

Abstract
We sought to develop a PET radioligand that would be useful for imaging human brain metabotropic subtype 1 receptors (mGluR1) in neuropsychiatric disorders and in drug development. 4-Fluoro-N-methyl-N-(4-(6-(methylamino)pyrimidin-4-yl)thiazol-2-yl)benzamide (FIMX, 11) was identified as having favorable properties for development as a PET radioligand. We developed a method for preparing [18F]11 in useful radiochemical yield and in high specific activity from [18F]fluoride ion and an N-Boc-protected (phenyl)aryliodonium salt precursor (15). In baseline experiments in rhesus monkey, [18F]11 gave high brain radioactivity uptake reflecting the expected distribution of mGluR1 with notably high uptake in cerebellum which became 47% lower by 120 min after radioligand injection. Pharmacological challenges demonstrated a very high proportion of the radioactivity in monkey brain to be bound specifically and reversibly to mGluR1. [18F]11 is concluded to be an effective PET radioligand for imaging mGluR1 in monkey brain and therefore merits further evaluation in human subjects.
INTRODUCTION
Glutamate is a major brain excitatory neurotransmitter that acts on a wide range of receptors that are categorized as either metabotropic (mGluR) or ionotropic (iGluR).1 Eight subtypes are recognized for mGluR and they fall into three groups based on their amino acid sequences and pharmacology.2,3 mGluR1, along with the mGluR5 subtype, belong to Group I, and in human brain are most abundant in cerebellum, hypothalamus, thalamus, basal ganglia, and hippocampus.4 They have been implicated in several neuropsychiatric disorders, including pain, cerebellar ataxia, extrapyramidal motor dysfunction, fear, anxiety, mood disorders, excitotoxicity, epilepsy, and schizophrenia.5–7 Consequently, mGluR1 are considered a target for drug development.8–10 A means to image and quantify human brain mGluR1 with PET would be valuable for better understanding the role of mGluR1 in neuropsychiatric disorders, and also for developing drugs intended to target these receptors. However, unlike for human brain mGluR5,11,12 satisfactory radioligands for quantifying human brain mGluR1 with PET are not yet established. Our ultimate goal is to develop such a radioligand.
Candidate ligands for PET imaging of brain receptors should present a variety of features, among which are importantly high affinity, high selectivity, moderate lipophilicity, and amenability to labeling with a positron-emitter, usually carbon-11 (t1/2 = 20.4 min) or fluorine-18 (t1/2 = 109.7 min).13–16 Previous efforts to develop a suitable mGluR1 PET radioligand have explored several candidates among a variety of structural classes.17–30 Some of these have been evaluated in rodents only. A few are reported to have been evaluated in primates, namely [11C]JNJ-16567083 ([11C]1), [18F]2, [11C]MMTP ([11C]3), [18F]MK-1312 ([18F]4), [18F]FPIT ([18F]5), [11C]LY2428703 ([11C]6), [18F]FITM ([18F]7) and [11C]8 (Figure 1). Only one of these, [11C]6, is known to have been further evaluated in human. Although [11C]6 showed promising imaging characteristics in rat28, this radioligand failed to give a receptor-specific signal in monkey or human subjects, primarily because of significantly lower effective binding potential (VS) for human mGluR1 (0.0161) than for rat mGluR1 (0.683).30 Vs is defined as fP × Bmax/KD, and although mGluR1 receptor density (Bmax) in human is similar to that in rat and higher than that in monkey,22,31 the plasma free fraction (fP) and affinity (1/KD) of 6 are each substantially lower for human than for either rat or monkey. Among other radioligands evaluated in monkey, [18F]7 appears to have been one of the most promising, as it displays high affinity (IC50 = 5.1 nM) and selectivity for binding to mGluR1, and moderate radioactivity uptake in monkey brain with high mGluR1-specific signal. Nevertheless, the reported radiosynthesis of [18F]723 requires heating of a base-sensitive nitro precursor at 180 °C and is known to be challenging.24 Moreover, washout of radioactivity from rat or monkey brain after intravenous administration of [18F]7 is quite slow and may hinder accurate quantification of mGluR1.23,26 A radioligand with faster brain kinetics in monkey is desirable.
Figure 1.

Candidate mGluR1 PET radioligands that have been evaluated in non-human primates.
The N-methyl analog of 7, which we dub FIMX (11) (Figure 2), has been reported to have threefold higher human mGluR1 affinity (IC50 = 1.8 nM), to retain high selectivity for binding to mGuR1 versus binding to mGluR5 in vitro;32 11 is also metabolized more extensively than 7 in human liver microsomes,32 which might also promote faster decrease of non-specific binding in brain. Moreover, we expected 11, having an N-methyl group in place of an N-isopropyl group, to have lower lipophilicity than 7. In view of these properties, we considered that labeling 11 with carbon-11 or fluorine-18 might provide an mGluR1 PET radioligand having improved features over [18F]7, including higher plasma free fraction, higher brain uptake, greater receptor-specific signal and faster brain kinetics.13–16,33 Here we report our development of a method for preparing [18F]11 in useful radiochemical yields, and an evaluation of this radioligand with PET, showing that [18F]11 is effective for imaging monkey brain mGluR1 and therefore warrants further evaluation in human subjects.
Figure 2.
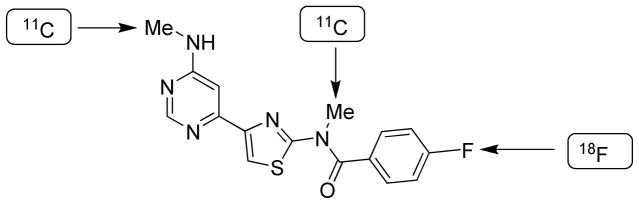
Structure of 11 (FIMX), and sites considered for labeling with a positron-emitter.
RESULTS AND DISCUSSION
Chemistry
The structure of 11 presents three sites that were initially considered for well-known methods of labeling with a positron-emitter (Figure 2), namely either of the two N-methyl groups for labeling by methylation with [11C]iodomethane,34 or the aryl fluoro substituent for labeling by aromatic nucleophilic substitution with [18F]fluoride ion.35 Our initial attempts at labeling 11 by methylation of either the secondary desmethyl amido analog or the N-desmethyl pyrimidinyl-amino analog with [11C]iodomethane under basic conditions did not give satisfactory yields or product purities (data not shown). Therefore, we focused on achieving the radiofluorination of 11. We first tried conventional substitutions in nitro and bromo analogs with [18F]fluoride ion, but no [18F]11 was formed, even under harsh conditions of high temperature and extended reaction times (data not shown). Our findings were consistent with the recognized difficulty of labeling the close analog 7 (Figure 1) by reaction of a nitro precursor with [18F]fluoride ion.23,24 The difficulty of this process for preparing [18F]7 or [18F]11 may be ascribed to the presence of only a weak electron-withdrawing para-N-methylamido group on the ring to be labeled with fluorine-18.35
We and others have studied the radiofluorination of diaryliodonium salts extensively,36–40 and have shown that this methodology enables introduction of cyclotron-produced no-carrier-added [18F]fluoride ion into aryl rings. Relatively few structurally complex radioligands have yet been prepared with this methodology.41–43 Nonetheless, we decided to explore this method for labeling 11, because of its notable success in labeling both electron-rich and electron-deficient arenes.36–40 We considered that the free amino group in 11 would need to be protected during synthesis of an iodonium salt precursor, and possibly during its radiofluorination to avoid detrimental interactions of the amino group with [18F]fluoride ion. We chose Boc as a protecting group, as this can be removed quite quickly in radiosyntheses with fluorine-18, usually under acidic conditions.44 Treatment of trialkylarylstannanes with Koser’s reagent (hydroxy(tosyloxy)iodobenzene) has been shown to be an effective method for producing functionalized diaryliodonium salts,39–43,45,46 and we considered this method for generating a desirable precursor. From this method, the diaryliodonium salt would have phenyl as one of the ring partners and tosylate as counterion. Based on experimental39,40 and theoretical studies47,48 on the aryl ring preference in the radiofluorination of diaryliodonium salts, we expected that radiofluorination would occur not at the phenyl ring but at the desired more electron-deficient ring. Moreover, the weakly nucleophilic tosylate ion, is well tolerated in the radiofluorination of diaryliodonium salts.39,40,42
The target precursor, (4-((4-(6-(tert-butoxycarbonyl(methyl)amino)pyrimidin-4-yl)thiazol-2-yl)(methyl)carbamoyl)phenyl)(phenyl)iodonium tosylate; 15), was successfully prepared in five steps from commercially available 4-(6-chloropyrimidin-4-yl)-N-methylthiazol-2-amine (Scheme 1). Thus, acylation of this starting material with 4-bromobenzoyl chloride gave 10, as previously reported.29 Subsequent amination of 10 gave 12 in quantitative yield, and then treatment of 12 with (Boc)2O in THF gave 13, also in excellent yield (96%). Treatment of 13 with (Bu3Sn)2 in the presence of tetrakis(triphenylphosphine)palladium(0) gave the tributylstannyl compound 14 in moderate yield (40%). Treatment of 14 with Koser’s reagent for 4 days at room temperature then gave the desired precursor 15 in 10 to 30% yields.
Scheme 1.

Synthesis of 11 and its radiolabeling precursor, 15.a
aReagents and conditions: (a) 4-fluorobenzoylchloride (for 9) or 4-bromobenzoyl chloride (for 10), Et3N, toluene, 100 °C, 4 h–overnight; (b) MeNH2, K2CO3, 1,4-dioxane, 80 °C, 1.5 h (for 11) or 85 °C, 5 h (for 12); (c) DMAP, (Boc)2O, THF, 70 °C, 1 h; (d) (Bu3Sn)2, Pd(PPh3)4, 1,4-dioxane, overnight; (e) Koser’s reagent, 4 d.
Here, we also synthesized 11 for use as a reference compound in chromatography and for more extensive pharmacological screening. This synthesis was achieved through acylation of 4-(6-chloropyrimidin-4-yl)-N-methylthiazol-2-amine with p-fluorobenzoyl chloride and subsequent amination (Scheme 1), as has been described previously but without full details.32
Pharmacological screen of 11
Ligand 11, has been reported32 to have a high affinity for human mGluR1, with an IC50 of 1.8 nM. We found 11 to have low affinity for a wide range of other mainly human receptors, transporters and binding sites, as represented by the following high Ki values: (2096 nM for mGluR5 rat brain, 2207 nM for 5-HT2A, 2536 nM for DAT, 7900 nM for mGluR5, and > 10,000 nM for 5-HT1A, 5-HT1B, 5-HT1D, 5-HT1E, 5-HT2A–2C, 5-HT3, 5-HT5A, 5-HT6, 5-HT7, α1A, α1B, α1D, α2A–α2C, β1–β3, rat BZP site, D1–D5, DAT, DOR, GABAA, H1, H3, KOR, M1–M5, MOR, NET, NMDA, SERT, TSPO, σ1 and σ2). Thus, 11 has excellent mGluR1-selectivity, as required for a prospective PET mGluR1 radioligand. Moreover, 11 was found to be an antagonist at both mGluR1 and mGluR5 when tested for stimulation of phosphoinositide hydrolysis49 using cloned rat receptors at a concentration of 10 μM.
Radiochemistry
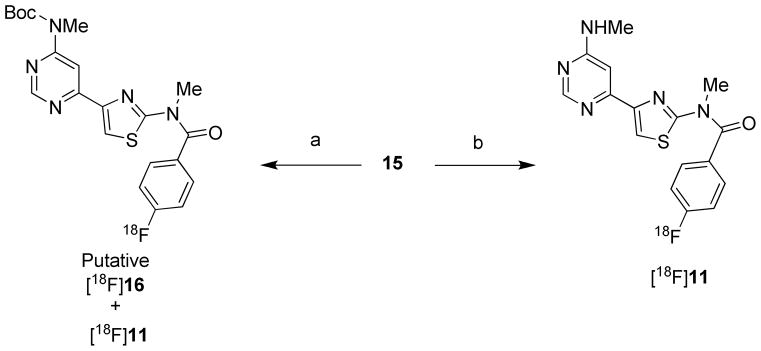
Polar and aprotic solvents, such as acetonitrile and DMF, have been used successfully for the radiofluorination of diaryliodonium salts.38–43 We attempted the synthesis of [18F]11 by treating 15 with cyclotron-produced [18F]fluoride ion in either of these solvents, and also in the higher boiling polar aprotic solvent DMSO. Reactions were conducted in the presence of cryptand (K 2.2.2), K2CO3, and also in most cases the free radical scavenger TEMPO (2,2,2-tetramethylpiperidine 1-oxyl), under various conditions with either thermal or microwave heating (Table 1). We expected that such reactions would produce the N-Boc-protected compound [18F]16 that would then need to be deprotected under acidic conditions to give [18F]11. TEMPO was often included because it has been shown to enhance yields in the radiofluorination of many diaryliodonium salts, presumably by hindering their free-radical decompositions.50 We found that TEMPO improved the incorporation of [18F]fluoride ion from 26 to 42% for reactions of 15 under thermal conditions (100 °C bath temperature, 5 min) in DMF (Table 1, entries 1 and 2). The dominant radioactive product from these reactions was very lipophilic and was presumed to be the expected intermediate [18F]16 (Scheme 2). In the presence of TEMPO, 3% of the initial fluorine-18 was incorporated into [18F]11, showing that some loss of the N-Boc protecting group had occurred. A microwave–promoted reaction, conducted over the temperature range 60–140 °C for 2 min in DMF, gave a very similar yield of [18F]16 to that from thermal conditions (100 °C for 5 min), but also increased the production of the desired [18F]11 to 11% (Table 1, entry 3). A microwave-promoted reaction in MeCN over the temperature range 61–113 °C for 2.5 min gave 30% incorporation of [18F]fluoride ion, but solely into [18F]16 (Table 1, entry 4). When using DMSO under microwave conditions, 49% of the [18F]fluoride ion was incorporated into organic products, specifically 9% into [18F]16 and 40% into the desired [18F]11 (Table 1, entry 5). Thus, DMF and DMSO gave similar incorporations of [18F]fluoride ion, but in DMSO the ratio of radioactive products was reversed in favor of the desired product [18F]11 over the intermediate [18F]16. The effectiveness of the radiofluorination of 15 in DMSO was unexpected in view of reports of low yields for the radiofluorination of some other diaryliodonium salts in DMSO.38,43 Increase of microwave reaction time in DMSO to 4 min (2 cycles of 2 min) reduced the yields of both [18F]16 and [18F]11 (Table 1, entry 6). Therefore, we initially selected DMSO as solvent and microwave heating for 2 min for the production of [18F]11 (Table 1, entry 7). Subsequently, we found that microwave irradiation for 2.5 min gave appreciably higher incorporations of [18F]fluoride ion into [18F]11 (38%; Table 1, entry 8), and these conditions then became standard for the production of [18F]11 for PET experiments. With these reaction conditions, followed by purification with single-pass reversed phase HPLC (see page S4 in Supporting Information), [18F]11 was obtained in useful radiochemical yield (20% decay-corrected), high radiochemical purity (96–100%) free of significant chemical impurities, and with specific activities of 2.84 ± 2.2 Ci/μmol (n = 7). Formulations of [18F]11 in sterile medium for injection were radiochemically stable for at least 4 hours.
Table 1.
Incorporation yields for reactions of [18F]fluoride ion with 15 under various conditions.a
| Entry | Conditions
|
Solvent | TEMPO (eq.) | [18F]F− incorporation (%)
|
|||
|---|---|---|---|---|---|---|---|
| Energy source | Temp. (°C) | Time (min) | Putative [18F]16 | [18F]11 | |||
| 1 | Thermal | 100 | 5 | DMF | -- | 26 | 0 |
| 2 | Thermal | 100 | 5 | DMF | 2 | 39 | 3 |
| 3 | μ-wave 100 W | 60–140 | 2 | DMF | 2 | 40 | 11 |
| 4 | μ-wave 90 W | 61–113 | 2.5 | MeCN | 4 | 30 | 0 |
| 5 | μ-wave 90 W | 60–149 | 2 | DMSO | 2 | 9 | 40 |
| 6 | μ-wave 90 W | 60–152 | 4 | DMSO | 2 | 3 | 29 |
| 7 | μ-wave 90 W | 60–149 | 2 | DMSO | 2 | n.m. | 18 (n = 2)b |
| 8 | μ-wave 90 W | 60–152 | 2.5 | DMSO | 2 | 0–14 | 38 (n =13)b |
Amounts of precursor were 1.4–1.9 mg, and reaction volume was 500 μL.
Products from reactions 7 and 8 were analyzed on a semi-preparative HPLC column, whereas other reactions were analyzed on an analytical column.
n.m. = not measured
Scheme 2.
Radiosynthesis of [18F]11.a
aReagents and conditions: (a) [18F]F−, K2CO3 / K 2.2.2, DMF, 100 °C, 5 min. (b) [18F]F−, K2CO3 / K 2.2.2, DMSO, microwave (90 W), 2–4 min.
The discovery that addition of acid was unnecessary for deprotection of [18F]16 to [18F]11 in DMSO under the preferred microwave conditions greatly simplified the production of [18F]11 in our upgraded Synthia51,52 apparatus. We reasoned that deprotection occurred through pyrolysis, and that this occurred more readily in DMSO than in acetonitrile or DMF under microwave conditions because the reaction mixture reached a higher temperature. We confirmed that exposure of neat solvents in the reactor to the microwave field used in the radiochemistry (90 W for 2.5 min), increased solvent temperatures at different rates such that their terminal temperatures were near their boiling points (189, 152 and 82 °C for DMSO, DMF and MeCN, respectively) (see page S5 in Supporting Information). We also observed decomposition of the precursor 15 at a temperature (180 °C) close to the boiling point of DMSO during an attempt to measure its melting point, consistent with likely loss of the N-Boc group with evolution of carbon dioxide.
Lipophilicities of 7 and 11
The cLogD values of 7 and 11 were calculated to be 2.77 and 2.25, respectively. The LogD of [18F]11 was measured to be 2.52 ± 0.04 (n = 6) which is within the desired range (2.0–3.0) for favorable radioligand properties, such as ability to pass the blood-brain barrier without consequent high non-specific binding, and without the generation of lipophilic radiometabolites in periphery or brain.13–16
PET Experiments in Monkey
Injection of [18F]11 into a rhesus monkey at baseline resulted in high brain radioactivity uptake. Regional peak uptakes were consistent with known mGluR1 density; maximal uptakes were highest in receptor-rich cerebellum (5.3 SUV at 12 min), moderate in thalamus (2.9 SUV), frontal cortex (2.7 SUV), hippocampus (2.5 SUV), putamen (2.5 SUV) and anterior cingulate (2.4 SUV), and lower in the rest of brain (Figure 3, panel A). Peak radioactivity uptakes in all regions were followed by a steady decrease in radioactivity level, as exemplified by receptor-rich cerebellum which decreased by 37% to 3.32 SUV at 90 min after injection, and by 47% to 2.8 SUV at the end of the 2-h scan. This decrease is approximately twice that reported previously for [18F]726, which was 17% at 90 min. In another experiment in the same monkey, in which the selective mGluR1 antagonist, JNJ-1625968553 (17 3,4-dihydro-2H-pyrano[2,3-b]quinolin-7-yl)-(cis-4-methoxycyclohexyl)-methanone; 3 mg/kg), was administered intravenously at 5 min before the injection of [18F]11, all regional time-activity curves were almost indistinguishable, with each showing a fast decline from a quick peak radioactivity level (Figure 3, panel B). The distribution of radioactivity across brain became low and uniform (Figure 4). This experiment indicated that a high level of brain radioactivity in the baseline experiment was due to specific binding to mGluR1 receptors. In an experiment in a second monkey, 17 (3 mg/kg) was administered intravenously at 30 min after administration of [18F]11, and this caused a rapid decrease in radioactivity at all mGluR1-containing regions, thereby confirming the reversibility of radioligand specific binding (Figure 3, panel C). In an experiment in a third monkey, the mGluR5-selective ligand, 3-((2-methyl-4-thiazolyl)ethynyl)pyridine (MTEP,18) was given at 30 min after administration of [18F]11, but this caused no change in the washout kinetics from any brain region (Figure 3, panel D); essentially, the data obtained were very similar to that in the baseline experiment in a different monkey. This experiment confirmed the specificity of [18F]11 for binding to mGluR1 versus mGluR5 in monkey brain, as expected from the in vitro binding data determined for cloned human receptors.32 No significant radioactivity uptake occurred in skull in any of the experiments, and radiodefluorination was therefore negligible (Figure 4)
Figure 3.

PET time-activity curves in selected brain regions of rhesus monkeys administered [18F]11 at: baseline (panel A); after preblock of mGluR1 with 17 (panel B); in an experiment in which 17 (3 mg/kg, i.v.) was administered at 30 min (panel C); and in an experiment in which the selective mGluR5 ligand 18 (5 mg/kg, i.v.) was administered at 30 min (Panel D).
Figure 4.
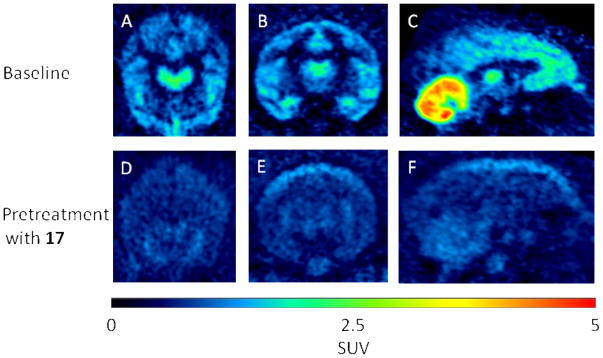
Brain PET images acquired as summed data from 0–120 min after intravenous injection of rhesus monkey with [18F]11 under baseline conditions (top row) or under conditions in which the same monkey was injected with the selective mGluR1 ligand 17 (3 mg/kg, i.v.) at 5 min before [18F]11 (bottom row). Panels A and D are transaxial, B and E coronal, and C and F sagittal images.
In Vitro Stability in Whole Blood and Plasma, and Plasma Free Fraction
[18F]11 was stable in vitro at room temperature for at least 30 min in monkey whole blood (98.1% unchanged) and plasma (98.9% unchanged). The plasma free fraction (fp) of [18F]11 for rhesus monkey was 0.0123 ± 0.0001 (n = 3). The fp value for human (0.0114 ± 0.0003, n = 3) was very similar and therefore is not expected to adversely affect the effective binding potential that might be obtained in PET imaging of human mGluR1 with this radioligand. These fp values are as would be expected for a compound with this measured LogD value.33
Emergence of Radiometabolites in Plasma
After intravenous injection of [18F]11 into monkey, radioactivity decreased rapidly in whole blood (Figure 5). Radioactivity concentrations in venous plasma were higher than in whole blood throughout the scanning period in accord with high binding to plasma proteins. HPLC analysis of venous plasma showed that [18F]11 represented 43 and 31% of total radioactivity at 12 and 31 min respectively. Thereafter, until the end of the 2-h scan, there was little further change in the percentage of radioactivity in plasma represented by [18F]11. At least five radiometabolites ([18F]A–[18F]E) were detected in plasma at 60 min after radioligand injection, and these were all less lipophilic than [18F]11, as judged by their shorter retention times in reversed phase HPLC (Figure 6). [18F]A–[18F]C were not well resolved from each other, but all were resolved from [18F]11. The distribution of radioactivity in plasma at this time-point was estimated to be: [18F]A,19%; [18F]B, 23%; [18F]C, 10%; [18F]D, 1%; [18F]E 21%; and [18F]11, 26%. Radiometabolites were not chemically identified but their lower lipophilicities relative to [18F]11 are expected to mitigate against significant brain entry that might contaminate receptor-specific signal. In this regard, the time-activity curves for monkey brain regions following intravenous injection of [18F]11 show continuous decline throughout the scanning period (Figure 3) and provide no evidence of significant ingress of radiometabolites.
Figure 5.

Time courses for total radioactivity in whole blood and venous plasma, and of [18F]11 in venous plasma.
Figure 6.
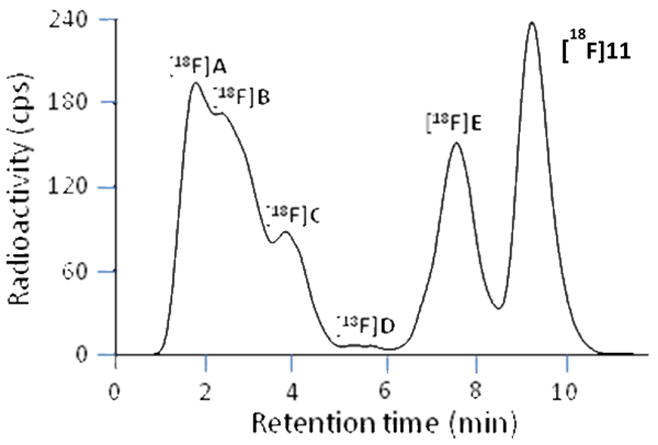
HPLC analysis of monkey plasma at 60 min after intravenous injection of [18F]11.
CONCLUSIONS
The highly selective mGluR1 radioligand [18F]11 may be produced from [18F]fluoride ion and the N-Boc-protected diaryliodonium salt precursor 15 in useful radiochemical yield. Intravenously injected [18F]11 gave high radioactivity uptake in rhesus monkey brain and a large proportion of this radioactivity was specifically and reversibly bound to mGluR1 receptors. [18F]11 appears to give much faster washout kinetics than the previously reported [18F]7. Thus, [18F]11 is an effective radioligand for imaging monkey brain with PET and therefore merits evaluation in human subjects.
EXPERIMENTAL
Materials
4-(6-Chloropyrimidin-4-yl)-N-methylthiazol-2-amine was purchased from Aquila Pharmatech LLC (Waterville, OH). The blocking agents 17 and 18 were purchased from Tocris Bioscience (Bristol, U.K.) and EMD Millipore (Billerica, MA), respectively. Other chemicals were purchased from Sigma-Aldrich Chemical Co. (Milwaukee, WI) and used as received.
General Methods
1H (400.13 MHz), 13C (100.62 MHz), and 19F (376.46 MHz) NMR spectra were recorded at room temperature on an Avance-400 spectrometer (Bruker, Billerica, MA). Chemical shifts are reported in δ units (ppm) downfield relative to the chemical shift for TMS. Abbreviations s, d, t, and bs denote singlet, doublet, triplet, and broad singlet, respectively. TLC was performed on silica gel layers (0.2 mm with fluorescent indicator; Polygram SIL G/UV254; Grace Davison Discovery Sciences; Deerfield, IL); compounds were visualized under UV light (λ = 254 nm).
HRMS data were acquired at the Mass Spectrometry Laboratory, University of Illinois at Urbana-Champaign (Urbana, IL), under electron ionization conditions using a double-focusing high-resolution mass spectrometer (Micromass; Waters, Milford, MA). Synthesized compounds were analyzed with LC-MS on an LCQ Deca instrument (Thermo Fisher Scientific Corp.; Waltham, MA). Gradient or isocratic LC was performed with binary solvents (A/B, 150 μL/min) composed of water-methanol-acetic acid (90: 10: 0.5 by vol.) (A) and methanol-acetic acid (100: 0.5, v/v) (B) on a Luna C18 column (3 μm, 50 mm × 2 mm; Phenomenex; Torrance, CA). For the analysis of compound 14, no LC column was used. Following electrospray ionization of the effluent, ions m/z 150–750 were acquired.
Melting points were measured with a Mel-Temp manual apparatus (Electrothermal, Thermo Fisher Scientific Corp.).
γ-Radioactivity from 18F was measured with a calibrated dose calibrator (Atomlab 300, Biodex Medical Systems, Shirley, NY) or a γ-counter (Wizard 3″, 1480 automatic γ-counter; PerkinElmer; Waltham, MA). Radioactivity measurements were corrected for physical decay. All radiochemistry was performed in a lead-shielded hot-cell for personnel protection from radiation.
Radioactive products were separated by HPLC on a Luna C18 column (5 μm, 10 × 250 mm; Phenomenex) eluted with 10 mM HCOONH4-MeOH at the stated compositions and flow rates. Eluates were monitored for radioactivity with a pin-diode detector (Bioscan Inc.; Washington, DC) and for UV absorbance at 230 nm with a System Gold 166 detector (Beckman Coulter Inc.; Indianapolis, IN).
The purity of compound 11 was assessed to be > 98% by reversed phase HPLC on a Luna C18 column (10 μm, 4.6 × 250 mm; Phenomenex) eluted with 70% A (0.1% aq.TFA)-30% B (MeCN) at 2 mL/min. [18F]11 was analyzed in the same manner. Eluates were monitored for UV absorbance at 230 nm (System Gold 166 detector, Beckman) and also when applicable for radioactivity (pin-diode detector, Bioscan). Samples were injected alone and then co-injected with the reference nonradioactive compound to check for co-elution. RCYs were calculated for labeled products isolated with HPLC.
Grouped quantitative data are expressed as mean ± SD.
Chemistry
N-(4-(6-Chloropyrimidin-4-yl)thiazol-2-yl)-4-fluoro-N-methylbenzamide (9)
4-Fluorobenzoyl chloride (232 mg; 1.47 mmol) was added to a suspension of 4-(6-chloropyrimidin-4-yl)-N-methylthiazol-2-amine (222 mg, 0.98 mmol) in toluene (5 mL) plus Et3N (594 mg, 5.88 mmol) under Ar. The mixture was stirred at 100 °C overnight, quenched with water, and finally extracted with CH2Cl2. The organic layer was dried over Na2SO4 and evaporated to remove solvent. Silica gel chromatography (CH2Cl2/Et3N, 1000: 1 v/v; then CH2Cl2/EtOAc, 4: 1 v/v) of the residue gave 9 as a light yellow solid (320 mg, 94%). Mp, 212–213 °C. 1H NMR (CDCl3): δ 3.77 (3 H, s), 7.20–7.24 (2 H, m), 7.62–7.65 (2 H, m), 8.04 (1 H, d, J = 0.8 Hz), 8.12 (1 H, s), 8.94 (1 H, d, J = 1.2 Hz). 13C NMR (CDCl3): δ 38.61, 115.91, 116.13, 117.37, 118.82, 130.04, 130.07, 130.17, 130.26, 146.14, 158.71, 160.42, 160.82, 162.25, 163.01, 165.53, 169.66. 19F NMR ((CD3)2SO): δ –107.25. LC-MS: m/z [M+H]+, 349.1. HRMS: calcd for C15H11N4OFSCl (M+ + H), 349.0326; found, 349.0328.
4-Fluoro-N-methyl-N-(4-(6-(methylamino)pyrimidin-4-yl)thiazol-2-yl)benzamide (11)
A mixture of 9 (400 mg, 1.2 mmol), K2CO3 (330 mg, 2.4 mmol), MeNH2 solution in ethanol (33%; 2.51 mL) in 1,4-dioxane (7 mL) was stirred at 80 °C for 1.5 h until TLC showed all starting material had reacted. The mixture was cooled to room temperature, quenched with water, and extracted four times with EtOAc. The organic layer was dried over MgSO4 and the solvent was removed under reduced pressure. The residue was redissolved in MeOH, precipitated with EtOAc followed by hexane, and then filtered to provide an almost white solid, which upon silica gel chromatography (CH2Cl2/MeOH, 20: 1 v/v) gave 11 as a white solid (98 mg, 29%). Mp, 187–189 °C. 1H NMR ((CD3)2SO): δ 2.85 (3 H, d, J = 4.4 Hz), 3.64 (3 H, s), 7.12 (1 H, bs), 7.37–7.42 (2 H, m), 7.54 (1 H, bs), 7.77–7.81 (2 H, m), 7.99 (1 H, s), 8.45 (1 H, s). 13C NMR ((CD3)2SO): δ 27.13, 38.12, 115.48, 115.70, 130.39, 130.48, 130.78, 130.81, 147.43, 158.33, 159.84, 162.02, 163.45, 164.49, 169.11. 19F NMR ((CD3)2SO): δ –109.04. LC-MS: m/z [M+H]+, 344.2. HRMS: calcd for C16H15N5OFS (M+ + H), 344.0981; found, 344.0979.
4-Bromo-N-methyl-N-(4-(6-(methylamino)pyrimidin-4-yl)thiazol-2-yl)benzamide (12)
A mixture of 1029 (40 mg, 98 μmol), K2CO3 (27 mg, 197 μmol) and MeNH2 solution in ethanol (33%; 0.35 mL) in 1,4-dioxane (3 mL) was stirred at 80 °C for 5 h. The mixture was then cooled to room temperature, quenched with water, and extracted three times with EtOAc. The organic layers were dried over MgSO4 and solvent was removed under reduced pressure. Silica gel chromatography (CH2Cl2/MeOH, 15: 1 v/v) of the residue gave 12 as a white solid (40 mg, 100%). Mp, 218–220 °C. 1H NMR ((CD3)2SO): δ 2.85 (3 H, d, J = 4.8 Hz), 3.62 (3 H, s), 7.12 (1 H, bs), 7.54 (1 H, bs), 7.64–7.67 (1 H, m), 7.75–7.78 (2 H, m), 7.99 (1 H, s), 8.45 (1 H, s). 13C NMR ((CD3)2SO): δ 38.00, 115.62, 124.38, 129.73, 131.56, 133.53, 147.41, 159.71, 169.10. LC-MS: m/z [M+H]+, 404.2. HRMS: calcd for C16H15N5OSBr (M+ + H), 404.0181; found, 404.0185.
tert-Butyl-6-(2-(4-bromo-N-methylbenzamido)thiazol-4-yl)pyrimidin-4-yl(methyl)carbamate (13)
A mixture of 12 (500 mg, 1.98 mmol), DMAP (890 mg, 9.9 mmol), and (Boc)2O (1.0 g, 6 mmol) in THF (30 mL) was stirred at 70 °C for 1 h until TLC showed all starting material had reacted. The solvent was removed under reduced pressure to give crude product that was then diluted with water and extracted with EtOAc. The organic layer was dried with MgSO4 and evaporated to dryness. Silica gel chromatography (hexane/EtOAc, 4: 1 v/v) of the residue gave 13 as a white solid (600 mg, 96%). Mp, 176–178 °C. 1H NMR (CDCl3): δ 1.59 (9 H, s), 3.49 (3 H, s), 3.75 (3 H, s), 7.46–7.49 (2 H, m), 7.64–7.67 (2 H, m), 8.01 (1 H, s), 8.55 (1 H, d, J = 1.2 Hz), 8.90 (1 H, d, J = 1.2 Hz). 13C NMR (CDCl3): δ 28.28, 33.31, 38.51, 82.56, 109.25, 116.82, 125.70, 129.28, 132.01, 133.10, 147.94, 153.61, 157.60, 158.71, 160.32, 161.89, 169.51. LC-MS: m/z [M+H]+, 503.9. HRMS: calcd for C21H23N5O3SBr (M+ + H), 504.0705; found, 504.0707.
tert-Butyl-methyl(6-(2-(N-methyl-4-(tributylstannyl)benzamido)thiazol-4-yl)pyrimidin-4-yl)carbamate (14)
Bu6Sn2 (633 mg, 1.09 mmol) and Pd(PPh3)4 (100 mg, 0.086 mmol) were added to a solution of 13 (500 mg, 0.99 mmol) in 1,4-dioxane (15 mL). The mixture was degassed for 30 min, heated at 85 °C for 5.5 h. The solvent dioxane was then removed under reduced pressure. Silica gel chromatography (hexane/EtOAc, 20: 1, then 6: 1 v/v) of the mixture then gave 14 as a colorless oil (280 mg, 40%). 1H NMR (CDCl3): δ 0.90 (9 H, t, J = 7.2 Hz), 1.09–1.13 (6 H, m), 1.32–1.37 (6 H, m), 1.53–1.59 (15 H, m), 3.49 (3 H, s), 3.78 (3 H, s), 7.50–7.52 (2 H, m), 7.59–7.61 (2 H, m), 8.00 (1 H, s), 8.55 (1 H, d, J = 1.2 Hz), 8.91 (1 H, d, J = 1.2 Hz). 13C NMR (CDCl3): δ 9.70, 13.67, 27.34, 28.28, 29.04, 33.29, 38.59, 82.54, 109.30, 116.66, 126.63, 133.57, 136.55, 147.13, 147.81, 153.61, 157.59, 158.84, 160.61, 161.89, 170.81. LC-MS: m/z [M+H]+, 716.0. HRMS: calcd for C33H50N5O3SSn (M+ + H), 716.2656; found, 716.2665.
(4-((4-(6-(tert-Butoxycarbonyl(methyl)amino)pyrimidin-4-yl)thiazol-2-yl)(methyl)carbamoyl)phenyl)(phenyl)iodonium tosylate (15)
Koser’s reagent (28 mg, 70 μmol) was added to a solution of 14 (50 mg, 70 μmol) in CH2Cl2 (2 mL). The mixture was stirred under Ar for 4 d, and then CH2Cl2 was removed under reduced pressure. The residue was then dissolved in MeOH. Et2O was added and the precipitate was filtered to give 15 as a white solid (12 mg, 21%). Mp, decomposed above 180°C. 1H NMR ((CD3)2SO): δ 1.54 (9 H, s), 2.28 (3 H, s), 3.39 (3 H, s), 3.57 (3 H, s), 7.10–7.12 (2 H, d, J = 8 Hz), 7.46–7.48 (2 H, d, J = 8 Hz), 7.56–7.59 (2 H, t, J = 7.6 Hz), 7.69–7.73 (1 H, t, J = 7.6 Hz), 7.83–7.85 (2 H, d, J = 8.4 Hz), 8.22 (1 H, s), 8.31–8.33 (2 H, d, J = 7.6 Hz), 8.40–8.42 (2 H, d, J = 8.4 Hz), 8.48 (1 H, s), 8.93 (1 H, s). 13C NMR ((CD3)2SO): δ 20.75, 27.75, 33.01, 37.96, 66.32, 82.45, 108.07, 116.60, 117.64, 118.45, 125.46, 128.00, 130.48, 131.86, 132.24, 135.26, 135.36, 137.52, 140.81, 145.76, 146.71, 152.87, 157.67, 157.98, 159.90, 161.26, 168.56. LC-MS: m/z [M+H]+, 628.0. HRMS: calcd for C27H27N5O3SI (M++H), 628.0879; found, 628.0880.
Comprehensive Screening of 11
Ligand 11 was screened for binding selectivity for mGluR1 versus a wide range of other mainly human brain binding sites, transporters and receptors by the National Institute of Mental Health (NIMH) Psychoactive Drug Screening Program (PDSP). The efficacy of 11 at mGluR1 and mGluR5 was also determined at 10 μM concentration. Detailed protocols can be found on the PDSP website: http://pdsp.med.unc.edu.
Radiochemistry
Radionuclide Production
No-carrier-added (NCA) [18F]fluoride ion was produced with the 18O(p,n)18F reaction, by irradiating 18O-enriched water (95 atom%; 1.8 mL) with a beam of protons (14.1 MeV; 20–25μA) from a PETtrace cyclotron (GE Medical Systems, Milwaukee, WI).
Preparation of [18F]Fluoride Ion Reagent
[18F]Fluoride ion reagent was prepared for reactions as follows. A glass V-vial (5 or 1 mL), equipped with a screw-on cap and liner (20/400 PTFE/silicone; Alltech Associates, Deerfield, IL), was loaded with an aliquot of a previously prepared solution (50 μL) of K2CO3 (10 mg) and K 2.2.2 (60 mg) in MeCN-H2O (9: 1, v/v; 2 mL). NCA [18F]fluoride ion (10–114 mCi) in [18O]H2O (15–600 μL) and then anhydrous MeCN (700 μL) were added to the vial. The solvent was evaporated off at 110 °C under a nitrogen stream (200 mL/min) vented at a reduced pressure. This azeotropic drying process was repeated three times.
Experimental Radiochemistry
Reactions were conducted by adding a solution of precursor 15 (1.5–1.9 mg) in MeCN, DMF or DMSO (0.5 mL) with or without TEMPO (0.6–2.2 mg) to the [18F]fluoride ion reagent, which was then heated thermally (100 °C) for 5 min or with microwaves (90 W; 60–150 °C) for a specified time between 2 and 4 min. At the end of the reaction the mixture was cooled and analyzed by HPLC. This was performed either on a Gemini-NX C18 column (5 μm, 4.6 × 250 mm; Phenomenex) eluted at 2 mL/min with a mixture of 50 mM NH4OAc (A) and MeOH (B) with B starting at 10%, increased to 80% over 6 min, kept at 80% for 6 min, increased to 100% over 1 min, and finally kept at 100% for 6 min, or on a Luna C18 column (5 μm, 10 × 250 mm; Phenomenex) eluted at 5 mL/min with a mixture of 10 mM HCOONH4 (C) and MeOH (B) with B started at 10%, increased to 60% over 20 min, and finally held at 60% for 25 min. Incorporation radiochemical yields were calculated from the HPLC radiochromatograms, without decay-correction.
Production of NCA [18F]11
Semi-automated radiosynthesis was conducted in an upgraded Synthia radiochemistry apparatus51 that had also been adapted to use microwave irradiation for the labeling reaction52. A solution of precursor 15 (1.4–1.9 mg) and TEMPO (0.7–2.2 mg) in DMSO (500 μL) was added to a still warm (~ 80 °C) radioactive V-vial containing dried [18F]fluoride ion reagent (135–260 mCi). The uncooled vial was transferred immediately to a microwave reactor (model 521; Resonance Instruments Inc., Skokie, IL) and heated (90 W, 2.5 min), giving an average final temperature of 179 °C. This reaction mixture was then cooled to rt, diluted with water (500 μL), and separated by HPLC on a semi-preparative size HPLC column, as described under ‘Experimental Radiochemistry’. Within a TRACERlab FX-FN box (GE; Milwaukee, WI), the fraction containing [18F]11 (tR = 33 min) was collected in a glass vial containing HPLC water (30 mL) and passed into a C18 Sep-pak (Waters Corp.) that had been activated by flushing first with ethanol (10 mL) and then water (10 mL). The [18F]11 was eluted from the Sep-pak with EtOH (1 mL) followed by saline for injection (2 mL) through a sterile filter (0.22 μm, Millipore MP; Waters Corp.) and into a sterile vial containing saline for injection (8 mL), with the vial vented through another sterile filter (0.22 μm; Millipore GV; Waters Corp.). The identity of [18F]11 (tR = 6.8 min) was confirmed by analytical radio-HPLC on a Luna C18 column (10 μm, 4.6 × 250 mm, Phenomenex) eluted at 2 mL/min with a mixture (3: 7, v/v) of MeCN and aq. TFA (0.1% v/v), and also by LC-MS of associated carrier (m/z = 344 [M+1]+). The radiochemical purity of [18F]11 was 96–100%. The radiochemical stability of the formulated dose was tested over 4 h at rt. The average decay-corrected radiochemical yield (RCY) of [18F]11 was 20 ± 6.2% (n = 7) from [18F]fluoride ion. The average specific activity of [18F]11 was 2.84 ± 2.20 Ci/μmol (n = 7) at end of synthesis.
Calculation and Measurement of LogD
cLogD (at pH 7.4) values for 7 and 11 were computed with Pallas 3.7 software (CompuDrug; Bal Harbor, FL). The value for the distribution coefficient of [18F]11 between n-octanol and sodium phosphate buffer (0.15 M, pH 7.4) was determined as described previously.33
Monkey PET Experiments
Monkey experiments were performed in compliance with the animal care requirements of our institution. Three rhesus monkeys (Macaca mulatta, 13.3, 7.0 and 8.1 kg) were used to acquire four PET scans. In a baseline experiment, [18F]11 was injected intravenously into the monkey (13.3 kg) as a bolus. In a receptor preblock experiment in the same monkey on a separate day, the selective mGluR1 ligand 17 (3 mg/kg), was administered intravenously 5 min before [18F]11. A challenge experiment was performed in a second monkey, in which 17 (3 mg/kg) was administered intravenously at 30 min after [18F]11 injection. In an experiment in a third monkey (8.1 kg) the selective mGluR5 ligand 18 (5 mg/kg) was administered intravenously at 30 min after injection of [18F]11. The injected activities were 4.6–6.58 mCi, and the specific activities at time of injection were 1.67–4.18 Ci/μmol.
PET scans
PET images were acquired with a microPET Focus 220 scanner (Siemens Medical Solution; Knoxville, TN) for 120 min in 33 frames, with scan durations ranging from 30 s to 5 min. Anesthesia was initiated with ketamine (10 mg/kg, i.m.) and then maintained with 1.6% isoflurane and 98.4% O2. The position of the head was fixed using a stereotaxic frame. Electrocardiogram, body temperature, heart and respiration rates were measured throughout the experiment. All PET images were corrected for attenuation and scatter.
Image analysis
Images were reconstructed using Fourier rebinning plus two-dimensional filtered back-projection. PET images were co-registered to a standardized monkey MRI template using SPM5 (Wellcome Trust Centre; London, UK). A set of 34 predefined brain regions of interest from the template were then applied to the co-registered PET image to obtain regional decay-corrected time–activity curves. Cerebellar grey matter, which is not included in the template, was delineated semi-automatically using isocontour regions of interest. Uptake of radioactivity in each region of interest was expressed in units of standardized uptake value, where SUV = [(% injected activity/cm3) × body weight (g))]/100.
Analysis of radiometabolites in plasma
Venous blood samples were collected at 10, 30, 60, and 120 min after intravenous injection of [18F]11 (5.24 mCi; 1.4 nmol/kg) into one monkey (7.0 kg). Plasma was separated and analyzed with radio-HPLC on a Novapak C18 column (4 μm, 100 × 8 mm; Waters Corp.) housed in a radial compression module (RCM-100) and eluted at 2.0 mL/min with MeOH: H2O: Et3N (65: 35: 0.1 by vol.), as described previously.54 Radioactivity levels in different blood components were expressed as SUV.
The stability of [18F]11 in vitro was determined by incubation in whole blood and plasma at room temperature for at least 30 min. The radioactive whole blood sample (200 μL) was mixed with distilled water (300 μL) at the end of the incubation for 30 s to lyse blood cells. 450-μL samples of the lysed cells and of the incubated radioactive plasma were each added to MeCN (720 μL) for deproteinization. The samples were then centrifuged (10,000 g) and the clear supernatant liquids analyzed with radio-HPLC. The precipitates were counted in a γ-counter to allow calculation of the extraction efficiency. The extraction efficiency of all samples was 91.5% ± 7.4% (n = 6). The percentage of unchanged radioligand in the analyte, as determined by radio-HPLC, was divided by the radiochemical purity of the radioligand to give the stabilities of the radioligand in whole blood and plasma.
Plasma free fractions were determined in monkey venous plasma and also in pooled human arterial plasma that had been stored at −70 °C, as described previously.55
Supplementary Material
Acknowledgments
This work is support by the Intramural Research Program of NIMH. We thank the NIH Clinical PET Center (Director: Dr. P. Herscovitch) for radioisotope production and the PDSP for performing binding assays. The PDSP is directed by Bryan L. Roth, Ph.D., with project officer Jamie Driscol (NIMH), at the University of North Carolina at Chapel Hill (Contract No. NO1MH32004).
ABBREVIATIONS USED
- BZP
benzodiazepine
- Boc
tert-butyloxycarbonyl
- H
histamine
- K 2.2.2
kryptofix 2.2.2
- KOR
κ-opiate receptor
- MOR
μ-opiate receptor
- PDSP
Psychoactive Drug Screening Program
- PTFE
poly(tetrafluoroethylene)
- NCA
no-carrier-added
- NET
noradrenalin transporter
- NIMH
National Institute of Mental Health
- RCY
decay-corrected radiochemical yield
- SUV
standardized uptake value
- TEMPO
2,2,6,6-tetramethylpiperidine 1-oxyl
- TSPO
18 kDa transporter protein
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information Available: Chromatograms for the HPLC separation of [18F]11 and analyses of 11 and [18F]11, plus temperature time-courses for different reaction solvents under microwave irradiation. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Kew JN, Kemp JA. Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology (Berl) 2005;179:4–29. doi: 10.1007/s00213-005-2200-z. [DOI] [PubMed] [Google Scholar]
- 2.Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Ann Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 3.Pin JP, Acher F. The metabotropoic glutamate receptors; structure, activation, mechanism and pharmacology. Curr Drug Target CNS Neurol Disord. 2002;1:297–317. doi: 10.2174/1568007023339328. [DOI] [PubMed] [Google Scholar]
- 4.Mutel V, Ellis GJ, Adam G, Chaboz S, Nilly A, Messer J, Bleuel Z, Metzler V, Malherbe P, Schlaeger EJ, Roughley BS, Faull RL, Richards JG. Characterization of [3H]quisqualate binding to recombinant rat metabotropic glutamate 1a and 5a receptors and to rat and human brain sections. J Neurochem. 2000;75:2590–2601. doi: 10.1046/j.1471-4159.2000.0752590.x. [DOI] [PubMed] [Google Scholar]
- 5.Schkeryantz JM, Kingston AE, Johnson MP. Prospects for metabotropic glutamate antagonists in the treatment of neuropathic pain. J Med Chem. 2007;50:2563–2568. doi: 10.1021/jm060950g. [DOI] [PubMed] [Google Scholar]
- 6.Wu WL, Burnett DA, Domalski M, Greenlee WJ, Li C, Bertorelli R, Freduzzi S, Lozza G, Veltri A, Reggiani A. Discovery of orally efficacious tetracyclic metabotropic glutamate receptor 1 (mGluR1) antagonists for the treatment of chronic pain. J Med Chem. 2007;50:5550–5553. doi: 10.1021/jm070590c. [DOI] [PubMed] [Google Scholar]
- 7.Bordi F, Ugolini A. Group I metabotropic glutamate receptors: implications for brain diseases. Prog Neurobiol. 1999;59:55–79. doi: 10.1016/s0301-0082(98)00095-1. [DOI] [PubMed] [Google Scholar]
- 8.Conn PJ. Physiological roles and therapeutic potential of metabotropic glutamate receptors. Ann N Y Acad Sci. 2003;1003:12–21. doi: 10.1196/annals.1300.002. [DOI] [PubMed] [Google Scholar]
- 9.Moghaddam B. Targeting metabotropic glutamate receptors for treatment of the cognitive symptoms of schizophrenia. Psychopharmacol (Berl) 2004;174:39–44. doi: 10.1007/s00213-004-1792-z. [DOI] [PubMed] [Google Scholar]
- 10.Spooren W, Ballard T, Gasparini F, Amalric M, Mutel V, Schreiber R. Insight into the function of Group I and Group II metabotropic glutamate (mGlu) receptors: behavioural characterization and implications for the treatment of CNS disorders. Behav Pharmacol. 2003;14:257–277. doi: 10.1097/01.fbp.0000081783.35927.8f. [DOI] [PubMed] [Google Scholar]
- 11.Brown AK, Kimura Y, Zoghbi SS, Siméon FG, Liow JS, Kreisl WC, Taku A, Fujita M, Pike VW, Innis RB. Metabotropic glutamate subtype 5 receptors are quantified in the human brain with a novel radioligand for PET. J Nucl Med. 2008;49:2042–2048. doi: 10.2967/jnumed.108.056291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong DF, Waterhouse R, Kuwabara H, Kim J, Brasic JR, Chamroonrat W, Stabins M, Holt DP, Dannals RF, Hamill TG, Mozley PD. F-18-FPEB, a PET radiopharmaceutical for quantifying metabotropic glutamate 5 receptors: a first-in-human study of radiochemical safety, biokinetics, and radiation dosimetry. J Nucl Med. 2013;54:388–396. doi: 10.2967/jnumed.112.107995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pike VW. Positron-emitting radioligands for studies in vivo — probes for human psychopharmacology. J Psychopharmacology. 1993;7:139–158. doi: 10.1177/026988119300700202. [DOI] [PubMed] [Google Scholar]
- 14.Laruelle M, Slifstein M, Huang Y. Relationships between radiotracer properties and image quality in molecular imaging of the brain with positron emission tomography. Mol Imaging Biol. 2003;5:363–375. doi: 10.1016/j.mibio.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 15.Patel S, Gibson R. In vivo site-directed radiotracers: a mini-review. Nucl Med Biol. 2008;35:805–815. doi: 10.1016/j.nucmedbio.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Pike VW. PET Radiotracers: crossing the blood-brain barrier and surviving metabolism. Trends Pharm Sci. 2009;30:431–440. doi: 10.1016/j.tips.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang Y, Narendran R, Bischoff F, Guo N, Zhu Z, Bae SA, Lesage AS, Laruelle M. A positron emission tomography radioligand for the in vivo labeling of metabotropic glutamate 1 receptor: (3-ethyl-2-[11C]methyl-6-quinolinyl)(cis-4-methoxycyclohexyl)methanone. J Med Chem. 2005;48:5096–5099. doi: 10.1021/jm050263+. [DOI] [PubMed] [Google Scholar]
- 18.Ohgami M, Haradahira T, Takai N, Zhang MR, Kawamura K, Yamaski T, Yanamoto K. [18F]FTIDC: a new PET radioligand for metabotropic glutamate receptor 1. Eur J Nucl Med Mol Imaging. 2009;36:S310. [Google Scholar]
- 19.Prabhakaran J, Majo VJ, Milak MS, Kassir SA, Palner M, Savenkova L, Mali P, Arango V, Mann JJ, Parsey RV, Kumar JS. Synthesis, in vitro and in vivo evaluation of [11C]MMTP: a potential PET ligand for mGluR1 receptors. Bioorg Med Chem Lett. 2010;20:3499–3501. doi: 10.1016/j.bmcl.2010.04.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yanamoto K, Konno F, Odawara C, Yamasaki T, Kawamura K, Hatori A, Yui J, Wakizaka H, Nengaki N, Takei M, Zhang MR. Radiosynthesis and evaluation of [11C]YM-202074 as a PET ligand for imaging the metabotropic glutamate receptor type 1. Nucl Med Biol. 2010;37:615–624. doi: 10.1016/j.nucmedbio.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 21.Fujinaga M, Yamasaki T, Kawamura K, Kumata K, Hatori A, Yui J, Yanamoto K, Yoshida Y, Ogawa M, Nengaki N, Maeda J, Fukumura T, Zhang MR. Synthesis and evaluation of 6-[1-(2-[18F]fluoro-3-pyridyl)-5-methyl-1H-1,2,3-triazol-4-yl]quinoline for positron emission tomography imaging of the metabotropic glutamate receptor type 1 in brain. Bioorg Med Chem. 2011;19:102–110. doi: 10.1016/j.bmc.2010.11.048. [DOI] [PubMed] [Google Scholar]
- 22.Hostetler ED, Eng W, Joshi AD, Sanabria-Bohóquez S, Kawamoto H, Ito S, O’Malley S, Krause S, Ryan C, Patel S, Williams M, Riffel K, Suzuki G, Ozaki S, Ohta H, Cook J, Burns HD, Hargreaves R. Synthesis, characterization, and monkey PET studies of [18F]MK-1312, a PET tracer for quantification of mGluR1 receptor occupancy by MK-5435. Synapse. 2011;65:125–135. doi: 10.1002/syn.20826. [DOI] [PubMed] [Google Scholar]
- 23.Yamasaki T, Fujinaga M, Yoshida Y, Kumata K, Yui JJ, Kawamura K, Hatori A, Fukumura T, Zhang MR. Radiosynthesis and preliminary evaluation of 4-[18F]fluoro-N-[4-[6-(isopropylamino)pyrimidin-4-yl]-1,3-thiazol-2-yl]-Nmethylbenzamide as a new positron emission tomography ligand for metabotropic glutamate receptor subtype 1. Bioorg Med Chem Lett. 2011;21:2998–3001. doi: 10.1016/j.bmcl.2011.03.046. [DOI] [PubMed] [Google Scholar]
- 24.Fujinaga M, Yamasaki T, Yui J, Hatori A, Xie L, Kawamura K, Asagawa C, Kumata K, Yoshida Y, Ogawa M, Nengaki N, Fukumura T, Zhang MR. Synthesis evaluation of novel radioligands for positron emission tomography imaging of metabotropic glutamate receptor subtype 1 (mGluR1) in rodent brain. J Med Chem. 2012;55:2342–2452. doi: 10.1021/jm201590g. [DOI] [PubMed] [Google Scholar]
- 25.Huang Y, Narendran R, Bischoff F, Guo N, Bae SA, Hwang DR, Lesage AS, Laruelle M. Synthesis and characterization of two PET radioligands for the metabotropic glutamate 1 (mGlu1) receptor. Synapse. 2012;66:1002–1014. doi: 10.1002/syn.21606. [DOI] [PubMed] [Google Scholar]
- 26.Yamasaki T, Fujinaga M, Maeda J, Kawamura K, Yui J, Hatori A, Yoshida Y, Nagai Y, Tokunaga M, Higuchi M, Suhara T, Fukumura T, Zhang MR. Imaging for metabotropic glutamate receptor subtype 1 in rat and monkey brains using PET with [18F]FITM. Eur J Nucl Med Mol Imaging. 2012;39:632–641. doi: 10.1007/s00259-011-1995-6. [DOI] [PubMed] [Google Scholar]
- 27.Fujinaga M, Maeda J, Yui J, Hatori A, Yamasaki T, Kawamura K, Kumta K, Yoshida Y, Nagai Y, Higiuchi M, Suhara T, Fukumura T. Characterization of 1-(2-[18F]fluoro-3-pyridyl)-4-(2-isopropyl-1-oxo-isoindoline-5-yl)-5-methyl-1H-1, 2, 3-triazole a PET ligand for imaging the metabotropc glutamate receptor type 1 in rat monkey brains. J Neurochem. 2012;121:115–124. doi: 10.1111/j.1471-4159.2011.07348.x. [DOI] [PubMed] [Google Scholar]
- 28.Zanotti-Fregonara P, Barth VN, Liow JS, Zoghbi SS, Clark DT, Rhoads E, Siuda E, Heinz BA, Nisenbaum E, Dressman B, Joshi E, Luffer-Atlas D, Fisher MJ, Masters JJ, Goebl N, Kuklish SL, Morse C, Tauscher J, Pike VW, Innis RB. Evaluation and in vitro in animals of a new 11C-labeled PET radioligand for metabotropic glutamate receptors 1 in brain. Eur J Nucl Med Mol Imaging. 2013;40:245–253. doi: 10.1007/s00259-012-2269-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fujinaga M, Yamasaki T, Maeda J, Yui J, Xie L, Nagai Y, Nengaki N, Hatori A, Kumata K, Kawamura K, Zhang MR. Development of N-[4-[6-(isopropylamino)pyrimidin-4-yl]1, 3-thiazol-2-yl}-N-methyl-4-[11C]methylbenzamide for positron emission tomography imaging of metabotropic glutaamte 1 receptor in monkey brain. J Med Chem. 2012;55:11042–11051. doi: 10.1021/jm301597s. [DOI] [PubMed] [Google Scholar]
- 30.Zanotti-Fregonara P, Barth VN, Zoghbi SS, Liow J-S, Nisenbaum E, Siuda E, Gladding RL, Rallis-Frutos D, Morse C, Tauscher J, Pike VW, Innis RB. 11C-LY2428703, a positron emission tomographic radioligand for the metabotropic glutamate receptor 1 is unsuitable for imaging in monkey and human brain. Eur J Nucl Med Mol Imaging Res. 2013;3:47, 1–9. doi: 10.1186/2191-219X-3-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lavreysen H, Pereira SN, Leysen JE, Langlois W, Lesgae AS. Metabotropic glutamate receptor 1 receptor distribution and occupancy in the rat brain. A quantitative autoradiographic study using [3H]R214127. Neuropharmacology. 2004;46:609–619. doi: 10.1016/j.neuropharm.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 32.Sato A, Nagatomi Y, Hirat Y, Ito S, Suzuki G, Kimura T, Maehara S, Hikichi H, Satow A, Hata M, Ohta H, Kawamoto H. Discovery and in vitro and in vivo profiles of 4-fluoro-N-[4-[6-(isopropylamino)pyrimidin-4-yl]-1, 3-thiazol-2-yl]-N-methylbenzamide as novel class of an orally active metabotropic glutamate receptor 1 (mGluR1) antagonist. Bioorg Med Chem Lett. 2009;19:5464–5468. doi: 10.1016/j.bmcl.2009.07.097. [DOI] [PubMed] [Google Scholar]
- 33.Zoghbi SS, Anderson KB, Jenko KJ, Luckenbaugh DA, Innis RB, Pike VW. On quantitative relationships between drug-like compound lipophilicity and plasma free fraction in monkey and human. J Pharm Sci. 2012;101:1028–1039. doi: 10.1002/jps.22822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elsinga PH. Radiopharmaceutical chemistry for positron emission tomography. Methods. 2002;27:208–217. doi: 10.1016/s1046-2023(02)00076-2. [DOI] [PubMed] [Google Scholar]
- 35.Cai LS, Lu SY, Pike VW. Chemistry with [18F]fluoride ion. Eur J Org Chem. 2008:2853–2873. [Google Scholar]
- 36.Pike VW, Aigbirhio FI. Reactions of cyclotron-produced [18F]fluoride with diaryliodonium salts — a novel single-step route to no-carrier-added [18F]fluoroarenes. J Chem Soc, Chem Commun. 1995:2215–2216. [Google Scholar]
- 37.Shah A, Pike VW, Widdowson DA. The syntheses of [18F]fluoroarenes from the reaction of cyclotron-produced [18F]fluoride ion with diaryliodonium salts. J Chem Soc, Perkin Trans. 1998;1:2043–2046. [Google Scholar]
- 38.Ross L, Ermert J, Hocke C, Coenen HH. Nucleophilic 18F-fluorination of heteroaromatic iodonium salts with no-carrier-added [18F]fluoride ion. J Am Chem Soc. 2007;129:8018–8025. doi: 10.1021/ja066850h. [DOI] [PubMed] [Google Scholar]
- 39.Chun JH, Lu S, Lee YS, Pike VW. Fast and high-yield micro-reactor syntheses of ortho-substituted [18F]fluoroarenes from reactions of [18F]fluoride ion with diaryliodonium salts. J Org Chem. 2010;75:3332–3338. doi: 10.1021/jo100361d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chun J-H, Lu SY, Pike VW. Radiosyntheses of meta-substituted [18F]fluoroarenes from [18F]fluoride ion and diaryliodonium tosylates within a microreactor. Eur J Org Chem. 2011:4439–4447. doi: 10.1002/ejoc.201100382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang MR, Kumata K, Suzuki K. A practical route for synthesizing a PET ligand containing [18F]fluorobenzene using reaction of diphenyliodonium salt with [18F]F−. Tetrahedron Lett. 2007;48:8632–8635. [Google Scholar]
- 42.Telu S, Chun JH, Siméon FG, Lu S, Pike VW. Effective syntheses of mGluR5 PET radioligands through the radiofluorination of diaryliodonium salts. Org Biomol Chem. 2011;9:6629–6638. doi: 10.1039/c1ob05555k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moon BS, Kil HS, Park JH, Kim JS, Park J, Chi DY, Lee BC, Kim SE. Facile aromatic radiofluorination of [18F]flumazenil from diaryliodonium salts with evaluation of their stability selectivity. Org Biomol Chem. 2011;9:8346–8355. doi: 10.1039/c1ob06277h. [DOI] [PubMed] [Google Scholar]
- 44.Chin FT, Morse CL, Shetty HU, Pike VW. Automated radiosynthesis of [18F]SPA-RQ for imaging human brain NK1 receptors with PET. J Label Compd Radiopharm. 2006;49:17–31. [Google Scholar]
- 45.Pike VW, Butt F, Shah A, Widdowson DA. Facile synthesis of substituted diaryliodonium tosylates by treatment of aryltributylstannanes with Koser’s reagent. J Chem Soc, Perkin Trans I. 1999:245–248. [Google Scholar]
- 46.Chun JH, Pike VW. Regiospecific syntheses of functionalized diaryliodonium tosylates via [hydroxy(tosyloxy)iodo]arenes generated in situ from (diacetoxyiodo)arenes. J Org Chem. 2012;77:1931–1938. doi: 10.1021/jo202517v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carroll MA, Martina–Santamaria S, Pike VW, Rzepa HS, Widdowson DA. An ab initio and MNDO-d SCF-MO computational study of stereoelectronic control in extrusion reactions of R2-I-F intermediates. J Chem Soc, Perkin Trans. 1999;2:2707–2714. [Google Scholar]
- 48.Martín-Santamaria S, Carroll MA, Pike VW, Rzepa HS, Widdowson DA. An ab initio and MNDO-d SCF-MPO computational study of extrusion reactions of R2I-F iodine(III) via dimeric, trimeric and tetrameric transition states. J Chem Soc, Perkin Trans. 2000;2:2158–2161. [Google Scholar]
- 49.Shi Q, Savage JE, Hufeisen SJ, Rauser L, Grajkowska E, Ernsberger P, Wroblewski JT, Nadeau JH, Roth BL. L-Homocysteine sulfinic acid and other acidic homocysteine derivatives are potent and selective metabotropic glutamate receptor agonists. J Pharmacol Exp Ther. 2003;305:131–142. doi: 10.1124/jpet.102.047092. [DOI] [PubMed] [Google Scholar]
- 50.Carroll MA, Smith G, Widdowson DA. Radical scavengers: a practical solution to the reproducibility issue in the fluoridation of diaryliodonium salts. J Fluorine Chem. 2007;128:127–132. [Google Scholar]
- 51.Smith D. [Last accessed 12th June 2013.];Synthia gets extreme makeover courtesy of National Instruments. http://sine.ni.com/cs/app/doc/p/id/cs-11319.
- 52.Lazarova N, Siméon FG, Musachio JL, Lu SY, Pike VW. Integration of a microwave reactor with Synthia to provide a fully automated radiofluorination module. J Label Compd Radiopharm. 2007;50:463–465. [Google Scholar]
- 53.Lavreysen H, Wouters R, Bischoff F, Nobrega Pereira S, Langlois X, Blokland S, Somers M, Dillen S, Lesage AS. JNJ16259685, a highly potent, selective and sytemically active mGlu1 receptor antagonist. Neuropharmacology. 2004;46:961–972. doi: 10.1016/j.neuropharm.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 54.Zoghbi SS, Shetty UH, Ichise M, Fujita M, Imaizumi M, Liow JS, Shah J, Musachio JL, Pike VW, Innis RB. PET imaging of the dopamine transporter with [18F]FECNT: a polar radiometabolite confounds brain radioligand measurements. J Nucl Med. 2006;47:520–527. [PubMed] [Google Scholar]
- 55.Gandelman MS, Baldwin RM, Zoghbi SS, Zea-Ponce Y, Innis RB. Evaluation of ultrafiltration for the free-fraction determination of single photon emission computed tomography (SPECT) radiotracers: β-CIT, IBF, and iomazenil. J Pharm Sci. 1994;83:1014–1019. doi: 10.1002/jps.2600830718. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.