

Abstract
Faithful transmission of genetic material is essential for the survival of all organisms. Eukaryotic chromosome segregation is driven by the kinetochore that assembles onto centromeric DNA to capture spindle microtubules and govern the movement of chromosomes. Its molecular mechanism has been actively studied in conventional model eukaryotes, such as yeasts, worms, flies and human. However, these organisms are closely related in the evolutionary time scale and it therefore remains unclear whether all eukaryotes use a similar mechanism. The evolutionary origins of the segregation apparatus also remain enigmatic. To gain insights into these questions, it is critical to perform comparative studies. Here, we review our current understanding of the mitotic mechanism in Trypanosoma brucei, an experimentally tractable kinetoplastid parasite that branched early in eukaryotic history. No canonical kinetochore component has been identified, and the design principle of kinetochores might be fundamentally different in kinetoplastids. Furthermore, these organisms do not appear to possess a functional spindle checkpoint that monitors kinetochore–microtubule attachments. With these unique features and the long evolutionary distance from other eukaryotes, understanding the mechanism of chromosome segregation in T. brucei should reveal fundamental requirements for the eukaryotic segregation machinery, and may also provide hints about the origin and evolution of the segregation apparatus.
Keywords: chromosome segregation, kinetochores, mitosis, CENP-A, Trypanosoma brucei, kinetoplastids
2. Introduction
The numerous organisms living on Earth are divided into three domains of life (Bacteria, Archaea and Eukaryota), and transmission of genetic information from generation to generation is essential for all. Regardless of cellular organization, this requires two processes; namely, the replication and segregation of chromosomes. Compared with the DNA replication machinery, which shares several common features [1,2], the segregation machinery appears much less conserved among the three domains of life. Here, we will focus on eukaryotic segregation mechanisms and refer readers to recent reviews on prokaryotic segregation processes [3–6].
3. Molecular mechanism of chromosome segregation revealed from studies of popular eukaryotes
During the last 40 years of research, basic mitotic mechanisms were elucidated using powerful model systems such as budding yeast, fission yeast, sea urchin, Xenopus egg extracts, worms, flies and mammalian tissue culture cells. The following picture has emerged from these studies (figure 1). The CDK/Cyclin complex drives cell cycle progression by promoting DNA replication and subsequent mitotic events through phosphorylation of hundreds of substrates [7–9]. Duplicated sister chromatids are held together by the cohesin complex [10,11]. This physical association enables cells to recognize which chromosomes to split during mitosis. Chromosome segregation depends on spindle microtubules and kinetochores: microtubules are dynamic polymers that consist of tubulin subunits [12,13], while a kinetochore is the macromolecular protein complex that assembles onto centromeric DNA [14,15]. During mitosis, kinetochores first form lateral attachments to microtubules, which are then converted to end-on attachments. This end-on attachment allows kinetochores to use the energy produced by the depolymerizing microtubules to move chromosomes [16–19]. Accurate chromosome segregation requires that a bipolar spindle is assembled and sister kinetochores form bi-oriented attachments to spindle microtubules emanating from opposite poles [20]. Attachment errors must be corrected to avoid mis-segregation [21]. To ensure high fidelity, cells possess a surveillance mechanism (the spindle checkpoint) that monitors the status of kinetochore–microtubule attachment and prevents cells from proceeding into anaphase in the presence of erroneous attachments [22,23]. Once all chromosomes have achieved proper bi-orientation, the spindle checkpoint is satisfied and the anaphase-promoting complex (APC/C) is activated [24–27]. This results in the activation of a protease called separase that cleaves the cohesin complex so that sister chromatids segregate away from each other [28]. The APC/C also promotes mitotic exit by degrading cyclins [29].
Figure 1.
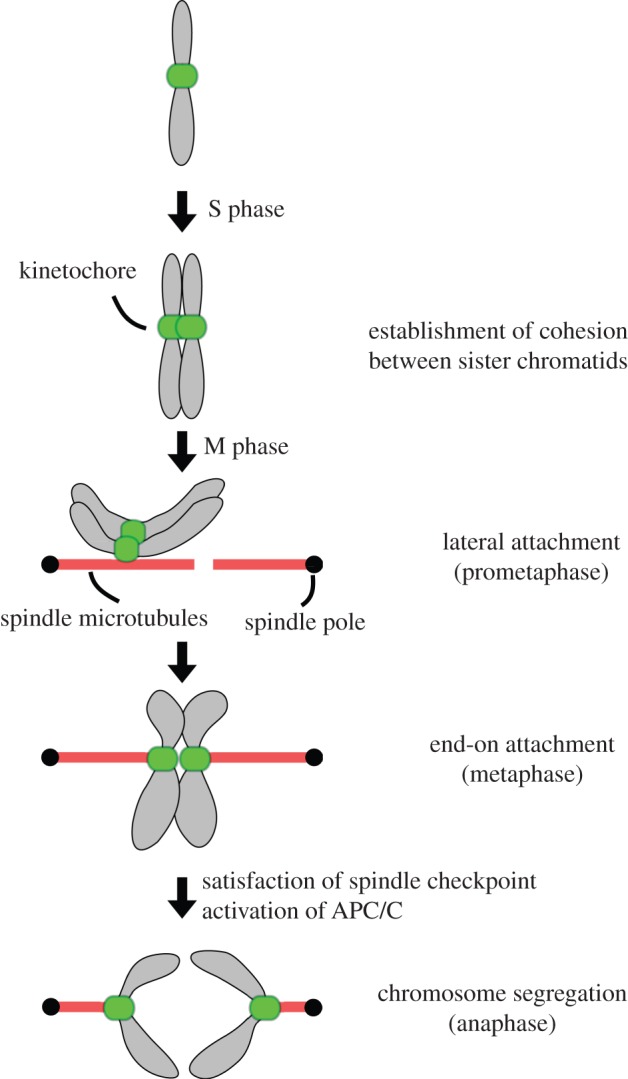
Mitotic chromosome segregation. Chromosomes are duplicated during S phase, and cohesion is established between sister chromatids. When cells enter mitosis, a bipolar spindle is assembled. Kinetochores initially form lateral attachments to spindle microtubules, which are then converted to end-on attachments. When all chromosomes form bi-oriented attachments (i.e. sister kinetochores attach to microtubules emanating from opposite poles), the spindle checkpoint is satisfied and the APC/C gets activated. This leads to the dissolution of cohesion so that the sister chromatids segregate away from each other.
4. What does ‘conserved from yeast to human’ actually mean?
The basic mitotic machinery appears well conserved among the popular model organisms mentioned earlier. When our favourite protein is conserved in both human and yeast, we often think that ‘this protein must be universally conserved across eukaryotes because human and yeast look very different!’. Is this a valid reasoning supported by scientific evidence?
According to the latest molecular phylogenetic tree, eukaryotes are divided into six supergroups (figure 2) [30–32]. The popular model organisms (human, fungi, worms, flies, frogs, etc.) all belong to the supergroup Opisthokonta, which means that these organisms are closely related in the evolutionary time scale. Therefore, even if a certain protein is conserved from yeast to human, the protein may be conserved only in the Opisthokonta supergroup, not in other supergroups. It is thus essential to examine eukaryotes with a wider evolutionary distance belonging to other supergroups if we want to reveal the extent of conservation in the eukaryotic kingdom.
Figure 2.

Current eukaryotic phylogenetic tree. In this unrooted tree, eukaryotes are divided into six supergroups, Opisthokonta, Amoebozoa, Excavata, Archaeplastida, SAR (stramenopiles, alveolates and rhizaria) and CCTH (cryptophytes, centrohelids, telonemids and haptophytes). Representative organisms whose draft genome sequences are available are shown as examples. The tree has been redrawn and modified from [30]. Branch lengths are arbitrary.
The availability of genome sequences now allows one to readily determine whether a protein of interest is present in distant eukaryotes using homology search programs such as BLAST [33] and HMMER [34]. A general bioinformatic assumption is that if amino acid sequences are similar, it is probably because the proteins possess a similar structure/function and a level of relatedness. However, it is important to keep in mind that a failure to detect putative homologues in fully sequenced genomes does not necessarily mean that the protein is truly absent. It is often the case that primary sequences have diverged too much to be recognized by homology search algorithms [35]. Conversely, it is also possible that even if proteins show a high level of conservation, they may function differently in different organisms owing to a different environment or other factors. For example, a highly conserved Cdc14 phosphatase plays critical roles in regulating late mitotic events in budding yeast, but not in many other eukaryotes [36]. Therefore, although bioinformatic analysis can provide a lot of information and insight, it is critical to validate the predictions experimentally.
5. Which evolutionarily distant organisms to study?
Understanding the extent of conservation throughout eukaryotes would be tremendously facilitated by studying the last eukaryotic common ancestor (LECA) from which all present eukaryotes diverged. However, it is not clear if such an organism exists today. As mentioned earlier, a current eukaryotic tree looks like figure 2, which is based on extensive genomic, ultrastructural and phylogenetic evidence [30–32]. The tree is unrooted because we still lack concrete views on the basal areas of eukaryotic evolution. Currently, there are several ideas proposed: rooting between unikont (Opisthokonta and Amoebozoa) and bikont (all other taxa) [37], between Opisthokonta and all other taxa [38,39], and between Archaeplastida and all other taxa [40]. In addition, based on the unique cytochrome c/c1 biogenesis [41,42], Cavalier-Smith [43] proposed that the root of the eukaryotic tree lies between Euglenozoa (or deep within the Euglenozoa tree) and all the rest of eukaryotes, which would place trypanosomes as one of the earliest branching organisms. More work is clearly needed to examine the validity and stability of these hypotheses. Regardless of the position of the root, however, it is clear that trypanosomes (Excavata) are evolutionary distant from commonly studied eukaryotes (Opisthokonta).
6. Trypanosoma brucei as a model to study chromosome segregation
We now introduce Trypanosoma brucei as an emerging model organism to examine the conservation/divergence of various biological processes, including chromosome segregation. Kinetoplastids are a group of unicellular flagellated eukaryotes, including parasitic trypanosomatids (e.g. T. brucei, Trypanosoma cruzi and Leishmania species) and free-living Bodonida (e.g. Bodo saltans). It is thought that the ancestor of trypanosomatids is the non-parasitic Bodonida [44–46]. Trypanosoma brucei is the causative agent of African sleeping sickness, which kills more than 10 000 people annually in sub-Saharan Africa [47,48], whereas T. cruzi and Leishmania species are responsible for Chagas disease and leishmaniasis, respectively. These parasites affect millions of people and animals in various parts of the world, so understanding the biology of these trypanosomatids has medical and economic relevance besides genuine scientific merits. Genome sequences are available for several species of Trypanosoma and Leishmania (from TriTrypDB; see http://tritrypdb.org) [49–53], as well as B. saltans (from Wellcome Trust Sanger Institute; see http://www.sanger.ac.uk) [54], which allows comparative studies among kinetoplastids to examine the evolution of parasitism as well as more generic biological questions.
6.1. Molecular tools
Among the kinetoplastid species, T. brucei is currently the most experimentally tractable organism. In addition to the genome sequence [49], many molecular tools are available (see table 1 for details). For example, efficient homologous recombination facilitates GFP-fusions for the examination of the cellular location of proteins [65], while inducible RNAi enables knockdown analysis to examine their function [75,76]. Genome-wide RNAi libraries are available [77,78,98]. Furthermore, the organism's doubling time is 6–9 h (cf. budding yeast, 2 h; fission yeast, 3 h; mammalian tissue culture, 24 h) and it thus takes only approximately 10 days to obtain clonal transfectants. Large-scale culture is also feasible [99], and one can readily perform affinity purifications (e.g. using TAP tag) to identify interacting proteins by mass spectrometry [82–86]. Although it may not be easy to arrest cells in mitosis owing to an apparent lack of the spindle checkpoint system (see below), it is possible to obtain synchronous cultures using hydroxyurea arrest and release [100] or a double elutriation method [101]. Armed with this powerful molecular toolkit, it is possible to address biological questions in T. brucei.
Table 1.
Examples of molecular tools in T. brucei. There are at least eight drugs for selection (G418, Hygromycin, Puromycin, Phleomycin, Blasticidin, Nourseothricin/ClonNAT, Ganciclovir and FOA). Cells are typically grown in semi-defined media (SDM-79 for procyclic form [55], HMI-9 for bloodstream form [56]). Procyclic form cells readily grow up to a density of 1 × 107 cells ml−1 (1 × 106 cells ml−1 for bloodstream form cells) and can be frozen for long-term storage in liquid nitrogen. A subspecies, Trypanosoma brucei brucei, cannot infect humans owing to its sensitivity to human lytic factor [57], and is used in many research laboratories. Various monoclonal antibodies are also available [58]. Genetic exchange occurs under special circumstances (in the tsetse fly [59–61]), but it is not a widely practicable technique. Differentiation of life cycles can be reproduced in vitro [62–64]. GFP, green fluorescent protein; TAP, tandem affinity purification; YFP, yellow fluorescent protein.
| techniques | references |
|---|---|
| epitope-tagging (e.g. TAP, FLAG, GFP and YFP) and gene deletion using homologous recombination | [65–69] |
| regulated gene expression using TetR and T7 RNA polymerase | [70–72] |
| Cre-Lox recombination | [73,74] |
| RNAi, genome-wide RNAi screening | [75–78] |
| fluorescence in situ hybridization | [79] |
| GFP tagging of chromosomes using LacO/LacI | [80,81] |
| affinity purification (immunoprecipitation, BioID) | [82–86] |
| chromatin immunoprecipitation (ChIP), ChIP-seq | [87,88] |
| microtubule drugs | [89–91] |
| live-cell imaging | [92–94] |
| stable isotope labelling by amino acids in cell culture | [95–97] |
6.2. Life cycle
Trypanosoma brucei transmits between tsetse flies (Glossina) and mammalian hosts, and undergoes a complicated life cycle (reviewed in [102,103]). It proliferates in the midgut of tsetse fly as a ‘procyclic form’. After migration to the salivary glands, it develops into proliferative ‘epimastigote forms’ and then to the non-proliferative ‘metacyclic form’, which is ready to transmit into mammalian hosts. Trypanosomes are introduced into mammalian hosts upon the bite of tsetse flies. Once in the mammalian hosts, they develop into proliferative ‘bloodstream slender form’ and non-proliferative ‘stumpy form’ parasites. Once stumpy form cells are taken up by tsetse flies, they develop into the proliferative procyclic form, completing the life cycle. Each life stage is associated with unique changes in cell morphology or expressed proteins [104]. Both procyclic form and bloodstream form cells are most often used in research laboratories because they are easily cultured in vitro.
6.3. Cell structure
Trypanosoma brucei has a long slender shape with a single flagellum attached to the cell body (figure 3) [105,106]. The cell shape is determined by the subpellicular microtubules that underlie the plasma membrane. These microtubules are equally spaced with defined polarity (plus end in the posterior end of the cell, and minus end towards the anterior end [89]) and are highly stable owing to numerous cross-links between them [107]. This microtubule array does not disassemble during cell division. Instead, new microtubules are added between the old ones, and the array is transmitted to daughter cells in a semi-conservative manner [108]. Unlike other eukaryotes, mitochondrion and Golgi are present as single-copy organelles located at specific positions. The kinetoplast (a large structure in the mitochondrion that contains the mitochondrial DNA) is physically attached to the basal body that locates at the base of a flagellum so that the segregation of mitochondrial DNA is coupled to that of basal bodies (figure 3) [109,110]. The single Golgi is also specifically located but the physical connection to other organelles or cytoskeleton has not been determined [92,111].
Figure 3.
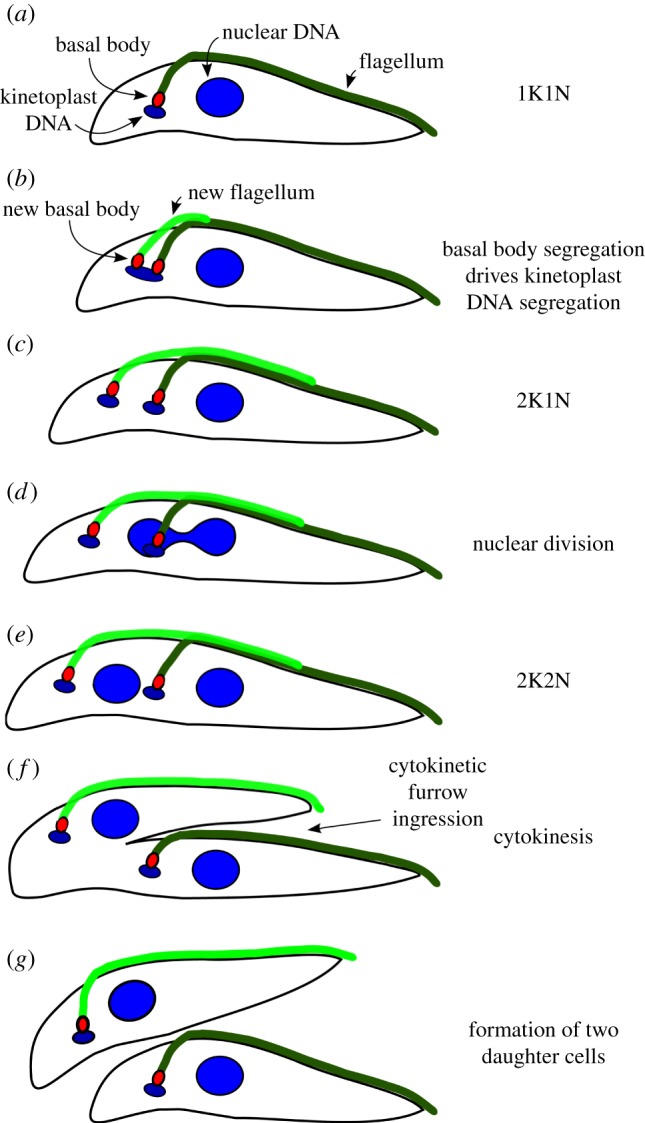
Diagram of the cell division cycle in T. brucei procyclic (insect) form cells. (a) G1 cells possess a single kinetoplast and nucleus (termed 1K1N) as well as an attached flagellum. (b) As the cell cycle progresses, a new basal body forms and nucleates a new flagellum. The nucleus is still in S phase when kinetoplast DNA shows an elongated morphology. (c) Segregation of basal bodies leads to the separation of attached kinetoplast. These cells are termed 2K1N. (d) Cells enter nuclear M phase, and chromosome segregation occurs. (e) Nuclear division is complete. These cells are termed 2K2N. (f) Cleavage furrow ingression occurs between the two flagella. (g) At the end of the cell cycle, two daughter cells are formed, and each cell inherits a single kinetoplast, nucleus and flagellum.
6.4. Chromosome structure
Trypanosoma brucei contains 11 diploid pairs of megabase chromosomes, as well as one to five intermediate chromosomes and approximately 100 minichromosomes of unknown ploidy [112] (figure 4a). These chromosomes are linear and have typical telomere repeats (TTAGGG) at the ends. Essentially, all the housekeeping genes are encoded in the megabase chromosomes and are transcribed as long polycistronic units with few exceptions [116–118]. The 26-Mb megabase chromosome genome contains approximately 9000 genes, including 1000 non-expressed variant surface glycoprotein (VSG) genes (most of which are pseudogenes [49]). Trypanosoma brucei lives extracellularly in the mammalian hosts and evades the immune response by means of antigenic variation [119]. Trypanosoma brucei expresses a single surface coat protein (variant surface glycoprotein, VSG) from one of approximately 15 expression sites (ESs), which locate proximal to the telomeres of megabase or intermediate chromosomes. Notably, expression of the VSGs is driven by RNA polymerase I from a special nuclear site, called the expression site body [80]. Although VSGs are highly immunogenic, T. brucei manages to escape the immune response by switching the expression of VSG up to once per 100 cell divisions [120–122]. This VSG switching often involves gene conversion of VSG cassettes into the active ES, creating and expressing a novel VSG gene that has not previously been seen by the immune system [123,124]. This intricate monoallelic expression and periodical switching of VSGs enable the parasites to evade the host immune response (reviewed in [125–127]), and it is thus difficult to develop effective vaccines. The parasite possesses approximately 100 minichromosomes that harbour additional VSG genes that serve as templates for recombination into one of the ESs [128]. Consistent with the concept that these small chromosomes are important for antigenic variation, they are segregated faithfully during cell division [129,130]. Minichromosomes are mostly composed of the 177 bp repeats of unknown function [131]. In addition to these linear chromosomes, circular DNA of up to 400 kb, called NR (NlaIII repeat) elements, are found in many strains, although their function remains unknown [132].
Figure 4.

Chromosome structure and organization. (a) Diagram of the three different types of chromosome in T. brucei. Essentially, all housekeeping genes are encoded in megabase chromosomes and are expressed in polycistronic transcription units. The centromere is located in a transcriptional strand-switch region of megabase chromosomes, while such a centromere appears absent from intermediate and minichromosomes. The core of minichromosomes consists of the 177 bp repeats and is constructed in a palindromic manner with a single inversion point in the centre. The complete intermediate chromosome structure is not known, but 177 bp repeats are present. (b) Diagram of the centromeric region of T. cruzi chromosome 3 and T. brucei chromosome 1, based on [113–115]. Various retro-elements are found in both species (e.g. INGI, DIRE, VIPER/SIRE and L1Tc). The T. brucei centromere additionally contains AT-rich repeats. Ribosomal RNA gene arrays are present on a subset of chromosomes. Note that these centromeric regions retain synteny in the two species that diverged more than 200 Myr ago.
6.5. Centromere structure
The centromere is the chromosomal locus where kinetochores assemble to mediate the interaction with microtubules. Despite its fundamental importance, centromere structures are highly divergent and three different types are found: the regional centromere, point centromere and holocentric [133]. It is thought that regional centromeres represent the ancestral form, whereas point centromeres and holocentricity are derived features [134,135]. Determining the position of a centromere can be done in several ways. (i) By mapping the region of a given chromosome that confers mitotic and/or meiotic stability (e.g. Saccharomyces cerevisiae and Arabidopsis [136,137]). (ii) By determining the DNA sequence that associates with kinetochore/centromere proteins. For example, CENP-A (Candida [138]) and topoisomerase II (human [139,140] and Plasmodium [141]) have been used. (iii) By mapping the position of primary constrictions using a fluorescence in situ hybridization approach [142].
The trypanosomatid centromere was first mapped in T. cruzi. Kelly and co-workers [113] performed a functional mapping (telomere-associated chromosome fragmentation) and delineated the 11–16 kb GC-rich regions that confer mitotic stability (figure 4b). The same regions also exhibit an increased topoisomerase II activity [114], strongly suggesting that these GC-rich regions represent the centromeres in T. cruzi. However, this is quite unusual because centromeric DNA is comparatively AT-rich in essentially all studied eukaryotes [143]. This unusual feature might be related to the absence of CENP-A (see below). The T. brucei centromere was subsequently mapped based on topoisomerase II activity. Unlike T. cruzi, its centromere contains AT-rich repetitive arrays (20–120 kb), which are restricted to a single site on each megabase chromosome [114,115] (figure 4b). The unit repeat size/sequence varies among chromosomes, although some chromosomes share similar sequences; for example, chromosomes 4, 5, 8, 9, 10 and 11 possess the almost identical sequence of 147 bp (the CIR147 repeat; see below), whereas chromosome 3 has a unique 120 bp sequence [114]. Although it remains to be shown that the AT-rich repeat region confers mitotic stability and centromere activity (i.e. binding of kinetochore proteins), the fact that only one region is found per chromosome and that the region is syntenic to the T. cruzi centromere (although the sequence is totally different) strongly suggests that it represents a centromere in T. brucei. Transposable elements are found at the centromere of both trypanosomes, as in many other eukaryotes [144]. In addition, rRNA gene arrays are present adjacent to the AT-rich repeats of chromosomes 1, 2, 3, 6 and 7, although their significance is currently unknown [114].
Repetitive sequences found at the centromere of many species are thought to promote the formation of heterochromatin using endogenous RNAi pathways [145,146]. Components of the RNAi machinery (Argonaute, Dicer) are present in T. brucei, and small RNAs are detected from the CIR147 repeats [147,148], which are present on the centromeric region of chromosomes 4, 5, 8, 9, 10 and 11 [114]. Interestingly, small RNAs are not found from the AT-rich repeats of the other centromeres (i.e. chromosomes 1, 2, 3, 6 and 7 [148]). However, we note that these centromeres contain rRNA gene arrays (see above), which may substitute the role of CIR147 repeats. Although it remains unknown whether conventional heterochromatin is formed at the centromere, knockdown of Argonaute leads to chromosome segregation defects in T. brucei [149]. Notably, repetitive sequences are not found at the centromeric region in T. cruzi [114], an organism that does not possess a functional RNAi pathway [150].
6.6. Cell cycle
Similar to other eukaryotes, the cell cycle of trypanosomes consists of distinct G1, S, G2 and M phases (reviewed in [151,152]). However, as mentioned earlier, trypanosomes possess two DNA-containing organelles (kinetoplast and nucleus), both of which must be segregated faithfully. During the cell cycle of procyclic form T. brucei, there are distinct replication and segregation periods for kinetoplast DNA and nuclear DNA (figure 3) [153–155]. The kinetoplast finishes its DNA replication first and the kinetoplast elongation/division occurs during the S phase of nuclear DNA. The nuclear division then occurs, followed by cytokinesis that results in the formation of two daughter cells that contain one kinetoplast and one nucleus. It remains unknown how these temporal orders are established and regulated.
Trypanosomes, like many other protists and fungi [156,157], do not disassemble their nuclear envelope during mitosis (a closed mitosis [158]), and a mitotic spindle forms within the nucleus. Many eukaryotes rely on two microtubule organizing centres (MTOCs) to nucleate a bipolar spindle [159,160]. Although no distinct structure such as a centrosome or a spindle pole body is detected in T. brucei, electron microscopy has visualized ring-like structures, inside the nucleus and close to the nuclear membrane, that appear to nucleate spindle microtubules during mitosis [161]. It appears probable that this is a truly intranuclear MTOC specific to the spindle because T. brucei is one of the organisms that lack a Brr6 domain protein that appears critical to the process of nuclear envelope fenestration in spindle morphogenesis [162]. This provides yet more evidence for a set of distinct and dispersed cytoplasmic and nuclear MTOCs in T. brucei whose differential activation will require regulation at specific points of the cell cycle [163]. A rhomboid-shaped bipolar spindle is initially assembled and converges into two poles at opposite ends of the nucleus. Later during mitosis, this focal organization is lost and the spindle becomes bifurcated at both ends [161]. Spindle pole-specific components have not been identified thus far, and the mechanism of bipolar spindle assembly remains largely unknown in trypanosomes.
6.7. Conserved mitotic players
Despite the long evolutionary distance, trypanosomes do possess a reasonable proportion of the basic mitotic machinery discovered in conventional model eukaryotes. This includes the CDK/Cyclin system [164,165], cohesin complex [166,167], separase [167], condensin complex [166], Aurora B [83,168,169], APC/C [170] and proteasome [171]. Therefore, the most basic cell cycle machinery appears to be conserved in these distant eukaryotes.
Similar to all other eukaryotes, tubulins are highly conserved in trypanosomes and are essential for the segregation of both large and small chromosomes [79]. Homologues of microtubule-associated proteins are also present, including XMAP215, EB1 and CLASP, although their relevance to mitotic events remains to be investigated. Similar to other eukaryotes, Kinesin-13 (a subfamily that includes MCAK that localizes at the inner centromere [172]) plays important roles in faithful chromosome segregation [173,174]. Polo-like kinase is also present, but it does not appear to play critical roles in chromosome segregation [175–177]. Some components of the nuclear pore complex have been detected at kinetochores in metazoans [178,179], and while nuclear pore components have also been identified in T. brucei [180], none have been detected at trypanosomatid kinetochores thus far.
7. What is unique?
7.1. Lack of conventional kinetochores? Absence of CENP-A
One of the most striking features in kinetoplastids is the failure to identify any homologous kinetochore protein by means of extensive bioinformatic analysis [49]. Indeed, no kinetochore protein has been identified in kinetoplastids to date. It is known that kinetochore proteins show a high degree of divergence even among the Opisthokonta supergroup [181,182], and it is thus possible that the primary sequence of kinetochore proteins in kinetoplastids have diverged too much to be detectable by currently available homology search algorithms. However, this possibility seems at odds with the finding that at least a few kinetochore components are readily identifiable in various eukaryotes from all the six supergroups, including Giardia and Trichomonas [181] (B.A. & K.G. 2013, unpublished data), organisms known to have evolved at faster rates than others [183].
Furthermore, trypanosomatids do not appear to possess a centromeric histone H3 variant (called CENP-A in human), which has a conserved histone fold domain and several unique features that distinguish it from canonical histone H3 [184]. Using this criterion, CENP-A candidates are readily identifiable in all sequenced eukaryotes except kinetoplastids (T. brucei, T. cruzi, Leishmania and B. saltans [143,184]). Trypanosoma brucei contains four canonical histones (H2A, H2B, H3 and H4) and four histone variants (H2AZ, H2Bv, H3v and H4v), as well as divergent H1 linker histones [185–187]. It is highly unlikely that H3v is a centromeric histone H3 variant; the gene is not essential for viability, and the protein is enriched at telomeres and transcription termination sites (although it is not known whether H3v is also enriched at centromeres [87,188]). Furthermore, none of the other histone variants (H2AZ, H2Bv, H4v) or histone modifications has been associated with centromeric function to date [87,189,190]. The absence of CENP-A in all sequenced kinetoplastids strongly implies its true absence, suggesting that their kinetochores may be different in a fundamental manner. It is essential to identify kinetochore components and examine whether kinetoplastid kinetochores are completely different or share any similarity with kinetochores of other eukaryotes.
Although no kinetochore-specific component is known, some proteins exhibit putative localization to kinetochores in addition to other locations. The Aurora B kinase, a component of the evolutionarily conserved chromosomal passenger complex, shows a dynamic localization pattern during mitosis in diverse eukaryotes [191]. It initially appears on chromatin at the onset of mitosis, localizes onto kinetochores during metaphase, and then moves onto the spindle midzone and cytokinetic furrow during anaphase. A similar localization pattern was observed for TbAUK1 (one of the three Aurora kinase homologues in T. brucei), which shows punctate signals on metaphase chromosomes, probably representing its kinetochore localization [83]. Microtubule-severing enzymes, Spastin and Fidgetin, also show dots in the nucleus (not cell cycle regulated) and may represent their kinetochore localization [192].
7.2. Insufficient number of kinetochores for chromosomes?
Although kinetochore proteins have not been identified in kinetoplastids, ultrastructural studies have detected electron-dense plaques within the nucleus [158,161,193]. These plaques are visible only in mitotic cells and appear to interact with spindle microtubules (up to four in T. brucei [161]), suggesting that they are probably kinetochores. However, there are several peculiarities with the structure. When these putative sister kinetochore pairs interact with microtubules from opposite poles (metaphase-like state), they exhibit a back-to-back configuration without distinct space between the two structures [161]. This contrasts with other eukaryotes that have a certain distance between sister kinetochore pairs. In human, this region is called the inner centromere where cohesins and chromosomal passenger complexes are enriched to mediate cohesion between sister chromatids and to promote attachment error correction [191,194]. The apparent lack of an inner centromere region could reflect a fundamental difference in centromere/kinetochore designs in T. brucei, and raises questions about where/how cohesins and passenger proteins are accumulated. In addition, the number of kinetochore-like plaques detected does not match the number of chromosomes in all trypanosomatids studied to date. Only up to eight plaques were visualized in T. brucei [193], an organism that contains 11 homologous (i.e. 22) megabase chromosomes and approximately 100 small chromosomes. Similarly, only 10 plaques were detected in T. cruzi (32 chromosomes [195]), and six in Leishmania (36 chromosomes [196]). Although it is possible that the discrepancy derives from experimental difficulties, a similar approach has detected 14 sister kinetochore pairs in Plasmodium falciparum [197], an organism that has 14 chromosomes [198]. It is therefore possible that kinetochores are assembled only on a subset of chromosomes or that centromeres of multiple chromosomes may cluster together to assemble a single kinetochore in trypanosomatids. It will be necessary to identify kinetochore proteins to gain insights into this enigma. Furthermore, in T. brucei, there are approximately 100 small chromosomes that appear to lack centromere activity [114], while the number of spindle microtubules is fewer than 100 [161]. Although several models have been proposed [193,199], the segregation mechanism of small chromosomes remains enigmatic. It is interesting to note that a similar phenomenon is observed in Ostreococcus tauri, the smallest known eukaryote [200]. Cryo-electron tomographic reconstitution visualized only approximately 10 spindle microtubules (note that kinetochore plaques were not visible in this study), although this organism contains 20 chromosomes. Conventional kinetochore proteins have been identified in O. tauri, so it will be important to reveal whether kinetochores are formed on all chromosomes, whether clustering of multiple kinetochores occur and how kinetochores interact with spindle microtubules.
7.3. Absence of the spindle checkpoint?
The spindle checkpoint is a surveillance mechanism that monitors the status of kinetochore–microtubule attachment and delays mitotic progression until all chromosomes achieve proper bi-orientation [201]. Although some organisms do not require the spindle checkpoint for their proliferation or development under normal conditions (e.g. budding yeast, fission yeast and flies [22,23,202,203]), its presence in diverse eukaryotes indicates that it is probably critical in the wild, where quality of life is not necessarily so assured. Spindle checkpoint components include Mad1, Mad2, Mad3 (BubR1), Bub1 and Bub3 [201]. It was proposed that Mad2 plays a crucial role in amplifying the checkpoint signal by undergoing conformational changes [204]. In trypanosomatids, only Mad2 can be identified by its primary sequence and the possession of a Mad2-like HORMA domain (Tb927.3.1750/TbMad2) [205]. TbMad2 is relatively well conserved (41% identity between T. brucei Tb927.3.1750/TbMad2 and human Mad2, 41% between S. cerevisiae and human, and 36% between T. brucei and S. cerevisiae). We found, however, that YFP-tagged TbMad2 in procyclic form cells shows a constitutive localization to the basal body area (figure 5), and does not show any kinetochore or nuclear signal during normal mitosis, nor even when spindle microtubules are disrupted by microtubule drugs (B.A. & K.G. 2013, unpublished data). Furthermore, a well-conserved Mad2-binding motif [206] is not present in the TbCdc20 protein, a critical target of the spindle checkpoint pathway in other eukaryotes. These observations suggest that TbMad2 is unlikely to be a functional homologue of the spindle checkpoint Mad2 protein despite the high level of sequence similarity.
Figure 5.
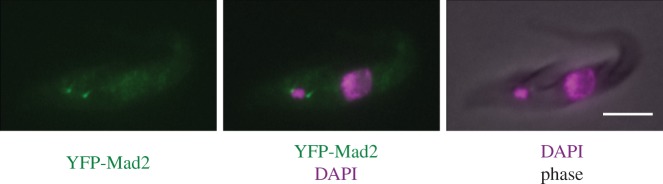
TbMad2 localizes at the basal body area. One allele of TbMad2 was endogenously tagged at the N-terminus with YFP. Similar results were obtained with C-terminally tagged Mad2. Cells were fixed with 4% formaldehyde and stained with DAPI. Scale bar, 5 µm.
Consistent with this possibility, there is no strong evidence that trypanosomatids possess a functional spindle checkpoint. Perturbation of spindle assembly does not prevent cells from undergoing cytokinesis [207], although the exact cell cycle state of the nucleus was not examined in this study. We therefore monitored the localization of AUK1 (an Aurora B homologue that shows dynamic localization patterns during mitosis), but did not obtain any evidence that cells are arrested in a pre-anaphase state in response to spindle damages (B.A. & K.G. 2013, unpublished data). Furthermore, inhibition of nuclear DNA replication prevents nuclear division, but cytokinesis still occurs [207]. These results suggest that cytokinesis occurs regardless of the state of nuclear DNA or bipolar spindle formation. This raises an important question: how are the cell cycle controls operating in this organism? One suggestion (as alluded to above) is that cells may monitor the state of basal bodies rather than nuclear DNA [207], an interesting possibility in the light of the evolutionary history of flagellated eukaryotes. It is thought that LECA possessed flagella and basal bodies in addition to the nucleus [208,209]. Because trypanosomes might be one of the earliest branching eukaryotes [43], it is interesting to speculate that an ancient function of the spindle checkpoint component Mad2 might have been to monitor the segregation of basal bodies/flagella, consistent with the TbMad2's localization to the basal body area (figure 5). Future studies are needed to reveal the function of Mad2 in trypanosomatids, which might provide hints about the origin of the spindle checkpoint system.
8. Perspectives: evolutionary cell biology
Studies in powerful model eukaryotes have led to an in-depth understanding of the mechanism of biological processes. Although it is essential to continue these efforts, it is also important to perform comparative studies to understand the extent of conservation/divergence across eukaryotes. This approach, termed ‘evolutionary cell biology’ [210], also aims to understand the design and working principles of fundamental biological processes, as well as to reveal their evolutionary history (e.g. centrioles/cilia/flagella [209], nucleus [211,212], cytoskeleton [213,214] and mitosis [215–218]). The goal of chromosome segregation is the partition of duplicated chromosomes. If there is a completely different way of achieving this task, understanding such a mechanism could provide insights about fundamental requirements for the process. Furthermore, if we are to obtain a complete understanding of the segregation machinery, we need to understand where it came from and how it evolved. Studying evolutionarily distant organisms is one way to obtain hints about the evolution of biological processes.
Here, we have focused mainly on the structure at the centre of the segregation mechanism (i.e. the kinetochore), but a lot of other mitotic processes deserve to be investigated as well. For example, the molecular mechanism of bipolar spindle assembly and cytokinesis remains obscure in trypanosomatids [219–221]. Furthermore, cells must coordinate various events in space and time. In T. brucei, mitochondrial DNA replication is achieved prior to the completion of nuclear DNA, but the molecular mechanism that facilitates this temporal periodic order is not known. Interestingly, in Cyanidioschyzon merolae (a red alga), the DNA replication of plastids and mitochondria also precedes that of the nucleus [222]. Future studies should reveal if similar regulatory principles operate in trypanosomatids. Regulating the position of the nucleus and other organelles relative to the site of the cytokinetic furrow is also critical to allow the accurate partition of segregated chromosomes [223,224]. Differential positioning of the cytokinetic furrow occurs in different life stages, although little is known about the molecular mechanism [225]. By addressing these questions, we should obtain better understanding of the mitotic mechanism in this distant eukaryotic parasite. Because T. brucei causes devastating African sleeping sickness disease, understanding its mechanism of chromosome segregation and the difference from the mechanism used by other organisms may also facilitate drug target identification, and therefore have great relevance for human and animal health.
9. Acknowledgements
We thank Robin Allshire and Andrea Musacchio for fruitful discussions. B.A. was supported by postdoctoral fellowships from the EMBO and Human Frontier Science Program. Research in the Gull laboratory is supported by the Wellcome Trust and BBSRC.
References
- 1.Robinson NP, Bell SD. 2005. Origins of DNA replication in the three domains of life. FEBS J. 272, 3757–3766 10.1111/j.1742-4658.2005.04768.x (doi:10.1111/j.1742-4658.2005.04768.x) [DOI] [PubMed] [Google Scholar]
- 2.Mott ML, Berger JM. 2007. DNA replication initiation: mechanisms and regulation in bacteria. Nat. Rev. Microbiol. 5, 343–354 10.1038/nrmicro1640 (doi:10.1038/nrmicro1640) [DOI] [PubMed] [Google Scholar]
- 3.Gerdes K, Howard M, Szardenings F. 2010. Pushing and pulling in prokaryotic DNA segregation. Cell 141, 927–942 10.1016/j.cell.2010.05.033 (doi:10.1016/j.cell.2010.05.033) [DOI] [PubMed] [Google Scholar]
- 4.Toro E, Shapiro L. 2010. Bacterial chromosome organization and segregation. Cold Spring Harb. Perspect. Biol. 2, a000349. 10.1101/cshperspect.a000349 (doi:10.1101/cshperspect.a000349) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalliomaa-Sanford AK, Rodriguez-Castañeda FA, McLeod BN, Latorre-Roselló V, Smith JH, Reimann J, Albers SV, Barillà D. 2012. Chromosome segregation in Archaea mediated by a hybrid DNA partition machine. Proc. Natl Acad. Sci. USA 109, 3754–3759 10.1073/pnas.1113384109 (doi:10.1073/pnas.1113384109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reyes-Lamothe R, Nicolas E, Sherratt DJ. 2012. Chromosome replication and segregation in bacteria. Annu. Rev. Genet. 46, 121–143 10.1146/annurev-genet-110711-155421 (doi:10.1146/annurev-genet-110711-155421) [DOI] [PubMed] [Google Scholar]
- 7.Hartwell LH, Culotti J, Pringle JR, Reid BJ. 1974. Genetic control of the cell division cycle in yeast. Science 183, 46–51 10.1126/science.183.4120.46 (doi:10.1126/science.183.4120.46) [DOI] [PubMed] [Google Scholar]
- 8.Evans T, Rosenthal ET, Youngblom J, Distel D, Hunt T. 1983. Cyclin: a protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell 33, 389–396 10.1016/0092-8674(83)90420-8 (doi:10.1016/0092-8674(83)90420-8) [DOI] [PubMed] [Google Scholar]
- 9.Nurse P. 1990. Universal control mechanism regulating onset of M-phase. Nature 344, 503–508 10.1038/344503a0 (doi:10.1038/344503a0) [DOI] [PubMed] [Google Scholar]
- 10.Michaelis C, Ciosk R, Nasmyth K. 1997. Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell 91, 35–45 10.1016/S0092-8674(01)80007-6 (doi:10.1016/S0092-8674(01)80007-6) [DOI] [PubMed] [Google Scholar]
- 11.Guacci V, Koshland D, Strunnikov A. 1997. A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell 91, 47–57 10.1016/S0092-8674(01)80008-8 (doi:10.1016/S0092-8674(01)80008-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borisy GG, Taylor EW. 1967. The mechanism of action of colchicine: binding of colchincine-3H to cellular protein. J. Cell Biol. 34, 525–533 10.1083/jcb.34.2.525 (doi:10.1083/jcb.34.2.525) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirschner M, Mitchison T. 1986. Beyond self-assembly: from microtubules to morphogenesis. Cell 45, 329–342 10.1016/0092-8674(86)90318-1 (doi:10.1016/0092-8674(86)90318-1) [DOI] [PubMed] [Google Scholar]
- 14.Brinkley BR, Stubblefield E. 1966. The fine structure of the kinetochore of a mammalian cell in vitro. Chromosoma 19, 28–43 10.1007/BF00332792 (doi:10.1007/BF00332792) [DOI] [PubMed] [Google Scholar]
- 15.Earnshaw WC, Rothfield N. 1985. Identification of a family of human centromere proteins using autoimmune sera from patients with scleroderma. Chromosoma 91, 313–321 10.1007/BF00328227 (doi:10.1007/BF00328227) [DOI] [PubMed] [Google Scholar]
- 16.Mandelkow EM, Mandelkow E, Milligan RA. 1991. Microtubule dynamics and microtubule caps: a time-resolved cryo-electron microscopy study. J. Cell Biol. 114, 977–991 10.1083/jcb.114.5.977 (doi:10.1083/jcb.114.5.977) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khodjakov A, Rieder CL. 1996. Kinetochores moving away from their associated pole do not exert a significant pushing force on the chromosome. J. Cell Biol. 135, 315–327 10.1083/jcb.135.2.315 (doi:10.1083/jcb.135.2.315) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grishchuk EL, Molodtsov MI, Ataullakhanov FI, McIntosh JR. 2005. Force production by disassembling microtubules. Nature 438, 384–388 10.1038/nature04132 (doi:10.1038/nature04132) [DOI] [PubMed] [Google Scholar]
- 19.Wang H-W, Nogales E. 2005. Nucleotide-dependent bending flexibility of tubulin regulates microtubule assembly. Nature 435, 911–915 10.1038/nature03606 (doi:10.1038/nature03606) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nicklas RB. 1997. How cells get the right chromosomes. Science 275, 632–637 10.1126/science.275.5300.632 (doi:10.1126/science.275.5300.632) [DOI] [PubMed] [Google Scholar]
- 21.Nicklas RB, Ward SC. 1994. Elements of error correction in mitosis: microtubule capture, release, and tension. J. Cell Biol. 126, 1241–1253 10.1083/jcb.126.5.1241 (doi:10.1083/jcb.126.5.1241) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li R, Murray AW. 1991. Feedback control of mitosis in budding yeast. Cell 66, 519–531 10.1016/0092-8674(81)90015-5 (doi:10.1016/0092-8674(81)90015-5) [DOI] [PubMed] [Google Scholar]
- 23.Hoyt MA, Totis L, Roberts BT. 1991. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell 66, 507–517 10.1016/0092-8674(81)90014-3 (doi:10.1016/0092-8674(81)90014-3) [DOI] [PubMed] [Google Scholar]
- 24.King RW, Peters JM, Tugendreich S, Rolfe M, Hieter P, Kirschner MW. 1995. A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell 81, 279–288 10.1016/0092-8674(95)90338-0 (doi:10.1016/0092-8674(95)90338-0) [DOI] [PubMed] [Google Scholar]
- 25.Sudakin V, Ganoth D, Dahan A, Heller H, Hershko J, Luca FC, Ruderman JV, Hershko A. 1995. The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol. Biol. Cell 6, 185–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Visintin R, Prinz S, Amon A. 1997. CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science 278, 460–463 10.1126/science.278.5337.460 (doi:10.1126/science.278.5337.460) [DOI] [PubMed] [Google Scholar]
- 27.Hwang LH, Lau LF, Smith DL, Mistrot CA, Hardwick KG, Hwang ES, Amon A, Murray AW. 1998. Budding yeast Cdc20: a target of the spindle checkpoint. Science 279, 1041–1044 10.1126/science.279.5353.1041 (doi:10.1126/science.279.5353.1041) [DOI] [PubMed] [Google Scholar]
- 28.Uhlmann F, Lottspeich F, Nasmyth K. 1999. Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature 400, 37–42 10.1038/21831 (doi:10.1038/21831) [DOI] [PubMed] [Google Scholar]
- 29.Glotzer M, Murray AW, Kirschner MW. 1991. Cyclin is degraded by the ubiquitin pathway. Nature 349, 132–138 10.1038/349132a0 (doi:10.1038/349132a0) [DOI] [PubMed] [Google Scholar]
- 30.Walker G, Dorrell RG, Schlacht A, Dacks JB. 2011. Eukaryotic systematics: a user's guide for cell biologists and parasitologists. Parasitology 138, 1638–1663 10.1017/S0031182010001708 (doi:10.1017/S0031182010001708) [DOI] [PubMed] [Google Scholar]
- 31.Adl SM, et al. 2012. The revised classification of eukaryotes. J. Eukaryot. Microbiol. 59, 429–514 10.1111/j.1550-7408.2012.00644.x (doi:10.1111/j.1550-7408.2012.00644.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katz LA. 2012. Origin and diversification of eukaryotes. Annu. Rev. Microbiol. 66, 411–427 10.1146/annurev-micro-090110-102808 (doi:10.1146/annurev-micro-090110-102808) [DOI] [PubMed] [Google Scholar]
- 33.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 10.1093/nar/25.17.3389 (doi:10.1093/nar/25.17.3389) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Finn RD, Clements J, Eddy SR. 2011. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 10.1093/nar/gkr367 (doi:10.1093/nar/gkr367) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dacks JB, Walker G, Field MC. 2008. Implications of the new eukaryotic systematics for parasitologists. Parasitol. Int. 57, 97–104 10.1016/j.parint.2007.11.004 (doi:10.1016/j.parint.2007.11.004) [DOI] [PubMed] [Google Scholar]
- 36.Mocciaro A, Schiebel E. 2010. Cdc14: a highly conserved family of phosphatases with non-conserved functions? J. Cell Sci. 123, 2867–2876 10.1242/jcs.074815 (doi:10.1242/jcs.074815) [DOI] [PubMed] [Google Scholar]
- 37.Stechmann A, Cavalier-Smith T. 2002. Rooting the eukaryote tree by using a derived gene fusion. Science 297, 89–91 10.1126/science.1071196 (doi:10.1126/science.1071196) [DOI] [PubMed] [Google Scholar]
- 38.Arisue N, Hasegawa M, Hashimoto T. 2005. Root of the Eukaryota tree as inferred from combined maximum likelihood analyses of multiple molecular sequence data. Mol. Biol. Evol. 22, 409–420 10.1093/molbev/msi023 (doi:10.1093/molbev/msi023) [DOI] [PubMed] [Google Scholar]
- 39.Katz LA, Grant JR, Parfrey LW, Burleigh JG. 2012. Turning the crown upside down: gene tree parsimony roots the eukaryotic tree of life. Syst. Biol. 61, 653–660 10.1093/sysbio/sys026 (doi:10.1093/sysbio/sys026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rogozin IB, Basu MK, Csürös M, Koonin EV. 2009. Analysis of rare genomic changes does not support the unikont–bikont phylogeny and suggests cyanobacterial symbiosis as the point of primary radiation of eukaryotes. Genome Biol. Evol. 1, 99–113 10.1093/gbe/evp011 (doi:10.1093/gbe/evp011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Allen JWA, Ferguson SJ, Ginger ML. 2008. Distinctive biochemistry in the trypanosome mitochondrial intermembrane space suggests a model for stepwise evolution of the MIA pathway for import of cysteine-rich proteins. FEBS Lett. 582, 2817–2825 10.1016/j.febslet.2008.07.015 (doi:10.1016/j.febslet.2008.07.015) [DOI] [PubMed] [Google Scholar]
- 42.Allen JWA. 2011. Cytochrome c biogenesis in mitochondria—Systems III and V. FEBS J. 278, 4198–4216 10.1111/j.1742-4658.2011.08231.x (doi:10.1111/j.1742-4658.2011.08231.x) [DOI] [PubMed] [Google Scholar]
- 43.Cavalier-Smith T. 2010. Kingdoms Protozoa and Chromista and the eozoan root of the eukaryotic tree. Biol. Lett. 6, 342–345 10.1098/rsbl.2009.0948 (doi:10.1098/rsbl.2009.0948) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simpson AGB, Lukes J, Roger AJ. 2002. The evolutionary history of kinetoplastids and their kinetoplasts. Mol. Biol. Evol. 19, 2071–2083 10.1093/oxfordjournals.molbev.a004032 (doi:10.1093/oxfordjournals.molbev.a004032) [DOI] [PubMed] [Google Scholar]
- 45.Simpson AGB, Stevens JR, Lukes J. 2006. The evolution and diversity of kinetoplastid flagellates. Trends Parasitol. 22, 168–174 10.1016/j.pt.2006.02.006 (doi:10.1016/j.pt.2006.02.006) [DOI] [PubMed] [Google Scholar]
- 46.Deschamps P, Lara E, Marande W, López-García P, Ekelund F, Moreira D. 2011. Phylogenomic analysis of kinetoplastids supports that trypanosomatids arose from within bodonids. Mol. Biol. Evol. 28, 53–58 10.1093/molbev/msq289 (doi:10.1093/molbev/msq289) [DOI] [PubMed] [Google Scholar]
- 47.Hotez PJ, Kamath A. 2009. Neglected tropical diseases in sub-saharan Africa: review of their prevalence, distribution, and disease burden. PLoS Negl. Trop. Dis. 3, e412. 10.1371/journal.pntd.0000412 (doi:10.1371/journal.pntd.0000412) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simarro PP, Cecchi G, Franco JR, Paone M, Diarra A, Ruiz-Postigo JA, Fèvre EM, Mattioli RC, Jannin JG. 2012. Estimating and mapping the population at risk of sleeping sickness. PLoS Negl. Trop. Dis. 6, e1859. 10.1371/journal.pntd.0001859 (doi:10.1371/journal.pntd.0001859) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berriman M, et al. 2005. The genome of the African trypanosome Trypanosoma brucei. Science 309, 416–422 10.1126/science.1112642 (doi:10.1126/science.1112642) [DOI] [PubMed] [Google Scholar]
- 50.El-Sayed NM, et al. 2005. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science 309, 409–415 10.1126/science.1112631 (doi:10.1126/science.1112631) [DOI] [PubMed] [Google Scholar]
- 51.El-Sayed NM, et al. 2005. Comparative genomics of trypanosomatid parasitic protozoa. Science 309, 404–409 10.1126/science.1112181 (doi:10.1126/science.1112181) [DOI] [PubMed] [Google Scholar]
- 52.Ivens AC, et al. 2005. The genome of the kinetoplastid parasite, Leishmania major. Science 309, 436–442 10.1126/science.1112680 (doi:10.1126/science.1112680) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peacock CS, et al. 2007. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat. Genet. 39, 839–847 10.1038/ng2053 (doi:10.1038/ng2053) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jackson AP, Quail MA, Berriman M. 2008. Insights into the genome sequence of a free-living kinetoplastid: Bodo saltans (Kinetoplastida: Euglenozoa). BMC Genomics 9, 594. 10.1186/1471-2164-9-594 (doi:10.1186/1471-2164-9-594) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brun R, Schönenberger M. 1979. Cultivation and in vitro cloning or procyclic culture forms of Trypanosoma brucei in a semi-defined medium. Short communication. Acta Trop. 36, 289–292 [PubMed] [Google Scholar]
- 56.Hirumi H, Hirumi K. 1994. Axenic culture of African trypanosome bloodstream forms. Parasitol. Today (Regul. Ed.) 10, 80–84 10.1016/0169-4758(94)90402-2 (doi:10.1016/0169-4758(94)90402-2) [DOI] [PubMed] [Google Scholar]
- 57.Vanhamme L, et al. 2003. Apolipoprotein L-I is the trypanosome lytic factor of human serum. Nature 422, 83–87 10.1038/nature01461 (doi:10.1038/nature01461) [DOI] [PubMed] [Google Scholar]
- 58.Woods A, Sherwin T, Sasse R, MacRae TH, Baines AJ, Gull K. 1989. Definition of individual components within the cytoskeleton of Trypanosoma brucei by a library of monoclonal antibodies. J. Cell Sci. 93, 491–500 [DOI] [PubMed] [Google Scholar]
- 59.Tait A, Turner CM. 1990. Genetic exchange in Trypanosoma brucei. Parasitol. Today (Regul. Ed.) 6, 70–75 10.1016/0169-4758(90)90212-M (doi:10.1016/0169-4758(90)90212-M) [DOI] [PubMed] [Google Scholar]
- 60.Gibson W, Garside L. 1991. Genetic exchange in Trypanosoma brucei brucei: variable chromosomal location of housekeeping genes in different trypanosome stocks. Mol. Biochem. Parasitol. 45, 77–89 10.1016/0166-6851(91)90029-6 (doi:10.1016/0166-6851(91)90029-6) [DOI] [PubMed] [Google Scholar]
- 61.Peacock L, Ferris V, Sharma R, Sunter J, Bailey M, Carrington M, Gibson W. 2011. Identification of the meiotic life cycle stage of Trypanosoma brucei in the tsetse fly. Proc. Natl Acad. Sci. USA 108, 3671–3676 10.1073/pnas.1019423108 (doi:10.1073/pnas.1019423108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brun R, Schönenberger M. 1981. Stimulating effect of citrate and cis-Aconitate on the transformation of Trypanosoma brucei bloodstream forms to procyclic forms in vitro. Z. Parasitenkd. 66, 17–24 10.1007/BF00941941 (doi:10.1007/BF00941941) [DOI] [PubMed] [Google Scholar]
- 63.Engstler M, Boshart M. 2004. Cold shock and regulation of surface protein trafficking convey sensitization to inducers of stage differentiation in Trypanosoma brucei. Genes Dev. 18, 2798–2811 10.1101/gad.323404 (doi:10.1101/gad.323404) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kolev NG, Ramey-Butler K, Cross GAM, Ullu E, Tschudi C. 2012. Developmental progression to infectivity in Trypanosoma brucei triggered by an RNA-binding protein. Science 338, 1352–1353 10.1126/science.1229641 (doi:10.1126/science.1229641) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kelly S, et al. 2007. Functional genomics in Trypanosoma brucei: a collection of vectors for the expression of tagged proteins from endogenous and ectopic gene loci. Mol. Biochem. Parasitol. 154, 103–109 10.1016/j.molbiopara.2007.03.012 (doi:10.1016/j.molbiopara.2007.03.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee MG, Van der Ploeg LH. 1990. Homologous recombination and stable transfection in the parasitic protozoan Trypanosoma brucei. Science 250, 1583–1587 10.1126/science.2177225 (doi:10.1126/science.2177225) [DOI] [PubMed] [Google Scholar]
- 67.Ten Asbroek AL, Ouellette M, Borst P. 1990. Targeted insertion of the neomycin phosphotransferase gene into the tubulin gene cluster of Trypanosoma brucei. Nature 348, 174–175 10.1038/348174a0 (doi:10.1038/348174a0) [DOI] [PubMed] [Google Scholar]
- 68.Bastin P, Bagherzadeh Z, Matthews KR, Gull K. 1996. A novel epitope tag system to study protein targeting and organelle biogenesis in Trypanosoma brucei. Mol. Biochem. Parasitol. 77, 235–239 10.1016/0166-6851(96)02598-4 (doi:10.1016/0166-6851(96)02598-4) [DOI] [PubMed] [Google Scholar]
- 69.Arhin GK, Shen S, Ullu E, Tschudi C. 2004. A PCR-based method for gene deletion and protein tagging in Trypanosoma brucei. Methods Mol. Biol. 270, 277–286 10.1385/1-59259-793-9:277 (doi:10.1385/1-59259-793-9:277) [DOI] [PubMed] [Google Scholar]
- 70.Wirtz E, Clayton C. 1995. Inducible gene expression in trypanosomes mediated by a prokaryotic repressor. Science 268, 1179–1183 10.1126/science.7761835 (doi:10.1126/science.7761835) [DOI] [PubMed] [Google Scholar]
- 71.Wirtz E, Leal S, Ochatt C, Cross GA. 1999. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 99, 89–101 10.1016/S0166-6851(99)00002-X (doi:10.1016/S0166-6851(99)00002-X) [DOI] [PubMed] [Google Scholar]
- 72.Poon SK, Peacock L, Gibson W, Gull K, Kelly S. 2012. A modular and optimized single marker system for generating Trypanosoma brucei cell lines expressing T7 RNA polymerase and the tetracycline repressor. Open Biol. 2, 110037. 10.1098/rsob.110037 (doi:10.1098/rsob.110037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Barrett B, LaCount DJ, Donelson JE. 2004. Trypanosoma brucei: a first-generation CRE-loxP site-specific recombination system. Exp. Parasitol. 106, 37–44 10.1016/j.exppara.2004.01.004 (doi:10.1016/j.exppara.2004.01.004) [DOI] [PubMed] [Google Scholar]
- 74.Scahill MD, Pastar I, Cross GAM. 2008. CRE recombinase-based positive-negative selection systems for genetic manipulation in Trypanosoma brucei. Mol. Biochem. Parasitol. 157, 73–82 10.1016/j.molbiopara.2007.10.003 (doi:10.1016/j.molbiopara.2007.10.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ngô H, Tschudi C, Gull K, Ullu E. 1998. Double-stranded RNA induces mRNA degradation in Trypanosoma brucei. Proc. Natl Acad. Sci. USA 95, 14 687–14 692 10.1073/pnas.95.25.14687 (doi:10.1073/pnas.95.25.14687) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang Z, Morris JC, Drew ME, Englund PT. 2000. Inhibition of Trypanosoma brucei gene expression by RNA interference using an integratable vector with opposing T7 promoters. J. Biol. Chem. 275, 40 174–40 179 10.1074/jbc.M008405200 (doi:10.1074/jbc.M008405200) [DOI] [PubMed] [Google Scholar]
- 77.Englund PT, Agbo EEC, Lindsay ME, Liu B, Liu Y, Motyka SA, Yildirir G, Zhao Z. 2005. RNAi libraries and kinetoplast DNA. Biochem. Soc. Trans. 33, 1409–1412 10.1042/BST20051409 (doi:10.1042/BST20051409) [DOI] [PubMed] [Google Scholar]
- 78.Alsford S, Turner DJ, Obado SO, Sanchez-Flores A, Glover L, Berriman M, Hertz-Fowler C, Horn D. 2011. High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res. 21, 915–924 10.1101/gr.115089.110 (doi:10.1101/gr.115089.110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ersfeld K, Gull K. 1997. Partitioning of large and minichromosomes in Trypanosoma brucei. Science 276, 611–614 10.1126/science.276.5312.611 (doi:10.1126/science.276.5312.611) [DOI] [PubMed] [Google Scholar]
- 80.Navarro M, Gull K. 2001. A pol I transcriptional body associated with VSG mono-allelic expression in Trypanosoma brucei. Nature 414, 759–763 10.1038/414759a (doi:10.1038/414759a) [DOI] [PubMed] [Google Scholar]
- 81.Landeira D, Bart J-M, Van Tyne D, Navarro M. 2009. Cohesin regulates VSG monoallelic expression in trypanosomes. J. Cell Biol. 186, 243–254 10.1083/jcb.200902119 (doi:10.1083/jcb.200902119) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schimanski B, Nguyen TN, Günzl A. 2005. Highly efficient tandem affinity purification of trypanosome protein complexes based on a novel epitope combination. Eukaryot. Cell 4, 1942–1950 10.1128/EC.4.11.1942-1950.2005 (doi:10.1128/EC.4.11.1942-1950.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li Z, Lee JH, Chu F, Burlingame AL, Günzl A, Wang CC. 2008. Identification of a novel chromosomal passenger complex and its unique localization during cytokinesis in Trypanosoma brucei. PLoS ONE 3, e2354. 10.1371/journal.pone.0002354 (doi:10.1371/journal.pone.0002354) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Acestor N, Zíková A, Dalley RA, Anupama A, Panigrahi AK, Stuart KD. 2011. Trypanosoma brucei mitochondrial respiratome: composition and organization in procyclic form. Mol. Cell. Proteomics 10, M110.006908. 10.1074/mcp.M110.006908 (doi:10.1074/mcp.M110.006908) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gheiratmand L, Brasseur A, Zhou Q, He CY. 2012. Biochemical characterization of the bi-lobe revealed a continuous structural network linking the bi-lobe to other single-copied organelles in Trypanosoma brucei. J. Biol. Chem. 288, 3489–3499 10.1074/jbc.M112.417428 (doi:10.1074/jbc.M112.417428) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Morriswood B, et al. 2012. Novel bilobe components in Trypanosoma brucei identified using proximity-dependent biotinylation. Eukaryot. Cell 12, 356–367 10.1128/EC.00326-12 (doi:10.1128/EC.00326-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Siegel TN, Hekstra DR, Kemp LE, Figueiredo LM, Lowell JE, Fenyo D, Wang X, Dewell S, Cross GAM. 2009. Four histone variants mark the boundaries of polycistronic transcription units in Trypanosoma brucei. Genes Dev. 23, 1063–1076 10.1101/gad.1790409 (doi:10.1101/gad.1790409) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tiengwe C, et al. 2012. Genome-wide analysis reveals extensive functional interaction between DNA replication initiation and transcription in the genome of Trypanosoma brucei. Cell Rep. 2, 185–197 10.1016/j.celrep.2012.06.007 (doi:10.1016/j.celrep.2012.06.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Robinson DR, Sherwin T, Ploubidou A, Byard EH, Gull K. 1995. Microtubule polarity and dynamics in the control of organelle positioning, segregation, and cytokinesis in the trypanosome cell cycle. J. Cell Biol. 128, 1163–1172 10.1083/jcb.128.6.1163 (doi:10.1083/jcb.128.6.1163) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rosenkranz V, Wink M. 2008. Alkaloids induce programmed cell death in bloodstream forms of trypanosomes (Trypanosoma b. brucei). Molecules 13, 2462–2473 10.3390/molecules13102462 (doi:10.3390/molecules13102462) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Giles NL, Armson A, Reid SA. 2009. Characterization of trifluralin binding with recombinant tubulin from Trypanosoma brucei. Parasitol. Res. 104, 893–903 10.1007/s00436-008-1271-2 (doi:10.1007/s00436-008-1271-2) [DOI] [PubMed] [Google Scholar]
- 92.He CY, Ho HH, Malsam J, Chalouni C, West CM, Ullu E, Toomre D, Warren G. 2004. Golgi duplication in Trypanosoma brucei. J. Cell Biol. 165, 313–321 10.1083/jcb.200311076 (doi:10.1083/jcb.200311076) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li Z, Umeyama T, Wang CC. 2008. The chromosomal passenger complex and a mitotic kinesin interact with the Tousled-like kinase in trypanosomes to regulate mitosis and cytokinesis. PLoS ONE 3, e3814. 10.1371/journal.pone.0003814 (doi:10.1371/journal.pone.0003814) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Price HP, MacLean L, Marrison J, O'Toole PJ, Smith DF. 2010. Validation of a new method for immobilising kinetoplastid parasites for live cell imaging. Mol. Biochem. Parasitol. 169, 66–69 10.1016/j.molbiopara.2009.09.008 (doi:10.1016/j.molbiopara.2009.09.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Butter F, Bucerius F, Michel M, Cicova Z, Mann M, Janzen CJ. 2012. Comparative proteomics of two life cycle stages of stable isotope-labeled Trypanosoma brucei reveals novel components of the parasite's host adaptation machinery. Mol. Cell. Proteomics 12, 172–179 10.1074/mcp.M112.019224 (doi:10.1074/mcp.M112.019224) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gunasekera K, Wüthrich D, Braga-Lagache S, Heller M, Ochsenreiter T. 2012. Proteome remodelling during development from blood to insect-form Trypanosoma brucei quantified by SILAC and mass spectrometry. BMC Genomics 13, 556. 10.1186/1471-2164-13-556 (doi:10.1186/1471-2164-13-556) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Urbaniak MD, Guther MLS, Ferguson MAJ. 2012. Comparative SILAC proteomic analysis of Trypanosoma brucei bloodstream and procyclic lifecycle stages. PLoS ONE 7, e36619. 10.1371/journal.pone.0036619 (doi:10.1371/journal.pone.0036619) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Alsford S, et al. 2012. High-throughput decoding of antitrypanosomal drug efficacy and resistance. Nature 482, 232–236 10.1038/nature10771 (doi:10.1038/nature10771) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rout MP, Field MC. 2001. Isolation and characterization of subnuclear compartments from Trypanosoma brucei: identification of a major repetitive nuclear lamina component. J. Biol. Chem. 276, 38 261–38 271 10.1074/jbc.M104024200 (doi:10.1074/jbc.M104024200) [DOI] [PubMed] [Google Scholar]
- 100.Chowdhury AR, Zhao Z, Englund PT. 2008. Effect of hydroxyurea on procyclic Trypanosoma brucei: an unconventional mechanism for achieving synchronous growth. Eukaryot. Cell 7, 425–428 10.1128/EC.00369-07 (doi:10.1128/EC.00369-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Archer SK, Inchaustegui D, Queiroz R, Clayton C. 2011. The cell cycle regulated transcriptome of Trypanosoma brucei. PLoS ONE 6, e18425. 10.1371/journal.pone.0018425 (doi:10.1371/journal.pone.0018425) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vickerman K. 1965. Polymorphism and mitochondrial activity in sleeping sickness trypanosomes. Nature 208, 762–766 10.1038/208762a0 (doi:10.1038/208762a0) [DOI] [PubMed] [Google Scholar]
- 103.Fenn K, Matthews KR. 2007. The cell biology of Trypanosoma brucei differentiation. Curr. Opin. Microbiol. 10, 539–546 10.1016/j.mib.2007.09.014 (doi:10.1016/j.mib.2007.09.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.MacGregor P, Szöőr B, Savill NJ, Matthews KR. 2012. Trypanosomal immune evasion, chronicity and transmission: an elegant balancing act. Nat. Rev. Microbiol. 10, 431–438 10.1038/nrmicro2779 (doi:10.1038/nrmicro2779) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sherwin T, Gull K. 1989. The cell division cycle of Trypanosoma brucei brucei: timing of event markers and cytoskeletal modulations. Phil. Trans. R. Soc. Lond. B 323, 573–588 10.1098/rstb.1989.0037 (doi:10.1098/rstb.1989.0037) [DOI] [PubMed] [Google Scholar]
- 106.Vaughan S, Gull K. 2008. The structural mechanics of cell division in Trypanosoma brucei. Biochem. Soc. Trans. 36, 421–424 10.1042/BST0360421 (doi:10.1042/BST0360421) [DOI] [PubMed] [Google Scholar]
- 107.Angelopoulos E. 1970. Pellicular microtubules in the family Trypanosomatidae. J. Protozool. 17, 39–51 [DOI] [PubMed] [Google Scholar]
- 108.Sherwin T, Gull K. 1989. Visualization of detyrosination along single microtubules reveals novel mechanisms of assembly during cytoskeletal duplication in trypanosomes. Cell 57, 211–221 10.1016/0092-8674(89)90959-8 (doi:10.1016/0092-8674(89)90959-8) [DOI] [PubMed] [Google Scholar]
- 109.Robinson DR, Gull K. 1991. Basal body movements as a mechanism for mitochondrial genome segregation in the trypanosome cell cycle. Nature 352, 731–733 10.1038/352731a0 (doi:10.1038/352731a0) [DOI] [PubMed] [Google Scholar]
- 110.Ogbadoyi EO, Robinson DR, Gull K. 2003. A high-order trans-membrane structural linkage is responsible for mitochondrial genome positioning and segregation by flagellar basal bodies in trypanosomes. Mol. Biol. Cell 14, 1769–1779 10.1091/mbc.E02-08-0525 (doi:10.1091/mbc.E02-08-0525) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Field H, Sherwin T, Smith AC, Gull K, Field MC. 2000. Cell-cycle and developmental regulation of TbRAB31 localisation, a GTP-locked Rab protein from Trypanosoma brucei. Mol. Biochem. Parasitol. 106, 21–35 10.1016/S0166-6851(99)00192-9 (doi:10.1016/S0166-6851(99)00192-9) [DOI] [PubMed] [Google Scholar]
- 112.El-Sayed NM, Hegde P, Quackenbush J, Melville SE, Donelson JE. 2000. The African trypanosome genome. Int. J. Parasitol. 30, 329–345 10.1016/S0020-7519(00)00015-1 (doi:10.1016/S0020-7519(00)00015-1) [DOI] [PubMed] [Google Scholar]
- 113.Obado SO, Taylor MC, Wilkinson SR, Bromley EV, Kelly JM. 2005. Functional mapping of a trypanosome centromere by chromosome fragmentation identifies a 16-kb GC-rich transcriptional ‘strand-switch’ domain as a major feature. Genome Res. 15, 36–43 10.1101/gr.2895105 (doi:10.1101/gr.2895105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Obado SO, Bot C, Nilsson D, Andersson B, Kelly JM. 2007. Repetitive DNA is associated with centromeric domains in Trypanosoma brucei but not Trypanosoma cruzi. Genome Biol. 8, R37. 10.1186/gb-2007-8-3-r37 (doi:10.1186/gb-2007-8-3-r37) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Echeverry MC, Bot C, Obado SO, Taylor MC, Kelly JM. 2012. Centromere-associated repeat arrays on Trypanosoma brucei chromosomes are much more extensive than predicted. BMC Genomics 13, 29. 10.1186/1471-2164-13-29 (doi:10.1186/1471-2164-13-29) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Clayton CE. 2002. Life without transcriptional control? From fly to man and back again. EMBO J. 21, 1881–1888 10.1093/emboj/21.8.1881 (doi:10.1093/emboj/21.8.1881) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Daniels J-P, Gull K, Wickstead B. 2010. Cell biology of the trypanosome genome. Microbiol. Mol. Biol. Rev. 74, 552–569 10.1128/MMBR.00024-10 (doi:10.1128/MMBR.00024-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ersfeld K. 2011. Nuclear architecture, genome and chromatin organisation in Trypanosoma brucei. Res. Microbiol. 162, 626–636 10.1016/j.resmic.2011.01.014 (doi:10.1016/j.resmic.2011.01.014) [DOI] [PubMed] [Google Scholar]
- 119.Vickerman K. 1978. Antigenic variation in trypanosomes. Nature 273, 613–617 10.1038/273613a0 (doi:10.1038/273613a0) [DOI] [PubMed] [Google Scholar]
- 120.Turner CM, Barry JD. 1989. High frequency of antigenic variation in Trypanosoma brucei rhodesiense infections. Parasitology 99, 67–75 10.1017/S0031182000061035 (doi:10.1017/S0031182000061035) [DOI] [PubMed] [Google Scholar]
- 121.Morrison LJ, Majiwa P, Read AF, Barry JD. 2005. Probabilistic order in antigenic variation of Trypanosoma brucei. Int. J. Parasitol. 35, 961–972 10.1016/j.ijpara.2005.05.004 (doi:10.1016/j.ijpara.2005.05.004) [DOI] [PubMed] [Google Scholar]
- 122.Lythgoe KA, Morrison LJ, Read AF, Barry JD. 2007. Parasite-intrinsic factors can explain ordered progression of trypanosome antigenic variation. Proc. Natl Acad. Sci. USA 104, 8095–8100 10.1073/pnas.0606206104 (doi:10.1073/pnas.0606206104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Robinson NP, Burman N, Melville SE, Barry JD. 1999. Predominance of duplicative VSG gene conversion in antigenic variation in African trypanosomes. Mol. Cell. Biol. 19, 5839–5846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Boothroyd CE, Dreesen O, Leonova T, Ly KI, Figueiredo LM, Cross GAM, Papavasiliou FN. 2009. A yeast-endonuclease-generated DNA break induces antigenic switching in Trypanosoma brucei. Nature 459, 278–281 10.1038/nature07982 (doi:10.1038/nature07982) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Navarro M, Peñate X, Landeira D. 2007. Nuclear architecture underlying gene expression in Trypanosoma brucei. Trends Microbiol. 15, 263–270 10.1016/j.tim.2007.04.004 (doi:10.1016/j.tim.2007.04.004) [DOI] [PubMed] [Google Scholar]
- 126.Horn D, McCulloch R. 2010. Molecular mechanisms underlying the control of antigenic variation in African trypanosomes. Curr. Opin. Microbiol. 13, 700–705 10.1016/j.mib.2010.08.009 (doi:10.1016/j.mib.2010.08.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rudenko G. 2011. African trypanosomes: the genome and adaptations for immune evasion. Essays Biochem. 51, 47–62 10.1042/bse0510047 (doi:10.1042/bse0510047) [DOI] [PubMed] [Google Scholar]
- 128.Sloof P, Menke HH, Caspers MP, Borst P. 1983. Size fractionation of Trypanosoma brucei DNA: localization of the 177-bp repeat satellite DNA and a variant surface glycoprotein gene in a mini-chromosomal DNA fraction. Nucleic Acids Res. 11, 3889–3901 10.1093/nar/11.12.3889 (doi:10.1093/nar/11.12.3889) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zomerdijk JC, Kieft R, Borst P. 1992. A ribosomal RNA gene promoter at the telomere of a mini-chromosome in Trypanosoma brucei. Nucleic Acids Res. 20, 2725–2734 10.1093/nar/20.11.2725 (doi:10.1093/nar/20.11.2725) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wickstead B, Ersfeld K, Gull K. 2003. The mitotic stability of the minichromosomes of Trypanosoma brucei. Mol. Biochem. Parasitol. 132, 97–100 10.1016/j.molbiopara.2003.08.007 (doi:10.1016/j.molbiopara.2003.08.007) [DOI] [PubMed] [Google Scholar]
- 131.Wickstead B, Ersfeld K, Gull K. 2004. The small chromosomes of Trypanosoma brucei involved in antigenic variation are constructed around repetitive palindromes. Genome Res. 14, 1014–1024 10.1101/gr.2227704 (doi:10.1101/gr.2227704) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Alsford NS, Navarro M, Jamnadass HR, Dunbar H, Ackroyd M, Murphy NB, Gull K, Ersfeld K. 2003. The identification of circular extrachromosomal DNA in the nuclear genome of Trypanosoma brucei. Mol. Microbiol. 47, 277–289 10.1046/j.1365-2958.2003.03266.x (doi:10.1046/j.1365-2958.2003.03266.x) [DOI] [PubMed] [Google Scholar]
- 133.Henikoff S, Ahmad K, Malik HS. 2001. The centromere paradox: stable inheritance with rapidly evolving DNA. Science 293, 1098–1102 10.1126/science.1062939 (doi:10.1126/science.1062939) [DOI] [PubMed] [Google Scholar]
- 134.Malik HS, Henikoff S. 2009. Major evolutionary transitions in centromere complexity. Cell 138, 1067–1082 10.1016/j.cell.2009.08.036 (doi:10.1016/j.cell.2009.08.036) [DOI] [PubMed] [Google Scholar]
- 135.Melters DP, Paliulis LV, Korf IF, Chan SWL. 2012. Holocentric chromosomes: convergent evolution, meiotic adaptations, and genomic analysis. Chromosome Res. 20, 579–593 10.1007/s10577-012-9292-1 (doi:10.1007/s10577-012-9292-1) [DOI] [PubMed] [Google Scholar]
- 136.Clarke L, Carbon J. 1980. Isolation of a yeast centromere and construction of functional small circular chromosomes. Nature 287, 504–509 10.1038/287504a0 (doi:10.1038/287504a0) [DOI] [PubMed] [Google Scholar]
- 137.Copenhaver GP, et al. 1999. Genetic definition and sequence analysis of Arabidopsis centromeres. Science 286, 2468–2474 10.1126/science.286.5449.2468 (doi:10.1126/science.286.5449.2468) [DOI] [PubMed] [Google Scholar]
- 138.Sanyal K, Baum M, Carbon J. 2004. Centromeric DNA sequences in the pathogenic yeast Candida albicans are all different and unique. Proc. Natl Acad. Sci. USA 101, 11 374–11 379 10.1073/pnas.0404318101 (doi:10.1073/pnas.0404318101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Floridia G, Zatterale A, Zuffardi O, Tyler-Smith C. 2000. Mapping of a human centromere onto the DNA by topoisomerase II cleavage. EMBO Rep. 1, 489–493 10.1093/embo-reports/kvd110 (doi:10.1093/embo-reports/kvd110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Spence JM, Fournier REK, Oshimura M, Regnier V, Farr CJ. 2005. Topoisomerase II cleavage activity within the human D11Z1 and DXZ1 alpha-satellite arrays. Chromosome Res. 13, 637–648 10.1007/s10577-005-1003-8 (doi:10.1007/s10577-005-1003-8) [DOI] [PubMed] [Google Scholar]
- 141.Kelly JM, McRobert L, Baker DA. 2006. Evidence on the chromosomal location of centromeric DNA in Plasmodium falciparum from etoposide-mediated topoisomerase-II cleavage. Proc. Natl Acad. Sci. USA 103, 6706–6711 10.1073/pnas.0510363103 (doi:10.1073/pnas.0510363103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Maloney KA, Sullivan LL, Matheny JE, Strome ED, Merrett SL, Ferris A, Sullivan BA. 2012. Functional epialleles at an endogenous human centromere. Proc. Natl Acad. Sci. USA 109, 13 704–13 709 10.1073/pnas.1203126109 (doi:10.1073/pnas.1203126109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Talbert PB, Bayes JJ, Henikoff S. 2009. Evolution of centromeres and kinetochores: a two-part fugue. In The kinetochore: (eds De Wulf P, Earnshaw WC.), pp. 1–37 New York, NY: Springer [Google Scholar]
- 144.Wong LH, Choo KHA. 2004. Evolutionary dynamics of transposable elements at the centromere. Trends Genet. 20, 611–616 10.1016/j.tig.2004.09.011 (doi:10.1016/j.tig.2004.09.011) [DOI] [PubMed] [Google Scholar]
- 145.Folco HD, Pidoux AL, Urano T, Allshire RC. 2008. Heterochromatin and RNAi are required to establish CENP-A chromatin at centromeres. Science 319, 94–97 10.1126/science.1150944 (doi:10.1126/science.1150944) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lejeune E, Bayne EH, Allshire RC. 2010. On the connection between RNAi and heterochromatin at centromeres. Cold Spring Harb. Symp. Quant. Biol. 75, 275–283 10.1101/sqb.2010.75.024 (doi:10.1101/sqb.2010.75.024) [DOI] [PubMed] [Google Scholar]
- 147.Patrick KL, Shi H, Kolev NG, Ersfeld K, Tschudi C, Ullu E. 2009. Distinct and overlapping roles for two Dicer-like proteins in the RNA interference pathways of the ancient eukaryote Trypanosoma brucei. Proc. Natl Acad. Sci. USA 106, 17 933–17 938 10.1073/pnas.0907766106 (doi:10.1073/pnas.0907766106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Tschudi C, Shi H, Franklin JB, Ullu E. 2012. Small interfering RNA-producing loci in the ancient parasitic eukaryote Trypanosoma brucei. BMC Genomics 13, 427. 10.1186/1471-2164-13-427 (doi:10.1186/1471-2164-13-427) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Durand-Dubief M, Absalon S, Menzer L, Ngwabyt S, Ersfeld K, Bastin P. 2007. The Argonaute protein TbAGO1 contributes to large and mini-chromosome segregation and is required for control of RIME retroposons and RHS pseudogene-associated transcripts. Mol. Biochem. Parasitol. 156, 144–153 10.1016/j.molbiopara.2007.07.016 (doi:10.1016/j.molbiopara.2007.07.016) [DOI] [PubMed] [Google Scholar]
- 150.Lye L-F, Owens K, Shi H, Murta SMF, Vieira AC, Turco SJ, Tschudi C, Ullu E, Beverley SM. 2010. Retention and loss of RNA interference pathways in trypanosomatid protozoans. PLoS Pathog. 6, e1001161. 10.1371/journal.ppat.1001161 (doi:10.1371/journal.ppat.1001161) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hammarton TC. 2007. Cell cycle regulation in Trypanosoma brucei. Mol. Biochem. Parasitol. 153, 1–8 10.1016/j.molbiopara.2007.01.017 (doi:10.1016/j.molbiopara.2007.01.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Li Z. 2012. Regulation of the cell division cycle in Trypanosoma brucei. Eukaryot. Cell 11, 1180–1190 10.1128/EC.00145-12 (doi:10.1128/EC.00145-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Woodward R, Gull K. 1990. Timing of nuclear and kinetoplast DNA replication and early morphological events in the cell cycle of Trypanosoma brucei. J. Cell Sci. 95, 49–57 [DOI] [PubMed] [Google Scholar]
- 154.Siegel TN, Hekstra DR, Cross GAM. 2008. Analysis of the Trypanosoma brucei cell cycle by quantitative DAPI imaging. Mol. Biochem. Parasitol. 160, 171–174 10.1016/j.molbiopara.2008.04.004 (doi:10.1016/j.molbiopara.2008.04.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gluenz E, Povelones ML, Englund PT, Gull K. 2011. The kinetoplast duplication cycle in Trypanosoma brucei is orchestrated by cytoskeleton-mediated cell morphogenesis. Mol. Cell. Biol. 31, 1012–1021 10.1128/MCB.01176-10 (doi:10.1128/MCB.01176-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pickett-Heaps J. 1974. The evolution of mitosis and the eukaryotic condition. Biosystems 6, 37–48 10.1016/0303-2647(74)90009-4 (doi:10.1016/0303-2647(74)90009-4) [DOI] [PubMed] [Google Scholar]
- 157.Heath IB. 1980. Variant mitoses in lower eukaryotes: indicators of the evolution of mitosis. Int. Rev. Cytol. 64, 1–80 10.1016/S0074-7696(08)60235-1 (doi:10.1016/S0074-7696(08)60235-1) [DOI] [PubMed] [Google Scholar]
- 158.Vickerman K, Preston TM. 1970. Spindle microtubules in the dividing nuclei of trypanosomes. J. Cell Sci. 6, 365–383 [DOI] [PubMed] [Google Scholar]
- 159.Tanenbaum ME, Medema RH. 2010. Mechanisms of centrosome separation and bipolar spindle assembly. Dev. Cell 19, 797–806 10.1016/j.devcel.2010.11.011 (doi:10.1016/j.devcel.2010.11.011) [DOI] [PubMed] [Google Scholar]
- 160.Nigg EA, Stearns T. 2011. The centrosome cycle: centriole biogenesis, duplication and inherent asymmetries. Nat. Cell Biol. 13, 1154–1160 10.1038/ncb2345 (doi:10.1038/ncb2345) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ogbadoyi E, Ersfeld K, Robinson D, Sherwin T, Gull K. 2000. Architecture of the Trypanosoma brucei nucleus during interphase and mitosis. Chromosoma 108, 501–513 10.1007/s004120050402 (doi:10.1007/s004120050402) [DOI] [PubMed] [Google Scholar]
- 162.Tamm T, Grallert A, Grossman EPS, Alvarez-Tabares I, Stevens FE, Hagan IM. 2011. Brr6 drives the Schizosaccharomyces pombe spindle pole body nuclear envelope insertion/extrusion cycle. J. Cell Biol. 195, 467–484 10.1083/jcb.201106076 (doi:10.1083/jcb.201106076) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gull K, Briggs L, Vaughan S. 2004. Basal bodies and microtubule organization in pathogenic protozoa. In Centrosomes in development and disease (ed. Nigg EA.), pp. 401–423 Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA [Google Scholar]
- 164.Hammarton TC, Clark J, Douglas F, Boshart M, Mottram JC. 2003. Stage-specific differences in cell cycle control in Trypanosoma brucei revealed by RNA interference of a mitotic cyclin. J. Biol. Chem. 278, 22 877–22 886 10.1074/jbc.M300813200 (doi:10.1074/jbc.M300813200) [DOI] [PubMed] [Google Scholar]
- 165.Tu X, Wang CC. 2004. The involvement of two cdc2-related kinases (CRKs) in Trypanosoma brucei cell cycle regulation and the distinctive stage-specific phenotypes caused by CRK3 depletion. J. Biol. Chem. 279, 20 519–20 528 10.1074/jbc.M312862200 (doi:10.1074/jbc.M312862200) [DOI] [PubMed] [Google Scholar]
- 166.Gluenz E, Sharma R, Carrington M, Gull K. 2008. Functional characterization of cohesin subunit SCC1 in Trypanosoma brucei and dissection of mutant phenotypes in two life cycle stages. Mol. Microbiol. 69, 666–680 10.1111/j.1365-2958.2008.06320.x (doi:10.1111/j.1365-2958.2008.06320.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bessat M, Ersfeld K. 2009. Functional characterization of cohesin SMC3 and separase and their roles in the segregation of large and minichromosomes in Trypanosoma brucei. Mol. Microbiol. 71, 1371–1385 10.1111/j.1365-2958.2009.06611.x (doi:10.1111/j.1365-2958.2009.06611.x) [DOI] [PubMed] [Google Scholar]
- 168.Tu X, Kumar P, Li Z, Wang CC. 2006. An aurora kinase homologue is involved in regulating both mitosis and cytokinesis in Trypanosoma brucei. J. Biol. Chem. 281, 9677–9687 10.1074/jbc.M511504200 (doi:10.1074/jbc.M511504200) [DOI] [PubMed] [Google Scholar]
- 169.Li Z, Umeyama T, Wang CC. 2009. The Aurora Kinase in Trypanosoma brucei plays distinctive roles in metaphase-anaphase transition and cytokinetic initiation. PLoS Pathog. 5, e1000575. 10.1371/journal.ppat.1000575 (doi:10.1371/journal.ppat.1000575) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kumar P, Wang CC. 2005. Depletion of anaphase-promoting complex or cyclosome (APC/C) subunit homolog APC1 or CDC27 of Trypanosoma brucei arrests the procyclic form in metaphase but the bloodstream form in anaphase. J. Biol. Chem. 280, 31 783–31 791 10.1074/jbc.M504326200 (doi:10.1074/jbc.M504326200) [DOI] [PubMed] [Google Scholar]
- 171.Mutomba MC, To WY, Hyun WC, Wang CC. 1997. Inhibition of proteasome activity blocks cell cycle progression at specific phase boundaries in African trypanosomes. Mol. Biochem. Parasitol. 90, 491–504 10.1016/S0166-6851(97)00197-7 (doi:10.1016/S0166-6851(97)00197-7) [DOI] [PubMed] [Google Scholar]
- 172.Wordeman L, Mitchison TJ. 1995. Identification and partial characterization of mitotic centromere-associated kinesin, a kinesin-related protein that associates with centromeres during mitosis. J. Cell Biol. 128, 95–104 10.1083/jcb.128.1.95 (doi:10.1083/jcb.128.1.95) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Chan KY, Matthews KR, Ersfeld K. 2010. Functional characterisation and drug target validation of a mitotic kinesin-13 in Trypanosoma brucei. PLoS Pathog. 6, e1001050. 10.1371/journal.ppat.1001050 (doi:10.1371/journal.ppat.1001050) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wickstead B, Carrington JT, Gluenz E, Gull K. 2010. The expanded Kinesin-13 repertoire of trypanosomes contains only one mitotic Kinesin indicating multiple extra-nuclear roles. PLoS ONE 5, e15020. 10.1371/journal.pone.0015020 (doi:10.1371/journal.pone.0015020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kumar P, Wang CC. 2006. Dissociation of cytokinesis initiation from mitotic control in a eukaryote. Eukaryot. Cell 5, 92–102 10.1128/EC.5.1.92-102.2006 (doi:10.1128/EC.5.1.92-102.2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hammarton TC, Kramer S, Tetley L, Boshart M, Mottram JC. 2007. Trypanosoma brucei Polo-like kinase is essential for basal body duplication, kDNA segregation and cytokinesis. Mol. Microbiol. 65, 1229–1248 10.1111/j.1365-2958.2007.05866.x (doi:10.1111/j.1365-2958.2007.05866.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.De Graffenried CL, Ho HH, Warren G. 2008. Polo-like kinase is required for Golgi and bilobe biogenesis in Trypanosoma brucei. J. Cell Biol. 181, 431–438 10.1083/jcb.200708082 (doi:10.1083/jcb.200708082) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Mishra RK, Chakraborty P, Arnaoutov A, Fontoura BMA, Dasso M. 2010. The Nup107-160 complex and gamma-TuRC regulate microtubule polymerization at kinetochores. Nat. Cell Biol. 12, 164–169 10.1038/ncb2016 (doi:10.1038/ncb2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wozniak R, Burke B, Doye V. 2010. Nuclear transport and the mitotic apparatus: an evolving relationship. Cell. Mol. Life Sci. 67, 2215–2230 10.1007/s00018-010-0325-7 (doi:10.1007/s00018-010-0325-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.DeGrasse JA, DuBois KN, Devos D, Siegel TN, Sali A, Field MC, Rout MP, Chait BT. 2009. Evidence for a shared nuclear pore complex architecture that is conserved from the last common eukaryotic ancestor. Mol. Cell. Proteomics 8, 2119–2130 10.1074/mcp.M900038-MCP200 (doi:10.1074/mcp.M900038-MCP200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Meraldi P, McAinsh AD, Rheinbay E, Sorger PK. 2006. Phylogenetic and structural analysis of centromeric DNA and kinetochore proteins. Genome Biol. 7, R23. 10.1186/gb-2006-7-3-r23 (doi:10.1186/gb-2006-7-3-r23) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Schleiffer A, Maier M, Litos G, Lampert F, Hornung P, Mechtler K, Westermann S. 2012. CENP-T proteins are conserved centromere receptors of the Ndc80 complex. Nat. Cell Biol. 14, 604–613 10.1038/ncb2493 (doi:10.1038/ncb2493) [DOI] [PubMed] [Google Scholar]
- 183.Philippe H, Lopez P, Brinkmann H, Budin K, Germot A, Laurent J, Moreira D, Müller M, Le Guyader H. 2000. Early-branching or fast-evolving eukaryotes? An answer based on slowly evolving positions. Proc. R. Soc. Lond. B 267, 1213–1221 10.1098/rspb.2000.1130 (doi:10.1098/rspb.2000.1130) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Malik HS, Henikoff S. 2003. Phylogenomics of the nucleosome. Nat. Struct. Mol. Biol. 10, 882–891 10.1038/nsb996 (doi:10.1038/nsb996) [DOI] [PubMed] [Google Scholar]
- 185.Alsford S, Horn D. 2004. Trypanosomatid histones. Mol. Microbiol. 53, 365–372 10.1111/j.1365-2958.2004.04151.x (doi:10.1111/j.1365-2958.2004.04151.x) [DOI] [PubMed] [Google Scholar]
- 186.Sullivan WJ, Jr, Naguleswaran A, Angel SO. 2006. Histones and histone modifications in protozoan parasites. Cell. Microbiol. 8, 1850–1861 10.1111/j.1462-5822.2006.00818.x (doi:10.1111/j.1462-5822.2006.00818.x) [DOI] [PubMed] [Google Scholar]
- 187.Postberg J, Forcob S, Chang W-J, Lipps HJ. 2010. The evolutionary history of histone H3 suggests a deep eukaryotic root of chromatin modifying mechanisms. BMC Evol. Biol. 10, 259. 10.1186/1471-2148-10-259 (doi:10.1186/1471-2148-10-259) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lowell JE, Cross GAM. 2004. A variant histone H3 is enriched at telomeres in Trypanosoma brucei. J. Cell Sci. 117, 5937–5947 10.1242/jcs.01515 (doi:10.1242/jcs.01515) [DOI] [PubMed] [Google Scholar]
- 189.Lowell JE, Kaiser F, Janzen CJ, Cross GAM. 2005. Histone H2AZ dimerizes with a novel variant H2B and is enriched at repetitive DNA in Trypanosoma brucei. J. Cell Sci. 118, 5721–5730 10.1242/jcs.02688 (doi:10.1242/jcs.02688) [DOI] [PubMed] [Google Scholar]
- 190.Figueiredo LM, Cross GAM, Janzen CJ. 2009. Epigenetic regulation in African trypanosomes: a new kid on the block. Nat. Rev. Microbiol. 7, 504–513 10.1038/nrmicro2149 (doi:10.1038/nrmicro2149) [DOI] [PubMed] [Google Scholar]
- 191.Carmena M, Wheelock M, Funabiki H, Earnshaw WC. 2012. The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nat. Rev. Mol. Cell Biol. 13, 789–803 10.1038/nrm3474 (doi:10.1038/nrm3474) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Casanova M, Crobu L, Blaineau C, Bourgeois N, Bastien P, Pagès M. 2009. Microtubule-severing proteins are involved in flagellar length control and mitosis in trypanosomatids. Mol. Microbiol. 71, 1353–1370 10.1111/j.1365-2958.2009.06594.x (doi:10.1111/j.1365-2958.2009.06594.x) [DOI] [PubMed] [Google Scholar]
- 193.Solari AJ. 1995. Mitosis and genome partition in trypanosomes. Biocell 19, 65–84 [PubMed] [Google Scholar]
- 194.Watanabe Y. 2010. Temporal and spatial regulation of targeting aurora B to the inner centromere. Cold Spring Harb. Symp. Quant. Biol. 75, 419–423 10.1101/sqb.2010.75.035 (doi:10.1101/sqb.2010.75.035) [DOI] [PubMed] [Google Scholar]
- 195.Solari AJ. 1980. The 3-dimensional fine structure of the mitotic spindle in Trypanosoma cruzi. Chromosoma 78, 239–255 10.1007/BF00328395 (doi:10.1007/BF00328395) [DOI] [PubMed] [Google Scholar]
- 196.Ureña F. 1986. Three-dimensional reconstructions of the mitotic spindle and dense plaques in three species of Leishmania. Z. Parasitenkd. 72, 299–306 10.1007/BF00928739 (doi:10.1007/BF00928739) [DOI] [PubMed] [Google Scholar]
- 197.Prensier G, Slomianny C. 1986. The karyotype of Plasmodium falciparum determined by ultrastructural serial sectioning and 3D reconstruction. J. Parasitol. 72, 731–736 10.2307/3281465 (doi:10.2307/3281465) [DOI] [PubMed] [Google Scholar]
- 198.Gerald N, Mahajan B, Kumar S. 2011. Mitosis in the human malaria parasite Plasmodium falciparum. Eukaryot. Cell 10, 474–482 10.1128/EC.00314-10 (doi:10.1128/EC.00314-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Gull K, Alsford S, Ersfeld K. 1998. Segregation of minichromosomes in trypanosomes: implications for mitotic mechanisms. Trends Microbiol. 6, 319–323 10.1016/S0966-842X(98)01314-6 (doi:10.1016/S0966-842X(98)01314-6) [DOI] [PubMed] [Google Scholar]
- 200.Gan L, Ladinsky MS, Jensen GJ. 2011. Organization of the smallest eukaryotic spindle. Curr. Biol. 21, 1578–1583 10.1016/j.cub.2011.08.021 (doi:10.1016/j.cub.2011.08.021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Musacchio A, Salmon ED. 2007. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 8, 379–393 10.1038/nrm2163 (doi:10.1038/nrm2163) [DOI] [PubMed] [Google Scholar]
- 202.He X, Patterson TE, Sazer S. 1997. The Schizosaccharomyces pombe spindle checkpoint protein mad2p blocks anaphase and genetically interacts with the anaphase-promoting complex. Proc. Natl Acad. Sci. USA 94, 7965–7970 10.1073/pnas.94.15.7965 (doi:10.1073/pnas.94.15.7965) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Buffin E, Emre D, Karess RE. 2007. Flies without a spindle checkpoint. Nat. Cell Biol. 9, 565–572 10.1038/ncb1570 (doi:10.1038/ncb1570) [DOI] [PubMed] [Google Scholar]
- 204.De Antoni A, et al. 2005. The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr. Biol. 15, 214–225 10.1016/j.cub.2005.01.038 (doi:10.1016/j.cub.2005.01.038) [DOI] [PubMed] [Google Scholar]
- 205.Aravind L, Koonin EV. 1998. The HORMA domain: a common structural denominator in mitotic checkpoints, chromosome synapsis and DNA repair. Trends Biochem. Sci. 23, 284–286 10.1016/S0968-0004(98)01257-2 (doi:10.1016/S0968-0004(98)01257-2) [DOI] [PubMed] [Google Scholar]
- 206.Luo X, Tang Z, Rizo J, Yu H. 2002. The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either Mad1 or Cdc20. Mol. Cell 9, 59–71 10.1016/S1097-2765(01)00435-X (doi:10.1016/S1097-2765(01)00435-X) [DOI] [PubMed] [Google Scholar]
- 207.Ploubidou A, Robinson DR, Docherty RC, Ogbadoyi EO, Gull K. 1999. Evidence for novel cell cycle checkpoints in trypanosomes: kinetoplast segregation and cytokinesis in the absence of mitosis. J. Cell Sci. 112, 4641–4650 [DOI] [PubMed] [Google Scholar]
- 208.Cavalier-Smith T. 2002. The phagotrophic origin of eukaryotes and phylogenetic classification of Protozoa. Int. J. Syst. Evol. Microbiol. 52, 297–354 [DOI] [PubMed] [Google Scholar]
- 209.Carvalho-Santos Z, Azimzadeh J, Pereira-Leal JB, Bettencourt-Dias M. 2011. Evolution: tracing the origins of centrioles, cilia, and flagella. J. Cell Biol. 194, 165–175 10.1083/jcb.201011152 (doi:10.1083/jcb.201011152) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Brodsky FM, Thattai M, Mayor S. 2012. Evolutionary cell biology: lessons from diversity. Nat. Cell Biol. 14, 651. 10.1038/ncb2539 (doi:10.1038/ncb2539) [DOI] [PubMed] [Google Scholar]
- 211.Wilson KL, Dawson SC. 2011. Evolution: functional evolution of nuclear structure. J. Cell Biol. 195, 171–181 10.1083/jcb.201103171 (doi:10.1083/jcb.201103171) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Field MC, Horn D, Alsford S, Koreny L, Rout MP. 2012. Telomeres, tethers and trypanosomes. Nucleus 3, 478–486 10.4161/nucl.22167 (doi:10.4161/nucl.22167) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Wickstead B, Gull K. 2011. The evolution of the cytoskeleton. J. Cell Biol. 194, 513–525 10.1083/jcb.201102065 (doi:10.1083/jcb.201102065) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Yutin N, Koonin EV. 2012. Archaeal origin of tubulin. Biol. Direct 7, 10. 10.1186/1745-6150-7-10 (doi:10.1186/1745-6150-7-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Drechsler H, McAinsh AD. 2012. Exotic mitotic mechanisms. Open Biol. 2, 120140. 10.1098/rsob.120140 (doi:10.1098/rsob.120140) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Frost A, et al. 2012. Functional repurposing revealed by comparing S. pombe and S. cerevisiae genetic interactions. Cell 149, 1339–1352 10.1016/j.cell.2012.04.028 (doi:10.1016/j.cell.2012.04.028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Vleugel M, Hoogendoorn E, Snel B, Kops GJPL. 2012. Evolution and function of the mitotic checkpoint. Dev. Cell 23, 239–250 10.1016/j.devcel.2012.06.013 (doi:10.1016/j.devcel.2012.06.013) [DOI] [PubMed] [Google Scholar]
- 218.Gourguechon S, Holt LJ, Cande WZ. 2013. The Giardia cell cycle progresses independently of the anaphase promoting complex. J. Cell Sci. 10.1242/jcs.121632 (doi:10.1242/jcs.121632) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Hammarton TC, Monnerat S, Mottram JC. 2007. Cytokinesis in trypanosomatids. Curr. Opin. Microbiol. 10, 520–527 10.1016/j.mib.2007.10.005 (doi:10.1016/j.mib.2007.10.005) [DOI] [PubMed] [Google Scholar]
- 220.Vaughan S. 2010. Assembly of the flagellum and its role in cell morphogenesis in Trypanosoma brucei. Curr. Opin. Microbiol. 13, 453–458 10.1016/j.mib.2010.05.006 (doi:10.1016/j.mib.2010.05.006) [DOI] [PubMed] [Google Scholar]
- 221.Farr H, Gull K. 2012. Cytokinesis in trypanosomes. Cytoskeleton (Hoboken) 69, 931–941 10.1002/cm.21074 (doi:10.1002/cm.21074) [DOI] [PubMed] [Google Scholar]
- 222.Kobayashi Y, Imamura S, Hanaoka M, Tanaka K. 2011. A tetrapyrrole-regulated ubiquitin ligase controls algal nuclear DNA replication. Nat. Cell Biol. 13, 483–487 10.1038/ncb2203 (doi:10.1038/ncb2203) [DOI] [PubMed] [Google Scholar]
- 223.Tyler KM, Matthews KR, Gull K. 2001. Anisomorphic cell division by African trypanosomes. Protist 152, 367–378 10.1078/1434-4610-00074 (doi:10.1078/1434-4610-00074) [DOI] [PubMed] [Google Scholar]
- 224.May SF, Peacock L, Almeida Costa CIC, Gibson WC, Tetley L, Robinson DR, Hammarton TC. 2012. The Trypanosoma brucei AIR9-like protein is cytoskeleton-associated and is required for nucleus positioning and accurate cleavage furrow placement. Mol. Microbiol. 84, 77–92 10.1111/j.1365-2958.2012.08008.x (doi:10.1111/j.1365-2958.2012.08008.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Sharma R, Peacock L, Gluenz E, Gull K, Gibson W, Carrington M. 2008. Asymmetric cell division as a route to reduction in cell length and change in cell morphology in trypanosomes. Protist 159, 137–151 10.1016/j.protis.2007.07.004 (doi:10.1016/j.protis.2007.07.004) [DOI] [PubMed] [Google Scholar]