr. (1988). Clinical correlations and prognosis based on serum autoantibodies in patients with systemic sclerosis. Arthritis Rheum. 31, 196–203 10.1002/art.1780310207 [DOI] [PubMed] [Google Scholar]
- Stock C. J., Sato H., Fonseca C., Banya W. A. S., Molyneaux P. L., Adamali H., et al. (2013). Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis. Thorax 68, 436–441 10.1136/thoraxjnl-2012-201786 [DOI] [PubMed] [Google Scholar]
- Surendran K., McCaul S. P., Simon T. C. (2002). A role for Wnt-4 in renal fibrosis. Am. J. Physiol. Renal Physiol. 282, F431–F441 [DOI] [PubMed] [Google Scholar]
- Tanjore H., Blackwell T. S., Lawson W. E. (2012). Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 302, L721–L729 10.1152/ajplung.00410.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanjore H., Cheng D.-S., Degryse A. L., Zoz D. F., Abdolrasulnia R., Lawson W. E., et al. (2011). Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J. Biol. Chem. 286, 30972–30980 10.1074/jbc.M110.181164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanjore H., Lawson W. E., Blackwell T. S. (2013). Endoplasmic reticulum stress as a pro-fibrotic stimulus. Biochim. Biophys. Acta 1832, 940–947 10.1016/j.bbadis.2012.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A. Q., Lane K., Phillips J., 3rd., Prince M., Markin C., Speer M., et al. (2002). Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am. J. Respir. Crit. Care Med. 165, 1322–1328 10.1164/rccm.200112-123OC [DOI] [PubMed] [Google Scholar]
- Thurm T., Kaltenborn E., Kern S., Griese M., Zarbock R. (2013). SFTPC mutations cause SP-C degradation and aggregate formation without increasing ER stress. Eur. J. Clin. Invest. 43, 791–800 10.1111/eci.12107 [DOI] [PubMed] [Google Scholar]
- Tsakiri K. D., Cronkhite J. T., Kuan P. J., Xing C., Raghu G., Weissler J. C., et al. (2007). Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. U.S.A. 104, 7552–7557 10.1073/pnas.0701009104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ham T. J., Kokel D., Peterson R. T. (2012). Apoptotic cells are cleared by directional migration and Elmo1- dependent macrophage engulfment. Curr. Biol. 22, 830–836 10.1016/j.cub.2012.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Moorsel C. H. M., van Oosterhout M. F. M., Barlo N. P., de Jong P. A., van der Vis J. J., Ruven H. J. T., et al. (2010). Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am. J. Respir. Crit. Care Med. 182, 1419–1425 10.1164/rccm.200906-0953OC [DOI] [PubMed] [Google Scholar]
- Vivante A., Mark-Danieli M., Davidovits M., Harari-Steinberg O., Omer D., Gnatek Y., et al. (2013). Renal hypodysplasia associates with a WNT4 variant that causes aberrant canonical WNT signaling. J. Am. Soc. Nephrol. 24, 550–558 10.1681/ASN.2012010097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyletal P., Bleyer A. J., Kmoch S. (2010). Uromodulin biology and pathophysiology—an update. Kidney Blood Press. Res. 33, 456–475 10.1159/000321013 [DOI] [PubMed] [Google Scholar]
- Wang X. M., Zhang Y., Kim H. P., Zhou Z., Feghali-Bostwick C. A., Liu F., et al. (2006). Caveolin-1: a critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J. Exp. Med. 203, 2895–2906 10.1084/jem.20061536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Kuan P. J., Xing C., Cronkhite J. T., Torres F., Rosenblatt R. L., et al. (2009). Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am. J. Hum. Genet. 84, 52–59 10.1016/j.ajhg.2008.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waseda Y., Yasui M., Nishizawa Y., Inuzuka K., Takato H., Ichikawa Y., et al. (2008). Angiotensin II Type 2 receptor antagonist reduces bleomycin-induced pulmonary fibrosis in mice. Respir. Res. 9:43 10.1186/1465-9921-9-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright F. A., Strug L. J., Doshi V. K., Commander C. W., Blackman S. M., Sun L., et al. (2011). Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nat. Genet. 43, 539–546 10.1038/ng.838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaguchi S., Ogawa Y., Shimmura S., Kawakita T., Hatou S., Satofuka S., et al. (2013). Angiotensin II Type 1 receptor antagonist attenuates lacrimal gland, lung, and liver fibrosis in a murine model of chronic graft-versus-host disease. PLoS ONE 8:e64724 10.1371/journal.pone.0064724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X., Gao J.-H., Chen Y., Song M., Liu X.-J. (2007). Preliminary linkage analysis and mapping of keloid susceptibility locus in a Chinese pedigree. Zhonghua Zheng Xing Wai Ke Za Zhi 23, 32–35 Available online at: http://www.ncbi.nlm.nih.gov/pubmed/?term=17393690 [PubMed] [Google Scholar]
- Yang J., Lin S.-C., Chen G., He L., Hu Z., Chan L., et al. (2013). Adiponectin promotes monocyte-to-fibroblast transition in renal fibrosis. J. Am. Soc. Nephrol. 24, 1644–1659 10.1681/ASN.2013030217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokohama S., Yoneda M., Haneda M., Okamoto S., Okada M., Aso K., et al. (2004). Therapeutic efficacy of an angiotensin II receptor antagonist in patients with nonalcoholic steatohepatitis. Hepatology 40, 1222–1225 10.1002/hep.20420 [DOI] [PubMed] [Google Scholar]
- Yoneda M., Hotta K., Nozaki Y., Endo H., Uchiyama T., Mawatari H., et al. (2009). Association between angiotensin II Type 1 receptor polymorphisms and the occurrence of nonalcoholic fatty liver disease. Liver Int. 29, 1078–1085 10.1111/j.1478-3231.2009.01988.x [DOI] [PubMed] [Google Scholar]
- Zain S. M., Mohamed Z., Mahadeva S., Rampal S., Basu R. C., Cheah P.-L., et al. (2013). Susceptibility and gene interaction study of the angiotensin II Type 1 receptor (AGTR1) gene polymorphisms with non-alcoholic fatty liver disease in a multi-ethnic population. PLoS ONE 8:e58538 10.1371/journal.pone.0058538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.-Y., Yu Q., Cheng T., Liao T., Nie C.-L., Wang A.-Y., et al. (2011). Role of caveolin-1 in the pathogenesis of tissue fibrosis by keloid-derived fibroblasts in vitro. Br. J. Dermatol. 164, 623–627 10.1111/j.1365-2133.2010.10111.x [DOI] [PubMed] [Google Scholar]
- Zhong Q., Zhou B., Ann D. K., Minoo P., Liu Y., Banfalvi A., et al. (2011). Role of endoplasmic reticulum stress in epithelial-mesenchymal transition of alveolar epithelial cells: effects of misfolded surfactant protein. Am. J. Respir. Cell Mol. Biol. 45, 498–509 10.1165/rcmb.2010-0347OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F., Wu B., Li P., Wang J., Tang H., Liu Y., et al. (2013). Association study confirmed susceptibility loci with keloid in the chinese han population. PLoS ONE 8:e62377 10.1371/journal.pone.0062377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zizzo G., Hilliard B. A., Monestier M., Cohen P. L. (2012). Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J. Immunol. 189, 3508–3520 10.4049/jimmunol.1200662 [DOI] [PMC free article] [PubMed] [Google Scholar]