

Abstract
Oxidative stress and angiogenic factors have been placed as the prime focus of scientific investigations after an establishment of link between vascular endothelial growth factor promoter (VEGF), hypoxia, and amyotrophic lateral sclerosis (ALS) pathogenesis. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter and mutant superoxide dismutase 1 (SOD1) which are characterised by atrophy and muscle weakness resulted in phenotype resembling human ALS in mice. This results in lower motor neurodegeneration thus establishing an important link between motor neuron degeneration, vasculature, and angiogenic molecules. In this review, we have presented human, animal, and in vitro studies which suggest that molecules like VEGF have a therapeutic, diagnostic, and prognostic potential in ALS. Involvement of vascular growth factors and hypoxia response elements also highlights the converging role of oxidative stress and neurovascular network for understanding and treatment of various neurodegenerative disorders like ALS.
1. Introduction
At the developmental stages, the establishment of a neurovascular network, outside CNS, is crucial to the subsequent brain and spinal cord development. Molecules deserving special attention in the course of development and maintenance of neurovasculature include VEGF (especially VEGF-A)/VEGF receptors, Notch, ephrin, semaphorins/plexin receptors, latent transforming growth factor β's [TGFβ's], and TGFβ receptors, αvβ8 integrin, neuropilins, and FGF1 [1–3]. Any dysregulation in the pathways having the above mentioned factors (responsible for angiogenesis) which contributes to the development of this communication network has serious consequences manifesting in the form of CNS disorders. Hence angiogenesis is required for vasculature development and is governed by the gene expression of vascular molecules [4]. Abnormal expression and reduced levels of VEGF have been explored to account for devastating disorders of the CNS, especially in studies focused on ALS, which is designated by motor neuron degeneration and is fatal in nature [5]. Genetic studies in a transgenic mouse and rat model of ALS with mutated superoxide dismutase 1 SOD1G93A have indicated that inhibition of hypoxia response element (HRE) in the VEGF gene promoter may promote motor neuron degeneration (since HRE is responsible for inducing angiogenesis through VEGF as shown in Figure 1) whereas administration of VEGF prolongs survival [6]. Hence, here we review the role of neurotrophic and angiogenic factors like VEGF in the pathogenesis of ALS.
Figure 1.
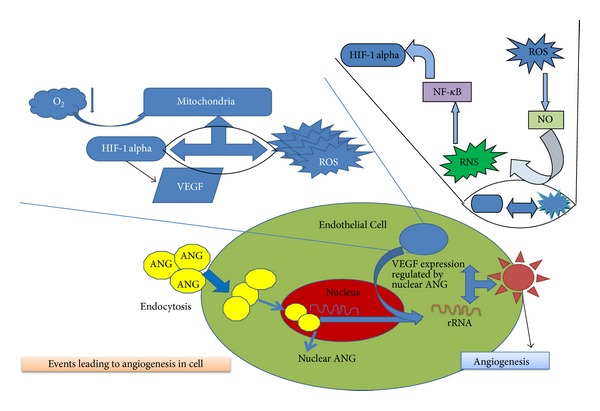
The role of hypoxia in stimulating the VEGF through an activation of HIF-1 alpha element. HIF-1 alpha gets activated in deficiency of oxygen in mitochondria leading to creation of oxidative stress. This involves the formation of reactive oxygen species which on reaction with free nitrogen forms NO ultimately leading to reactive nitrogen species (RNS). This RNS further activates NF-κB pathway which ultimately leads to activation of HIF-1 alpha factor. The activated form of HIF-1 alpha further leads to VEGF activation thus leading to angiogenesis.
2. ALS: A Fatal Disease of the Motor Neurons
Motor neuron disease (MND) defines conglomerate of related and progressive degenerative disorders characterized by selective degeneration of upper motor and lower motor neuron located in the motor cortex and brain stem and spinal cord, respectively [4]. The disease may either affect lower motor neuron (progressive muscular atrophy) or upper motor neurons (primary lateral sclerosis) or both upper/lower motor neurons (amyotrophic lateral sclerosis); however, careful pathological and clinical studies in MND have shown that extra-motor parts of the central nervous system are also affected. ALS is the most severe MND where selective degeneration of motor neurons leads to atrophy of voluntary muscles followed by paralysis and may prove fatal [5]. Mechanisms of selective degeneration of motor neurons in ALS are obscure. Largely, ALS symptoms include weakness of muscles, especially those in the hands, arms, and legs with or without dysarthria and dysphagia. Fasciculation or muscle twitching is also an important clinical finding [7].
3. ALS: Contributing Factors
ALS occurs in both sporadic and familial form at an incidence varying between 0.4 and 2.6 for every 100,000 individuals and a prevalence rate of 4–6 per 100,000 population per year [8]. The etiology of ALS has been elusive and believed to be multifactorial. Though causes of most cases of ALS are unknown, major factors include genetic factors like point mutations in superoxide dismutase 1 (SOD1) gene accounting for around 20% of familial ALS (fALS) cases [9]. The purely lower motor neuron (LMN) degeneration variant of ALS shows missense mutations in CHMP2B (charged multivesicular protein 2B; involved in cellular transport). In 10% cases of ALS, patients with CHMP2B mutations are shown to have lower motor neuron degeneration. Apart from this, other genes like vesicle-associated membrane protein B (VAPB) (which is involved in providing unfolded protein response to endoplasmic reticulum), senataxin (SETX) (gene present in central nervous system involving brain and spinal cord as well as muscle and play major role in DNA repair to maintain integrity of cell), and dynactin 1 (involved in cellular transport during cell division and specially in axonal transport of nerve cells) with mutations have been shown to play role in aggregate formation and hampering the normal activity of the motor neurons thus contributing to the pathogenesis of ALS overall in subject's body [10–14]. Genes encoding angiogenin (ANG) having missense mutations have also been involved in the pathogenesis of ALS. Angiogenin, like VEGF, is produced in response to hypoxia and plays a role in neovascularisation as shown in Figure 1. Its importance further stems from the fact that it can regulate the expression of VEGF [15, 16]. Hypoxia takes place when oxygen availability is low in cell due to which the mitochondria produces ROS species which in turn reacts with nitric oxide (NO) to produce reactive nitrogen species RNS and activates HIF-α pathway through NF-κB pathway resulting in stimulation of VEGF. The expression of this VEGF is dependent on the nucleolar ANG which directly helps in stimulating the proliferation of epithelial cells and helps in angiogenesis [17]. However, this hypothesis raises a question whether angiogenin crosses the blood brain barrier or is retained in cerebrospinal fluid [18].
Apart from genetic factors, the presence of insoluble intracellular protein aggregates in motor neurons and reactive astrocytes are considered as the hallmarks for the disease (Figure 3). [19]. The other factors include glutamate toxicity [20], lack of trophic growth factors [6, 21], autoimmunity [22], toxin [23], and susceptibility of motor neurons to neuro-degeneration because of their large size and high energy demands [24].
Figure 3.
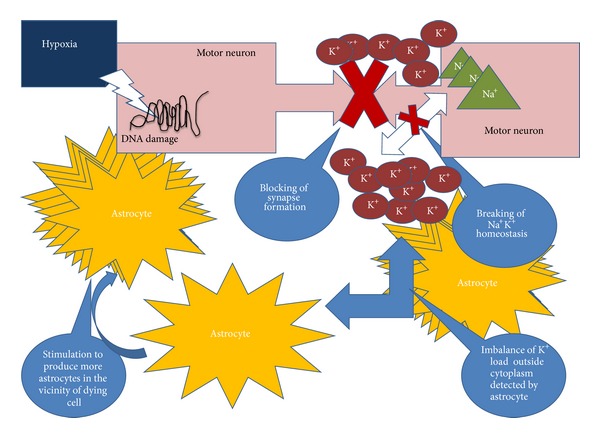
Hypoxia induced mobilisation of astrocytes. Astrogliosis is the result of aggressive increase of astrocytes number in the vicinity of damaged neuron cell. Synapse formation is hampered when there is neuronal damage thus leading to breakdown of Na+K+ homeostasis. This K+ concentration is detected by the astrocytes.
Currently, there is no treatment that could substantially alleviate the disease burden because of incomplete understanding of ALS etiology. Food and Drug Administration (FDA) has approved only single drug for the treatment of ALS, a glutamate antagonist that is Riluzole [25, 26]. Riluzole has also been studied as a potential inhibitor of VEGF induced endothelial cell proliferation under both in vitro and in vivo conditions [27]. Its neuroprotective effect via sodium channel blockage is brought about by the fact that this mechanism increases resistance to hypoxia through a reduction in energy demands (a decreased cerebral glucose consumption) [28].
4. VEGF: The Neurotrophic and Angiogenic Family
VEGFA gene in humans is positioned at chromosome 6p21.3 with eight exons and is expressed as several isoforms of different amino acid chain lengths because of alternative splicing (VEGF 121, VEGF 145, VEGF 165, VEGF 183, VEGF 189, VEGF 206) [29] that differ in their ability to bind heparin, neuropilin-1 (NP-1), and neuropilin-2 (NP-2). Two classes of receptors for VEGF are the tyrosine kinase and the nontyrosine kinase receptors. VEGFR1 (Flt-1 (fms-related tyrosine kinase 1)), VEGFR2 (KDR/Flk-1 (kinase insert domain receptor/fetal liver kinase-1), and VEGFR-3 (Flt-4) are three structurally related receptors present in tyrosine kinase class V, whereas neuropilin-1 (NP-1) and neuropilin-2 (NP-2) are part of nontyrosine kinase receptors. VEGF binds to NP 1 and 2 and VEGFR1 and 2 but not to VEGFR-3 as the latter one is not a receptor for VEGF. Studies indicate that for transmission of critical angiogenic signals in response to VEGF VEGFR2 plays the role of key mediator [30]. However in case of VEGFR1 the major function is prevention of VEGF binding to VEGFR1 thought to be done by a virtue of “decoy receptor” to negatively regulate angiogenesis [31]. Neuropilins (NP1 and 2) whose primary location is in central nervous system are described as receptor for collapsin/semaphorin family, which are responsible for controlling neuronal cell guidance [32, 33]. For VEGF165 and a coreceptor of VEGFR2 Neuropilin-1 (NP-1), it is a specific receptor whereas Neuropilin-2 (NP-2) binds VEGF165 and VEGF145 in isoform specific manner. VEGF is the part of genes which accommodate placental growth factor (PLGF), VEGFB, VEGFC, VEGFD, and VEGFE including VEGF-A, out of which lymphatic vessels development is affected by VEGF-C [34]. Recent evidence from studies also indicates that neural cells are directly affected by VEGF-A, VEGF-B and VEGF-C [35]. In ALS, VEGF has been studied as an important member of gene families impacting the pathology of disease.
5. VEGF: Molecular Risk Factor in ALS
The lack of trophic (growth) factors has been hypothesized as probable cause of ALS. Since growth factors are neurotrophic and help in growth, survival, and maintenance of neuronal cells. The hypoxia response brings together a cascade of events involving angiogenic and inflammatory factors (Figures 1 and 3). Studies have focussed on predicting/correlating disease state with changing levels of such factors in body fluids even though these have been conducted utilising heterogeneous controls.
VEGF and its receptors are reported to be localised in neurons and astrocytes [36, 37] which, in case of ischemia or spinal cord injuries, provides neuroprotection and stimulates neuronal growth. Decreased VEGF levels may impair perfusion and induce ischemia of motor neurons, other than depriving cells of important survival and neuroprotective signals which are VEGF dependent [6].
Cronin et al. reported elevated levels of serum angiogenin, but no change in serum VEGF levels was observed. The authors also failed to observe any correlation between serum angiogenin and VEGF levels [16]. In another study, the patients with limb onset and long duration of ALS showed higher concentration of CSF VEGF as compared to those with bulbar onset of ALS and patients with short duration illness, respectively [38]. It may be possible that significant increase in cerebrospinal fluid (CSF) VEGF levels may have protective role against over-excitation of motor neurons (excitotoxicity). This overexitation may be mediated by excessive accumulation of glutamate at synaptic cleft in patients with limb onset of ALS and those with long duration of the disease, since it was suggested that the increased levels of VEGF account for a compensatory mechanism and may be required to stabilize neuronal excitation [39]. The rationale was further supported by Bogaert et al. who reported that VEGF protects motor neuron against excitotoxicity by upregulating Glutamate receptor 2 [40]. Significantly, lower baseline CSF VEGF levels in case of patients with ALS in comparison to normal controls and neurologic controls during early phase of disease have been observed, suggesting the possible link of ALS pathogenesis with VEGF gene regulation [41].
Moreau et al. demonstrated that hypoxaemic ALS patients had lower VEGF levels in CSF from normoxaemic ALS patients. This happened due to an early defect in hypoxia induced factor-1 (HIF-1) mediated regulation of VEGF. In contrast, higher levels of VEGF in CSF were demonstrated in hypoxaemic neurological controls than normoxaemic neurological controls. Hypoxaemia severity in ALS is explained by dysregulation of VEGF in ALS. This association of VEGF expression and hypoxia (Figure 1) in ALS introduced a concept of incongruous response [42]. Nagata et al. failed to reproduce the above results as no significant difference was observed in CSF VEGF levels between ALS patients, normal controls, and controls with other neurological disorders [43]. It was argued by Cronin and coworkers that the conflicting reports of elevated, normal, and decreased VEGF might have resulted from different study designs and ELISA kit employed with varying diagnostic criteria of ALS patients, diverse clinical details of ALS patients including definite and probable forms of disease [16]. In a unique histochemical study, a markedly elevated level of VEGF was detected in the skin of ALS patients when compared with normal subjects suggesting a positive correlation of VEGF levels in skin and severity of ALS patients [44]. The finding suggests systemic dysregulation of VEGF expression in ALS. Recently, it has been observed that elevated levels of VEGFA in CSF, serum, and peripheral blood mononuclear cells may account for substantially prolonged life span of Indian ALS patients as compared to their Western counterparts [45–47]. Surprisingly, longer survival is shown in Indian ALS patients after onset (~9 year) of ALS [45, 46, 48, 49]. Further, reduced levels of soluble VEGFR1 (sVEGFR1), an inhibitory receptor of VEGF, have been observed in these patients, supporting the neurotropic nature of VEGF [50]. However, these results need confirmation in comparable Caucasian ALS population.
6. ALS: VEGF and Oxidative Stress
Lowering of VEGF levels places neural tissue at the risk of limited perfusion thus making way for motor neuron degeneration [51]. This degeneration is a direct consequence of the fact that the deficient oxygen and glucose levels created as a result of decreased vascular perfusion can hardly meet the energy demands of motor neurons [52]. Oxidative stress due to hypoperfusion has been reported in cases of other neurodegenerative disorders such as Alzheimer's disease [53]. Oxidative stress is one of the outcomes of hypoperfusion apart from energy failure as blood is known to carry several vital components essential for cell survival including glucose and ferritin. As glucose is able to readily cross blood brain barrier (BBB), the deficiency of blood flow leads to reduced supply of glucose to brain resulting in limited energy production for cells. Similarly, the deficiency of ferritin, which is responsible for binding of free iron, results in formation of reactive oxygen species as shown in Figure 2 [54]. At least one study has reported that the variable levels of VEGF lead to altered ferritin levels [55]. Therefore, it is safe to say that oxidative stress deserves special significance in the pathogenesis of neurodegenerative diseases like ALS since motor neurons are particularly susceptible to oxidative damage.
Figure 2.
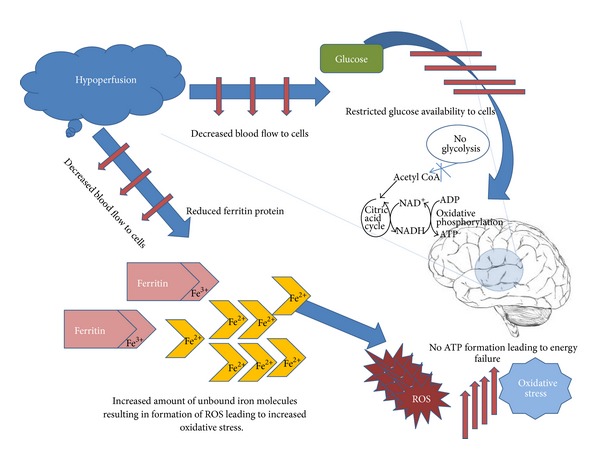
Role of hypoperfusion in elevation of oxidative stress and energy failure. As hypoperfusion reduces blood flow towards cells resulting in reduced ferritin Fe3+ protein, it releases unbound iron Fe2+ molecules resulting in formation of ROS thus increasing the oxidative stress. Hypoperfusion also leads to unavailability of glucose to brain cells thus leading to energy failure.
This significance is born out of the fact that the first evidence of association between ALS pathology and VEGF came when Oosthuyse et al. created homozygous VEGF (VEGF δ/δ) knock-in mice by introducing homozygous mutation of hypoxia response element (HRE) in the VEGF gene promoter to study angiogenic property of VEGF. They observed that almost 60% of mice did not survive before or around birth due to vasculature aberrations in lungs. The 40% who survived began to develop symptoms like classical ALS around five months of age [6]. This unusual finding compelled researchers to explore significance of growth factors in pathology of ALS utilising a variety of tools such as those discussed below.
6.1. Autopsy Based Studies
Spinal cord tissue analysis of ALS patients has revealed elevated dendritic cell marker transcripts (like CD83) and monocytic/macrophage/microglial transcripts [56], expression of cyclooxygenase-2 (COX-2) [57], connective tissue growth factor (CTGF) [58], monocyte chemoattractant protein-1 (MCP1) [56] and VEGF receptor (VEGFR)-1 [59], and activity of glutamate dehydrogenase (GDH) accompanied by reduced levels of glutamate and aspartate [60].
The increase in CTGF expression is explained by the fact that CTGF plays an important role in astrogliosis which is often seen as a consequence of hypoxic conditions and is therefore a pathological hallmark of ALS [58]. As depicted in Figure 3 astrogliosis is the result of aggressive increase of astrocytes number in the vicinity of damaged neuron cell. Hypoxia generally induces damage in the DNA of the neuronal cells. Since the neuronal damage has taken place its normal activity of synapse formation is hampered affecting the Na+K+ activity in those cells leading to breakdown of Na+K+ homeostasis. This change in balance of K+ concentration is detected by the astrocytes. This alteration results in the activation of astrocytes by initiation of clustering around the damaged cells in order to restore the functioning of those damaged cells [61–63].
Gliosis is also related to the enhanced GDH activity as reported by Malessa et al. [60]. The function of the GDH is to enhance the availability of the glutamate. This glutamate further acts as neurotransmitter or gliotransmitters since it increases the availability of Ca+ required by glial cells to perform their normal function of providing protection, nutrition, and avoiding accumulation of any chemicals involved in synapse formation which may later lead to toxication of neuron cell. Recruitment of glial cells to the site of damage may be considered as the body's primary response to save the dying neurons [64, 65], and thus the fact may be related to the point of association of enhanced GDH activity to gliosis. The authors also suggested a disturbance in cholinergic transmission in ALS spinal cord thus contributing to the reduced amino acid levels [60]. Glutamate and aspartate amino acids are linked with the neurotransmitters in the body. They are mainly the excitatory neurotransmitters, which utilise the Na+K+ pump to maintain their flow to the postsynaptic cleft during the nerve transmission. Li and Zhuo demonstrated that cholinergic transmitters play a role in inhibiting the glutamate based transmission. Release of acetylcholine leads to the activation of the muscarinic receptors, resulting in an inhibition of AMPA receptors (also called as glutamate receptors), and it increases the nonavailability of glutamate. This evidence also supports the fact mentioned in the above study that disturbance in cholinergic transmission may lead to reduced amino acid levels [66].
VEGF was first measured in spinal cord and serum of ALS patients by Nygren and colleagues. Authors did not observe any significant alteration in spinal cord VEGF levels, but they were able to observe higher serum VEGF levels in ALS patients in comparison to controls similar to those later reported by Gupta et al. in case of Indian ALS patients [46]. Considering the higher levels of VEGF in serum suggests that the cells other than central nervous system or which are not part of CNS are involved. In case of ALS skeletal muscles are the most affected region of body. Regional ischemia, a condition in which the blood supply is halted in specific region of brain, has been reported in case of ALS [67]. Rissanen et al. observed higher levels of VEGF in skeletal muscles with acute phase of ischemia [68]. Thus, it was hypothesized that VEGF is expressed in skeletal muscles in response to hypoxia and the increase was also reflected in serum [69].
The autopsy samples depict the terminal stage of the disease and provide a reliable proof of the disease and its signatures [70].
6.2. Muscle Biopsy Based Studies
In contrast to the increased cyclooxygenase (COX) activity in spinal cord of ALS patients, as discussed above, Crugnola et al. reported COX deficiencies in 46% patients, based on their histochemical analysis of muscle specimens. Moreover, molecular studies and biochemical analysis on the selected specimens displaying severe COX deficiencies even correlated with mutations in SOD1 and TARDBP genes and mitochondrial DNA defects thus pointing towards the secondary nature of COX deficiencies in the pathogenesis of ALS in light of the genetic nature of defects [71]. This is also confirmed by the findings of Vielhaber et al. who observed mitochondrial DNA damage in skeletal muscle, along with lowered levels of mitochondrial Mn-SOD [72]. The specific nature of mitochondrial dysfunction is further revealed by studying mitochondrial markers like citrate synthase and succinate dehydrogenase in muscle, histochemically. However, such a study by Krasnianski et al. revealed that one cannot narrow down the observed mitochondrial changes to only depict ALS but in fact view them as an indication of other neurogenic atrophies too [73]. In view of neurotrophic support provided by muscle tissue, the findings by Küst et al. depicted enhanced expression of nerve growth factor (NGF) and neurotrophins such as brain-derived neurotrophic factor (BDNF), in postmortem bicep tissue of ALS patients. Even so, externally administered neurotrophins have not shown promising results in human trials or animal models of ALS [74].
6.3. Polymorphism Based Studies
Increased oxidative stress implies consequent increased oxidative damage for motor neuronal DNA. Such oxidative damage of DNA is driven by the base excision repair (BER) system. One such product of oxidative damage of DNA is 8-hydroxy-2′-deoxyguanosine (8-OHdG) which is regulated by two enzymes, namely, human 8-oxoguanine DNA glycosylase 1 (hOGG1) and apurinic/apyrimidinic endonuclease APE1. Consequently, mutations and polymorphisms in coding area of genes coding for both of these enzymes are of interest to researchers. Concurrent oxidative stress conditions and a faulty DNA repair system are a risk factor for motor neurons.
In most studies concerning hOGG1 Ser326Cys polymorphism levels of 8-OHdG are taken into account as 8-OHdG is the product of DNA oxidation [75]. A study conducted by Chen et al. showed the reduced activity of hOGG1 in patients with 326 CC polymorphisms (P = 0.02) as compared to those with 326 SC polymorphisms (P = 0.05) [76]. Similar observations were made by authors in current study. In a Caucasian study, Coppedè et al. studied the distribution of allele frequencies and genotypes in sALS patients and controls for the hOGG1 Ser326Cys polymorphism in sALS patients and controls. The authors reported a significantly increased sALS risk associated with a combined Ser326Cys + Cys326Cys genotype. However, the Ser326Cys genotype showed nonsignificant results predicting that the hOGG1 Ser326Cys polymorphism in patient also pose a risk factor for ALS. Ser326Cys polymorphism takes place when at exon 7, position 1245 C to G substitution occurs and as a result S is substituted to C in codon 326.
Another interesting observation (though not significant as the test group of subjects used for the study was small, more significant results can be obtained if the study with large number of patients is conducted) in the above study was the fact that sALS patients as opposed to those bearing one or two copies of the 326Cys mutant allele bearing the Ser326Ser genotype displayed lower levels of AOPP (advanced oxidation protein products; believed to be stable markers of oxidative damage to proteins) [77]. Since abnormal levels of VEGF are implicated as risk factor in ALS, it is evident that mice with hypoxia response element deletion in vascular endothelial growth factor gene develop features reminiscent of ALS [5] although no spontaneous mutations have been observed in HRE in ALS patients [78, 79]. Large family-based and case-control cohort of North American white subjects (n = 1,603) were studied for the association of sALS with promoter polymorphisms of three VEGF genes. VEGF promoter polymorphisms do not find their casual role in ALS in light of absence of their association with sALS [80]. Risk of developing ALS has been associated to VEGF due to alterations in sequence in the promoter region of gene. In The Netherlands, 373 patients with sporadic ALS along with 615 matched healthy controls were found to have VEGF promoter haplotypes. No significant association between the previously reported at-risk haplotypes and ALS was found [81]. However, in some studies ALS has been found to be associated with VEGF C2578A polymorphism. In a study of Chinese population by Zhang et al. 115 sALS patients with 200 healthy individuals were analyzed for C2578A polymorphism (by amplifying 2705 to 2494 bps of VEGF gene promoter). Reports were in disagreement to previous studies from Caucasian populations as Chinese population did not fall susceptible for ALS due to C2578A polymorphism (attributing the effect to different genetic background in Chinese population) [82]. No significant association of ALS with three common VEGF variations [-2578C/A, -1154G/A, and -634G/C] in original form or in haplotype combination in a recent meta-analysis study comprising of over 7000 individuals involving three North American population and eight European populations was reported. However, in males -2578AA genotype increased the risk of ALS in subgroup analyses by gender [83] in contrast to a German study which suggested that risk of ALS in case of female patients might be higher as the VEGF role might be gender dependent [84]. Oates and Pamphlett did not observe any alteration of functioning of motor neurons by epigenetic transcriptional silencing of VEGF gene by methylation [85]. Additionally, screening of regulatory sequences of VEGFR2 found no association of polymorphism of VEGFR2 gene with risk of ALS [86]. Although association of VEGF with ALS has been well established by culture and animal studies, evidence from genetic studies in human cohorts suggests only a minor association between VEGF and the risk of developing ALS.
The role of VEGF involvement in ALS is questioned due to lack of association of VEGF genotypes and haplotypes in large meta-analysis study. Possibilities for VEGF role in predisposed patients to ALS cannot be ruled out. More studies are needed to discern the actual role of VEGF in pathogenesis of ALS.
6.4. Animal Model Based Studies
6.4.1. Primates
The concept of utilizing the cytotoxic properties of the extract obtained from the spinal cord of ALS patients was applied by Zil'ber et al. as early as in 1963 so as to reproduce the disease in rhesus monkeys. The authors could only conclude to a viral nature of this disease, but at the same time they recognised that the high incidence previously reported in the Chamorro tribe of Guam suggested a unique basis [87]. Another study conducted on rhesus monkeys attempted to validate the efficacy of bovine SOD as a therapeutic agent to compensate for the functions of the mutated form of the enzyme. SOD being a locally acting enzyme was administered intrathecally and intraventricularly so as to bypass the blood brain barrier. The injected bSOD showed commendable tolerance though its clearance was slower when compared with results obtained from rats. But the therapy when administered into a late stage FALS patient did not show promising results [88].
6.4.2. Rodents
The neuroprotective effect of VEGF suggests that exogenous VEGF administration may prevent degeneration of motor neuron. In a SOD1Gly93Ala rat model of ALS, it was shown that onset of paralysis was delayed by 17 days, improved motor performance, and extended lifespan by 22 days due to intracerebroventricular (i.c.v.) delivery of recombinant (VEGF). The study demonstrated the high scale effect in animal models of ALS achieved by protein delivery [89]. Intrathecal transplantation of human neural stem cells overexpressing VEGF increased the duration of survival of a transgenic ALS mouse model [90]. Similarly, mice after spinal cord ischemia showed susceptibility to paralysis in nervous tissue with reduced VEGF-A expression levels whereas after treatment with VEGF-A showed protective effect against ischemic motor neuron death [91]. These results unveil a therapeutic potential of VEGF for degenerating motor neurons in case of human ALS. In similar study authors investigating the protective role of VEGF during ischemia has shown to reduce infarct size, improve neurological performance, and enhance the survival of newborn neurons in the dentate gyrus and subventricular zone in adult rat brain with focal cerebral ischemia. Thus, VEGF shows acute neuroprotective effect, and prolongs survival of new neurons in the ischemic brain [92].
Zheng et al. demonstrated for the first time in Cu/Zn SOD1 transgenic mouse model of ALS that VEGF delayed diseased symptoms progression and prolonged survival, suggesting the importance of VEGF or related compounds in the treatment of ALS patients [93]. Rats having VEGF treatment showed significantly improved performance up to 6 weeks after spinal cord contusion injury compared with control animals. Furthermore, the group showed that VEGF treated animals had increased amount of spared tissue in the lesion centre with higher blood vessel density in parts of the wound area compared to controls, proving neurogenic and angiogenic capacity of VEGF [94]. Enhanced expression of VEGF by intramuscular administration of zinc finger transcription factor in SOD1 rats has been shown to improve functional disability [95]. Nitric oxide is known to decrease pressure in blood vessels [96], and it is possible that low VEGF adversely affects vasculature via changing the amount of nitric oxide released from endothelial cells, which further impairs perfusion and causes ischemic damage of motor neurons [91]. Moreover, decreased flow of blood has been observed in patients with ALS [97]. Both mechanisms may contribute to adult-onset progressive degeneration of motor neurons, muscle weakness, paralysis, and death, a typical feature of amyotrophic lateral sclerosis. It was earlier demonstrated that exposure to low levels of lead prolongs survival of ALS transgenic mouse, possibly mediated by upregulation of VEGF, which in turn reduces astrocytosis [98]. In another case retrograde delivery of lentivirus into mouse model of ALS prolonged survival in animals. Authors reported that lentivirus helped in stimulation of VEGF levels during diseased condition in animals [99]. Although in ALS animal models VEGF delivery has been successful, dose of delivery of VEGF should be adequately optimized to prevent adverse effects on the vascular system. It is possible that levels of VEGF higher than a certain threshold value may increase leakiness of blood vessels and modulate permeability of blood brain barrier [100] and therefore result in intrathecal accumulation of fluid. The presence of the blood breakdown product hemosiderin in and around spinal cord motor neurons supports increased leakiness and malformed blood vessels in ALS mouse models [101].
It must be noted that a drawback with using SOD1 based transgenic models is that SOD1 gene mutations represent only 20% of cases of familial ALS, which themselves represent just 10% of the total ALS cases. Therefore, remaining 90% of ALS cases, sporadic in nature, are difficult to mimic using such animal models [70].
6.5. Cell Culture Based Studies
Owing to a translational gap from animal models of ALS to humans, in vitro investigations utilising human motor neurons and astrocytes purified from the human embryonic spinal cord anterior horns allow for greater manipulations and are therefore a critical tool in discerning mechanisms pertaining to motor neuron degeneration in ALS [102].
The mRNA level of VEGF has been an important marker to analyse the role of VEGF in ALS. Destabilization and downregulation of VEGF mRNA with concomitant loss of protein expression in glial cells expressing mutant SOD1 in vitro are in consensus with many reports on the role of reduced VEGF expression in ALS pathogenesis [103]. In contrast, it was reported that hypoxia induced proteins bind and stabilize VEGF mRNA transcript resulting in increased expression of VEGF as a compensatory protective mechanism in later stages of disease [104].
The potential role of VEGF in preventing cell death by SOD-1 mutation has been studied in NSC-34 motor neuron cell line from mouse. Infection by adenovirus containing mutant Gly93Ala-SOD1 was shown to increase cell death and cellular oxidative stress. However, VEGF showed a dose dependent resistance to oxidative damage from hydrogen peroxide, TNF-alpha, and mutant Gly93Ala-SOD1 in NSC-34 cells treated with VEGF. Both phosphoinositide-3-kinase (PI3-K) and mitogen activated protein kinase (MAPK) activities in mouse NSC-34 motor neuron-like cells were activated by VEGF [105]. Recently, a culture study using primary culture of SOD1 mutated rat motor neurons has shown that decrease in VEGF before or during motor neuron degeneration amplifies the risk of mutated SOD1 induced toxicity in motor neurons [106]. Thus, the in vitro study shows VEGF as an antiapoptotic molecule. Overexpression of VEGF in the hippocampus using recombinant adeno associated virus vector in adult rats has been reported to result in improved cognition in association with approximately 2-fold increase in neurogenesis. Moreover, environmental induction of neurogenesis is completely blocked RNA interference based inhibition of VEGF expression. This data supports a model whereby VEGF acting via kinase insert domain receptor (KDR) is a mediator of the effect of the environment on neurogenesis and cognition [107]. Meng et al. investigated in vitro the proliferation and differentiation of subventricular zone neural progenitors of adult mouse by virtue of direct effect of VEGF. Downregulation of endogenous VEGF receptors 1 and 2, in association with reduced neural progenitor cell proliferation and enhanced neuronal differentiation, was reported as a result of high dose (500 ng/mL) of VEGF, whereas endogenous VEGF receptors 1 and 2 were significantly upregulated without increased proliferation and differentiation at low dose (50 ng/mL) of VEGF. Above given experiments suggest that VEGF regulates neurogenesis and its high dose enhances adult neural progenitor cell differentiation into neurons showing exogenous VEGF to exert a biphasic effect on the expression of endogenous VEGF receptors [108]. It has been shown that VEGF induces differentiation of stem cells in endothelial cells which in turn secrete various neurotrophic factors and infers a novel mechanism of neuroprotection by VEGF [109]. Apart from VEGF, recently, VEGFB was shown to protect cultured primary motor neurons. Further, it was observed that mutated SOD1 ALS mouse without VEGFB gene developed more severe form of ALS than ALS mouse with VEGFB [110].
7. VEGF in Blood Brain Barrier (BBB) and Blood Spinal Cord Barrier (BSCB)
Blood brain barrier (BBB) is the only checkpoint that stops inflammatory agents to reach central nervous system (CNS), as it contains a balanced interaction of microvascular endothelial cells and other components such as astrocytes, pericytes, neurons, and basement membrane. These components are collectively called as neovascular unit NVU. Tight junctions among NVU make the entry of undesirable components restricted to CNS [111]. BBB breakdown may lead to disruption of various biochemical reactions or may lead to accumulation of various inflammatory proteins that may aggravate the disease conditions of CNS [112–114]. Similarly, blood brain and spinal cord barrier which can be a morphological isotype for BBB performs same function in separating the spinal cord from all harmful components that may lead to diseased conditions of nervous system [115]. It has been observed that in case of human and animal model studies both the infiltration of brain and spinal cord with T cell, dendritic cells, or IgG have resulted in degeneration of motor neurons [116]. Claudins play a major role in forming tight junctions in the body among the cells to function as a barrier or act as a filter for these inflammatory factors to enter CNS [117]. Earlier studies have shown that astrocytes produce certain chemokines which play a role in attracting the dendritic cells to the CNS [118]. Recently, a link between the reactive astrocytes and disruption of these barriers has been reported. Argaw et al. tried to examine a link between astrocyte derived VEGFA and BBB permeability. Astrocytic expression of HIF-alpha and VEGFA leads to downregulation of claudins CLN-5 and their regulatory protein OCLN [119]. VEGFA, by the virtue of tyrosine phosphorylation, downregulates the expression of CLN ultimately resulting in disruption of permeability barrier. VEGF induces the migration among the endothelial cells and increasing the permeability to CNS [120] (Figure 4). However, theis link of VEGF is conflicting with the earlier reports in this paper regarding the protective role of VEGF in ALS pathogenesis.
Figure 4.
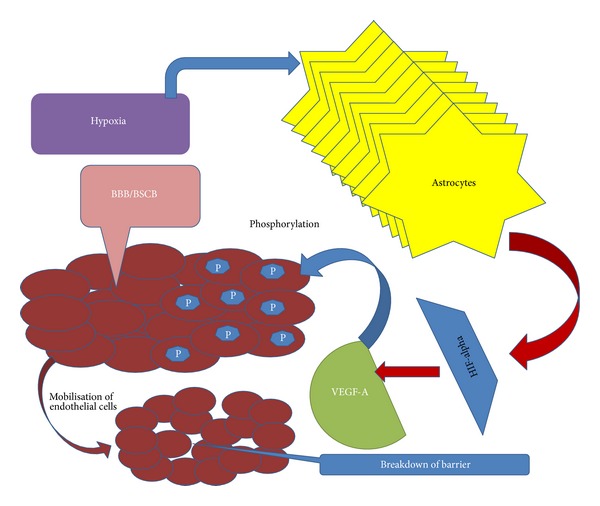
VEGF and permeability of blood brain barrier. Astrocytic expression of HIF-alpha and VEGFA leads to downregulation of claudins CLN-5 and their regulatory protein OCLN. VEGFA, by the virtue of tyrosine phosphorylation, downregulates the expression of CLN ultimately resulting in disruption of permeability barrier. VEGF induces the migration among the endothelial cells and increases the permeability to CNS.
8. Natural Products and Regulation of VEGF Expression
Naturally occurring compounds are also in current focus to examine their role in VEGF expression. The mechanism has been postulated to be common in all cases for those which are known to be responsible for increase in the expression of VEGF. All of them have been shown to affect the HIF pathway inducing the expression of VEGF. It is not clear how these natural compounds can be successfully translated for clinical use in near future which will need more studies. Certain extracts like turmeric, gigko biloba, and ginseng have been shown in mice studies to delay the disease onset or prolong survival in mice studies. However, recently a group from China reported a component Baicalin in the roots of plant Scutellaria baicalensis which enhances the expression of VEGF [121]. Although the HIF expression was less as compared to VEGF, authors reported that other transcription factors such as oestrogen-related receptors (EERs) exert their effects via VEGF promoters. Peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α), an important molecule independent of activator, is shown to interact with ERRα [122, 123]. These PGC-1α are shown to enhance expression of VEGF in cultured muscle cells in vivo in HIF independent pathway [121, 124]. In contrast, grape seed extract (GSE) is known for its antitumor properties and is shown useful in case of breast, lung, skin, or gastrointestinal cancer [125–127]. Lu and group recently showed that GSE reduced the VEGF expression by inhibiting the HIF expression in human breast tissue cancer cells. Authors argued that it involved the blockade of HIF expression by inhibiting AKT-3 pathway normally known for supporting the cell survival [128]. Apart from these other natural components have been shown to provide nonsatisfactory results in certain trials conducted in different human population. Vitamin E the most commonly studied antioxidant has been implicated with role of slowing down the disease progression in its severe form. Desnuelle C and colleagues conducted a study in French population of ALS patients. 289 patients were recruited for the study. All of them were randomly assigned the dose of Vitamin E and were assessed after every 3 months. The results did not show the effect in survival of muscle cells, except for the fact that patients who were administered the Vitamin E stayed in the milder form of disease for longer time [129]. Another study done in German population with high dose of Vitamin-E (5000 mg/day) showed ineffective results in comparison to the placebo effect [130]. However, in one of the meta-analyses of 23 studies published in year 2008, it was stated that antioxidants whether in combination or during individual administration do not show effective results [131]. Creatine one of the sports supplement has been known to increase the muscle strength. One study that came up in 1999 was conducted in animal model of ALS. Transgenic mice of ALS was administered with creatine dose. Authors reported that creatine was helpful in saving the mice neurons from dying in the age of 120 days. The group reported that creatine was also helpful in saving the mice from oxidative stress as well [132]. Later in 2004 a translational study performed with the same idea in human subjects demonstrated totally opposite results. 175 probable laboratory supported ALS patients were administered the 10 gm dose of creatine daily. The study showed no effect on survival rate neither it helped in reviving the rate of functional activities in patients [133]. Cannabinoid another naturally produced chemical present in humans as well as animals was studied by a group in 2004 in ALS mice model. They reported that Cannabinoid helped in prolonging survival of animals. Authors also reported reduced oxidative damage in spinal cord cell cultures of ALS mice and showed that it acts as an antiexcitotoxic agent in vitro [134]. Similar results have been shown in case of synthetically produced chemical called as cannabinol with dosage of 5 mg/kg/day for over a period of 12 weeks although no effect was there on survival [135]. Details for the functioning of these agents have not been mentioned but all of them report to choose or affect the oxidative stress pathway, although the oxidative stress based stimulation pathway by these compounds for VEGF cannot be ignored. Several studies report a common path for VEGF enhanced expression, but validity of usefulness of these natural components in case of ALS still needed to be studied.
9. Concluding Remarks
The human, animal, and culture studies have shown that VEGF could be a promising therapeutic target in ALS. Upregulation of VEGF by different means such as genetic engineering, transplantation of stem cells overexpressing VEGF, and/or direct infusion of VEGF may rescue the damage of motor neurons and enhance the survival of patients with ALS either by increasing blood perfusion or direct neuroprotective effect on motor neurons. However, additional blinded preclinical studies of VEGF, particularly among primates, are still needed in ALS and other neurodegenerative disorders including Alzheimer's and Parkinson's disease before starting clinical trials. Regardless of the conflicting reports describing the role of oxidative stress and role of VEGF in various ALS investigations, both human and in vivo studies suffer from longitudinal analysis including the prospective nutritional interventional studies. Besides, the patient oriented genetic profiling studies have failed to include large cohort of homogeneous populations thus impacting the understanding of the demographic-SNP link in motor neuron degeneration. Nonpharmacological therapeutic approaches in ALS have not been adequately addressed and need new research focus for development of therapeutics.
Acknowledgments
Akshay Anand and Keshav Thakur are the first authors. The authors thank Dr. Peter Carmeliet and Dr. Strokebaum for reviewing the paper and providing their expert comments.
References
- 1.James JM, Gewolb C, Bautch VL. Neurovascular development uses VEGF-A signaling to regulate blood vessel ingression into the neural tube. Development. 2009;136(5):833–841. doi: 10.1242/dev.028845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bautch VL, James JM. Neurovascular development: the beginning of a beautiful friendship. Cell Adhesion and Migration. 2009;3(2):199–204. doi: 10.4161/cam.3.2.8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCarty JH. Integrin-mediated regulation of neurovascular development, physiology and disease. Cell Adhesion and Migration. 2009;3(2):211–215. doi: 10.4161/cam.3.2.7767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lambrechts D, Carmeliet P. VEGF at the neurovascular interface: therapeutic implications for motor neuron disease. Biochimica et Biophysica Acta. 2006;1762(11-12):1109–1121. doi: 10.1016/j.bbadis.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 5.Dugdale DC, Hoch DB, Zieve D. Amyotrophic Lateral Sclerosis. A.D.A.M. Medical Encyclopedia; 2010. [Google Scholar]
- 6.Oosthuyse B, Moons L, Storkebaum E, et al. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nature Genetics. 2001;28(2):131–138. doi: 10.1038/88842. [DOI] [PubMed] [Google Scholar]
- 7.Mills KR. Characteristics of fasciculations in amyotrophic lateral sclerosis and the benign fasciculation syndrome. Brain. 2010;133(11):3458–3469. doi: 10.1093/brain/awq290. [DOI] [PubMed] [Google Scholar]
- 8.Román GC. Neuroepidemiology of amyotrophic lateral sclerosis: clues to aetiology and pathogenesis. Journal of Neurology Neurosurgery and Psychiatry. 1996;61(2):131–137. doi: 10.1136/jnnp.61.2.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 10.Cox LE, Ferraiuolo L, Goodall EF, et al. Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS) PLoS ONE. 2010;5(3, article e9872) doi: 10.1371/journal.pone.0009872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chance PF, Rabin BA, Ryan SG, et al. Linkage of the gene for an autosomal dominant form of juvenile amyotrophic lateral sclerosis to chromosome 9q34. The American Journal of Human Genetics. 1998;62(3):633–640. doi: 10.1086/301769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen YZ, Bennett CL, Huynh HM, et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4) The American Journal of Human Genetics. 2004;74(6):1128–1135. doi: 10.1086/421054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishimura AL, Mitne-Neto M, Silva HCA, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. The American Journal of Human Genetics. 2004;75(5):822–831. doi: 10.1086/425287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puls I, Jonnakuty C, LaMonte BH, et al. Mutant dynactin in motor neuron disease. Nature Genetics. 2003;33(4):455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- 15.Fernández-Santiago R, Hoenig S, Lichtner P, et al. Identification of novel Angiogenin (ANG) gene missense variants in German patients with amyotrophic lateral sclerosis. Journal of Neurology. 2009;256(8):1337–1342. doi: 10.1007/s00415-009-5124-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cronin S, Greenway MJ, Ennis S, et al. Elevated serum angiogenin levels in ALS. Neurology. 2006;67(10):1833–1836. doi: 10.1212/01.wnl.0000244466.46020.47. [DOI] [PubMed] [Google Scholar]
- 17.Kishimoto K, Yoshida S, Ibaragi S, et al. Hypoxia-induced up-regulation of angiogenin, besides VEGF, is related to progression of oral cancer. Oral Oncology. 2012;48(11):1120–1127. doi: 10.1016/j.oraloncology.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 18.Lambrechts D, Lafuste P, Carmeliet P, Conway EM. Another angiogenic gene linked to amyotrophic lateral sclerosis. Trends in Molecular Medicine. 2006;12(8):345–347. doi: 10.1016/j.molmed.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 19.Barbeito LH, Pehar M, Cassina P, et al. A role for astrocytes in motor neuron loss in amyotrophic lateral sclerosis. Brain Research Reviews. 2004;47(1–3):263–274. doi: 10.1016/j.brainresrev.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 20.Plaitakis A, Caroscio JT. Abnormal glutamate metabolism in amyotrophic lateral sclerosis. Annals of Neurology. 1987;22(5):575–579. doi: 10.1002/ana.410220503. [DOI] [PubMed] [Google Scholar]
- 21.Kaspar BK, Lladó J, Sherkat N, Rothstein JD, Gage FH. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301, article 5634:839–842. doi: 10.1126/science.1086137. [DOI] [PubMed] [Google Scholar]
- 22.Niebroj-Dobosz I, Jamrozik Z, Janik P, Hausmanowa-Petrusewicz I, Kwieciński H. Anti-neural antibodies in serum and cerebrospinal fluid of amyotrophic lateral sclerosis (ALS) patients. Acta Neurologica Scandinavica. 1999;100(4):238–243. doi: 10.1111/j.1600-0404.1999.tb00387.x. [DOI] [PubMed] [Google Scholar]
- 23.Murch SJ, Cox PA, Banack SA, Steele JC, Sacks OW. Occurrence of β-methylamino-L-alanine (BMAA) in. ALS/PDC patients from Guam. Acta Neurologica Scandinavica. 2004;110(4):267–269. doi: 10.1111/j.1600-0404.2004.00320.x. [DOI] [PubMed] [Google Scholar]
- 24.Shaw PJ, Eggett CJ. Molecular factors underlying selective vulnerability of motor neurons to neurodegeneration in amyotrophic lateral sclerosis. Journal of Neurology. 2000;247(supplement 1):I17–I27. doi: 10.1007/BF03161151. [DOI] [PubMed] [Google Scholar]
- 25.Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. The New England Journal of Medicine. 1994;330(9):585–591. doi: 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]
- 26.Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. The Lancet. 1996;347(9013):1425–1431. doi: 10.1016/s0140-6736(96)91680-3. [DOI] [PubMed] [Google Scholar]
- 27.Yoo MH, Hyun HJ, Koh JY, Yoon YH. Riluzole inhibits VEGF-induced endothelial cell proliferation in vitro and hyperoxia-induced abnormal vessel formation in vivo . Investigative Ophthalmology and Visual Science. 2005;46(12):4780–4787. doi: 10.1167/iovs.05-0376. [DOI] [PubMed] [Google Scholar]
- 28.Obrenovitch TP. Amyotrophic lateral sclerosis, excitotoxicity and riluzole. Trends in Pharmacological Sciences. 1998;19(1):9–11. doi: 10.1016/s0165-6147(97)01124-3. [DOI] [PubMed] [Google Scholar]
- 29.Takahashi H, Shibuya M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clinical Science. 2005;109(3):227–241. doi: 10.1042/CS20040370. [DOI] [PubMed] [Google Scholar]
- 30.Shalaby F, Rossant J, Yamaguchi TP, et al. Failure of blood-island formation and vasculogenesis in Flk-1 deficient mice. Nature. 1995;376(6535):62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 31.Hiratsuka S, Minowa O, Kuno J, Noda T, Shibuya M. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(16):9349–9354. doi: 10.1073/pnas.95.16.9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fujisawa H. Roles of neuropilinneuropilin and plexin in the development of nervous system. Tanpakushitsu kakusan koso. 1997;42(3, supplement 1):584–588. [PubMed] [Google Scholar]
- 33.Neufeld G, Cohen T, Shraga N, Lange T, Kessler O, Herzog Y. The neuropilins: multifunctional semaphorin and VEGF receptors that modulate axon guidance and angiogenesis. Trends in Cardiovascular Medicine. 2002;12(1):13–19. doi: 10.1016/s1050-1738(01)00140-2. [DOI] [PubMed] [Google Scholar]
- 34.Olsson A-K, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling—in control of vascular function. Nature Reviews Molecular Cell Biology. 2006;7(5):359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 35.Raab S, Plate KH. Different networks, common growth factors: shared growth factors and receptors of the vascular and the nervous system. Acta Neuropathologica. 2007;113(6):607–626. doi: 10.1007/s00401-007-0228-3. [DOI] [PubMed] [Google Scholar]
- 36.Lennmyr F, Ata KA, Funa K, Olsson Y, Terént A. Expression of vascular endothelial growth factor (VEGF) and its receptors (Flt-1 and Flk-1) following permanent and transient occlusion of the middle cerebral artery in the rat. Journal of Neuropathology and Experimental Neurology. 1998;57(9):874–882. doi: 10.1097/00005072-199809000-00009. [DOI] [PubMed] [Google Scholar]
- 37.Yang X, Cepko CL. Flk-1, a receptor for vascular endothelial growth factor (VEGF), is expressed by retinal progenitor cells. Journal of Neuroscience. 1996;16(19):6089–6099. doi: 10.1523/JNEUROSCI.16-19-06089.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iłzecka J. Cerebrospinal fluid vascular endothelial growth factor in patients with amyotrophic lateral sclerosis. Clinical Neurology and Neurosurgery. 2004;106(4):289–293. doi: 10.1016/j.clineuro.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 39.McCloskey DP, Hintz TM, Scharfman HE. Modulation of vascular endothelial growth factor (VEGF) expression in motor neurons and its electrophysiological effects. Brain Research Bulletin. 2008;76(1-2):36–44. doi: 10.1016/j.brainresbull.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bogaert E, van Damme P, Poesen K, et al. VEGF protects motor neurons against excitotoxicity by upregulation of GluR2. Neurobiology of Aging. 2010;31(12):2185–2191. doi: 10.1016/j.neurobiolaging.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 41.Devos D, Moreau C, Lassalle P, et al. Low levels of the vascular endothelial growth factor in CSF from early ALS patients. Neurology. 2004;62(11):2127–2129. doi: 10.1212/01.wnl.0000129913.44351.a3. [DOI] [PubMed] [Google Scholar]
- 42.Moreau C, Devos D, Brunaud-Danel V, et al. Paradoxical response of VEGF expression to hypoxia in CSF of patients with ALS. Journal of Neurology, Neurosurgery and Psychiatry. 2006;77(2):255–257. doi: 10.1136/jnnp.2005.070904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagata T, Nagano I, Shiote M, et al. Elevation of MCP-1 and MCP-1/VEGF ratio in cerebrospinal fluid of amyotrophic lateral sclerosis patients. Neurological Research. 2007;29(8):772–776. doi: 10.1179/016164107X229795. [DOI] [PubMed] [Google Scholar]
- 44.Suzuki M, Watanabe T, Mikami H, et al. Immunohistochemical studies of vascular endothelial growth factor in skin of patients with amyotrophic lateral sclerosis. Journal of the Neurological Sciences. 2009;285(1-2):125–129. doi: 10.1016/j.jns.2009.06.021. [DOI] [PubMed] [Google Scholar]
- 45.Gupta PK, Prabhakar S, Abburi C, Sharma NK, Anand A. Vascular endothelial growth factor-A and chemokine ligand (CCL2) genes are upregulated in peripheral blood mononuclear cells in Indian amyotrophic lateral sclerosis patients. Journal of Neuroinflammation. 2011;8, article 114 doi: 10.1186/1742-2094-8-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gupta PK, Prabhakar S, Sharma S, Anand A. Vascular endothelial growth factor-A (VEGF-A) and chemokine ligand-2 (CCL2) in amyotrophic lateral sclerosis (ALS) patients. Journal of Neuroinflammation. 2011;8, article 97 doi: 10.1186/1742-2094-8-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gupta PK, Prabhakar S, Sharma S, Anand A. A predictive model for amyotrophic lateral sclerosis (ALS) diagnosis. Journal of the Neurological Sciences. 2012;312(1-2):68–72. doi: 10.1016/j.jns.2011.08.021. [DOI] [PubMed] [Google Scholar]
- 48.Nalini A, Thennarasu K, Gourie-Devi M, Shenoy S, Kulshreshtha D. Clinical characteristics and survival pattern of 1,153 patients with amyotrophic lateral sclerosis: experience over 30 years from India. Journal of the Neurological Sciences. 2008;272(1-2):60–70. doi: 10.1016/j.jns.2008.04.034. [DOI] [PubMed] [Google Scholar]
- 49.Bradley WG. Commentary on Professor Stephen Hawking's disability advice. Annals of Neurosciences. 2009;16:101–102. [Google Scholar]
- 50.Anand A, Gupta PK, Sharma NK, Prabhakar S. Soluble VEGFR1 (sVEGFR1) as a novel marker of amyotrophic lateral sclerosis (ALS) in the North Indian ALS patients. The European Journal of Neurology. 2012;19(5):788–792. doi: 10.1111/j.1468-1331.2011.03548.x. [DOI] [PubMed] [Google Scholar]
- 51.Skene JHP, Cleveland DW. Hypoxia and lou gehrig. Nature Genetics. 2001;28(2):107–108. doi: 10.1038/88805. [DOI] [PubMed] [Google Scholar]
- 52.Storkebaum E, Lambrechts D, Carmeliet P. VEGF: once regarded as a specific angiogenic factor, now implicated in neuroprotection. BioEssays. 2004;26(9):943–954. doi: 10.1002/bies.20092. [DOI] [PubMed] [Google Scholar]
- 53.Aliev G, Smith MA, Obrenovich ME, de la Torre JC, Perry G. Role of vascular hypoperfusion-induced oxidative stress and mitochondria failure in the pathogenesis of Alzheimer disease. Neurotoxicity Research. 2003;5(7):491–504. doi: 10.1007/BF03033159. [DOI] [PubMed] [Google Scholar]
- 54.Orino K, Lehman L, Tsuji Y, Ayaki H, Torti SV, Torti FM. Ferritin and the response to oxidative stress. Biochemical Journal. 2001;357(1):241–247. doi: 10.1042/0264-6021:3570241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harned J, Ferrell J, Lall MM, et al. Altered ferritin subunit composition: change in iron metabolism in lens epithelial cells and downstream effects on glutathione levels and VEGF secretion. Investigative Ophthalmology and Visual Science. 2010;51(9):4437–4446. doi: 10.1167/iovs.09-3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Henkel JS, Engelhardt JI, Siklós SL, et al. Presence of dendritic cells, MCP-1, and activated Microglia/Macrophages in amyotrophic Lateral sclerosis spinal cord tissue. Annals of Neurology. 2004;55(2):221–235. doi: 10.1002/ana.10805. [DOI] [PubMed] [Google Scholar]
- 57.Maihöfner C, Probst-Cousin S, Bergmann M, Neuhuber W, Neundörfer B, Heuss D. Expression and localization of cyclooxygenase-1 and -2 in human sporadic amyotrophic lateral sclerosis. The European Journal of Neuroscience. 2003;18(6):1527–1534. doi: 10.1046/j.1460-9568.2003.02879.x. [DOI] [PubMed] [Google Scholar]
- 58.Spliet WGM, Aronica E, Ramkema M, Aten J, Troost D. Increased expression of connective tissue growth factor in amyotrophic lateral sclerosis human spinal cord. Acta Neuropathologica. 2003;106(5):449–457. doi: 10.1007/s00401-003-0741-y. [DOI] [PubMed] [Google Scholar]
- 59.Spliet WGM, Aronica E, Ramkema M, et al. Immunohistochemical localization of vascular endothelial growth factor receptors-1, -2 and -3 in human spinal cord: altered expression in amyotrophic lateral sclerosis. Neuropathology and Applied Neurobiology. 2004;30(4):351–359. doi: 10.1111/j.1365-2990.2003.00543.x. [DOI] [PubMed] [Google Scholar]
- 60.Malessa S, Leigh PN, Bertel O, Sluga E, Hornykiewicz O. Amyotrophic lateral sclerosis: glutamate dehydrogenase and transmitter amino acids in the spinal cord. Journal of Neurology Neurosurgery and Psychiatry. 1991;54(11):984–988. doi: 10.1136/jnnp.54.11.984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends in Neurosciences. 2009;32(12):638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barres BA. The mystery and magic of Glia: a perspective on their roles in health and disease. Neuron. 2008;60(3):430–440. doi: 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 63.Sofroniew MV. Reactive astrocytes in neural repair and protection. Neuroscientist. 2005;11(5):400–407. doi: 10.1177/1073858405278321. [DOI] [PubMed] [Google Scholar]
- 64.Martineau M, Baux G, Mothet J-P. Gliotransmission at central glutamatergic synapses: D-serine on stage. Journal of Physiology. 2006;1:211–217. doi: 10.1016/j.jphysparis.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 65.Zhang Q, Haydon PG. Roles for gliotransmission in the nervous system. Journal of Neural Transmission. 2005;112(1):121–125. doi: 10.1007/s00702-004-0119-x. [DOI] [PubMed] [Google Scholar]
- 66.Li P, Zhuo M. Cholinergic, noradrenergic, and serotonergic inhibition of fast synaptic transmission in spinal lumbar dorsal horn of rat. Brain Research Bulletin. 2001;54(6):639–647. doi: 10.1016/s0361-9230(01)00470-1. [DOI] [PubMed] [Google Scholar]
- 67.Karpati G, Klassen G, Tanser P. The effects of partial chronic denervation on forearm metabolism. Canadian Journal of Neurological Sciences. 1979;6(2):105–112. doi: 10.1017/s0317167100119481. [DOI] [PubMed] [Google Scholar]
- 68.Rissanen TT, Vajanto I, Hiltunen MO, et al. Expression of vascular endothelial growth factor and vascular endothelial growth factor receptor-2 (KDR/Fik-1) in ischemic skeletal muscle and its regeneration. The American Journal of Pathology. 2002;160(4):1393–1403. doi: 10.1016/S0002-9440(10)62566-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nygren I, Larsson A, Johansson A, Askmark H. VEGF is increased in serum but not in spinal cord from patients with amyotrophic lateral sclerosis. NeuroReport. 2002;13(17):2199–2201. doi: 10.1097/00001756-200212030-00007. [DOI] [PubMed] [Google Scholar]
- 70.Vijayalakshmi K, Alladi PA, Sathyaprabha TN, Subramaniam JR, Nalini A, Raju TR. Cerebrospinal fluid from sporadic amyotrophic lateral sclerosis patients induces degeneration of a cultured motor neuron cell line. Brain Research. 2009;1263:122–133. doi: 10.1016/j.brainres.2009.01.041. [DOI] [PubMed] [Google Scholar]
- 71.Crugnola V, Lamperti C, Lucchini V, et al. Mitochondrial respiratory chain dysfunction in muscle from patients with amyotrophic lateral sclerosis. Archives of Neurology. 2010;67(7):849–854. doi: 10.1001/archneurol.2010.128. [DOI] [PubMed] [Google Scholar]
- 72.Vielhaber S, Kunz D, Winkler K, et al. Mitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosis. Brain. 2000;123(7):1339–1348. doi: 10.1093/brain/123.7.1339. [DOI] [PubMed] [Google Scholar]
- 73.Krasnianski A, Deschauer M, Neudecker S, et al. Mitochondrial changes in skeletal muscle in amyotrophic lateral sclerosis and other neurogenic atrophies. Brain. 2005;128(8):1870–1876. doi: 10.1093/brain/awh540. [DOI] [PubMed] [Google Scholar]
- 74.Küst BM, Copray JCWM, Brouwer N, Troost D, Boddeke HWGM. Elevated levels of neurotrophins in human biceps brachii tissue of amyotrophic lateral sclerosis. Experimental Neurology. 2002;177(2):419–427. doi: 10.1006/exnr.2002.8011. [DOI] [PubMed] [Google Scholar]
- 75.Valavanidis A, Vlachogianni T, Fiotakis C. 8-hydroxy-2′ -deoxyguanosine (8-OHdG): a critical biomarker of oxidative stress and carcinogenesis. Journal of Environmental Science and Health C. 2009;27(2):120–139. doi: 10.1080/10590500902885684. [DOI] [PubMed] [Google Scholar]
- 76.Chen S-K, Hsieh WA, Tsai M-H, et al. Age-associated decrease of oxidative repair enzymes, human 8-oxoguanine DNA glycosylases (hOGG1), in human aging. Journal of Radiation Research. 2003;44(1):31–35. doi: 10.1269/jrr.44.31. [DOI] [PubMed] [Google Scholar]
- 77.Coppedè F, Mancuso M, Gerfo AL, et al. Association of the hOGG1 Ser326Cys polymorphism with sporadic amyotrophic lateral sclerosis. Neuroscience Letters. 2007;420(2):163–168. doi: 10.1016/j.neulet.2007.04.067. [DOI] [PubMed] [Google Scholar]
- 78.Brockington A, Kirby J, Eggitt D, et al. Screening of the regulatory and coding regions of vascular endothelial growth factor in amyotrophic lateral sclerosis. Neurogenetics. 2005;6(2):101–104. doi: 10.1007/s10048-004-0201-4. [DOI] [PubMed] [Google Scholar]
- 79.Gros-Louis F, Laurent S, Lopes AAS, et al. Absence of mutations in the hypoxia response element of VEGF in ALS. Muscle and Nerve. 2003;28(6):774–775. doi: 10.1002/mus.10498. [DOI] [PubMed] [Google Scholar]
- 80.Chen W, Saeed M, Mao H, et al. Lack of association of VEGF promoter polymorphisms with sporadic ALS. Neurology. 2006;67(3):508–510. doi: 10.1212/01.wnl.0000227926.42370.04. [DOI] [PubMed] [Google Scholar]
- 81.van Vught PWJ, Sutedja NA, Veldink JH, et al. Lack of association between VEGF polymorphisms and ALS in a Dutch population. Neurology. 2005;65(10):1643–1645. doi: 10.1212/01.wnl.0000184514.39853.56. [DOI] [PubMed] [Google Scholar]
- 82.Zhang Y, Zhang H, Fu Y, et al. VEGF C2578A polymorphism does not contribute to amyotrophic lateral sclerosis susceptibility in sporadic Chinese patients. Amyotrophic Lateral Sclerosis. 2006;7(2):119–122. doi: 10.1080/14660820600600657. [DOI] [PubMed] [Google Scholar]
- 83.Lambrechts D, Poesen K, Fernández-Santiago R, et al. Meta-analysis of vascular endothelial growth factor variations in amyotrophic lateral sclerosis: increased susceptibility in male carriers of the -2578AA genotype. Journal of Medical Genetics. 2009;46(12):840–846. doi: 10.1136/jmg.2008.058222. [DOI] [PubMed] [Google Scholar]
- 84.Fernández-Santiago R, Sharma M, Mueller JC, et al. Possible gender-dependent association of vascular endothelial growth factor (VEGF) gene and ALS. Neurology. 2006;66(12):1929–1931. doi: 10.1212/01.wnl.0000219756.71928.25. [DOI] [PubMed] [Google Scholar]
- 85.Oates N, Pamphlett R. An epigenetic analysis of SOD1 and VEGF in ALS. Amyotrophic Lateral Sclerosis. 2007;8(2):83–86. doi: 10.1080/17482960601149160. [DOI] [PubMed] [Google Scholar]
- 86.Brockington A, Wokke B, Nixon H, Hartley JA, Shaw PJ. Screening of the transcriptional regulatory regions of vascular endothelial growth factor receptor 2 (VEGFR2) in amyotrophic lateral sclerosis. BMC Medical Genetics. 2007;8, article 23 doi: 10.1186/1471-2350-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zil'ber LA, Bajdakova ZL, Gardašjan AN, Konovalov NV, Bunina TL, Barabadze EM. Study of the etiology of amyotrophic lateral sclerosis. Bulletin of the World Health Organization. 1963;29(4):449–456. [PMC free article] [PubMed] [Google Scholar]
- 88.Smith RA, Balis FM, Ott KH, Elsberry DD, Sherman MR, Saifer MGP. Pharmacokinetics and tolerability of ventricularly administered superoxide dismutase in monkeys and preliminary clinical observations in familial ALS. Journal of the Neurological Sciences. 1995;129(supplement 1):13–18. doi: 10.1016/0022-510x(95)00051-3. [DOI] [PubMed] [Google Scholar]
- 89.Storkebaum E, Lambrechts D, Dewerchin M, et al. Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nature Neuroscience. 2005;8(1):85–92. doi: 10.1038/nn1360. [DOI] [PubMed] [Google Scholar]
- 90.Hwang DH, Lee HJ, Park IH, et al. Intrathecal transplantation of human neural stem cells overexpressing VEGF provide behavioral improvement, disease onset delay and survival extension in transgenic ALS mice. Gene Therapy. 2009;16(10):1234–1244. doi: 10.1038/gt.2009.80. [DOI] [PubMed] [Google Scholar]
- 91.Lambrechts D, Storkebaum E, Morimoto M, et al. VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nature Genetics. 2003;34(4):383–394. doi: 10.1038/ng1211. [DOI] [PubMed] [Google Scholar]
- 92.Sun Y, Jin K, Xie L, et al. VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. Journal of Clinical Investigation. 2003;111(12):1843–1851. doi: 10.1172/JCI17977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zheng C, Nennesmo I, Fadeel B, Henter JI. Vascular endothelial growth factor prolongs survival in a transgenic mouse model of ALS. Annals of Neurology. 2004;56(4):564–567. doi: 10.1002/ana.20223. [DOI] [PubMed] [Google Scholar]
- 94.Widenfalk J, Lipson A, Jubran M, et al. Vascular endothelial growth factor improves functional outcome and decreases secondary degeneration in experimental spinal cord contusion injury. Neuroscience. 2003;120(4):951–960. doi: 10.1016/s0306-4522(03)00399-3. [DOI] [PubMed] [Google Scholar]
- 95.Kliem MA, Heeke BL, Franz CK, et al. Intramuscular administration of a VEGF zinc finger transcription factor activator (VEGF-ZFP-TF) improves functional outcomes in SOD1 rats. Amyotrophic Lateral Sclerosis. 2011;12(5):331–339. doi: 10.3109/17482968.2011.574142. [DOI] [PubMed] [Google Scholar]
- 96.Silva DF, Porto DL, Araújo IGA, et al. Endothelium-derived nitric oxide is involved in the hypotensive and vasorelaxant effects induced by discretamine in rats. Pharmazie. 2009;64(5):327–331. [PubMed] [Google Scholar]
- 97.Kobari M, Obara K, Watanabe S, Dembo T, Fukuuchi Y. Local cerebral blood flow in motor neuron disease: correlation with clinical findings. Journal of the Neurological Sciences. 1996;144(1-2):64–69. doi: 10.1016/s0022-510x(96)00151-7. [DOI] [PubMed] [Google Scholar]
- 98.Barbeito AG, Martinez-Palma L, Vargas MR, et al. Lead exposure stimulates VEGF expression in the spinal cord and extends survival in a mouse model of ALS. Neurobiology of Disease. 2010;37(3):574–580. doi: 10.1016/j.nbd.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Azzouz M, Ralph GS, Storkebaum E, et al. VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature. 2004;429, article 6990:413–417. doi: 10.1038/nature02544. [DOI] [PubMed] [Google Scholar]
- 100.Ay I, Francis JW, Brown RH., Jr. VEGF increases blood-brain barrier permeability to Evans blue dye and tetanus toxin fragment C but not adeno-associated virus in ALS mice. Brain Research. 2008;1234:198–205. doi: 10.1016/j.brainres.2008.07.121. [DOI] [PubMed] [Google Scholar]
- 101.Zhong Z, Deane R, Ali Z, et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nature Neuroscience. 2008;11(4):420–422. doi: 10.1038/nn2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Silani V, Braga M, Ciammola A, Cardin V, Scarlato G. Motor neurones in culture as a model to study ALS. Journal of Neurology. 2000;247(supplement 1):I28–I36. doi: 10.1007/s004150050554. [DOI] [PubMed] [Google Scholar]
- 103.Lu L, Zheng L, Viera L, et al. Mutant Cu/Zn-superoxide dismutase associated with amyotrophic lateral sclerosis destabilizes vascular endothelial growth factor mRNA and downregulates its expression. Journal of Neuroscience. 2007;27(30):7929–7938. doi: 10.1523/JNEUROSCI.1877-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Scandurro AB, Beckman BS. Common proteins bind mRNAs encoding erythropoietin, tyrosine hydroxylase, and vascular endothelial growth factor. Biochemical and Biophysical Research Communications. 1998;246(2):436–440. doi: 10.1006/bbrc.1998.8639. [DOI] [PubMed] [Google Scholar]
- 105.Li B, Xu W, Luo C, Gozal D, Liu R. VEGF-induced activation of the PI3-K/Akt pathway reduces mutant SOD1-mediated motor neuron cell death. Molecular Brain Research. 2003;111(1-2):155–164. doi: 10.1016/s0169-328x(03)00025-1. [DOI] [PubMed] [Google Scholar]
- 106.Lunn JS, Sakowski SA, Kim B, Rosenberg AA, Feldman EL. Vascular endothelial growth factor prevents G93A-SOD1-induced motor neuron degeneration. Developmental Neurobiology. 2009;69(13):871–884. doi: 10.1002/dneu.20747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.During MJ, Cao L. VEGF, a mediator of the effect of experience on hippocampal neurogenesis. Current Alzheimer Research. 2006;3(1):29–33. doi: 10.2174/156720506775697133. [DOI] [PubMed] [Google Scholar]
- 108.Meng H, Zhang Z, Zhang R, et al. Biphasic effects of exogenous VEGF on VEGF expression of adult neural progenitors. Neuroscience Letters. 2006;393(2-3):97–101. doi: 10.1016/j.neulet.2005.09.044. [DOI] [PubMed] [Google Scholar]
- 109.Rizvanov AA, Kiyasov AP, Gaziziov IM, et al. Human umbilical cord blood cells transfected with VEGF and L1CAM do not differentiate into neurons but transform into vascular endothelial cells and secrete neuro-trophic factors to support neuro-genesis-a novel approach in stem cell therapy. Neurochemistry International. 2008;53(6–8):389–394. doi: 10.1016/j.neuint.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 110.Poesen K, Lambrechts D, van Damme P, et al. Novel role for vascular endothelial growth factor (VEGF) receptor-1 and its ligand VEGF-B in motor neuron degeneration. Journal of Neuroscience. 2008;28(42):10451–10459. doi: 10.1523/JNEUROSCI.1092-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Blood-brain barrier: structural components and function under physiologic and pathologic conditions. Journal of Neuroimmune Pharmacology. 2006;1(3):223–236. doi: 10.1007/s11481-006-9025-3. [DOI] [PubMed] [Google Scholar]
- 112.Schluesener HJ, Sobel RA, Linington C, Weiner HL. A monoclonal antibody against a myelin oligodendrocyte glycoprotein induces relapses and demyelination in central nervous system autoimmune disease. Journal of Immunology. 1987;139(12):4016–4021. [PubMed] [Google Scholar]
- 113.Germanò A, Caffo M, Angileri FF, et al. NMDA receptor antagonist felbamate reduces behavioral deficits and blood-brain barrier permeability changes after experimental subarachnoid hemorrhage in the rat. Journal of Neurotrauma. 2007;24(4):732–744. doi: 10.1089/neu.2006.0181. [DOI] [PubMed] [Google Scholar]
- 114.Graesser D, Solowiej A, Bruckner M, et al. Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1-deficient mice. Journal of Clinical Investigation. 2002;109(3):383–392. doi: 10.1172/JCI13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bartanusz V, Jezova D, Alajajian B, Digicaylioglu M. The blood-spinal cord barrier: morphology and clinical implications. Annals of Neurology. 2011;70(2):194–206. doi: 10.1002/ana.22421. [DOI] [PubMed] [Google Scholar]
- 116.Garbuzova-Davis S, Saporta S, Haller E, et al. Evidence of compromised blood-spinal cord barrier in early and late symptomatic SOD1 mice modeling ALS. PLoS ONE. 2007;2(11, article e1205) doi: 10.1371/journal.pone.0001205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wolburg H, Wolburg-Buchholz K, Kraus J, et al. Localization of claudin-3 in tight junctions of the blood-brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. Acta Neuropathologica. 2003;105(6):586–592. doi: 10.1007/s00401-003-0688-z. [DOI] [PubMed] [Google Scholar]
- 118.Ambrosini E, Remoli ME, Giacomini E, et al. Astrocytes produce dendritic cell-attracting chemokines in vitro and in multiple sclerosis lesions. Journal of Neuropathology and Experimental Neurology. 2005;64(8):706–715. doi: 10.1097/01.jnen.0000173893.01929.fc. [DOI] [PubMed] [Google Scholar]
- 119.Argaw AT, Asp L, Zhang J, et al. Astrocyte derived VEGF-A drives blood brain barrier disruption in CNS inflammatory disease. Journal of Clinical Investigation. 2012;122(7):2454–2468. doi: 10.1172/JCI60842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Esser S, Lampugnani MG, Corada M, Dejana E, Risau W. Vascular endothelial growth factor induces VE-cadherin tyrosine. Journal of Cell Science. 1998;111(13):1853–1865. doi: 10.1242/jcs.111.13.1853. [DOI] [PubMed] [Google Scholar]
- 121.Zhang K, Lu J, Mori T, et al. Baicalin increases VEGF expression and angiogenesis by activating the ERRα/PGC-1α pathway. Cardiovascular Research. 2011;89(2):426–435. doi: 10.1093/cvr/cvq296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor α in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Molecular and Cellular Biology. 2000;20(5):1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Arany Z. PGC-1 coactivators and skeletal muscle adaptations in health and disease. Current Opinion in Genetics and Development. 2008;18(5):426–434. doi: 10.1016/j.gde.2008.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Arany Z, Foo SY, Ma Y, et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1α . Nature. 2008;451, article 7181:1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- 125.Singh RP, Tyagi AK, Dhanalakshmi S, Agarwal R, Agarwal CN. Grape seed extract inhibits advanced human prostate tumor growth and angiogenesis and upregulates insulin-like growth factor binding protein-3. International Journal of Cancer. 2004;108(5):733–740. doi: 10.1002/ijc.11620. [DOI] [PubMed] [Google Scholar]
- 126.Kaur M, Singh RP, Gu M, Agarwal R, Agarwal C. Grape seed extract inhibits in vitro and in vivo growth of human colorectal carcinoma cells. Clinical Cancer Research. 2006;12(20):6194–6202. doi: 10.1158/1078-0432.CCR-06-1465. [DOI] [PubMed] [Google Scholar]
- 127.Arii M. Chemopreventive effect of grape seed extract on intestinal carcinogenesis in the APCMin mouse. Proceedings of the American Association for Cancer Research. 1998;39, article 20 [Google Scholar]
- 128.Lu J, Zhang K, Chen S, Wen W. Grape seed extract inhibits VEGF expression via reducing HIF-1α protein expression. Carcinogenesis. 2009;30(4):636–644. doi: 10.1093/carcin/bgp009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Desnuelle C, Dib M, Garrel C, Favier A. A double-blind, placeho-controlled randomized clinical trial of α-tocopherol (vitamin E) in the treatment of amyotrophic lateral sclerosis. ALS riluzole-tocopherol Study Group. Amyotrophic Lateral Sclerosis and Other Motor Neuron Disorders. 2001;2(1):9–18. doi: 10.1080/146608201300079364. [DOI] [PubMed] [Google Scholar]
- 130.Graf M, Ecker D, Horowski R, et al. High dose vitamin E therapy in amyotrophic lateral sclerosis as add-on therapy to riluzole: results of a placebo-controlled double-blind study. Journal of Neural Transmission. 2005;112(5):649–660. doi: 10.1007/s00702-004-0220-1. [DOI] [PubMed] [Google Scholar]
- 131.Orrell RW, Lane RJM, Ross M. A systematic review of antioxidant treatment for amyotrophic lateral sclerosis/motor neuron disease. Amyotrophic Lateral Sclerosis. 2008;9(4):195–211. doi: 10.1080/17482960801900032. [DOI] [PubMed] [Google Scholar]
- 132.Klivenyi P, Ferrante RJ, Matthews RT, et al. Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nature Medicine. 1999;5(3):347–350. doi: 10.1038/6568. [DOI] [PubMed] [Google Scholar]
- 133.Groeneveld GJ, Veldink JH, van der Tweel I, et al. A randomized sequential trial of creatine in amyotrophic lateral sclerosis. Annals of Neurology. 2003;53(4):437–445. doi: 10.1002/ana.10554. [DOI] [PubMed] [Google Scholar]
- 134.Raman C, McAllister SD, Rizvi G, Patel SG, Moore DH, Abood ME. Amyotrophic lateral sclerosis: delayed disease progression in mice by treatment with a cannabinoid. Amyotrophic Lateral Sclerosis and Other Motor Neuron Disorders. 2004;5(1):33–39. doi: 10.1080/14660820310016813. [DOI] [PubMed] [Google Scholar]
- 135.Weydt P, Hong S, Witting A, Möller T, Stella N, Kliot M. Cannabinol delays symptom onset in SOD1 (G93A) transgenic mice without affecting survival. Amyotrophic Lateral Sclerosis and Other Motor Neuron Disorders. 2005;6(3):182–184. doi: 10.1080/14660820510030149. [DOI] [PubMed] [Google Scholar]