

Highlights
► The structures of oestrogen-metabolising enzymes are discussed and compared. ► Substrate recognition and reaction mechanisms of these enzymes are discussed. ► The impact of structure on inhibitor design is briefly mentioned.
Abbreviations: 17β-HSD, 17β-hydroxysteroid dehydrogenase; COMT, catechol-O-methyl transferase; DHEA(S), dehydroepiandrosterone (sulfate); DHETNA, O5′-[9-(3,17β-dihydroxy-1,3,5(10)-estratrien-16β-yl)-nonanoyl]adenosine; DNC, 3,5-dinitrocatechol; E1(S), estrone (sulfate); E2(S), estradiol (sulfate); E3, estriol; E4, estetrol; ER, estrogen receptor; E2B, 3-(((8R,9S,13S,14S,16R,17S)-3,17-dihydroxy-13-methyl-7,8,9,11,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-16-yl)methyl)benzamide; FAD/FMN, flavin adenine dinucleotide/flavin mononucleotide; FG, formylglycine; HFG(S), hydroxyformylglycine (sulfate); mb-COMT, membrane-bound COMT; NADP+, nicotinamide adenine dinucleotide phosphate (oxidised); NADPH, nicotinamide adenine dinucleotide phosphate (reduced); PAP, 3′-phosphoadenosine-5′-phosphate; PAPS, 3′-phosphoadenosine-5′-phosphosulfate; s-COMT, soluble COMT; SAM, S-adenosyl methionine; SDR, short-chain dehydrogenase/reductase
Keywords: Oestrogen, Protein structure, Reaction mechanism, Aromatase, Sulfatase, Sulfotransferase, 17β-Hydroxysteroid dehydrogenase
Abstract
Many enzymes catalyse reactions that have an oestrogen as a substrate and/or a product. The reactions catalysed include aromatisation, oxidation, reduction, sulfonation, desulfonation, hydroxylation and methoxylation. The enzymes that catalyse these reactions must all recognise and bind oestrogen but, despite this, they have diverse structures. This review looks at each of these enzymes in turn, describing the structure and discussing the mechanism of the catalysed reaction. Since oestrogen has a role in many disease states inhibition of the enzymes of oestrogen metabolism may have an impact on the state or progression of the disease and inhibitors of these enzymes are briefly discussed.
This article is part of a Special Issue entitled ‘CSR 2013’.
1. Introduction
Estrogens have many roles in the body including, but not limited to, in reproduction and the menstrual cycle [1–5], many breast cancers [6], the development of osteoarthritis [7], the prevention of heart disease [8], neuroprotection during cerebral ischaemia [9] and in multiple sclerosis [10], appetite and eating behaviour [11], fat metabolism [12], schizophrenia [13], autoimmunity [14], and auditory and visual processing [15]. Estrogens exert their effects in several ways [16–18]. In the ‘classic genome response’ oestrogen binds to specific intracellular oestrogen receptors (ERα and ERβ) that, subsequent to oestrogen binding, dimerise and translocate to the nucleus, where they modulate the transcription of target genes that contain oestrogen-responsive elements in their promoters. However, these same oestrogen receptors have also been shown (a) to bind to other transcription factors thus influencing the expression of genes that do not contain oestrogen-responsive elements in their promoters, and (b) to engage signal transduction pathways (that may include, but are not limited to, the activation of protein kinases), thus modulating cellular responses to oestrogen. Signal transduction pathways can also be activated by oestrogen binding to cell surface membrane bound receptors [16–18].
The three most common estrogens are estrone (E1), estradiol (E2) and estriol (E3). A fourth oestrogen is estetrol (E4). Estradiol is the most potent. Estrone and estradiol are synthesised by the aromatisation of androstenedione and testosterone, respectively (Fig. 1). They can also be interconverted by the action of 17β-hydroxysteroid dehydrogenases (17β-HSDs). Estriol is synthesised from estrone via a 16α-hydroxyestrone intermediate. Although, in some tissues, estrogens can be made on demand, oestrogen can be stored in the form of estrone sulfate. This is synthesised from estrone by the action of oestrogen sulfotransferase with estrone being regenerated by the steroid sulfatase-catalysed hydrolysis of estrone sulfate. A review of many aspects of human steroidogenesis is available [19]. Estrogens are eliminated from the body mainly as the sulfated and glucuronidated derivatives [20, and references therein]. The first step in synthesising these conjugates is the generation of the hydroxylated derivatives. Hydroxylation occurs primarily at the 2-, 4- and 16-positions. The hydroxyl group can then be sulfated, glucuronidated or methylated.
Fig. 1.

Pathways of oestrogen metabolism. Not all pathways are shown. There are tissue-specific variations in synthetic pathways: see [19]. The A-ring hydroxylated compounds are converted to the methoxy compounds by catechol O-methyltransferases. The structures of estetrol and ethinylestradiol are shown, as is the steroid ring labelling and atom numbering system.
Because there is little published work on estetrol it is discussed only briefly here. It is synthesised in the foetal liver, but its function is presently unknown. Several possible biosynthetic routes have been proposed starting from various androgens, estrogens, and their 3-sulfated derivatives, with the hydroxylations occurring in various different orders and, in the case of the androgens, the aromatisation occurring either before or after the hydroxylations. Evidence suggests that estetrol is made through multiple biosynthetic routes. See [21–29] for details. Estetrol will not be discussed further herein.
Given the role of estrogens in preventing, causing and exacerbating disease a good knowledge of how estrogens are synthesised and metabolised may help in the understanding and treatment of disease. This review looks in turn at each of the enzymes involved in oestrogen metabolism in terms of their structure and reaction mechanism, before a final section compares the various enzymes. Inhibition of the enzymes of oestrogen metabolism impacts on the amount of oestrogen in the body and this has been the subject of multiple therapeutic approaches leading to clinically active agents [30,31]. Also, synthetic estrogens, e.g., ethinylestradiol (Fig. 1), have been widely employed in hormone replacement therapy [32] and, combined with a progestin, contraception [33,34]. Although inhibitors of these enzymes and synthetic estrogens are not the focus of this review they are briefly discussed, mainly through references to recent reviews. Unless otherwise stated, all discussions below refer to the human enzymes.
2. Methods
All dockings of ligands into binding sites were carried out using the GOLD software running in Gold Suite v5.0.1. All other manipulations of molecules, including building ligands and preparing proteins for docking runs, were carried out using the Schrödinger software running under Maestro 9.2.109. The protein crystal structures were downloaded from the PDB and run through the Protein Preparation Wizard. Hydrogens were added assuming a neutral pH and the resulting structures were put through a brief minimisation procedure. Visual inspection of these structures was used to identify those residues that form substrate binding sites. All figures of proteins and ligands were prepared using PyMOL (Delano Scientific). Figures of reaction schemes and molecules were prepared using ChemBioDraw Ultra 12.0. Multalin was used to perform sequence alignments (http://multalin.toulouse.inra.fr/multalin/multalin.html).
3. Oestrogen sulfotransferase
Oestrogen sulfotransferase (EC 2.8.2.4, SULT1E1) is a cytosolic enzyme that catalyzes the sulfonation of estrogens, utilising 3′-phosphoadenosine-5′-phosphosulfate (PAPS) as the sulfate-donating cofactor [35,36] (Fig. 2). This enzyme has a high affinity for estrogens with maximal activity at about Km = 20 nM [37]. There are two other sulfotransferases in the SULT1 class that catalyse the sulfonation of estrogens: SULT1A1 and SULT1A3 (both EC 2.8.2.1, phenol sulfotransferase). However, these enzymes have much lower affinities for estrogens, with maximal activity at about Km = 25 μM. For all three human proteins the amino acid sequence is available, as is that of murine SULT1E1 (Fig. 3): SULT1A1 and SULT1A3 are 92.5% identical, and both are approximately 50% identical to SULT1E1. The sequence of murine SULT1E1 is 75% identical to that of the human protein.
Fig. 2.

(A) The reactions catalysed by oestrogen sulfotransferase. (B) The cofactor, before and after reaction.
Fig. 3.

Sequences of sulfotransferases. Human SULT1E1 (oestrogen sulfotransferase) UniProt P49888. Murine SULT1E1 (oestrogen sulfotransferase) UniProtP49891. Human SULT1A1 (phenol sulfotransferase) UniProt P50225. Human SULT1A3 (phenol sulfotransferase) UniProt P50224. Residues identical across all four proteins are highlighted in yellow. Other residues identical to the human SULT 1E1 are highlighted in cyan. Secondary structural elements, as identified by PyMOL (Delano Scientific) in the 1G3M structure of human SULT1E1, are shown in bold: the numbering of these elements is consistent with that of [49]. H – helix, S – sheet.
Available from the PDB are two crystal structures of human SULT1E1, three of murine SULT1E1, two of human SULT1A3, and eight of human SULT1A1 (Table 1). Given the high level of sequence identity it is not surprising that the three-dimensional structures are superimposable. The protein is a single domain globular structure (Fig. 4).
Table 1.
The crystal structures of sulfotransferases. The mutations are all remote from the substrate and cofactor binding sites and are not expected to have any influence on binding or catalysis.
| Protein | PDB code | Ligands | Mutations | Reference |
|---|---|---|---|---|
| Human 1E1 | 1HY3 | PAPS | V269E | [38] |
| 1G3M | PAP, polychlorinated biphenol | – | [39] | |
| Murine | 1AQU | PAP, estradiol | – | [40] |
| 1AQY | PAP | – | [40] | |
| 1BO6 | PAP, vanadate | – | [41] | |
| Human 1A1 | 1LS6 | PAP, p-nitrophenol | – | [46] |
| 1Z28 | PAP | – | [45] | |
| 2D06 | PAP, estradiol | – | [44] | |
| 3U3J | PAP | R213H, V223M | [47] | |
| 3U3K | PAP, naphthalene-2-ol | R213H, V223M | [47] | |
| 3U3M | PAP, 3-cyano-7-hydroxycoumarin | R213H, V223M | [47] | |
| 3U3O | PAP, 3-cyano-7-hydroxycoumarin | R213H, V223M | [47] | |
| 3U3R | PAP, p-nitrophenol | R213H, V223M, D249G | [47] | |
| Human 1A3 | 1CJM | Sulphate | – | [42] |
| 2A3R | PAP, dopamine | – | [43] | |
Fig. 4.

The three-dimensional structure of oestrogen sulfotransferase. The 1G3M (SULT1E1) structure [39] is shown. The cofactor PAPS (purple carbons) is taken from the 1HY3 structure [38], and the steroid (pink carbons) is from the 2D06 structure [44]. The helices, H0-H17 are identified, as are the sheets Sa-Sd. Sheets Se and Sf are hidden behind H3, H11 and H15. The flexible loops involved in binding the substrate and cofactor are identified thus: Loop 1, purple; Loop 2, brown; Loop 3, dark blue.
Thirty-one crystal structures of twelve different human sulfotransferases have been the subject of a recent review [48] that covers substrate specificity and mechanisms of action. An older review covering sulfotransferase structure and inhibitors is available [49].
Fig. 5 shows the structure of the binding site of human SULT1E1. To generate Fig. 5A the reaction product was docked into the 1HY3 structure after the sulfate had been removed from the cofactor. To generate Fig. 5B the substrate was docked into the 1HY3 structure. In both instances, subsequent to docking, the protein ligand complex was put through 1000 rounds of minimisation prior to preparing the figures. A comparison of the two structures (Fig. 5C) shows that three of the water molecules have different positions but the ligand moves only slightly, and the surrounding residues barely move at all.
Fig. 5.

The binding site of oestrogen sulfotransferase. (A) Estrone sulfate (pink carbons) and 3′-phosphoadenosine-5′-phosphate (purple carbons) in the binding site. The red spheres are the oxygen atoms of water molecules in the binding site. (B) Estrone (cyan carbons) and 3′-phosphoadenosine-5′-phosphosulfate (purple carbons) in the binding site. The brown spheres are the oxygen atoms of water molecules in the binding site. (C) An overlay of A and B, showing the short distance the sulfate has to move during the reaction.
The substrate and cofactor binding site takes the form of a tube, open at both ends, running through the core of the protein. The residues forming the substrate binding site are predominantly hydrophobic and enclose the substrate quite closely. These residues (Y20, F23, P46, F75, F80, C83, K85, M89, K105, H107, F138, F141, V145, A146, G147, H148, Y168, Y239, L242, I246 and M247) are mainly from the three flexible loops (Fig. 4), loop 1 (between helices 5 and 6), loop 2 (between helices 8 and 9), and loop 3 (between helices 15 and 16), but also helices H1 and H8 and sheet Sd. A particularly close interaction is that of the hydrogens attached to C6 and C7 in the B-ring with the face of the Y20 ring. The side chain of F141 points directly at the A-ring of the substrate to have a face–edge π–π interaction. K105 has a hydrogen bond to the oxygen at the 3-position. These interactions position the substrate hydroxyl so that it can act as the acceptor in the sulfate transfer.
The cofactor is enclosed by nineteen residues (K47, S48, G49, T50, T51, W52, K105, H107, R129, S137, Y192, T226, F228, M231, F254, M255, R256, K257 and G258) that are predominantly in helices 3, 8 and 15, and loop 3. The PAPS adenine moiety has a face–face π–π stacking interaction with W52 and hydrogen bonds to Y192 and T226. The ribose-3-phosphate has hydrogen bond interactions with R129, K257, G258 and R256, and the 5-phosphate with G49 and T51. The sulfate is held in position by interaction with K47 and K105. There are four water molecules that may play a role in catalysis and/or that interact with the substrate or cofactor.
A comparison of all human sulfotransferase structures suggests that the binding of substrate and PAPS can occur in a random order, and that substrate binding may be cooperative with prior cofactor binding, but not vice versa [50]. It seems likely that loop 3 must move to enable both substrate and cofactor to bind. The ends of this loop (helices 15 and 16) are in the vicinity of the cofactor: if the entire loop opens as if the ends of the loop act as a hinge then the prior binding of the cofactor may position the remainder of the loop to more easily bind the substrate. This is supported by crystal structures of SULT1A1 in complex with PAP and substrate where the binding site is very similar to that observed when just PAP is bound [47]. (The conformation of this loop subsequent to PAPS binding is also thought to influence substrate binding in human SULT2A1 [51].) There are some reports that at high substrate concentrations sulfotransferases, particularly SULT1A1, suffer from substrate inhibition [46,50]. Whether this is of relevance in vivo is undetermined.
Three conserved residues, K47, H107 and S137, are thought to be involved in the reaction mechanism (Fig. 6) [38,52]. S137 forms a hydrogen bond to K47, thus preventing the side chain nitrogen of K47 from interacting with the bridging oxygen. This prevents PAPS hydrolysis. Upon substrate binding, the catalytic base H107 abstracts a proton from the substrate hydroxyl, thus enabling nucleophilic attack at the sulphur atom in PAPS. The consequent accumulation of partial negative charge on the bridging oxygen of PAPS disrupts the interaction between S137 and K47, thus enabling K47 to interact with the 5′-phosphate. This assists in the dissociation of the sulfyryl group and completes the catalytic reaction cycle [52,53]. The sulfonation takes place via an in-line displacement mechanism though whether the mechanism is associative (SN2-like) or dissociative (SN1-like) is not yet clear [53,54].
Fig. 6.

The oestrogen sulfotransferase reaction mechanism. (A) The apoenzyme. (B) The enzyme-PAPS–substrate complex. (C) The intermediate. (D) The enzyme–PAP–product complex.
Adapted from [52].
4. Steroid sulfatase
Steroid sulfatase (EC 3.1.6.2, aryl sulfatase C, steryl-sulfatase) catalyzes the conversion of estrone sulfate to estrone (Fig. 1), and dehydroepiandrosterone sulfate (DHEAS) to dehydroepiandrosterone (DHEA) (the reverse of the reaction shown in Fig. 2). The protein is found in the lumen of the endoplasmic reticulum and, in terms of tissue distribution, is probably ubiquitous at low levels, with greater amounts in those tissues associated with reproduction. The biology of steroid sulfatase has been reviewed [55] as has the role of steroid sulfatase in oestrogen metabolism [56].
The gene has been cloned and sequenced, the protein expressed and purified [57,58] and shown to consist of 583 residues (Fig. 7). The structure of the protein has been solved (PDB code 1P49: [59]) and shown to have a globular head (approximately 65 Å × 50 Å × 45 Å) and a hydrophobic tail about 40 Å long (Fig. 8). This tail is embedded in the lumenal membrane of the endoplasmic reticulum. The crystal structure shows that several residues undergo post-translational glycosylation. Another post-translational modification is that of C75 to formylglycine (FG) (Fig. 9) [60]. This is further modified, by hydration, to form the gem-diol hydroxyformylglycine (HFG), though the resting state of the enzyme appears to be hydroxyformylglycine sulfate (HFGS) with the sulfate linked to the pro-(S) hydroxyl group.
Fig. 7.

The sequence of steroid sulfatase (UniProt P08842). Residue C75 (highlighted) undergoes post-translational modification to formylglycine [60].
Fig. 8.

The three-dimensional structure of steroid sulfatase. PDB code 1P49[59]. (A) A cartoon of the structure coloured blue at the N-terminus through to red at the C-terminus. Residue HFGS75 and the calcium ion are in the middle of the structure. (B) The surface of the protein showing the hydrophobic nature of the membrane-embedded tail at the top.
Fig. 9.
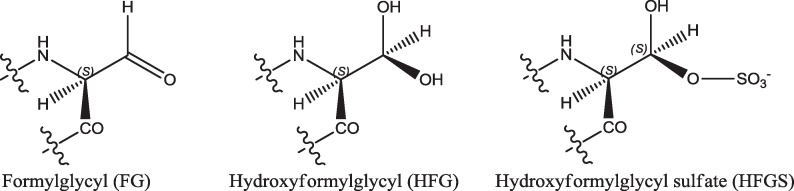
The structures of formylglycyl, hydroxyformylglycyl and hydroxyformylglycyl sulfate residues.
Visual inspection of the structure shows that a number of hydrogen bonds help position the HFGS and, specifically, the side chain in the binding site (Fig. 10). The HFGS backbone carbonyl may be involved in a hydrogen bond with the backbone NH of R79, and the HFGS side chain hydroxyl may form a hydrogen bond to the side chain of K134 and H136. There are four or, possibly, five interactions of the sulfate with the protein that may serve to position the sulfate during the reaction. The first of these four is an internal hydrogen bond from the HFGS backbone NH, and the other four involve the side chains of H290, K134 and, possibly, K368. A calcium ion in the binding site has an interaction with one of the HFGS sulfate oxygens. The calcium ion also has charge–charge interactions with the side chains of D35, D36, D342 and Q343. The substrate binding site is formed by residues L74, HFGS75, R98, T99, G100, V101, L103, L164, T165, L167, R168, V177, F178, T180, G181, H290, T291, H346, E349, K368, N447, T484, H485, V486, F488 and C489.
Fig. 10.

The interactions of the calcium ion and HFGS75 in the binding site of steroid sulfatase. Hydrogen bonds involving HFGS75 are shown in orange. Interactions with the calcium ion are shown in black, except for the interaction with HFGS75 which is shown in blue. Based on the 1P49 structure [59]. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
One proposed mechanism for the steroid sulfatase-catalysed reaction is shown in Fig. 11 [61]. In Step I FG75 is activated by a water molecule to form the gem-diol. In step II the substrate sulphur undergoes nucleophilic attack by the pro-(S) hydroxyl following its activation by Ca2+. This results in the sulfate moiety being covalently linked to FG75 and the unconjugated substrate being released with the sulfate being replaced by a hydrogen abstracted from H290. In step III the pro-(R) hydroxyl of HFGS75 is deprotonated by H136 and the ester bond is broken by attack of the activated free hydroxyl. The sulfate is released and FG75 is regenerated. Release of the sulfate may require an additional water molecule as a nucleophile. This water may accompany the incoming substrate, shielding the charged sulfate during its passage into the substrate binding site.
Fig. 11.

Proposed catalytic mechanism of steroid sulfatase.
Adapted from [61].
The reaction site lies at the base of a long narrow pocket that opens beside the hydrophobic tail. This suggests that the substrate and product may have to travel through the endoplasmic reticulum membrane in order to enter and leave the substrate binding site. The binding site itself is formed predominantly from hydrophobic residues (Fig. 12). The crystal structure has two water molecules in the substrate binding site, one of them putatively being the catalytic water, but the other being a crystallographic water interacting with the sulfate of HFGS75. This second water has to be removed prior to performing docking experiments to determine how the substrate and product might fit into the binding site.
Fig. 12.

The steroid sulfatase binding site. (A) The crystal structure of the steroid sulfatase substrate binding site. HFGS75 is shown with purple carbons. The calcium ion is the yellow sphere. The putative catalytic water is the red sphere. The brown sphere is the only other water present in the substrate binding site in the crystal structure. For both water molecules only the oxygen is shown. (B) Estrone docked into the substrate binding site of steroid sulfatase. The water in the substrate binding site was removed prior to docking: it occupies the space taken by the estrone hydroxyl. (C) Estrone sulfate docked into the substrate binding site of steroid sulfatase. Prior to docking the sulfate was removed from residue 75 to leave hydroxyformylglycine. (D) An overlay of B and C showing the identical docked pose of the estrone core and how little the sulfate has to move during the reaction. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Inhibition of steroid sulfatase (together with the inhibition of aromatase – see below) should reduce the amount of oestrogen in the body, particularly in target tumour tissue, and be beneficial in a range of hormone-dependent diseases. The development of steroid sulfatase inhibitors has been the subject of a number of recent reviews [62–66].
5. 17β-Hydroxysteroid dehydrogenases
There are fourteen different vertebrate enzymes classified as 17β-hydroxysteroid dehydrogenases of which twelve have been found in human tissues [67]. Despite their name the preferred substrate of some of these enzymes is other than steroids and, when the substrate is a steroid, the reaction may be either an oxidation or a reduction depending on the cofactor and cellular localisation [68]. 17β-Hydroxysteroid dehydrogenase type 1 (EC 1.1.1.62, 17β-HSD1) catalyses the conversion of estrone to estradiol, and of 16α-hydroxyestrone to estriol (Fig. 1). The reverse reactions are catalysed by 17β-HSD2 which also facilitates the conversion of testosterone to androstenedione, the reverse reaction of which is catalysed by 17β-HSD3.
All three enzymes are members of the short-chain dehydrogenase/reductase (SDR) structural family. The aligned sequences are shown in Fig. 13. Listed in Table 2 are the twenty-two publicly available structures of 17β-HSD1. Most of these structures have a ligand in the substrate binding site: the structures of these ligands are shown in Fig. 14. The gene can be translated into 328 amino acids but the mature protein has lost the initiating methionine and, for historical reasons, is often (but not always) numbered A1–Q327. Herein the A1–Q327 numbering is used. There are no publicly available crystal structures of either 17β-HSD2 or 17β-HSD3, but the overall 3D structure of both is believed to be similar to that of 17β-HSD1 (Fig. 15).
Fig. 13.

Sequence alignment of 17β-hydroxysteroid dehydrogenases. An alignment of the 17β-HSD1 (UniProt ID code P14061), 17β-HSD2 (UniProt ID code P37059) and 17β-HSD3 (UniProt ID code P37058) sequences. Identical residues are highlighted in yellow. Those residues in 17β-HSD1 forming the substrate binding site, as identified by visual inspection of the structure, are shown in bold, and those forming the NADPH binding site are shown in red. Secondary structure elements in 17β-HSD1 are identified as H – helix and S – sheet. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Table 2.
The crystal structures of 17β-HSD1. No papers describing the 3DEY, 3KLP and 3KM0 structures have been published. The structures of the ligands are shown in Fig. 14.
| PDB code | Mutated residues | Ligands | Reference |
|---|---|---|---|
| 1A27 | – | NADP, estradiol | [69] |
| 1BHS | – | – | [70] |
| 1DHT | – | 5α-Dihydrotestosterone | [71] |
| 1EQU | – | NADP, equilin | [72] |
| 1FDS | – | Estradiol | [73] |
| 1FDT | – | NADP, estradiol | [73] |
| 1FDU | H221L | NADP, estradiol | [74] |
| 1FDV | H221L | NAD | [74] |
| 1FDW | H221Q | Estradiol | [74] |
| 1I5R | – | DHETNAa | [75] |
| 1IOL | D112E | Estradiol | [76] |
| 1JTV | – | Testosterone | [77] |
| 1QYV | – | NADP | [78] |
| 1QYW | – | NADP, 5α-androstan-3,17-dione, | [78] |
| 1QYX | – | NADP, 4-androstene-3,17-dione | [78] |
| 3DEY | – | 5α-Dihydrotestosterone | – |
| 3DHE | – | 3β-Hydroxy-5-androsten-17-one | [71] |
| 3HB4 | – | E2Bb | [79] |
| 3HB5 | – | NADP, E2Bb | [79] |
| 3KLM | – | 5α-Dihydrotestosterone | [80] |
| 3KLP | – | (3α,8α,17β)-androst-5-ene-3,17-diol | – |
| 3KM0 | – | NADP, 5α-androstan-3β,17β-diol | – |
DHETNA: O5′-[9-(3,17β-dihydroxy-1,3,5(10)-estratrien-16β-yl)-nonanoyl]adenosine.
E2B: 3-(((8R,9S,13S,14S,16R,17S)-3,17-dihydroxy-13-methyl-7,8,9,11,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-16-yl)methyl)benzamide.
Fig. 14.

The structures of the ligands in the crystal structures of 17β-HSD1. DHETNA: O5′-[9-(3,17β-dihydroxy-1,3,5(10)-estratrien-16β-yl)-nonanoyl]adenosine. E2B: 3-(((8R,9S,13S,14S,16R,17S)-3,17-dihydroxy-13-methyl-7,8,9,11,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-16-yl)methyl)benzamide.
Fig. 15.

The structure of 17β-HSD1. A cartoon of the 17β-HSD1 structure coloured blue at the N-terminus through to red at the C-terminus. The secondary structure elements are labelled as in Fig. 13: H – helix and S – sheet. NADPH is shown with purple carbons and estradiol with pink carbons (based on the 1A27 structure [69]). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The protein is built around a core of seven parallel β-strands with at least one helix between successive strands. This makes up the Rossman fold typical of nucleotide binding proteins. The loop between sheet 1 and helix 1 has the GxxxGxG motif (residues G9–G15, where G is glycine and x any other residue) that is common to oxidation/reduction enzymes that bind nicotinamide cofactors. S142, at the end of sheet 5, and Y155 and K159, in helix 9, have been shown to be essential for activity [81].
The substrate binding site is formed by residues G94, L95, L96, S142, V143, G144, M147, L149, P150, N152, Y155, C185, G186, P187, F192, M193, V196, Y218, H221, S222, V225, F226, F259, L262, M279, E282 and V283. Of these twenty-six residues only fourteen are in secondary structural elements, i.e., helices or sheets, with the remainder being in loops between the structural elements. One of these loops, the substrate binding loop comprising residues H189–V196, is flexible enough that it is observed in two conformations in the 1FDT structure. This loop probably has to move to allow the substrate into the binding site. The following residues form the NADPH binding site: T8, G9, C10, S11, S12, G13, I14, G15, R37, L64, D65, V66, R67, C89, N90, A91, G92, L93, V113, T140, G141, S142, Y155, K159, C185, V188, H189, T190, A191, F192, M193 and K195.
The hydride transfer reactions catalysed by these enzymes are intrinsically reversible but, in vivo, are effectively unidirectional due to the relative concentrations of NADPH and NADP+ [82]. Based on the measurement of isotope exchange rates between substrate-product pairs the reaction catalysed by 17β-HSD1 has been reported to be a random order bi–bi mechanism [83] (Fig. 16). However, a dynamics study based on crystal structures of the apo enzyme and binary and ternary complexes has suggested that there is a preferred order with NADPH binding before the substrate, and the NADP+ being released before the product [84]. In the reaction the pro-(S) hydride is transferred from NADPH to the alpha face of the estrone C17. A hydride is then transferred to the C17 oxygen, stabilised by interaction with the hydroxyl of S142, from the hydroxyl of Y155. There is then postulated to be a hydride transfer network involving the NADPH ribose hydroxyl, the K159 amino moiety and a water molecule that results in the formation of a hydronium ion [84] (Fig. 17). Work with the 17β-HSD from the filamentous fungus Cochliobolus lunatus has shown that cofactor dissociation is the slowest step of the reaction and that the catalytic activity might be modulated by differences in the conformation of the substrate binding loop consequent upon cofactor and substrate binding [85]. Enzymes of the SDR family are typically active as dimers or tetramers, and work with the C. lunatus enzyme suggests that dimerisation may be necessary for activity [86].
Fig. 16.
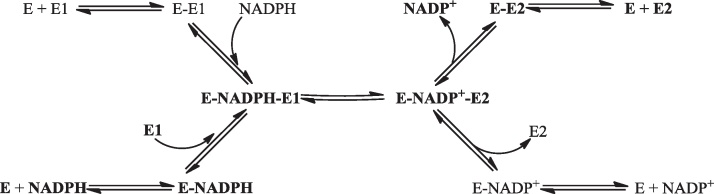
The proposed random order bi–bi mechanism of 17β-HSD1. The mechanism believed to be preferred is shown in bold [83,84]. E – enzyme, E1 – estrone, E2 – estradiol.
Fig. 17.

The substrate binding site of 17β-HSD1. The nucleotide binding GxxxGxG motif and the residues involved in catalysis are shown in green. The NADPH is in purple and the estradiol in pink. Secondary structure elements are shown in grey. Some of the possible hydrogen bonds are shown by the black dashed lines. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Of the estrogens estradiol is the most potent and the inhibition of estradiol production is beneficial in the treatment of oestrogen-dependent diseases. Inhibition of 17β-HSD1 should prevent estradiol production, though the concomitant inhibition of aromatase (see below) and steroid sulfatase is probably necessary for greater reduction of estradiol levels. A variety of steroidal and non-steroidal inhibitors have been synthesised that have IC50 values in the low nanomolar range. For recent reviews of these enzymes and their inhibitors see [87–89].
6. Aromatase (cytochrome P450 19A1, oestrogen synthase)
Aromatase (cytochrome P450 19A1, cytochrome P450AROM, oestrogen synthase) catalyzes the aromatisation of the ‘A’ ring of androstendione to produce estrone and the aromatisation of the ‘A’ ring of testosterone to produce estradiol (Fig. 1). The enzyme consists of 503 amino acids (Fig. 18) and an iron-containing haem group (protoporphyrin IX; Fig. 19). The protein is glycosylated on N12 [90]. Other post-translational modifications are performed by the tyrosine kinases c-Src [91] and PTP1B [92]: phosphorylation of Y361 by c-Src increases aromatase activity. The enzyme is found in the cytoplasm bound to the endoplasmic reticulum with the N-terminal residues probably penetrating through the membrane and the glycosylated residue in the lumen of the endoplasmic reticulum. However, it has been suggested that the N-terminal residues are required not so much for membrane association as for orientation [93]. There is evidence to suggest that aromatase forms dimers or higher oligomers [93,94]. The proposed mode of oligomerisation may be a further means of regulating enzyme activity as it would prevent the phosphorylation of Y361 [94].
Fig. 18.

The sequence of aromatase (UniProt ID code P11511). H – helix and S – sheet.
Fig. 19.

The structure of ligands in the aromatase binding site. The protoporphyrin is in all the crystal structures of aromatase.
There are five publicly available crystal structures of human placental aromatase: PDB code 3EQM [95,96] has androstenedione in the substrate binding site, as does 3S79 [97]; the inhibitor exemestane (Fig. 19) is in the 3S7S structure [97]; the 4GL5 and 4GL7 structures [97] both have O-linked alkyne chains attached to the 6-position of the steroid ring. All five structures are of residues S45–N496. They reveal a predominantly helical single domain protein with the ligand tightly enclosed in a largely hydrophobic pocket, situated deep within the protein, that is formed by residues R115, I133, F134, F221, W224, E302, I305, A306, D309, T310, V369, V370, L372, V373, M374, L477 and S478, and the haem (Fig. 20). There is a hydrogen bond between the backbone NH of M374 and the ligand carbonyl at the 17-position and another between the side chain of D309 and the ligand carbonyl at the 3-position. In the 3EQM and 3S79 structures the androstenedione has an identical pose, as do the haems and the residues forming the binding site. The B, C and D rings of exemestane overlay the equivalent rings in androstenedione but, because of the additional double bond in the A ring of exemestane, the A ring is slightly distorted relative to the androstenedione A ring. The C6-methylidene carbon is accommodated by slight shifts (less than 1 Å) in the position of T310 and, to a lesser extent, S478. Despite the greater size of the 6-substituent in the 4GL5 and 4GL7 structures there is no further significant movement of any of the amino acid residues in the substrate binding site.
Fig. 20.

The aromatase substrate binding site. (A) The haem-capped cavity tightly enclosing androstenedione. (B) The residues forming the substrate binding site.
The aromatisation occurs in a three-step mechanism [98,99] (Fig. 21). Two consecutive hydroxylations generate, first, a hydroxyl at C19 and, second, a gem-diol intermediate that dehydrates to leave an aldehyde [100]. The ketone at the 3-position then enolises and the aldehyde is cleaved from C10 and ejected as methanoic (formic) acid [101]. This implies that the aromatase catalytic centre can catalyse two different types of reaction (Eqs. (1) and (2)). Note that the first hydroxylation of C19, whilst leaving the absolute configuration of C10 unchanged, does change the stereochemistry of C10 from R to S.
| (1) |
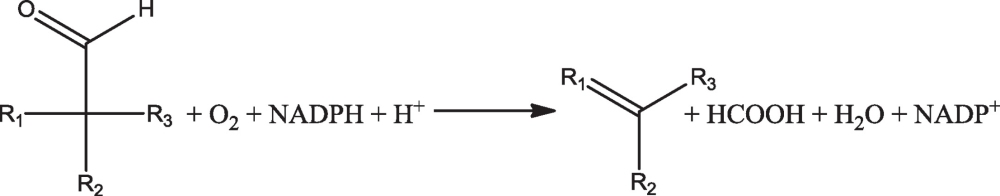 |
(2) |
Fig. 21.

The mechanism of the aromatase-catalysed reaction. Carbons 3, 10 and 19 are identified as is the stereochemistry of C10.
Residues involved in the catalytic act probably include A306, D309 and T310. The binding site opens to the ER membrane so the substrate has to traverse the membrane in order to enter the binding site. NADPH does not bind directly to aromatase. Two electrons are transferred from NADPH to the aromatase haem group via NADPH cytochrome P450 reductase, an FAD/FMN-containing protein [102,103].
Atomic force microscopy has been used to observe aromatase dimers in membranes, and possibly higher order interactions [93]. Computational modelling of aromatase in membranes suggests that a dynamic quaternary organisation together with fluctuations of the active site, the cavity enclosing the haem and the substrate access channel are necessary for activity [104,105].
The inhibition of aromatase is a suitable treatment for a number of clinical conditions that are caused or aggravated by the overproduction of oestrogen. Aromatase inhibitors such as anastrozole, letrozole and exemestane (Fig. 22) have found roles in the treatment of breast cancer [106], ovarian cancer [107] and growth maximisation in puberty in children with short stature [108], and it has been suggested that aromatase inhibitors may have a role to play in treating some cases of lung cancer [109].
Fig. 22.
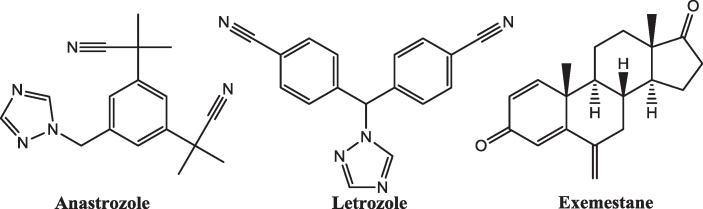
The structure of some clinically used aromatase inhibitors.
7. Enzymes of steroid hydroxylation
Aromatase, cytochrome P450 19A1, catalyzes the hydroxylation of testosterone and androstenedione at C19 prior to aromatising the A-ring. Other cytochrome P450s catalyse hydroxylations at other sites [110], most commonly the 2-, 4- and 16-positions. It has been argued that which cytochrome is involved with which hydroxylation is dependent on both the body tissue and the substrate concentration [111].
Cytochrome P450s 1A1, 2C19, 3A4 and 3A5 are amongst those reported to catalyse hydroxylation at the 16α-position [111,112]. This generates 16α-hydroxyestrone which can then be converted to estriol by 17β-HSD1. Reported to be involved in both 2- and 4-hydroxylations are cytochrome P450s 1A1, 1A2 and 1B1, though the relative importance of these might depend upon the tissue, and others are probably involved [111]. In the PDB are the structures of more than 500 cytochrome P450s, over 100 of which are of human proteins. The structures of CYPs 1A2, 1B1 and 3A4 are briefly discussed herein.
There is one crystal structure of human CYP 1A2, PDB code 2HI4 [113], and one of CYP 1B1, PDB code 3PM0 [114]. The structure of both is based on that of the canonical P450 fold – twelve α-helices and four β-sheets, designated A-L and 1–4, respectively – with a few additional helices. Both have the same haem group, protoporphyrin IX, as that in aromatase (Fig. 19). In both enzymes the substrate binding site is a narrow pocket that tightly encloses a molecule of α-naphthoflavone, with many of the residues forming the binding sites being the same in the two proteins. In the following listing of these residues the numbers for CYP 1A2 are given first, with the numbers for the equivalent residues in CYP 1B1 in brackets. One face of the ligand is bordered by I117 (V126), T118 (S127), S122 (S131), T124 (A133), F125 (F134), T223 (N228), F226 (F231), V227 (G232), F256 (L264), N257 (N265), L382 (V395), T385 (T398), I386 (I399), L497 (L509) and T498 (T510). F226 (F231) forms a face-to-face π–π stacking interaction with the ligand. The other face of the ligand is contacted by V220 (L225), F260 (F268), N312 (T325), D313 (D326), F315 (F328), G316 (G329), A317 (A330), F319 (Q332), D320 (D333) and T321 (T334).
Since both enzymes can hydroxylate estrone at either the 2- or 4-position there must be sufficient flexibility in the substrate binding site, despite the restrictiveness observed in the crystal structures, to allow the substrate to bind in different poses to present either the 2- or 4-positions to the catalytic centre. That such flexibility of substrate binding exists is demonstrated by the fact that the pose of the α-naphthoflavone in one structure is the opposite way round to that in the other structure (Fig. 23).
Fig. 23.

CYP 1A2 and CYP 1B1 substrate binding site structures. Overlay of CYP 1A2 (PDB code 2HI4[113]) and CYP 1B1 (PDB code 3PM0[114]) demonstrating the similarity of the residues forming the substrate binding site and the different pose of the bound ligand, α-naphthoflavone. To avoid obscuring the ligand residues in the foreground are shown as lines rather than sticks. CYP 1A2 – green protein/haem, yellow ligand. CYP 1B1 – blue protein/haem, purple ligand. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
There are nine publicly available structures of CYP 3A4 (Table 3). This enzyme catalyzes hydroxylation at the 16α-position. In the various structures there are seven different structurally diverse ligands (Fig. 24), though one of these, progesterone, is not in the substrate binding site but in a groove on the surface. To accommodate these ligands there is considerable variability in the shape of the substrate binding site (Fig. 25). This variability shows that there is plenty of space to accommodate estrone such that it presents the 16-position for hydroxylation.
Table 3.
The crystal structures of cytochrome P450 3A4. The structures of the ligands are shown in Fig. 22.
| PDB code | Ligands | Reference |
|---|---|---|
| 1TQN | – | [115] |
| 1W0E | – | [116] |
| 1W0F | Progesteronea | [116] |
| 1W0G | Metyrapone | [116] |
| 2J0D | Erythromycin A | [117] |
| 2V0M | Ketoconazoleb | [117] |
| 3NXU | Ritonavir | [118] |
| 3TJS | Desthiazolylmethyloxycarbonyl ritonavir | [119] |
| 3UA1 | Bromoergocryptine | [120] |
The progesterone is not in the substrate binding site, but in a groove on the surface of the protein.
There are two molecules of ketoconazole in the substrate binding site.
Fig. 24.

The structures of the ligands in the crystal structures of cytochrome P450 3A4.
Fig. 25.

The CYP450 3A4 substrate binding site. (A) Ritonavir in the substrate binding site of CYP450 3A4. Taken from the 3NXU crystal structure [118]. (B) The variability in the shape of the substrate binding site. All nine structures are superimposed and equivalent residues coloured the same. The haem and ritonavir from the 3NXU structure are shown. In both figures the haem is shown in purple and the ritonavir in cyan. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
8. Catechol O-methyl transferase (COMT)
The 2- and 4-hydroxy groups on the oestrogen A ring can be converted to methoxy groups by catechol O-methyl transferase (COMT; EC 2.1.1.6; reviewed in [121]) though it should be noted that hydroxylation and ketonisation of estrogens is not limited to these positions: Zhu and Conney [122] list hydroxylations at the 1-, 2-, 4-, 6-, 7-, 11-, 14-, 15-, 16-, 17- and 18-positions. This enzyme is regarded as a phase II drug metabolising enzyme that catalyzes a detoxifying step in drug metabolism [123]. Hydroxylation and methylation are the first steps in the oestrogen degradative and excretory pathways, though the hydroxylated and methoxylated compounds may have roles beyond being mere waste products: see, e.g., [124].
COMT exists in two forms, soluble (s-COMT, 221 residues) and membrane bound (mb-COMT, 271 residues) that differ only in the presence of an additional fifty residues at the N-terminal end of the membrane-bound form (Fig. 26). The residue numbering for s-COMT, 1–221, will be used herein. Residue 108 is a site of a common polymorphism: it can be either valine or methionine. The rarer methionine polymorphism has been associated with a variety of neuropsychiatric disorders [125], increased risk of breast cancer [126,127], lower pain threshold [128] and improved cognitive function involving working memory [129]. These different effects of the polymorphs may be due to the slightly greater enzyme activity of the valine variant [130–132] and/or the greater range of conformational states of the methionine variant observed in molecular dynamics simulations [133] that were suggested by the greater instability (with respect to temperature, autoxidation and denaturants) of the methionine variant (see, e.g., [134] and references therein).
Fig. 26.

The sequence of human COMT. UniProt ID code P21964. The fifty residues present in mb-COMT but absent in s-COMT are shown in italics. The residue numbering, 1-271, is for mb-COMT: s-COMT is numbered 1–221 (shown in brackets) starting from residue 51 of mb-COMT. Highlighted is residue 158 (mb-COMT) or 108 (s-COMT) that is the site of a V/M polymorphism. Secondary structure elements are labelled: H – helix; S – sheet.
There are three crystal structures of human s-COMT, one of which is of the methionine variant (3BWY; [135]) and the others of the valine variant (3BWM; [136]: 3A7E; unpublished). All three have the methyl group donor, S-adenosyl methionine (SAM), and an inhibitor, 3,5-dinitrocatechol (DNC), bound (Fig. 27). The structure of s-COMT is one of two sets of α-helices sandwiching a seven-stranded β-sheet core (Fig. 28). A shallow cleft on the protein surface is occupied by SAM, DNC and a magnesium ion. The SAM binding pocket has a base formed by the C-terminal ends of β-sheets a–d with the walls of the pocket formed by helix 6 and the loops between helix 2 and helix 3, β-sheet a and helix 4, and β-sheet d and helix 7. The SAM methyl group is directed towards the substrate binding site and the catechol oxygen to be methylated.
Fig. 27.

Ligands in the crystal structures of COMT. (A) The structure of S-adenosyl methionine (SAM). (B) 3,5-dinitrocatechol (DNC).
Fig. 28.

The structure of s-COMT. A cartoon of the s-COMT structure coloured blue at the N-terminus through to red at the C-terminus. The secondary structure elements are labelled as in Fig. 27: H – helix and S – sheet. SAM and DNC are shown with purple carbons. The yellow sphere is a magnesium ion. Based on the 3BWM structure [136]. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The methylation reaction is dependent on an initial strict sequential order of reactant binding. SAM binding is followed by the binding of a magnesium ion which is followed by the catechol entering the binding site [130,136]. This order is necessary because the binding of one changes the shape of the binding site so that the next can bind. If reactant binding does not occur in this order a dead-end complex is formed. The magnesium ion is held in place by coordination with three acidic residues, D141, D169 and E199 the latter two of which are oriented by interaction with K46 (Fig. 29). The oxygen acting as the methyl recipient is activated by K144 which acts as a general base in the methylation. In a direct bimolecular transfer the SAM methyl group is transferred from the sulphur to the catechol hydroxyl oxygen through an SN2-like transition state [137].
Fig. 29.

The COMT substrate binding site. SAM is shown with purple carbons, DNC with cyan carbons. Five amino acid side chains are shown with green carbons. The interaction of K144 with a potential substrate is shown by the orange dashed line. Interactions between three acid groups (D141, D169 and E199) and the metal ion are shown by the black dashed lines. The interactions of D169 and E199 with K46 are shown by the pink dashed lines. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
COMT inhibitors have been developed primarily for the treatment of Parkinson's disease [138,139].
9. Discussion
Sulfotransferase, sulfatase, 17β-HSD1 and aromatase all have to recognise estrone (as either a substrate or a product of the catalysed reaction) and at least one other steroid. Despite this common recognition requirement there is no structural similarity between the four proteins in terms of the secondary and tertiary structure and little similarity in the structure of the oestrogen binding sites (Fig. 30). The possible exception to this assertion is, perhaps unsurprisingly given that they catalyse opposite reactions, sulfatase and sulfotransferase. These two enzymes both have a flat surface against which the substrate A and B rings can nestle. This flat surface is formed by L103 in sulfatase and V145 in sulfotransferase (Fig. 31). Both enzymes also have a histidine and two lysines adjacent to the cofactor sulfate group – H136, K134 and K368 in sulfatase, and H107, K47 and K105 in sulfotransferase – possibly suggesting some degree of similarity in the mechanism of the sulfate transfer reactions.
Fig. 30.

Structure comparison of four oestrogen binding proteins. Shown with estrone (cyan) in the substrate binding site and in the same orientation: (A) aromatase (haem in purple); (B) 17β-HSD1 (NADP in purple); (C) sulfotransferase (PAPS in purple); (D) sulfatase (HFGS75 in purple). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 31.

Overlay of the sulfotransferase and sulfatase oestrogen binding sites. Comparison of sulfotransferase (protein: pink carbons, PAPS: purple carbons) and sulfatase (protein: light green carbons, HFGS75: dark green carbons). Estrone is shown with cyan carbons. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Since estrogens have an important role to play in hormone-dependent cancers the inhibition of the enzymes responsible for producing estrogens is equally important. Historically, most effort has been focused on the development of aromatase and sulfatase inhibitors [140], but more recently considerable effort has been put into the development of 17β-HSD1 inhibitors [87–89]. Aromatase is the only enzyme capable of aromatising the ‘A’ ring thus converting androgens to estrogens. The inhibition of aromatase will, therefore, prevent oestrogen production. However, stores of estrone sulfate (accumulated from the circulation throughout the body at higher levels in breast cancer tissue than normal tissue [141,142]) can be converted to estrone by sulfatase and then to estradiol by 17β-HSD1. With levels of sulfatase and 17β-HSD1 being higher in cancerous tissue than non-cancerous tissue this has been reported to be the main route of oestrogen production in cancerous breast tissue [143–145; see, also, 146] which might suggest that the inhibition of aromatase alone would be unlikely to be a successful treatment in more than a minority of cases. However, aromatase inhibitors such as letrozole, anastrozole and exemestane have proven to be highly effective treatments for many breast cancers [106].
The different binding site structures of the various oestrogen metabolising enzymes mean that specific inhibitors can be designed. A more recent approach, however, is to design molecules that inhibit multiple drug targets, and the first reports of single molecule dual aromatase-sulfatase inhibitors have been published [147–149]. Inhibitors of other combinations of enzymes are feasible and the development of these will be facilitated by structure-based design. Inhibition of aromatase already reduces circulating levels of estradiol to below detectable levels. The concurrent inhibition of sulfatase and also, perhaps, 17β-HSD1 should cut the localised in situ production of estradiol in tumour tissue. By inhibiting aromatase, sulfatase and 17β-HSD1 all oestrogen production should be stopped. This may, however, have adverse consequences as estrogens are known to have protective effects on the heart [150]: the incidence of cardiovascular disease in postmenopausal women is three times that of premenopausal women of the same age [151]. This is due, at least in part, to the increased level of aldosterone [152] which is synthesised by CYP11B2 (aldosterone synthase). The simultaneous inhibition of CYP19 (aromatase) and CYP11B2 should reduce the levels of both oestrogen and aldosterone thus having the desired anti-cancer and anti-cardiovascular disease effects. The first dual inhibitors of CYP19 and CYP11B2 have recently been reported [8].
Acknowledgements
This research was supported by the Wellcome Trust through Programme grant no (082837 to BVLP) and by Wellcome VIP funding.
Contributor Information
Mark P. Thomas, Email: m.thomas@bath.ac.uk.
Barry V.L. Potter, Email: b.v.l.potter@bath.ac.uk.
References
- 1.Carreau S., Bouraima-Lelong H., Delalande C. Estrogen, a female hormone involved in spermatogenesis. Advances in Medical Sciences. 2012;57:31–36. doi: 10.2478/v10039-012-0005-y. [DOI] [PubMed] [Google Scholar]
- 2.Pepe G.J., Albrecht E.D. Actions of placental and fetal adrenal steroid hormones in primate pregnancy. Endocrine Reviews. 1995;16:608–648. doi: 10.1210/edrv-16-5-608. [DOI] [PubMed] [Google Scholar]
- 3.Albrecht E.D., Pepe G.J. Estrogen regulation of placental angiogenesis and fetal ovarian development during primate pregnancy. International Journal of Developmental Biology. 2010;54:397–407. doi: 10.1387/ijdb.082758ea. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kamel R.M. The onset of human parturition. Archives of Gynecology and Obstetrics. 2010;281:975–982. doi: 10.1007/s00404-010-1365-9. [DOI] [PubMed] [Google Scholar]
- 5.Silberstein S.D., Merriam G.R. Physiology of the menstrual cycle. Cephalalgia. 2000;20:148–154. doi: 10.1046/j.1468-2982.2000.00034.x. [DOI] [PubMed] [Google Scholar]
- 6.Chang M. Dual roles of estrogen metabolism in mammary carcinogenesis. BMB Reports. 2011;44:423–434. doi: 10.5483/BMBRep.2011.44.7.423. [DOI] [PubMed] [Google Scholar]
- 7.Linn S., Murtaugh B., Casey E. Role of sex hormones in the development of osteoarthritis. PM&R. 2012;4:S169–S173. doi: 10.1016/j.pmrj.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 8.Hu Q., Yin L., Hartmann R.W. Selective dual inhibitors of CYP19 and CYP11B2: targeting cardiovascular diseases hiding in the shadow of breast cancer. Journal of Medicinal Chemistry. 2012;55:7080–7089. doi: 10.1021/jm3004637. [DOI] [PubMed] [Google Scholar]
- 9.van der Spuy W.J., Pretorius E. Interrelation between inflammation, thrombosis and neuroprotection in cerebral ischemia. Reviews in the Neurosciences. 2012;23:269–278. doi: 10.1515/revneuro-2012-0028. [DOI] [PubMed] [Google Scholar]
- 10.Kipp M., Amor S., Krauth R., Beyer C. Multiple sclerosis: neuroprotective alliance of estrogen-progesterone and gender. Frontiers in Neuroendocrinology. 2012;33:1–16. doi: 10.1016/j.yfrne.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 11.Hirschberg A.L. Sex hormones, appetite and eating behaviour in women. Maturitas. 2012;71:248–256. doi: 10.1016/j.maturitas.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 12.Oosthuyse T., Bosche A.N. Oestrogen's regulation of fat metabolism during exercise and gender specific effects. Current Opinion in Pharmacology. 2012;12:363–371. doi: 10.1016/j.coph.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 13.Kulkarni J., Gavrilidis E., Worsley R., Hayes E. Role of estrogen treatment in the management of schizophrenia. CNS Drugs. 2012;26:549–557. doi: 10.2165/11630660-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 14.Lateef A., Petri M. Hormone replacement and contraceptive therapy in autoimmune diseases. Journal of Autoimmunity. 2012;38:J170–J176. doi: 10.1016/j.jaut.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 15.Pinaud R., Tremere L.A. Control of central auditory processing by a brain-generated oestrogen. Nature Reviews Neuroscience. 2012;13:521–527. doi: 10.1038/nrn3291. [DOI] [PubMed] [Google Scholar]
- 16.Marino M., Galluzzo P., Ascenzi P. Estrogen signalling pathways to impact gene transcription. Current Genomics. 2006;7:497–508. doi: 10.2174/138920206779315737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Safe S., Kim K. Non-classical genomic estrogen receptor (ER)/specificity protein and ER/activating protein-1 signalling pathways. Journal of Molecular Endocrinology. 2008;41:263–275. doi: 10.1677/JME-08-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maggiolini M., Picard D. The unfolding stories of GPR30, a new membrane-bound estrogen receptor. Journal of Endocrinology. 2010;204:105–114. doi: 10.1677/JOE-09-0242. [DOI] [PubMed] [Google Scholar]
- 19.Miller W.L., Auchus R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocrine Research. 2011;32:81–151. doi: 10.1210/er.2010-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Z., Kanjo Y., Mizutani S. Urinary excretion rates of natural estrogens and androgens from humans, and their occurrence and fate in the environment: a review. Science of the Total Environment. 2009;47:4975–4985. doi: 10.1016/j.scitotenv.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 21.Schwers J., Eriksson G., Diczfalusy E. 15α-Hydroxylation: a new pathway of estrogen metabolism in the human fetus and newborn. Biochimica et Biophysica Acta. 1965;100:313–316. doi: 10.1016/0304-4165(65)90464-2. [DOI] [PubMed] [Google Scholar]
- 22.Schwers J., Govaerts-Videtsky M., Wiqvist N., Diczfalusy E. Metabolism of oestrone sulphate by the previable human foetus. Acta Endocrinologica. 1965;50:597–610. doi: 10.1530/acta.0.0500597. [DOI] [PubMed] [Google Scholar]
- 23.Mancuso S., Benagiano G., Dell’Acqua S., Shapiro M., Wiqvist N., Diczfalusy E. Studies on the metabolism of C-19 steroids in the human foeto-placental unit. 4. Aromatisation and hydroxylation products formed by previable foetuses perfused with androstenedione and testosterone. Acta Endocrinologica. 1968;57:208–227. doi: 10.1530/acta.0.0570208. [DOI] [PubMed] [Google Scholar]
- 24.YoungLai E.V., Solomon S. Formation of estra-1,3,5(10)-triene-3,15α,16α,17β-tetrol (estetrol) and estra-1,3,5(10)-triene-3,15α,17β-triol from neutral precursors. Journal of Clinical Endocrinology and Metabolism. 1968;28:1611–1617. doi: 10.1210/jcem-28-11-1611. [DOI] [PubMed] [Google Scholar]
- 25.YoungLai E.V., Bowman J.M., Solomon S. Formation of ring-D-hydroxylated estrogens from C19 precursors administered to the fetus in utero. Canadian Journal of Biochemistry. 1969;47:25–29. doi: 10.1139/o69-005. [DOI] [PubMed] [Google Scholar]
- 26.Schut H.A.J., Bowman J.M., Solomon S. Precursor role of 15α-hydroxyestradiol and 15α-hydroxyandrostenedione in the formation of estetrol. Canadian Journal of Biochemistry. 1978;56:101–106. doi: 10.1139/o78-016. [DOI] [PubMed] [Google Scholar]
- 27.Cantineau R., Kremers P., De Graeve J., Gielen J.E., Lambotte R. 15-And 16-hydroxylations of androgens and estrogens in the human fetal liver: a critical step in estetrol biosynthesis. Journal of Steroid Biochemistry. 1985;22:195–201. doi: 10.1016/0022-4731(85)90112-8. [DOI] [PubMed] [Google Scholar]
- 28.Coelingh Bennink H.J.T, Holinka C.F., Diczfalusy E. Estetrol review: profile and potential clinical applications. Climacteric. 2008;11(Suppl. 1):47–58. doi: 10.1080/13697130802073425. [DOI] [PubMed] [Google Scholar]
- 29.Holinka C.F., Diczfalusy E., Coelingh Bennink H.J.T. Estetrol: a unique steroid in human pregnancy. Journal of Steroid Biochemistry and Molecular Biology. 2008;110:138–143. doi: 10.1016/j.jsbmb.2008.03.027. [DOI] [PubMed] [Google Scholar]
- 30.Glück S., Gorouhi F. Clinical and economic benefits of aromatase inhibitor therapy in early-stage breast cancer. American Journal of Health-System Pharmacy. 2011;68:1699–1706. doi: 10.2146/ajhp100492. [DOI] [PubMed] [Google Scholar]
- 31.Palmieri C., Januszewski A., Stanway S., Coombes R.C. Irosustat: a first-generation steroid sulfatase inhibitor in breast cancer. Expert Review of Anticancer Therapy. 2011;11:179–183. doi: 10.1586/era.10.201. [DOI] [PubMed] [Google Scholar]
- 32.Hickey M., Elliott J., Davison S.L. Hormone replacement therapy. BMJ. 2012;344:e763. doi: 10.1136/bmj.e763. [DOI] [PubMed] [Google Scholar]
- 33.Shulman L.P. The state of hormonal contraception today: benefits and risks of hormonal contraceptives: combined estrogen and progestin contraceptives. American Journal of Obstetrics and Gynecology. 2011;205(4 Suppl.):S9–S13. doi: 10.1016/j.ajog.2011.06.057. [DOI] [PubMed] [Google Scholar]
- 34.Fruzzetti F., Trémollieres F., Bitzer J. An overview of the development of combined oral contraceptives containing estradiol: focus on estradiol valerate/dienogest. Gynecological Endocrinology. 2012;28:400–408. doi: 10.3109/09513590.2012.662547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song W.-C. Biochemistry and reproductive endocrinology of estrogen sulfotransferase. Annals of the New York Academy of Sciences. 2001;948:43–50. doi: 10.1111/j.1749-6632.2001.tb03985.x. [DOI] [PubMed] [Google Scholar]
- 36.Falany C.N. Enzymology of human cytosolic sulfotransferases. FASEB Journal. 1997;11:206–216. doi: 10.1096/fasebj.11.4.9068609. [DOI] [PubMed] [Google Scholar]
- 37.Falany C.N., Krasnykh V., Falany J.L. Bacterial expression and characterization of a cDNA for human liver estrogen sulfotransferase. Journal of Steroid Biochemistry and Molecular Biology. 1995;52:529–539. doi: 10.1016/0960-0760(95)00015-r. [DOI] [PubMed] [Google Scholar]
- 38.Pedersen L.C., Petrotchenko E., Shevtsov S., Negishi M. Crystal structure of the human estrogen sulfotransferase-PAPS complex. Evidence for catalytic role of Ser137 in the sulfyryl transfer reaction. Journal of Biological Chemistry. 2002;277:17928–17932. doi: 10.1074/jbc.M111651200. [DOI] [PubMed] [Google Scholar]
- 39.Shevtsov S., Petrotchenko E.V., Pedersen L.C., Negishi M. Crystallographic analysis of a hydroxylated polychlorinated biphenyl (OH-PCB) bound to the catalytic estrogen binding site of human estrogen sulfotransferase. Environmental Health Perspectives. 2003;111:884–888. doi: 10.1289/ehp.6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kakuta Y., Pedersen L.G., Carter C.W., Negishi M., Pedersen L.C. Crystal structure of estrogen sulphotransferase. Natural Structural Biology. 1997;4:904–908. doi: 10.1038/nsb1197-904. [DOI] [PubMed] [Google Scholar]
- 41.Kakuta Y., Petrotchenko E.V., Pedersen L.C., Negishi M. The sulfuryl transfer mechanism. Crystal structure of a vanadate complex of estrogen sulfotransferase and mutational analysis. Journal of Biological Chemistry. 1998;273:27325–27330. doi: 10.1074/jbc.273.42.27325. [DOI] [PubMed] [Google Scholar]
- 42.Bidwell L.M., McManus M.E., Gaedigk A., Kakuta Y., Negishi M., Pedersen L., Martin J.L. Crystal structure of human catecholamine sulfotransferase. Journal of Molecular Biology. 1999;293:521–530. doi: 10.1006/jmbi.1999.3153. [DOI] [PubMed] [Google Scholar]
- 43.Lu J.-H, Li H. -T., Liu M. -C., Zhang J. -P., Li M., An X. -M., Chang W. -R. Crystal structure of human sulfotransferase SULT1A3 in complex with dopamine and 3’-phospho-adenosine-5′-phosphate. Biochemical and Biophysical Research Communications. 2005;335:417–423. doi: 10.1016/j.bbrc.2005.07.091. [DOI] [PubMed] [Google Scholar]
- 44.Gamage N.U., Tsvetanov S., Duggleby R.G., McManus M.E., Martin J.L. The structure of human SULT1A1 crystallized with estradiol. An insight into active site plasticity and substrate inhibition with multi-ring substrates. Journal of Biological Chemistry. 2005;280:41482–41486. doi: 10.1074/jbc.M508289200. [DOI] [PubMed] [Google Scholar]
- 45.Lu J., Li H., Zhang J., Li M., Liu M.-Y., An X., Liu M.-C., Chang W. Crystal structures of SULT1A2 and SULT1A1*3: insights into the substrate inhibition and the role of Tyr149 in SULT1A2. Biochemical and Biophysical Research Communications. 2010;396:429–434. doi: 10.1016/j.bbrc.2010.04.109. [DOI] [PubMed] [Google Scholar]
- 46.Gamage N.U., Duggleby R.G., Barnett A.C., Tresillian M., Latham C.F., Liyou N.E., McManus M.E., Martin J.L. Structure of a human carcinogen-converting enzyme, SULT1A1. Structural and kinetic implications of substrate inhibition. Journal of Biological Chemistry. 2003;278:7655–7662. doi: 10.1074/jbc.M207246200. [DOI] [PubMed] [Google Scholar]
- 47.Berger I., Guttman C., Amar D., Zarivach R., Aharoni A. The molecular basis for the broad substrate specificity of human sulfotransferase 1A1. PLoS ONE. 2011;6:e26794. doi: 10.1371/journal.pone.0026794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dong D., Ako R., Wu B. Crystal structures of human sulfotransferases: insights into the mechanisms of action and substrate selectivity. Expert Opinion On Drug Metabolism & Toxicology. 2012;8:635–646. doi: 10.1517/17425255.2012.677027. [DOI] [PubMed] [Google Scholar]
- 49.Rath V.L., Verdugo D., Hemmerich S. Sulfotransferase structural biology and inhibitor discovery. Drug Discovery Today. 2004;9:1003–1011. doi: 10.1016/S1359-6446(04)03273-8. [DOI] [PubMed] [Google Scholar]
- 50.Allali-Hassani A., Pan P.W., Dombrovski L., Najmanovich R., Tempel W., Dong A., Loppnau P., Martin F., Thornton J., Edwards A.M., Bochkarev A., Plotnikov A.N., Vedadi M., Arrowsmith C.H. Structural and chemical profiling of the human cytosolic sulfotransferases. PLoS Biology. 2007;5:e97. doi: 10.1371/journal.pbio.0050097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cook I., Wang T., Falany C.N., Leyh T.S. A nucleotide-gated molecular pore selects sulfotransferase substrates. Biochemistry. 2012;51:5674–5683. doi: 10.1021/bi300631g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Negishi M., Pedersen L.G., Petrotchenko E., Shevtsov S., Gorokhov A., Kakuta Y., Pedersen L.C. Structure and function of sulfotransferases. Archives of Biochemistry and Biophysics. 2001;390:149–157. doi: 10.1006/abbi.2001.2368. [DOI] [PubMed] [Google Scholar]
- 53.Gamage N.U., Barnett A., Hempel N., Duggleby R.G., Windmill K.F., Martin J.L., McManus M.E. Human sulfotransferases and their role in chemical metabolism. Toxicological Sciences. 2006;90:5–22. doi: 10.1093/toxsci/kfj061. [DOI] [PubMed] [Google Scholar]
- 54.Chapman E., Best M.D., Hanson S.R., Wong C.-H. Sulfotransferases: structure, mechanism, biological activity, inhibition, and synthetic utility. Angewandte Chemie. International Ed. In English. 2004;43:3526–3548. doi: 10.1002/anie.200300631. [DOI] [PubMed] [Google Scholar]
- 55.Reed M.J., Purohit A., Woo L.W.L., Newman S.P., Potter B.V.L. Steroid sulfatase: molecular biology, regulation, and inhibition. Endocrine Reviews. 2005;26:171–202. doi: 10.1210/er.2004-0003. [DOI] [PubMed] [Google Scholar]
- 56.Purohit A., Woo L.W.L., Potter B.V.L. Steroid sulfatase: a pivotal player in estrogen synthesis and metabolism. Molecular and Cellular Endocrinology. 2011;340:154–160. doi: 10.1016/j.mce.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 57.Stein C., Hille A., Seidel J., Rijnbout S., Waheed A., Schmidt B., Geuze H., von Figura K. Cloning and expression of human steroid-sulfatase. Membrane topology, glycosylation, and subcellular distribution in BHK-21 cells. Journal of Biological Chemistry. 1989;264:13865–13872. [PubMed] [Google Scholar]
- 58.Hernandez-Guzman F.G., Higashiyama T., Osawa Y., Ghosh D. Purification, characterization and crystallization of human placental estrone/dehydroepiandrosterone sulfatase, a membrane-bound enzyme of the endoplasmic reticulum. Journal of Steroid Biochemistry and Molecular Biology. 2001;78:441–450. doi: 10.1016/s0960-0760(01)00119-4. [DOI] [PubMed] [Google Scholar]
- 59.Hernandez-Guzman F.G., Higashiyama T., Pangborn W., Osawa Y., Ghosh D. Structure of human estrone sulfatase suggests functional roles of membrane association. Journal of Biological Chemistry. 2003;278:22989–22997. doi: 10.1074/jbc.M211497200. [DOI] [PubMed] [Google Scholar]
- 60.Schmidt B., Selmer T., Ingendoh A., von Figura K. A novel amino acid modification in sulfatases that is defective in multiple sulfatase deficiency. Cell. 1995;82:271–278. doi: 10.1016/0092-8674(95)90314-3. [DOI] [PubMed] [Google Scholar]
- 61.Ghosh D. Human sulfatases: a structural perspective to catalysis. Cellular and Molecular Life Sciences. 2007;64:2013–2022. doi: 10.1007/s00018-007-7175-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Geisler J., Sasano H., Chen S., Purohit A. Steroid sulfatase inhibitors: promising new tools for breast cancer therapy? Journal of Steroid Biochemistry and Molecular Biology. 2011;125:39–45. doi: 10.1016/j.jsbmb.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 63.Maltais R., Poirier D. Steroid sulfatase inhibitors: a review covering the promising 2000–2010 decade. Steroids. 2011;76:929–948. doi: 10.1016/j.steroids.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 64.Woo L.W.L, Purohit A., Potter B.V.L. Development of steroid sulfatase inhibitors. Molecular and Cellular Endocrinology. 2011;340:175–185. doi: 10.1016/j.mce.2010.12.035. [DOI] [PubMed] [Google Scholar]
- 65.Purohit A., Foster P.A. Steroid sulfatase inhibitors for estrogen- and androgen-dependent cancers. Journal of Endocrinology. 2012;212:99–110. doi: 10.1530/JOE-11-0266. [DOI] [PubMed] [Google Scholar]
- 66.S.J. Williams, Sulfatase inhibitors: a patent review, Expert Opinion on Therapeutic Patents, in press, http://dx.doi.org/10.1517/13543776.2013.736965. [DOI] [PubMed]
- 67.Moeller G., Adamski J. Integrated view on 17beta-hydroxysteroid dehydrogenases. Molecular and Cellular Endocrinology. 2009;301:7–19. doi: 10.1016/j.mce.2008.10.040. [DOI] [PubMed] [Google Scholar]
- 68.Moeller G., Adamski J. Multifunctionality of human 17β-hydroxysteroid dehydrogenases. Molecular and Cellular Endocrinology. 2006;248:47–55. doi: 10.1016/j.mce.2005.11.031. [DOI] [PubMed] [Google Scholar]
- 69.C. Mazza, Human type 1 17β-hydroxysteroid dehydrogenase: site directed mutagenesis and X-ray crystallography structure-function analysis, Thesis, Universite Joseph Fourier, 1997.
- 70.Ghosh D., Pletnev V.Z., Zhu D.-W., Wawrzak Z., Daux W.L., Pangborn W., Labrie F., Lin S.-X. Structure of human estrogenic 17β-hydroxysteroid dehydrogenase at 2.20 Å resolution. Structure. 1995;3:503–513. doi: 10.1016/s0969-2126(01)00183-6. [DOI] [PubMed] [Google Scholar]
- 71.Han Q., Campbell R.L., Gangloff A., Huang Y.-W., Lin S.-X. Dehydroepiandrosterone and dihydrotestosterone recognition by human estrogenic 17β-hydroxysteroid dehydrogenase. C-18/C-19 steroid discrimination and enzyme-induced strain. Journal of Biological Chemistry. 2000;275:1105–1111. doi: 10.1074/jbc.275.2.1105. [DOI] [PubMed] [Google Scholar]
- 72.Sawicki M., Erman M., Puranen T., Vihko P., Ghosh D. Structure of the ternary complex of human 17β-hydroxysteroid dehydrogenase type 1 with 3-hydroxyestra-1,3,5,7-tetraen-17-one (equilin) and NADP+ Proceedings of the National Academy of Sciences of the United States of America. 1999;96:840–845. doi: 10.1073/pnas.96.3.840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Breton R., Housset D., Mazza C., Fontecilla-Camps J.C. The structure of a complex of human 17β-hydroxysteroid dehydrogenase with estradiol and NADP+ identifies two principal targets for the design of inhibitors. Structure. 1996;4:905–915. doi: 10.1016/s0969-2126(96)00098-6. [DOI] [PubMed] [Google Scholar]
- 74.Mazza C., Breton R., Housset D., Fontecilla-Camps J.C. Unusual charge stabilization of NADP+ in 17β-hydroxysteroid dehydrogenase. Journal of Biological Chemistry. 1998;273:8145–8152. doi: 10.1074/jbc.273.14.8145. [DOI] [PubMed] [Google Scholar]
- 75.Qiu W., Campbell R.L., Gangloff A., Dupuis P., Boivin R.P., Tremblay M.R., Poirier D., Lin S.-X. A concerted, rational design of type 1 17β-hydroxysteroid dehydrogenase inhibitors: estradiol-adenosine hybrids with high affinity. FASEB Journal. 2002;16:1829–1831. doi: 10.1096/fj.02-0026fje. [DOI] [PubMed] [Google Scholar]
- 76.Azzi A., Rehse P.H., Zhu D.-W., Campbell R.L., Labrie F., Liu S.-X. Crystal structure of human estrogenic 17β-hydroxysteroid dehydrogenase complexed with 17β-estradiol. Natural Structural Biology. 1996;3:665–668. doi: 10.1038/nsb0896-665. [DOI] [PubMed] [Google Scholar]
- 77.Gangloff A., Shi R., Nahoum V., Lin S.-X. Pseudo-symmetry of C19-steroids, alternative binding orientations and multispecificity in human estrogenic 17β-hydroxysteroid dehydrogenase. FASEB Journal. 2003;17:274–276. doi: 10.1096/fj.02-0397fje. [DOI] [PubMed] [Google Scholar]
- 78.Shi R., Lin S.-X. Cofactor hydrogen bonding onto the protein main chain is conserved in the short chain dehydrogenase/reductase family and contributes to nicotinamide orientation. Journal of Biological Chemistry. 2004;279:16778–16785. doi: 10.1074/jbc.M313156200. [DOI] [PubMed] [Google Scholar]
- 79.Mazumdar M., Fournier D., Zhu D.-W., Cadot C., Poirier D., Lin S.-X. Binary and ternary crystal structure analyses of a novel inhibitor with 17β-HSD type 1: a lead compound for breast cancer therapy. Biochemical Journal. 2009;424:357–366. doi: 10.1042/BJ20091020. [DOI] [PubMed] [Google Scholar]
- 80.Aka J.A., Mazumdar M., Chen C.-Q., Poirier D., Lin S.-X. 17β-hydroxysteroid dehydrogenase type 1 stimulates breast cancer by dihydrotestosterone inactivation in addition to estradiol production. Molecular Endocrinology. 2010;24:832–845. doi: 10.1210/me.2009-0468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Puranen T., Poutanen M., Ghosh D., Vihko P., Vihko R. Characterization of structural and functional properties of human 17β-hydroxysteroid dehydrogenase type 1 using recombinant enzymes and site-directed mutagenesis. Molecular Endocrinology. 1997;11:77–86. doi: 10.1210/mend.11.1.9872. [DOI] [PubMed] [Google Scholar]
- 82.Sherbet D.P., Guryev O.L., Papari-Zareei M., Mizrachi D., Rambally S., Akbar S., Auchus R.J. Biochemical factors governing the steady-state estrone/estradiol ratios catalysed by human 17β-hydroxysteroid dehydrogenases types 1 and 2 in HEK-293 cells. Endocrinology. 2009;150:4154–4162. doi: 10.1210/en.2008-1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Betz G. Reaction mechanism of 17β-estradiol dehydrogenase determined by equilibrium rate exchange. Journal of Biological Chemistry. 1971;246:2063–2068. [PubMed] [Google Scholar]
- 84.Negri M., Recanatini M., Hartmann R.W. Insights in 17β-HSD1 enzyme kinetics and ligand binding by dynamic motion investigation. PLoS ONE. 2010;5:e12026. doi: 10.1371/journal.pone.0012026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.A. Cassetta, I. Krastanova, K. Kristan, M. Brunskole Švegelj, D. Lamba, T. Lanišnik Rižner, J. Stojan, Insights into subtle conformational differences in the substrate-binding loop of fungal 17β-hydroxysteroid dehydrogenase: a combined structural and kinetic approach. http://dx.doi.org/10.1042/BJ20110567. [DOI] [PubMed]
- 86.Kristan K., Deluca D., Adamski J., Stojan J., Rižner T.L. Dimerization and enzymatic activity of fungal 17β-hydroxysteroid dehydrogenase from the short-chain dehydrogenase/reductase superfamily. BMC Biochemistry. 2005;6:28. doi: 10.1186/1471-2091-6-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Day J.M., Tuthill H.J., Purohit A. 17β-Hydroxysteroid dehydrogenase inhibitors. Minerva Endocrinologica. 2010;35:87–108. [PubMed] [Google Scholar]
- 88.Poirier D. 17β-Hydroxysteroid dehydrogenase inhibitors: a patent review. Expert Opinion on Therapeutic Patents. 2010;20:1123–1145. doi: 10.1517/13543776.2010.505604. [DOI] [PubMed] [Google Scholar]
- 89.Marchais-Oberwinkler S., Henn C., Möller G., Klein T., Negri M., Oster A., Spadaro A., Werth R., Wetzel M., Xu K., Frotscher M., Hartmann R.W., Adamski J. 17β-Hydroxysteroid dehydrogenases (17β-HSDs) as therapeutic targets: protein structures, functions, and recent progress in inhibitor development. Journal of Steroid Biochemistry and Molecular Biology. 2011;125:66–82. doi: 10.1016/j.jsbmb.2010.12.013. [DOI] [PubMed] [Google Scholar]
- 90.Shimozawa O., Sakaguchi M., Ogawa H., Harada N., Mihara K., Omura T. Core glycosylation of cytochrome P-450(arom). Evidence for localization of N terminus of microsomal cytochrome P-450 in the lumen. Journal of Biological Chemistry. 1993;268:21399–21402. [PubMed] [Google Scholar]
- 91.Catalano S., Barone I., Giordano C., Rizza P., Qi H., Gu G., Malivindi R., Bonofiglio D., Andò S. Rapid estradiol/ERα signaling enhances aromatase activity in breast cancer cells. Molecular Endocrinology. 2009;23:1634–1645. doi: 10.1210/me.2009-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Barone I., Giordano C., Malivindi R., Lanzino M., Rizza P., Casaburi I., Bonofiglio D., Catalano S., Andò S. Estrogens and PTP1B function in a novel pathway to regulate aromatase enzymatic activity in breast cancer cells. Endocrinology. 2012;153:5157–5166. doi: 10.1210/en.2012-1561. [DOI] [PubMed] [Google Scholar]
- 93.Praporski S., Ng S.M., Nguyen A.D., Corbin C.J., Mechler A., Zheng J., Conley A.J., Martin L.L. Organization of cytochrome P450 enzymes involved in sex steroid synthesis. Protein–protein interactions in lipid membranes. Journal of Biological Chemistry. 2009;284:33224–33232. doi: 10.1074/jbc.M109.006064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ghosh D., Jiang W., Lo J., Egbuta C. Higher order organization of human placental aromatase. Steroids. 2011;76:753–758. doi: 10.1016/j.steroids.2011.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ghosh D., Griswold J., Erman M., Pangborn W. Structural basis for androgen specificity and oestrogen synthesis in human aromatase. Nature. 2009;457:219–224. doi: 10.1038/nature07614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ghosh D., Griswold J., Erman M., Pangborn W. X-ray structure of human aromatase reveals an androgen-specific active site. Journal of Steroid Biochemistry and Molecular Biology. 2010;118:197–202. doi: 10.1016/j.jsbmb.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ghosh D., Lo J., Morton D., Valette D., Xi J., Griswold J., Hubbell S., Egbuta C., Jiang W., An J., Davis H.M.L. Novel aromatase inhibitors by structure-guided design. Journal of Medicinal Chemistry. 2012;55:8464–8476. doi: 10.1021/jm300930n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sohl C.D., Guengerich F.P. Kinetic analysis of the three-step steroid aromatase reaction of human cytochrome P450 19A1. Journal of Biological Chemistry. 2010;285:17734–17743. doi: 10.1074/jbc.M110.123711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Akhtar M., Wright J.N., Lee-Robichaud P. A review of mechanistic studies on aromatase (CYP19) and 17α-hydroxylase-17,20-lyase (CYP17) Journal of Steroid Biochemistry and Molecular Biology. 2011;125:2–12. doi: 10.1016/j.jsbmb.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 100.Akhtar M., Skinner S.J.M. The intermediary role of a 19-oxoandrogen in the biosynthesis of oestrogen. Biochemical Journal. 1968;109:318–321. doi: 10.1042/bj1090318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Akhtar M., Calder M.R., Corina D.L., Wright J.N. Mechanistic studies on C-19 demethylation in oestrogen biosynthesis. Biochemical Journal. 1982;201:569–580. doi: 10.1042/bj2010569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang M., Roberts D.L., Paschke R., Shea T.M., Masters B.S.S., Kim J.-J.P. Three-dimensional structure of NADPH-cytochrome P450 reductase: prototype for FMN- and FAD-containing enzymes. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:8411–8416. doi: 10.1073/pnas.94.16.8411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hong Y., Rashid R., Chen S. Binding features of steroidal and non-steroidal inhibitors. Steroids. 2011;76:802–806. doi: 10.1016/j.steroids.2011.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jiang W., Ghosh D. Motion and flexibility in human cytochrome P450 aromatase. PLos ONE. 2012;7:e32565. doi: 10.1371/journal.pone.0032565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sgrignani J., Magistrato A. Influence of the membrane lipophilic environment on the structure and on the access/egress routes of the human aromatase enzyme. A computational study. Journal of Chemical Information and Modeling. 2012;52:1595–1606. doi: 10.1021/ci300151h. [DOI] [PubMed] [Google Scholar]
- 106.Obiorah I., Jordan V.C. Progress in endocrine approaches to the treatment and prevention of breast cancer. Maturitas. 2011;70:315–321. doi: 10.1016/j.maturitas.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.AlHilli M.M., Long H.L., Podratz K.C., Bakkum-Gamez J.N. Aromatase inhibitors in the treatment of recurrent ovarian granulosa cell tumors: brief report and review of the literature. Journal of Obstetrics and Gynaecology Research. 2012;38:340–344. doi: 10.1111/j.1447-0756.2011.01698.x. [DOI] [PubMed] [Google Scholar]
- 108.Mauras N. Strategies for maximising growth in puberty in children with short stature. Pediatric Clinics of North America. 2011;58:1167–1179. doi: 10.1016/j.pcl.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 109.Verma M.K., Miko Y., Sasano H. Aromatase in human lung carcinoma. Steroids. 2011;76:759–764. doi: 10.1016/j.steroids.2011.02.020. [DOI] [PubMed] [Google Scholar]
- 110.Martucci C.P., Fishman J. P450 enzymes of estrogen metabolism. Pharmacology & Therapeutics. 1993;57:237–257. doi: 10.1016/0163-7258(93)90057-k. [DOI] [PubMed] [Google Scholar]
- 111.Cribb A.E., Knight M.J., Dryer D., Guernsey J., Hender K., Tesch M., Saleh T.M. Role of polymorphic human cytochrome P450 enzymes in estrone oxidation. Cancer Epidemiology, Biomarkers and Prevention. 2006;15:551–558. doi: 10.1158/1055-9965.EPI-05-0801. [DOI] [PubMed] [Google Scholar]
- 112.Huang Z., Guengerich F.P., Kaminsky L.S. 16α-Hydroxylation of estrone by human cytochrome P4503A4/5. Carcinogenesis. 1998;19:867–872. doi: 10.1093/carcin/19.5.867. [DOI] [PubMed] [Google Scholar]
- 113.Sansen S., Yano J.K., Reynald R.L., Schoch G.A., Griffin K.J., Stout C.D., Johnson E.F. Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P450 1A2. Journal of Biological Chemistry. 2007;282:14348–14355. doi: 10.1074/jbc.M611692200. [DOI] [PubMed] [Google Scholar]
- 114.Wang A., Savas U., Stout C.D., Johnson E.F. Structural characterization of the complex between α-naphthoflavone and human cytochrome P450 2B1. Journal of Biological Chemistry. 2011;286:5736–5743. doi: 10.1074/jbc.M110.204420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yano J.K., Wester M.R., Schoch G.A., Griffin K.J., Stout C.D., Johnson E.F. The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-Å resolution. Journal of Biological Chemistry. 2004;279:38091–38094. doi: 10.1074/jbc.C400293200. [DOI] [PubMed] [Google Scholar]
- 116.Williams P.A., Cosme J., Vinković D.M., Ward A., Angove H.C., Day P.J., Vonrhein C., Tickle I.J., Johti H. Crystal structures of human cytochrome P450 3A4 bound to methyrapone and progesterone. Science. 2004;305:683–686. doi: 10.1126/science.1099736. [DOI] [PubMed] [Google Scholar]
- 117.Ekroos M., Sjögren T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:13682–13687. doi: 10.1073/pnas.0603236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sevrioukova I.F., Poulos T.L. Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:18422–18427. doi: 10.1073/pnas.1010693107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sevrioukova I.F., Poulos T.L. Interaction of human cytochrome P4503A4 with ritonavir analogs. Archives of Biochemistry and Biophysics. 2012;520:108–116. doi: 10.1016/j.abb.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sevrioukova I.F., Poulos T.L. Structural and mechanistic insights into the interaction of cytochrome P4503A4 with bromoergocryptine, a type 1 ligand. Journal of Biological Chemistry. 2012;287:3510–3517. doi: 10.1074/jbc.M111.317081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Männistö P.T., Kaakkola S. Catechol-O-methyltransferase (COMT): biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacological Reviews. 1999;51:593–628. [PubMed] [Google Scholar]
- 122.Zhu B.T., Conney A.H. Functional role of estrogen metabolism in target cells: review and perspective. Carcinogenesis. 1998;19:1–27. doi: 10.1093/carcin/19.1.1. [DOI] [PubMed] [Google Scholar]
- 123.Jancova P., Anzenbacher P., Anzenbacherova E. Phase II drug metabolizing enzymes. Biomedical Papers of the Medical Faculty of the University Palacky, Olomouc, Czech Republic. 2010;154:103–116. doi: 10.5507/bp.2010.017. [DOI] [PubMed] [Google Scholar]
- 124.Mueck A.O., Seeger H. 2-Methoxyestradiol – biology and mechanism of action. Steroids. 2010;75:625–631. doi: 10.1016/j.steroids.2010.02.016. [DOI] [PubMed] [Google Scholar]
- 125.Hosák L. Role of the COMT gene Val158Met polymorphism in mental disorders: a review. European Psychiatry. 2007;22:276–281. doi: 10.1016/j.eurpsy.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 126.Dawling S., Roodi N., Mernaugh R.L., Wang X., Parl F.F. Catechol-O-methyltransferase (COMT)-mediated metabolism of catechol estrogens: comparison of wild-type and variant COMT isoforms. Cancer Research. 2001;61:6716–6722. [PubMed] [Google Scholar]
- 127.Yim D.-S., Park S.K., Yoo K.-Y., Yoon K.-S., Chung H.H., Kang H.J., Ahn S.-H., Noh D.-Y., Choe K.-J., Jang I.-J., Shin S.-G., Strickland P.T., Hirvonen A., Kang D. Relationship between the Val158Met polymorphism of catechol O-methyltransferase and breast cancer. Pharmacogenetics. 2001;11:279–286. doi: 10.1097/00008571-200106000-00001. [DOI] [PubMed] [Google Scholar]
- 128.Zubieta J.-K., Heitzeg M.M., Smith Y.R., Bueller J.A., Xu K., Xu Y., Koeppe R.A., Stohler C.S., Goldman D. COMT val158met genotype affects μ-opioid neurotransmitter responses to a pain stressor. Science. 2003;299:1240–1243. doi: 10.1126/science.1078546. [DOI] [PubMed] [Google Scholar]
- 129.Goldberg T.E., Egan M.F., Gscheidle T., Coppola R., Weickert T., Kolachana B.S., Goldman D., Weinberger D.R. Executive subprocesses in working memory: relationship to catechol-O-methyltransferase Val158Met genotype and schizophrenia. Archives of General Psychiatry. 2003;60:889–896. doi: 10.1001/archpsyc.60.9.889. [DOI] [PubMed] [Google Scholar]
- 130.Lotta T., Vidgren J., Tilgmann C., Ulmanen I., Melén K., Julkunen I., Taskinen J. Kinetics of human soluble and membrane-bound catechol O-methyltransferase: a revised mechanism and description of the thermolabile variant of the enzyme. Biochemistry. 1995;34:4202–4210. doi: 10.1021/bi00013a008. [DOI] [PubMed] [Google Scholar]
- 131.Lachman H.M., Morrow B., Shprintzen R., Veit S., Parsia S.S., Faedda G., Goldberg R., Kucherlapati R., Papolos D.F. Association of codon 108/158 catechol-O-methyltransferase gene polymorphism with the psychiatric manifestations of velo-cardio-facial syndrome. American Journal of Medical Genetics. 1996;67:468–472. doi: 10.1002/(SICI)1096-8628(19960920)67:5<468::AID-AJMG5>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 132.Chen J., Lipska B.K., Halim N., Ma Q.D., Matsumoto M., Melhem S., Kolachana B.S., Hyde T.M., Herman M.M., Apud J., Egan M.F., Kleinman J.E., Weinberger D.R. Functional analysis of genetic variation in catechol-O-methyltransferase (COMT): effects on mRNA, protein, and enzyme activity in post-mortem human brain. American Journal of Human Genetics. 2004;75:807–821. doi: 10.1086/425589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rutherford K., Bennion B.J., Parson W.W., Daggett V. The 108M polymorph of human catechol O-methyltransferase is prone to deformation at physiological temperatures. Biochemistry. 2006;45:2178–2188. doi: 10.1021/bi051988i. [DOI] [PubMed] [Google Scholar]
- 134.Rutherford K., Alphandéry E., McMillan A., Daggett V., Parson W.W. The V108M mutation decreases the structural stability of catechol O-methyltransferase. Biochimica et Biophysica Acta. 2008;1784:1098–1105. doi: 10.1016/j.bbapap.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 135.Rutherford K., Le Trong I., Stenkamp R.E., Parson W.W. Crystal structures of human 108V and 108M catechol O-methyltransferase. Journal of Molecular Biology. 2008;380:120–130. doi: 10.1016/j.jmb.2008.04.040. [DOI] [PubMed] [Google Scholar]
- 136.Tsao D., Diatchenko L., Dokholyan N.V. Structural mechanism of S-adenosyl methionine binding to catechol O-methyltransferase. PLoS ONE. 2011;6:e24287. doi: 10.1371/journal.pone.0024287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Woodard R.W., Tsai M. -D., Floss H.G., Crooks P.A., Coward J.K. Stereochemical course of the transmethylation catalyzed by catechol O-methyltransferase. Journal of Biological Chemistry. 1980;255:9124–9127. [PubMed] [Google Scholar]
- 138.Learmouth D.A., Kiss L.E., Soares-da-Silva P. The chemistry of catechol-O-methyltransferase inhibitors. International Review of Neurobiology. 2010;95:119–162. doi: 10.1016/B978-0-12-381326-8.00006-5. [DOI] [PubMed] [Google Scholar]
- 139.Nissinen E., Männistö P.T. Biochemistry and pharmacology of catechol-O-methyltransferase inhibitors. International Review of Neurobiology. 2010;95:73–118. doi: 10.1016/B978-0-12-381326-8.00005-3. [DOI] [PubMed] [Google Scholar]
- 140.Hong Y., Chen S. Aromatase, estrone sulfatase, and 17β-hydroxysteroid dehydrogenase: structure-function studies and inhibitor development. Molecular and Cellular Endocrinology. 2011;340:120–126. doi: 10.1016/j.mce.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.van Landeghem A.A.J, Poortman J., Nabuurs M., Thijssen J.H.H. Endogenous concentration and subcellular distribution of androgens in normal and malignant human breast tissue. Cancer Research. 1985;45:2907–2912. [PubMed] [Google Scholar]
- 142.Pasqualini J.R., Chetrite G., Blacker C., Feinstein M.C., Delalonde L., Talbi M., Maloche C. Concentrations of estrone, estradiol, and estrone sulfate and evaluation of sulfatase and aromatase activities in pre- and postmenopausal breast cancer patients. Journal of Clinical Endocrinology and Metabolism. 1996;81:1460–1464. doi: 10.1210/jcem.81.4.8636351. [DOI] [PubMed] [Google Scholar]
- 143.Gunnarsson C., Olsson B.M., Stål O. Abnormal expression of 17β-hydroxysteroid dehydrogenases in breast cancer predicts late recurrence. Cancer Research. 2001;61:8448–8451. [PubMed] [Google Scholar]
- 144.Pasqualini J.R. The selective estrogen enzyme modulators in breast cancer: a review. Biochimica et Biophysica Acta. 2004;1654:123–143. doi: 10.1016/j.bbcan.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 145.Vihko P., Herrala A., Härkönen P., Isomaa V., Kaija H., Kurkela R., Pulkka A. Control of cell proliferation of steroids: the role of 17HSDs. Molecular and Cellular Endocrinology. 2006;248:141–148. doi: 10.1016/j.mce.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 146.Simpson E.R. Sources of estrogen and their importance. Journal of Steroid Biochemistry and Molecular Biology. 2003;86:225–230. doi: 10.1016/s0960-0760(03)00360-1. [DOI] [PubMed] [Google Scholar]
- 147.Woo L.W.L, Sutcliffe O.B., Bubert C., Grasso A., Chander S.K., Purohit A., Reed M.J., Potter B.V.L. First dual aromatase-steroid sulfatase inhibitors. Journal of Medicinal Chemistry. 2003;46:3193–3196. doi: 10.1021/jm034033b. [DOI] [PubMed] [Google Scholar]
- 148.Woo L.W.L., Bubert C., Sutcliffe O.B., Smith A., Chander S.K., Mahon M.F., Purohit A., Reed M.J., Potter B.V.L. Dual aromatase-steroid sulfatase inhibitors. Journal of Medicinal Chemistry. 2007;50:3540–3560. doi: 10.1021/jm061462b. [DOI] [PubMed] [Google Scholar]
- 149.Woo L.W.L., Jackson T., Putey A., Cozier G., Leonard P., Acharya K.R., Chander S.K., Purohit A., Reed M.J., Potter B.V.L. Highly potent first examples of dual aromatase-steroid sulfatase inhibitors based on a biphenyl template. Journal of Medicinal Chemistry. 2010;53:2155–2170. doi: 10.1021/jm901705h. [DOI] [PubMed] [Google Scholar]
- 150.Beer S., Reincke M., Kral M., Callies F., Strömer H., Dienesch C., Steinhauer S., Ertl G., Allolio B., Neubauer S. High-dose 17β-estradiol treatment prevents development of heart failure post-myocardial infarction in the rat. Basic Research in Cardiology. 2007;102:9–18. doi: 10.1007/s00395-006-0608-1. [DOI] [PubMed] [Google Scholar]
- 151.Witteman J.C.M., Grobbee D.E., Kok F.J., Hofman A., Valkenburg H.A. Increased risk of atherosclerosis in women after the menopause. BMJ. 1989;298:642–644. doi: 10.1136/bmj.298.6674.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Macova M., Armando I., Zhou J., Baiardi G., Tyurmin D., Larrayoz-Roldan I.M., Saavedra J.M. Estrogen reduces aldosterone, upregulates adrenal angiotensin II AT2 receptors and normalizes adrenomedullary Fra-2 in ovariectomized rats. Neuroendocrinology. 2008;88:276–286. doi: 10.1159/000150977. [DOI] [PMC free article] [PubMed] [Google Scholar]