

Abstract
A new series of derivatives of 3-(1,4-diazabicyclo[3.2.2]nonan-4-yl)dibenzo[b,d]thiophene 5,5-dioxide with high binding affinities and selectivity for α7-nicotinic acetylcholine receptors (α7-nAChRs) (Ki = 0.4 – 20 nM) has been synthesized for PET imaging of α7-nAChRs. Two radiolabeled members of the series [18F]7a (Ki = 0.4 nM) and [18F]7c (Ki = 1.3 nM) were synthesized. [18F]7a and [18F]7c readily entered the mouse brain and specifically labeled α7-nAChRs. The α7-nAChR selective ligand 1 (SSR180711) blocked the binding of [18F]7a in the mouse brain in a dose-dependent manner. The mouse blocking studies with non-α7-nAChR CNS drugs demonstrated that [18F]7a is highly α7-nAChR selective. In agreement with its binding affinity the binding potential of [18F]7a (BPND = 5.3 – 8.0) in control mice is superior to previous α7-nAChR PET radioligands. Thus, [18F]7a displays excellent imaging properties in mice and has been chosen for further evaluation as a potential PET radioligand for imaging of α7-nAChR in non-human primates.
Introduction
Cerebral neuronal nicotinic cholinergic receptors (nAChRs) are ligand-gated ion channels composed of α (i.e., α2–α10) and β (i.e., β2–β4) subunits that can assemble in multiple combinations of pentameric structures. Among the many nAChRs subtypes in the human CNS, heteropentameric α4β2-nAChRs and homopentameric α7-nAChRs are predominant.1, 2 α7-nAChRs are composed of five identical α7 subunits, and each subunit provides an orthosteric binding site for its neurotransmitter, acetylcholine.3 Many lines of evidence associate α7-nAChRs with the pathophysiology of a variety of disorders such as schizophrenia and AD, anxiety, depression, traumatic brain injury, multiple sclerosis, inflammation and drug addiction.4–12
Clinical experiments with α7-nAChR agonists have demonstrated that selective activation of the receptor is a viable approach towards improving cognitive performance in patients with schizophrenia.13, 14
Because of the importance of the α7-nAChR in human neurophysiology and as a potential drug target, synthesis and pre-clinical examination of α7-nAChR subtype selective compounds receive substantial interest in industry and academia.9, 14 A number of α7-nAChR drugs are currently in various stages of the development for treatment of a variety of disorders including schizophrenia, AD, multiple sclerosis, depression, asthma and type-2 diabetes.15–17
In vivo imaging and quantification of α7-nAChR binding in humans would provide a significant advance in the understanding of α7-nAChR-related CNS disorders and could also facilitate novel α7-nAChR drug development. Positron emission tomography (PET) is the most advanced technique to quantify neuronal receptors and their occupancy in vivo, and the development of a suitable PET radiotracer for α7-nAChRs would be of particular interest.
Many lead structures of α7-nAChR ligands have been identified within various structural classes. A number of these ligands have been radiolabeled for PET ([18F], [11C]) and single-photon emission computed tomography (SPECT) ([123I]) (Table 1) and studied in mice, pigs and non-human primates as potential α7- nAChR probes.18–29 Most of these radioligands entered the animal brain, but manifested relatively low specific binding (see for review30–32) and insufficient BPND values (BPND < 1) (Table 1). [11C]CHIBA-1001 is the only α7-nAChR PET radioligand so far that has been studied in human subjects,25 but it also exhibits low specific binding (Table 1).
Table 1.
In vitro properties and binding potential in cortex (BPND) of the previously published PET/SPECT radioligands for imaging of α7-nAChR.
| Radioligand | α7-nAChR, Ki, nM |
BPNDa | References | |
|---|---|---|---|---|
| Mice | Monkey or pig |
|||
 |
0.26 | 0.6 | - | 18 |
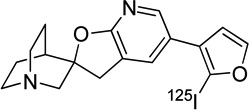
 |
n/a | ~ 0 | - | 21 |
 |
10.8 | 0.2 – 0.5 | 0.3 | 22 |
 |
11 | 0.6 – 0.7 | 0.5 | 22 |
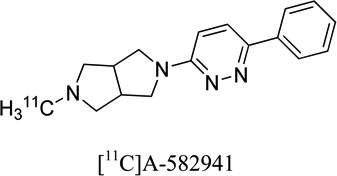
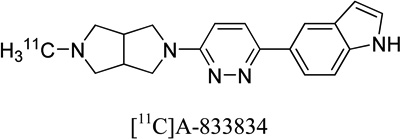 |
0.24, 1.53 | 0.5 | - | 23 |
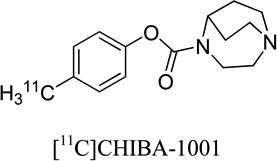 |
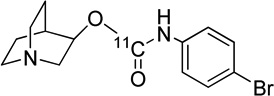
46, 120, 193 | ~ 0.1 | 0.6 | 19, 25, 38, 39 |
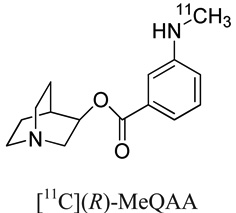 |
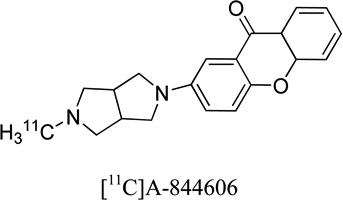
40.6 | 0.4 | 0.4 | 20 |
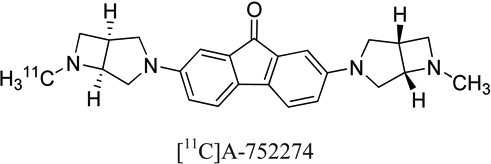 |
0.092 | low brain uptake | low brain uptake | 23 |
 |
0.5 – 0.6 | 1.9 | - | 24 |
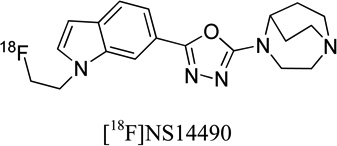 |
2.5 | low brain uptake | - | 28 |
 |
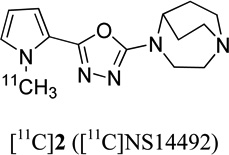
24.9 | - | 0.7 | 19 |
 |
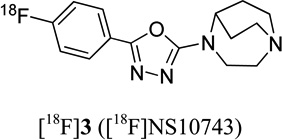
2.2 | - | ~ 1 | 26 |
 |
11.6 | 0.4 | 0.8 | 29 |
 |
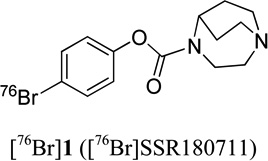
0.2 | 0.8 | - | 27, 40 |
Due to the exceptionally low concentration (Bmax) of cerebral α7-nAChR binding sites in the human (5–15 fmol/mg protein)33 and animal brain (1.5 – 12 fmol/mg tissue),34, 35 a PET radioligand with high specific brain uptake for this receptor subtype must exhibit very high binding affinity and selectivity, along with other important properties (e.g., lipophilicity, polar surface area, suitability for radiolabeling) in an appropriate range (see for details30, 32, 36, 37). The general aptness of a PET radioligand for quantitative imaging studies is defined by a conventional criterion Bmax/KD ≥ 10.37 This equation predicts that a picomolar range of the binding affinity is required for a good α7-nAChR PET radioligand (KD ≤ 0.15 – 1.2 nM), whereas the most previously published α7-nAChR radioligands exhibited nanomolar binding affinities (Table 1). It is noteworthy, however, that the inhibition binding assays of the published compounds have been performed under a variety of assay conditions and, thus, the values of Ki listed in Table 1 may not be directly comparable to one another (see Discussion for details).
Recently Abbott Laboratories has reported 3-(1,4-diazabicyclo[3.2.2]nonan-4-yl)dibenzo[b,d]thiophene 5,5-dioxide 5 (Figure 2) as an α7-nAChR selective antagonist with extraordinarily high binding affinity, Ki = 0.023 nM.43 We envisioned the synthesis of fluoro-derivatives of 5 producing a new set of compounds with similar or even better binding affinities with the potential for radiolabeling with [18F] for PET imaging. The rationale for the design of fluorine-bearing analogs of compound 5 has been strengthened by reports that fluorine substituents can increase the metabolic stability and the rate and extent of blood-brain barrier penetration of radiotracers.44
Figure 2.

3-(1,4-diazabicyclo[3.2.2]nonan-4-yl)dibenzo[b,d]thiophene 5,5-dioxide 5, an α7-nAChR antagonist with very high binding affinity43 that was used as the lead compound in this report.
In this study, we describe the design, synthesis and in vitro and in vivo characterization in mice of a series of high α7-nAChR binding affinity derivatives of 5 as potential probes for PET imaging of α7-nAChR receptor.
Results and Discussion
Chemistry
Synthesis of α7-nAChR ligands
The fluoro-derivatives 7a–e of 3-(1,4-diazabicyclo[3.2.2]nonan-4-yl)-dibenzo[b,d]thiophene 5,5-dioxide 5 were synthesized via the Buchwald-Hartwig cross-coupling reaction between the respective fluoro-bromo compounds 6a–e with 1,4-diazabicyclo[3.2.2]nonane (Scheme 1).
Scheme 1.

(a) Pd2(dba)3, rac-BINAP, toluene, 1,4-diazabicyclo[3.2.2]nonane, Cs2CO3, 85 °C, 24 h.
The nitro-derivatives of (1,4-diazabicyclo[3.2.2]nonan-4-yl)-dibenzo[b,d]thiophene 5,5-dioxide 10 and 11 were synthesized similarly starting with respective nitro-bromo-dibenzothiophene derivatives 8 and 9 (Scheme 2). Reduction of nitro groups in 10 and 11 with iron powder gave corresponding anilines 12 and 13 in high yield. Diazotization-iodination of 12 and 13 yielded corresponding iodides 14 and 15 (Scheme 2).
Scheme 2.

(a) Pd2(dba)3, rac-BINAP, toluene, 1,4-diazabicyclo[3.2.2]nonane, Cs2CO3, 85 °C, 24 h; (b) iron powder, NH4Cl, THF, MeOH, water, 80 °C, 3 h; (c) (i) 4N H2SO4, CH3CN, NaNO2, −5 °C, 30 min; (ii) NaI, CuI, water 70 °C, 30 min.
Synthesis of intermediate compounds
The synthesis of intermediate fluoro-bromide 6a was performed in four steps (Scheme 3). Coupling of commercially available 4-bromo-2-fluoro-nitrobenzene 16 and 2-fluoro-thiophenol 17 gave nitrodiaryl thioether 18 that was reduced to aniline 19. Aniline 19 was treated with sodium nitrite at 0 °C in the presence of hydrochloric acid and sodium tetrafluoroborate to yield a corresponding diazonium tetrafluoroborate derivative (not shown). The intramolecular deazotisation/cyclization of the diazonium salt in the presence of copper (I) oxide and 0.1 N sulfuric acid afforded fluoro-bromo-dibenzothiophene derivative 20, that in turn was oxidized with hydrogen peroxide to 6a in high yield (Scheme 3).
Scheme 3.

Reagents and conditions: (a) Cs2CO3, DMF, 5 h; (b) iron powder, NH4Cl, THF, MeOH, water, 80 °C, 3 h; (c)(i) NaNO2, 50% HCl, 0–5 °C, 30 min; (ii) NaBF4, 0–5 °C, 30 min; (iii) Cu2O, 0.1 N H2SO4, 35–40 °C 30 min. (d) 30% H2O2, acetic acid, 60 °C, 24 h.
The fluoro-bromo isomers 6b and 6c were synthesized in four steps via the commercially available dibenzo[b,d]thiophene 5,5-dioxide 21 and 2-nitrodibenzo[b,d]thiophene 22, respectively (Scheme 4). In brief, nitration of compound 21 and oxidation of compound 22 gave compounds 23 and 24, respectively. Bromination of compounds 23 and 24 provided mono-bromo derivatives 25 and 9 that sequentially were reduced to anilines 26 and 27, respectively, in high yields. The anilines 26 and 27 were converted to fluorides 6b and 6c in moderate yields by the Schiemann reaction via the corresponding intermediate diazonium fluoroborates (structures not shown). The diazonium salts precipitated in the reaction mixture and were isolated by filtration in high yields.
Scheme 4.

Reagents and conditions: (a) H2SO4, HNO3, 0°C; (b) 30% H2O2, acetic acid, 60 °C, 24 h; (c) NBS, conc. H2SO4 24 h; (d) SnCl2•2H2O, 37% HCl, HOAc, 100 °C, 60 min or iron powder, NH4Cl, THF, MeOH, water, 80 °C, 3 h; (f) (i) 48% HBF4, 0–5 °C 10 min; (ii) NaNO2, 0–5 °C, 1 h; (iii) xylene, 135 °C, 30 min.
The brominated isomers 6d and 6e were prepared by bromination of 4-fluorodibenzo[b,d]thiophene 5,5-dioxide 29 starting with 4-fluorodibenzo[b,d]thiophene 28.45 Oxidation of 28 with hydrogen peroxide gave dioxide 29 in nearly quantitative yield. Bromination of 29 with one equivalent NBS in H2SO4 afforded two isomeric bromides: 6d as the main product in 24% yield as well as 6e as a minor product in 13% yield. A substantial amount of compound 29 (about 50%) was recovered from the reaction mixture. Isomers 6d and 6e were readily separated by silica gel chromatography.
3-Bromo-6-nitrodibenzo[b,d]thiophene 5,5-dioxide 8 was synthesized in two steps: (1) oxidation of 4-nitrodibenzo[b,d]thiophene 3046 gave 4-nitrodibenzo[b,d]thiophene 5,5-dioxide 31 in 90% yield; (2) bromination of compound 31 provided compound 8 as the only product in 77% yield. (Scheme 6)
Scheme 6.
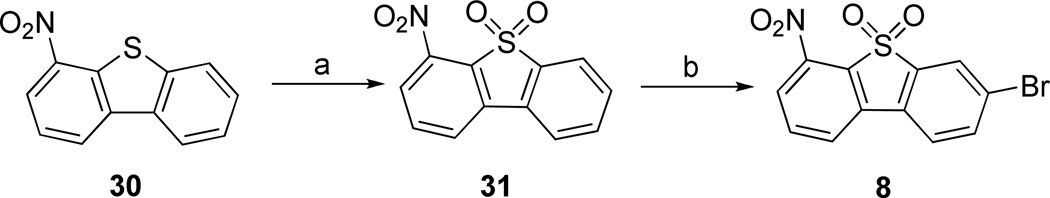
Reagents and conditions: (a) 30% H2O2, acetic acid, 60 °C, 24 h; (b) NBS, H2SO4, 24 h.
In vitro inhibition binding assay
The results of the α7-nAChR in vitro inhibition binding assays for compounds 7a–e, 10, 11, 14 and 15 are shown in Table 2. In order to determine α7-nAChR selectivity of new compounds vs. other nAChR subtypes, binding assays for the main cerebral heteromeric nAChR subtypes (α2β2-, α2β4-, α3β2-, α3β4-, α4β2-, α4β4-) were also performed (Table 2). In addition, because α7-nAChR shares 30% homology with the 5-HT3 receptor and first generation α7-nAChR radioligands exhibited low α7-nAChR/5-HT3 selectivity,27 the in vitro binding affinity at the 5-HT3 receptor was also determined for selected compounds of our series (Table 2).
Table 2.
Inhibition in vitro binding affinities (Ki, nM) of the new series 7a–e, 10, 11, 14 and 15 and reference compounds toward α7-nAChR, heteromeric nAChR subtypes and 5-HT3.
| Compound | α7-nAChRa | Heteromeric nAChR subtypes b | 5-HT3 c | Selectivity | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| α2β2 | α2β4 | α3β2 | α3β4 | α4β2 | α4β4 | α7/α4β2 | α7/5HT3 | |||
| 7a | 0.37, 0.45 | >10000 | 4000 | 1000 | 709 | 562 | 1000 | 230 | 1370 | 561 |
| 7b | 1.02, 1.37 | ntd | nt | nt | nt | nt | nt | nt | - | - |
| 7c | 1.32, 1.35 | 1000 | 8000 | 2000 | 5000 | 885 | 3000 | 505 | 663 | 378 |
| 7d | 1.83, 2.45 | 292 | 838 | 678 | 3000 | 141 | 1000 | nt | 66 | - |
| 7e | 17.8, 20.3 | >10000 | 562 | 2000 | 261 | 4000 | 251 | nt | 210 | - |
| 10 | 0.34, 0.35 | nt | nt | nt | nt | nt | nt | nt | - | - |
| 11 | 3.41, 6.21 | nt | nt | nt | nt | nt | nt | nt | - | - |
| 14 | 0.93, 1.93 | nt | nt | nt | nt | nt | nt | nt | - | - |
| 15 | 6.46, 8.77 | 784 | 6000 | 1000 | 9000 | 477 | 5000 | nt | 63 | - |
Rat cortical membranes, radiotracer [125I]α-bungarotoxin (0.1 nM), KD = 0.7 nM
Inhibition in vitro binding assay of all heteromeric nAChR subtypes was performed with stably transfected HEK293 cells and [3H]epibatidine (0.5 nM), KD = 0.021 nM (α2β2-nAChR), KD = 0.084 nM (α2β4-nAChR), KD = 0.034 nM (α3β2-nAChR), KD = 0.29 nM (α3β4-nAChR), KD = 0.046 nM (α4β2-nAChR), KD = 0.094 nM (α4β4-nAChR).55
Human 5-HT3 recombinant/HEK293 cells, radiotracer [3H]GR65630 (0.35 nM), KD = 0.5 nM
nt = not tested
The α7-nAChR assays for 7a–e, 10, 11, 14 and 15 were performed using a commercial assay consisting of rat cortical membranes (rich in α7-nAChR) in competition against 0.1 nM [125I]α-bungarotoxin, an α7-nAChR antagonist with a KD of 0.7 nM. These assays were performed independently in duplicate, each twice (Table 2). Assays for two reference compounds, methyllycaconitine (MLA), a conventional reference α7-nAChR antagonist, and compound 5,43 a lead of our series, were also performed (Table 3).
Table 3.
Inhibition in vitro binding affinities (Ki, nM) of reference compounds toward α7-nAChR. The binding assay conditions are the same as those in the Table 2.
| Compound | α7-nAChR |
|---|---|
| MLA | 2.91 ± 0.76 (n = 9) |
| 2 | 20.4 |
| 3 | 38.0 |
| 4 | 3.3 |
| 5 | 0.30, 0.50 |
The new series of fluoro isomers 7a–d exhibited high binding affinity at α7-nAChRs with Ki values in the range of 0.3 – 2.5 nM, whereas the binding affinity of isomer 7e was lower (Table 2). The Ki values of the fluoro derivatives 7a–d (Table 2) were better than that of the conventional reference α7-nAChR ligand MLA (Table 3). Among all fluoro isomers compound 7a manifested the best α7-nAChR binding affinity that was an order of magnitude better than MLA and at least comparable to the non-fluorinated lead 5 (Tables 2, 3).
Within the series 7a–e, two fluoro derivatives 7a and 7c were selected for further evaluation. This selection was based on the high α7-nAChR binding affinity and selectivity of 7a and 7c (see Table 2) and the suitability of these compounds for radiolabeling with [18F]. The radiolabeling of [18F]7a and [18F]7c was anticipated to be accomplished by a direct nucleophilic substitution (SNAr) with [18F]fluoride via the nitro 10 and 11 or iodo derivatives 14 and 15, respectively. The leaving nitro- in 10 and 11 or iodo-groups in 14 and 15 are activated for SNAr fluorination by the powerful electron-withdrawing SO2Ar on the ortho and para positions, respectively.47–50 We did not find in the literature an example of fluorination of nitro- or iododibenzothiophene 5,5-dioxides, but the structural analog of 11, 4,4'-sulfonylbis(p-nitrobenzene), has been converted to the corresponding fluoro-derivative with good yield.51
The fluoro derivative 7b that also exhibited high α7-nAChR binding affinity was not selected for further studies because the activating SO2Ar was located on the meta position to the leaving group and direct radiolabeling of [18F]7b via its nitro or iodo-derivative was less likely. A detailed search of the literature for SNAr reactions for 3-nitrodibenzo[b,d]thiophene 5,5-dioxide, a potential precursor for [18F]7b, or its structural analogs (1-nitro-3-(phenylsulfonyl)benzene, etc.) did not reveal any previous publications.
The potential precursors 10, 11, 14 and 15 for 18F-fluorination of [18F]7a and [18F]7c were studied in the same α7-nAChR inhibition binding assay. The nitro compounds 10 and 11 exhibited comparable α7-nAChR binding affinities to those of the corresponding fluorides 7a and 7c, whereas the binding affinities of iodo-derivatives 14 and 15 were lower.
Currently, there is no conventional in vitro competition binding assay for α7-nAChR. Different research groups use different radioligands ([125I]α-bungarotoxin, [3H]α-bungarotoxin, [3H]MLA, [125I]iodo-MLA, [3H]A-585539, etc) and different sources of receptor tissue (cell lines, brain, adrenal glands) under different conditions for this assay.26, 29, 52–54 It is not surprising that the difference in the Ki values for the same compound under different assay conditions can exceed an order of magnitude.53, 54 Therefore, a direct comparison of Ki values of the previously published α7-nAChR ligands with compounds of our new series is not practical.
For the purpose of comparison, we determined the Ki values of three most recently published α7-nAChR PET radioligands [11C]2,26 [18F]3 29 and [18F]427 (Table 3) under the same assay conditions as that of our series (Table 2). It was noteworthy that the α7-nAChR binding affinities of the best compounds of our series 7a and 7c were substantially better than those of the previous radioligands.
Heteromeric nAChR subtypes assays (α2β2-, α2β4-, α3β2-, α3β4-, α4β2-, α4β4-nAChR) were performed in our labs using membrane preparations from HEK293 cells expressing the transfected nAChR under test in competition with 0.5 nM [3H]epibatidine to investigate the specificity of the ligand for each receptor (Table 2).
The heteromeric nAChR Ki values of the tested compounds 7a, 7c–7e and 15 were substantially greater than the corresponding α7-nAChR Ki’s, indicating a high α7-/heteromeric-nAChR subtype selectivity of all studied compounds (Table 2). Thus, the fluoro isomer 7a with the best α7-nAChR binding affinity also manifested an excellent selectivity versus heteromeric nAChR including the main cerebral subtype α4β2-nAChR (Table 2). Interestingly, the α7/α4β2 selectivity of iodo derivative 15 is ten times lower than the corresponding fluoro derivative 7c.
5-HT3 assay. The in vitro binding affinity of the most promising members of the series, compounds 7a and 7c, at the 5-HT3 receptor were determined commercially using membrane preparations from HEK293 cells expressing transfected human 5-HT3R in competition with 0.35 nM [3H]GR65630, a 5-HT3R antagonist with a KD of 0.5 nM. The assay demonstrated that fluoro compounds 7a and 7c manifest relatively low 5-HT3 binding affinities and they are highly α7-nAChR/5HT3 selective (Table 2).
Lipophilicity of 7a and 7c
Lipophilicity (logD7.4) is considered an important property of CNS radioligand because it has been linked to the blood-brain barrier permeability and non-specific binding.35, 37, 38 The lipophilicity values for 7a and 7c (logD7.4 = 2.0) were calculated with ACD Labs Structure Designer Suite (ACD Labs, Toronto) and fall within the conventional range for CNS PET radioligands.
Radiochemistry
We radiolabeled the fluoro isomers 7a and 7c that exhibited highest binding affinity within the series with fluorine-18. The radiosyntheses were performed remotely in one step by 1,10-diaza-4,7,13,16,21,24-hexaoxabicyclo[8.8.8]hexacosane (Kryptofix-222®) - assisted radiofluorination of the respective nitro- 10 and 11 (Scheme 7) or iodo-precursors 14 and 15 using a radiochemistry synthesis module (Microlab, GE) followed by the semi-preparative HPLC separation and formulation of [18F]7a and [18F]7c as sterile apyrogenic solutions in 7% ethanolic saline.
Scheme 7.

Radiosynthesis of [18F]7a and [18F]7c
It is noteworthy that the radiotracer product yields from iodo-precursors 14 and 15 were substantially lower than those of the nitro-precursors 10 and 11. The conventional Kryptofix-222/potassium carbonate assisted radiofluorination of both iodo-derivatives 14 and 15 in DMSO at 130–180 °C produced [18F]7a and [18F]7c with radiochemical yields below 0.5% and this radiosynthesis pathway was not optimized further (not shown).
The radiofluorination of nitro-derivatives 10 or 11 (Scheme 7) in the presence of Kryptofix-222/potassium carbonate at 160 °C produced [18F]7a or [18F]7c in a slightly better yield (2–3%). Further optimization of this radiosynthesis suggested that both final products [18F]7a or [18F]7c rapidly decomposed in the DMSO reaction solution in the presence of highly basic K2CO3, but the radiochemical yield was improved if the less basic potassium oxalate was used. In the presence of potassium oxalate, the final products [18F]7a and [18F]7c were prepared under similar reaction conditions with comparable radiochemical yields of 16 ± 6% (n=14) (nondecay-corrected), with specific radioactivities in the range of 330 – 1260 GBq/µmol (9–34 Ci/µmol), and a radiochemical purity greater than 99%. The nitro-precursors 10 and 11 that exhibited substantial α7-nAChR binding affinity (Table 2) were fully separated by preparative HPLC and were not detected by analytical HPLC in the final products [18F]7a and [18F]7c (see Table 6 for the HPLC details).
Table 6.
HPLC conditions for [18F]7a and [18F]7c
| Column | Mobile phase | Flow rate, mL/min |
Product retention time, min |
Nitro precursor retention time, min |
|
|---|---|---|---|---|---|
| [18F]7a, preparative | XBridge C18 column, 10 µm (250 × 10 mm) | CH3OH/CH3CN/H2O/Me3N 260:120:620:2 | 12 | 32 | 21 |
| [18F]7a, analytical | XBridge C18 column, 5 µm (250 × 4.6 mm) | CH3CN/H2O/Et3N 390:610:1 | 2 | 7.4 | 5.5 |
| [18F]7c, preparative | XBridge C18 column, 10 µm (150 × 10 mm) | CH3CN/H2O/NH3 280:720:1 | 10 | 20 | 27 |
| [18F]7c, analytical | XBridge C18 column, 3.5 µm (100 × 4.6 mm) | CH3CN/H2O/NH3 380:620:1 | 2 | 3.4 | 5.2 |
Biodistribution Studies of [18F]7a and [18F]7c in mice
Baseline studies in mice
Radioligands [18F]7a and [18F]7c were evaluated in mice as potential PET tracers for imaging α7-nAChRs. After intravenous injection, [18F]7a and [18F]7c exhibited robust initial brain uptake followed by washout. The highest accumulation of radioactivity of both radioligands occurred in the superior/inferior colliculus, hippocampus and frontal cortex. Moderate uptake was observed in thalamus and striatum and the lowest radioactivity was seen in cerebellum (Figs. 4–6). This distribution of radioactivity was similar to the previously published in vitro data on the distribution of α7-nAChRs in rodents.56, 57 The clearance rate of [18F]7a and [18F]7c from cerebellum was higher than that from any other regions studied. The ratios of tissues to cerebellum increased steadily over the 90 minutes, reaching values of 10 for [18F]7a and 4.5 for [18F]7c. The better ratios for compound [18F]7a vs. [18F]7c are in agreement with in vitro α7-nAChR binding affinity of these compounds (Table 2).
Figure 4.

Regional distribution of [18F]7a (Left) and [18F]7c (Right) in CD-1 mice. Data: mean %injected dose/g tissue ± SD (n = 3). Abbreviations: Coll = superior and inferior colliculus; Hipp = hippocampus; FrCtx = frontal cortex; Rest = rest of brain; Th = thalamus; Str = striatum; CB = cerebellum
Figure 6.

Blocking of [18F]7a and [18F]7c with α7-nAChR – selective ligands in CD-1 mice. Left panel: Dose dependent blockade of [18F]7a (0.07 mCi, specific radioactivity = 7900 mCi/µmol, i.v.) accumulation by intravenous co-injection with 1 (doses 0.02 mg/kg, 0.2 mg/kg, 1 mg/kg, 3 mg/kg) in the mouse brain regions 90 min after the injection. *P ≤ 0.01, significantly different from controls (ANOVA). Right panel: Dose dependent blockade of [18F]7c (0.07 mCi, specific radioactivity = 11000 mCi/µmol, i.v.) accumulation by intravenous co-injection with 5 (doses 0.001 mg/kg, 0.0045 mg/kg, 0.014 mg/kg) in the mouse brain regions 90 min after the injection. *P < 0.01, significantly different from controls; and **P = 0.06, insignificantly different from control (ANOVA). Data: mean %injected dose/g tissue ± SD (n=3). Abbreviations: Coll = superior and inferior colliculus; Hipp = hippocampus; Ctx = cortex; Str = striatum; Th = thalamus; Rest = rest of brain; CB = cerebellum.
Specificity and selectivity of [18F]7a and [18F]7c binding in the mouse brain
A conventional in vivo blockade methodology with CNS drugs is used here for demonstration of specificity and selectivity at the α7-nAChR receptor in the mouse brain. A self-blockade study with a non-radioactive form of a radioligand estimates whether or not the binding is specific. A blockade study with a drug that is highly selective at the target binding site is expected to show the selectivity and specificity of the radioligand binding. A dose-escalation blockade with such a target selective drug provides further evidence of the radioligand specificity and selectivity and it is useful for demonstration of the radioligand suitability for evaluation of conventional drug candidates. In addition, blockade with CNS drugs that do not bind at the target site provide more evidence of the radioligand selectivity versus other cerebral binding sites.
Self-blockade studies of [18F]7a with 7a (Fig. 5-Left) and [18F]7c with 7c (Fig. 5-Right) demonstrated a reduction of the radioligand uptake in most brain regions except the cerebellum, a region with low density of α7-nAChRs. The studies showed that accumulation of [18F]7a and [18F]7c radioactivity in the mouse brain was specific. When the specific binding of the radioligands in the hippocampus and colliculus was estimated by using the radioactivity concentration in the blocked cerebellum as nonspecific binding, the specific binding value amounted to 94% and 80% and the baseline-to-blockade ratio in the α7-nAChR-rich regions was 13 and 5 for [18F]7a and [18F]7c, respectively. This result also demonstrated that [18F]7a exhibited a higher level of specificity and greater uptake in the mouse brain versus [18F]7c.
Figure 5.

Self-blockade study of [18F]7a and [18F]7c in CD-1 mice. Left panel: Inhibition of [18F]7a (0.07 mCi, specific radioactivity = 9200 mCi/µmol, i.v.) accumulation by intravenous co-injection with 7a (0 mg/kg (white) and 0.3 mg/kg (black)) in the mouse brain regions 90 min after the injection. *P < 0.01, significantly different from controls; **P = 0.04, insignificantly different from controls (ANOVA). Right panel: Inhibition of [18F]7c (0.07 mCi, specific radioactivity = 12000 mCi/µmol, i.v.) accumulation by intravenous co-injection with 7c (0 mg/kg (white) and 0.2 mg/kg (black)) in the mouse brain regions 90 min after the injection. *P ≤ 0.01, **P = 0.015, significantly different from controls; and ***P = 0.5, insignificantly different from controls (ANOVA). Data: mean %injected dose/g tissue ± SD (n=3). Abbreviations: Coll = superior and inferior colliculus; Hipp = hippocampus; FrCtx = frontal cortex; Str = striatum; Rest = rest of brain, CB = cerebellum.
Neither behavioral nor locomotor activity changes were observed in the mice in the blockade studies with 7a (0.3 mg/kg, i.v.) or 7c (0.2 mg/kg, i.v.).
Blocking with selective α 7-nAChR ligands. A blockade study of [18F]7a with 1, a selective α7-nAChR partial agonist with a Ki of 22 nM,58 showed a dose dependent blockade in all regions studied. However, in the α7-nAChR-poor cerebellum, the blockade was significant only with the highest dose of 1 (3 mg/kg) (Fig. 6-Left). A similar dose-response study was performed with [18F]7c using compound 5, a selective α7-nAChR antagonist, as a blocker (Fig. 6-Right). These studies confirmed that the in vivo binding of [18F]7a and [18F]7c was specific and mediated by α7-nAChR. The dose-escalation response demonstrated that both radioligands are suitable tools for evaluation of new α7-nAChR drug candidates.
It is noteworthy that the doses of 1 that significantly blocked the [18F]7a binding in CD1 mice were comparable to the doses of 1 that significantly improved cognitive deficit in the various rodent models of schizophrenia.59, 60 This finding suggests that [18F]7a is suitable radioprobe for in vivo studies in mice with pharmacologically relevant doses of α7-nAChR drugs.
Because the lowest regional uptake of [18F]7a and [18F]7c was seen in the cerebellum the regional BPND values in mice were approximated for a single time point measurement (90 min) as BPND = (regional uptake/cerebellum uptake) – 142 (Table 4). The substantially higher BPND values for [18F]7a are in agreement with greater binding affinity of this compound versus [18F]7c (Table 2; also see Fig. 9). The BPND values for both radioligands [18F]7a and [18F]7c were superior to all previously published α7-nAChR PET radioligands (Table 1)
Table 4.
Approximate BPND values (unitless) of [18F]7a and [18F]7c in the mouse brain regions. Data: mean ± SD (n = 6)
| Region | Coll | Hipp | Ctx |
|---|---|---|---|
| Compound | |||
| [18F]7a | 8.0 ± 1.6 | 5.5 ± 1.7 | 5.3 ± 1.2 |
| [18F]7c | 2.0 ± 0.5 | 3.1 ± 0.7 | 2.0 ± 0.3 |
Abbreviations: Coll = superior and inferior colliculus; Hipp = hippocampus; Ctx = cortex
Figure 9.

Correlation of the BPNDcortex (unitless) vs. 1/Ki (nM−1) of α7-nAChR PET radioligands [11C]2, [18F]3, [18F]4, [18F]7a and [18F]7c (y = 1.91x + 0.52, R2 = 0.98). The BPND values are shown in the Tables 1 and 3; the SD values are available for [18F]7a and [18F]7c only. All Ki values were obtained in this study under the same binding assay conditions (Tables 2 and 3).
Blocking with nicotine and α4β2-nAChR selective cytisine. The blockade of [18F]7a in CD1 mouse brain with cytisine, a partial nicotinic agonist selective for α4β2-nAChR and other β2/β4-containing heteromeric nAChR subtypes while exhibiting low α7-nAChR binding affinity,52, 55, 61 showed insignificant reduction of radioactivity accumulation in all regions studied (Fig. 7). This result demonstrated that [18F]7a manifested insignificant binding at α4β2-nAChRs in the mouse brain.
Figure 7.

Blockade of [18F]7a accumulation in CD-1 mouse brain regions by injection of cytisine (1 mg/kg, s.c.) and nicotine (5 mg/kg, s.c.) (both 90 min after the injection). Data: mean %injected dose/g tissue ± SD (n = 3). Abbreviations: Coll = superior and inferior colliculus; Hipp = hippocampus; Ctx = cortex; CB = cerebellum; Rest = rest of brain. The effect of cytisine was insignificant in all regions studied (P > 0.05, asterisk is not shown). The difference between control and nicotine was significant (*P ≤ 0.01) in all regions, except CB (**P = 0.9) (ANOVA). The study demonstrates that [18F]7a does not bind in vivo at the main cerebral α4β2-nAChR subtype and it is suitable for nicotine blockade studies.
The blockade study of [18F]7a with nicotine that binds at all nAChR subtypes including α7-nAChR52 showed significant blockade in all regions, except the nAChR-poor cerebellum. This suggests that [18F]7a can be used for nicotine addiction or smoking studies in mice. The lesser blockade of [18F]7a with nicotine (Fig. 7) in comparison with 1 (Fig. 6) is due to the rather modest binding affinity of nicotine at α7-nAChR (Ki = 610 nM).52
Blocking with non-a7-nAChR CNS ligands. For determination of in vivo selectivity of [18F]7a for α7-nAChRs vs. several major CNS receptor systems, we compared the regional distribution (Fig. 8) of the radiotracer in control CD-1 mice vs. mice preinjected with various CNS active drugs or the positive control 1 (see Table 5 for the drug list). None of the drugs except 1 reduced accumulation of radioactivity when compared with controls (Fig. 8). The absence of blockade with the 5-HT3-selective drug ondansetron was especially remarkable because α7-nAChR ligands often bind to this receptor subtype. This finding suggests that in the mouse brain the radioligand [18F]7a was selective for α7-nAChRs versus several major cerebral binding sites.
Figure 8.

Effect of various CNS drugs (Table 5) on accumulation of [18F]7a in CD-1 mouse brain regions 90 min after injection of tracer expressed as %ID/g tissue. Abbreviations: Coll = superior and inferior colliculus; Hipp = hippocampus; Ctx = cortex; CB = cerebellum; REST = rest of brain. Data are mean ± SD (n=3). *P < 0.01, significantly different from controls. Columns that do not include the asterisk are insignificantly different from controls (P > 0.05) (ANOVA, single-factor analysis). The graph demonstrates that unlike the positive control (1) all non-α7-nAChR CNS drugs do not have an effect on the cerebral uptake of [18F]7a and the radiotracer is α7-nAChR selective in vivo.
Table 5.
CNS drugs (2 mg/kg, s.c.) for α7-nAChR selectivity studies in mice
| Drug | Target receptor | Dose (mg/kg) | Time of administration before radiotracer, min |
|---|---|---|---|
| 1 | Selective α7-nAChR partial agonist | 2 | 10 |
| Ondansetron | Selective 5-HT3 antagonist | 2 | 10 |
| SCH23390 | D1- and D5-antagonist and 5-HT1C/2C agonist | 2 | 10 |
| Spiperone | D2-like and 5-HT2A receptor antagonist | 2 | 10 |
| Ketanserin | 5-HT2/5-HT2C antagonist | 2 | 10 |
| Naltrindole | Selective δ-opioid antagonist | 2 | 10 |
5-HT = 5-hydroxytryptoamine (serotonin)
Comparison of imaging properties of [18F]7a and [18F]7c with previous α7-nAChR PET radioligands
Binding potential (BPND), a measure of in vivo specific binding and one of the most important imaging characteristics of a PET radioligand, is defined as the ratio of Bmax (receptor density) to KD (radioligand equilibrium dissociation constant) or the product of Bmax and binding affinity.41, 62 Therefore, the binding affinities (1/Ki) of α7-nAChR radioligands should correlate linearly with their BPND values.
The comparison of all previously published α7-nAChR radioligands (Table 1) revealed little correlation between 1/Ki and BPND (R2 = 0.05, not shown). It was likely that the lack of correlation was due to the wide variability in binding assay conditions for these compounds when performed by various research groups (see Discussion above).
When the α7-nAChR binding assay for the radioligands is performed under the same assay conditions (Tables 2, 3), the binding affinities 1/Ki correlate linearly (Fig. 9) with the cortical BPND’s of [18F]7a and [18F]7c (Table 4) and [11C]2, [18F]3 and [18F]4 (Table 1). This finding may explain why the specific binding of the very high affinity radioligands [18F]7a and [18F]7c is superior to the previous α7-nAChR radioligands with lower binding affinities. This result emphasizes further the importance of high binding affinity for the imaging properties of α7-nAChR radioligands.
Conclusion
A series of 3-(1,4-diazabicyclo[3.2.2]nonan-4-yl)dibenzo[b,d]thiophene 5,5-dioxide derivatives with high binding affinities for α7-nAChRs (Ki = 0.4 – 20 nM) has been synthesized with potential application for PET imaging of α7-nAChRs. Two members of the series, 7a and 7c, with the best α7-nAChR binding affinities (Ki = 0.4 and 1.3 nM, respectively) and high selectivity vs. other nicotinic subtypes and 5-HT3, were radiolabeled with 18F.
[18F]7a and [18F]7c readily entered the mouse brain and specifically and selectively labeled cerebral α7-nAChR receptors. The binding potential (BPND) values in mouse cortex of [18F]7a, [18F]7c and previously published α7-nAChR radioligands correlated linearly with their binding affinities (1/Ki) when the binding affinity values were determined under the same assay conditions. In agreement with the binding affinity of [18F]7a its BPND value in mice was substantially better than those of the previous α7-nAChR radioligands.
The best PET radioligand of this new series [18F]7a exhibits excellent α7-nAChR imaging properties in the mouse brain. Therefore, [18F]7a holds promise as a highly specific PET radioligand for quantification of α7-nAChR receptors and further evaluation of this radioligand in baboon PET studies is underway.
Experimental Section
All reagents were used directly as obtained commercially unless otherwise noted. Reaction progress was monitored by TLC using silica gel 60 F254 (0.040−0.063 mm) with detection by UV. All moisture-sensitive reactions were performed under an argon atmosphere using oven-dried glassware and anhydrous solvents. Column flash chromatography was carried out using E. Merck silica gel 60F (230–400 mesh). Analytical thin-layer chromatography (TLC) was performed on aluminum sheets coated with silica gel 60 F254 (0.25 mm thickness, E. Merck, Darmstadt, Germany). Melting points were determined with a Fisher-Johns apparatus and were not corrected. 1H NMR spectra were recorded with a Bruker-400 NMR spectrometer at nominal resonance frequencies of 400 MHz, in CDCl3 or DMSO-d6 (referenced to internal Me4Si at δH 0 ppm). The chemical shifts (δ) were expressed in parts per million (ppm). First order J values were given in Hertz. Splitting patterns are described as singlet (s), doublet (d), triplet (t), quartet (q), and broad (br). High resolution mass spectra were recorded utilizing electrospray ionization (ESI) at the University of Notre Dame Mass Spectrometry facility. All compounds that were tested in the biological assays were analyzed by combustion analysis (CHN) to confirm the purity >95%. Elemental analyses were determined by Galbraith Laboratories, Inc. (Knoxville, TN). The HPLC system consisted of two Waters model 600 pumps, two Rheodyne model 7126 injectors, an in-line Waters model 441 UV detector (254 nm), and a single sodium iodide crystal flow radioactivity detector. All HPLC chromatograms were recorded with Varian Galaxy software (version 1.8). The analytical and semi-preparative chromatographies were performed using Waters XBridge C-18 10 µm columns (analytical 4.6 × 250 mm and preparative 10 × 250 mm).
A dose calibrator (Capintec 15R) was used for all radioactivity measurements. Radiofluorination was performed with a modified GE MicroLab radiochemistry box.
Chemistry
Typical procedure for reduction of nitro derivatives to anilines 12, 13, 19, 26, 27
A mixture of nitro compound (1 mmol), iron powder (4 mmol), ammonium chloride (1.2 mmol) in methanol (6 mL), THF (6 mL) and water (2 mL) was heated to reflux (80 °C) for 3 hours. The resulted mixture was diluted with ethanol and concentrated and dried under vacuum. The residue was purified by silica gel column chromatography (CHCl3/i-PrOH/Et3N 10:1:0.1 to 10:30:4) to give the corresponding aniline derivative.
Typical procedure for bromination
N-bromosuccinimide (NBS) (1 mmol) was added to a solution of the starting 1,4-dibenzothiophene derivative (1 mmol) in concentrated H2SO4 (3.6 mL) at room temperature. After 24 h, the solution was carefully poured into ice/water. The solids were filtered and washed with water and methanol. The obtained solids were recrystallized from 95% EtOH to afford the bromo compounds.
Typical procedure for oxidation of 1,4-dibenzothiophene derivatives
1,4-Dibenzothiophene derivative (1 mmol) was dissolved in glacial acetic acid (2.8 mL) at room temperature. Aqueous hydrogen peroxide (30%, 1.4 mL) was added in small portions to the stirred solution. The addition of H2O2 resulted initially in some precipitation. The mixture was stirred at 60 °C for 24 h, then cooled to room temperature. The solid was filtered off, sequentially washed with 70% aqueous acetic acid, then 30% aqueous acetic acid, then water and dried to afford the title compound.
3-Bromo-6-fluorodibenzo[b,d]thiophene 5,5-dioxide (6a)
The typical procedure for oxidation of 1,4-dibenzothiophene was followed, starting with 20 (600 mg, 2.13 mmol). The title compound 6a (648 mg, 97%) was obtained as white crystals. 1H NMR (CDCl3, 400 MHz) δ 7.97 (s, 1H), 7.81 (dd, J=12.0, 1.8 Hz, 1H), 7.68 (d, J=8.0 Hz, 1H), 7.66 (dd, J=8.0, 4.0 Hz, 1H), 7.59 (d, J=8.0 Hz, 1H), 7.24 (t, J=8.0 Hz, 1H).
3-Bromo-7-fluorodibenzo[b,d]thiophene 5,5-dioxide (6b)
A mixture of 26 (620 mg, 2 mmol) and 48% tetrafluoroboric acid (HBF4) (4 mL) was stirred at 0–5 °C for 10 min. A cold solution of sodium nitrite (204 mg in 0.8 mL of water, 3 mmol) was added dropwise with stirring. After stirring for 1 h at 0–5 °C the precipitated intermediate diazonium tetrafluoroborate was collected by filtration, washed with cold tetrafluoroboric acid (5%) and water, Et2O and dried under vacuum. The diazonium tetrafluoroborate was boiled in xylene (135 °C) for 120 min. The solvent was evaporated under reduced pressure. The residue was extracted with mixture of chloroform and water. The chloroform layer was separated and concentrated. The residue was chromatographed on silica gel using hexanes-EtOAc (4:1) as eluent to give 6b as a pale yellow solid (330 mg, 53%). 1H NMR (CD3Cl, 400 MHz) δ 7.96 (d, J=2.0 Hz, 1H), 7.81-7.77 (m, 2H), 7.64 (d, J=8.0 Hz, 1H), 7.54 (dd, J=8.0, 4.0 Hz, 1H), 7.40 – 7.35 (m, 1H)
7-Bromo-2-fluorodibenzo[b,d]thiophene 5,5-dioxide (6c)
A mixture of 27 (310 mg, 1 mmol) and 48% tetrafluoroboric acid (HBF4) (2 mL) was stirred at 0 –5 °C for 10 min. A cold solution of sodium nitrite (102 mg, 1.5 mmol) in 0.4 mL of water was added dropwise with stirring. After stirring for 1 h at 0 –5 °C, the precipitated diazonium tetrafluoroborate was collected by filtration, washed with cold tetrafluoroboric acid (5%) and water, Et2O and dried under vacuum. The diazonium tetrafluoroborate was boiled in xylene (135 °C) for 30 min. The solvent was evaporated under reduced pressure. The residue was treated with chloroform and water. The chloroform layer was separated and concentrated. The residue was chromatographed on silica gel using hexanes-EtOAc (4:1) as eluent to give 6c as a pale yellow solid (156 mg, 50%). 1H NMR (DMSO-d6, 400 MHz) δ 8.38 (d, J=4.0 Hz, 1H), 8.23-8.18 (m, 2H), 8.13-8.07 (m, 2H), 7.54 (t, J=8.0 Hz, 1H).
3-Bromo-4-fluorodibenzo[b,d]thiophene 5,5-dioxide (6d) and 1-bromo-4-fluorodibenzo[b,d]thiophene 5,5-dioxide (6e)
The typical procedure for bromination was followed, starting with 29 (905 mg, 3.86 mmol). Separation of the crude reaction product by silica gel chromatography using hexanes/ethyl acetate (5:2) yielded two isomers 6d (285 mg, 0.91 mmol, 23.6%) and 6e (160 mg, 0.51 mmol, 13%). The isomer 6e was in the first chromatography fraction, whereas 6d was in the second fraction.
6d: Rf=0.31 (hexanes/EtOAc 2:1); 1H NMR (CDCl3, 400 MHz) δ 7.86-7.80 (m, 3H), 7.70 (t, J=8.0 Hz, 1H), 7.63 (d, J=8.0 Hz, 1H), 7.49 (d, J=8.0 Hz, 1H).
6e : Rf=0.5 (hexanes/EtOAc 2:1); 1H NMR (CDCl3, 400 MHz) δ 8.94 (d, J=8.0 Hz, 1H), 7.90 (d, J=8.0 Hz, 1H), 7.83-7.79 (m, 1H), 7.75 (dd, J=8.0, 4.0 Hz, 1H), 7.67 (d, J=8.0 Hz, 1H), 7.11 (t, J=8.0 Hz, 1H).
Typical procedure for Buchwald-Hartwig cross-coupling reaction
3-(1,4-Diazabicyclo[3.2.2]nonan-4-yl)-6-fluorodibenzo[b,d]thiophene 5,5-dioxide (7a)
A catalyst solution was prepared by mixing tris(dibenzylideneacetone)dipalladium (Pd2(dba)3; 58 mg, 0.063 mmol; Aldrich) and racemic BINAP (39 mg, 0.125 mmol; Strem) in toluene (4 mL) and heating the mixture to 90 °C for 15 min. The solution was cooled, and then added to a mixture of 1,4-diazabicyclo[3.2.2]nonane (200 mg, 1.58 mmol) and 6a (0.492 g, 1.58 mmol), in toluene (12 mL). Cs2CO3 (766 mg, 2.4 mmol; Aldrich) was then added, and the reaction mixture was flushed with nitrogen and heated overnight at 80–85 °C. After cooling to room temperature, the mixture was concentrated and purified by silica gel flash chromatography (CHCl3:i-PrOH:Et3N 10:1:0.2). The title compound 7a (227 mg, 40% yield) was obtained as yellow solid. 1H NMR (DMSO-d6, 400 MHz) δ 7.89 (d, J=8.0 Hz, 1H), 7.77 (d, J=8.0 Hz, 1H), 7.73 (t, J=8.0 Hz, 1H), 7.29-7.24 (m, 2H), 7.12 (d, J=8.0 Hz, 1H), 4.19 (s, 1H), 3.70-3.67 (m, 2H), 2.98-2.91 (m, 4H), 2.88-2.82 (m, 2H), 1.99 (m, 2H), 1.72-1.66 (m, 2H); HRMS calculated for C19H20FN2O2S ([M+H]) 359.1224; found, 359.1240.
Preparation of 7a•p-TSA salt
A mixture of 7a (30 mg, 0.084 mmol) and p-toluenesulfonic acid monohydrate (19 mg, 0.099 mmol) was stirred in EtOAc-EtOH (2 mL, 10:1) at room temperature for 2 h. The resulting solid was collected, washed with EtOAc-EtOH (2 mL, 10:1) and EtOAc (3 mL) and dried under vacuum to afford the title compound as a yellow solid (32 mg, 72% yield). 1H NMR (DMSO-d6, 400 MHz) δ 10.10 (s, 1H), 7.99 (d, J=8.0 Hz, 1H), 7.85 (d, J=8.0 Hz, 1H), 7.79-7.73 (m, 1H), 7.49-7.45 (m, 3H), 7.32 (t, J=8.0 Hz, 1H), 7.25 (dd, J=8.0, 4.0 Hz, 1H), 7.12 (br s, 1H), 7.10 (br s, 1H), 4.47 (s, 1H), 3.95-3.93 (m, 2H), 3.49-3.39 (m, 6H), 2.29 (s, 3H), 2.19 (m, 2H), 2.05 (m, 2H); Elemental Analysis for C26H27FN2O5S2, Calcd: C, 58.85; H, 5.13; N, 5.28; Found: C, 58.57; H, 5.04; N, 5.18.
4-(7-Fluorodibenzo[b,d]thiophen-3-yl)-1,4-diazabicyclo[3.2.2]nonane 5,5-dioxide (7b)
The typical procedure for Buchwald-Hartwig cross-coupling reaction was followed, starting with 6b (0.2 g, 0.64 mmol), the title compound 7b was obtained as a yellow solid (104 mg, 0.29 mmol, 45% yield). 1H NMR (DMSO-d6, 400 MHz) δ 7.99 (dd, J=8.0, 4.0 Hz, 1H), 7.89 (dd, J=8.0, 3.0 Hz, 1H), 7.86 (d, J=8.0 Hz, 1H), 7.55 (m, 1H), 7.25 (d, J=4.0 Hz, 1H), 7.11 (dd, J=8.0, 4.0 Hz, 1H), 4.17 (s, 1H), 3.66 (m, 2H), 2.99-2.91 (m, 3H), 2.87-2.82 (m, 3H), 2.00-1.97 (m, 2H), 1.71-1.65 (m, 2H). Elemental Analysis for C19H19FN2O2S • 0.1 H2O, Calcd: C, 62.37; H, 5.17; N, 7.58; Found: C, 62.25; H, 5.44; N, 7.19.
7-(1,4-Diazabicyclo[3.2.2]nonan-4-yl)-2-fluorodibenzo[b,d]thiophene 5,5-dioxide (7c)
The typical procedure for Buchwald-Hartwig cross-coupling reaction was followed, starting with 6c (0.226 g, 0.72 mmol) and the title compound 7c (140 mg, 54% yield) was obtained as a yellow solid. 1H NMR (DMSO-d6, 400 MHz) δ 7.93-7.87 (m, 3H), 7.26-7.21 (m, 2H), 7.12 (d, J=8.0 Hz, 1H), 4.20 (s, 1H), 3.71-3.68 (m, 2H), 3.01-2.83 (m, 6H), 2.00 (br s, 2H), 1.73-1.67 (m, 2H); HRMS calculated for C19H20FN2O2S ([M+H]) 359.1224; found, 359.1241.
TSA salt: 1H NMR (DMSO-d6, 400 MHz) δ 10.08 (s, 1H), 8.00-7.94 (m, 3H), 7.48 (d, J=8.0 Hz, 1H), 7.43 (m, 2H), 7.32-7.25 (m, 2H), 7.12 (br, 1H), 7.10 (br, 1H), 4.48 (s, 1H), 3.94 (m, 2H), 3.47-3.38 (m, 6H), 2.29 (s, 3H), 2.19 (m, 2H), 2.06-1.99 (m, 2H); Elemental Analysis for C26H27FN2O5S2 •0.75 H2O, Calcd: C, 57.39; H, 5.28; N, 5.15; Found: C, 57.22; H, 5.11; N, 5.12.
3-(1,4-Diazabicyclo[3.2.2]nonan-4-yl)-4-fluorodibenzo[b,d]thiophene 5,5-dioxide (7d)
The typical procedure for Buchwald-Hartwig cross-coupling reaction was followed, starting with 6d (0.246 g, 0.78 mmol) and the title compound 7d was obtained as yellow solid (170 mg, 0.47 mmol, 60% yield). Free base: 1H NMR (DMSO-d6, 400 MHz) δ 8.06 (d, J=8.0 Hz, 1H), 7.91 (d, J=8.0 Hz, 1H), 7.81 (d, J=8.0 Hz, 1H), 7.75 (t, J=8.0 Hz, 1H), 7.54 (t, J=8.0 Hz, 1H), 7.36 (t, J=8.0 Hz, 1H), 3.82 (s, 1H), 3.43-3.40 (m, 2H), 3.03-3.00 (m, 2H), 2.93-2.89 (m, 4H), 2.00-1.97 (m, 2H), 1.74-1.66 (m, 2H); HRMS calculated for C19H20FN2O2S ([M+H]) 359.1224; found, 359.1246; TSA salt: 1H NMR (DMSO-d6, 400 MHz) δ 10.16 (s, 1H), 8.12 (br s, 1H), 7.94-7.90 (m, 2H), 7.78 (d, J=8.0 Hz, 1H), 7.59 (d, J=8.0 Hz, 1H), 7.50-7.40 (m, 3H), 7.12 (m, 2H), 4.01 (s, 1H), 3.54-3.38 (m, 6H), 2.30 (s, 3H), 2.19 (s, 2H), 2.07 (s, 2H), 1.09-1.03 (m, 2H); Elemental Analysis for C26H27FN2O5S2 •0.5 H2O, Calcd: C, 57.87; H, 5.23; N, 5.19; Found: C, 58.21; H, 5.56; N, 4.88.
1-(1,4-Diazabicyclo[3.2.2]nonan-4-yl)-4-fluorodibenzo[b,d]thiophene 5,5-dioxide (7e)
The typical procedure for Buchwald-Hartwig cross-coupling reaction was followed, starting with 6e (0.112 g, 0.36 mmol). The title compound 7e was obtained as a yellow solid (52 mg, 0.15 mmol, 40% yield). 1H NMR (CDCl3, 400 MHz) δ 8.50 (d, J=8.0 Hz, 1H), 7.84 (d, J=8.0 Hz, 1H), 7.68 (t, J=8.0 Hz, 1H), 7.55 (t, J=8.0 Hz, 1H), 7.43 (dd, J=8.0, 4.0 Hz, 1H), 7.14 (t, J=8.0 Hz, 1H), 3.66-3.63 (m, 1H), 3.29-3.21 (m, 5H), 3.14-3.09 (m, 2H), 2.16-2.10 (m, 2H), 1.87-1.71 (m, 3H); HRMS calculated for C19H20FN2O2S ([M+H]) 359.1224; found, 359.1215. Elemental Analysis for C19H19FN2O2S •1.5 H2O, Calcd: C, 59.20; H, 5.75; N, 7.27; Found: C, 58.90; H, 5.76; N, 7.10.
3-Bromo-6-nitrodibenzo[b,d]thiophene 5,5-dioxide (8)
The typical procedure for bromination was followed, starting with 31 (1.96 g, 7.5 mmol) and compound 8 was obtained as a pale brown solid (1.73 g, 77%). 1H NMR (DMSO-d6, 400 MHz) δ 8.70 (d, J=8.0 Hz, 1H), 8.45-8.43 (m, 2H), 8.28 (d, J=8.0, Hz, 1H), 8.14-8.09 (m, 2H). HRMS calculated for C12H6BrNNaO4S ([M+Na]+) 361.9093; found, 361.9080.
7-Bromo-2-nitrodibenzo[b,d]thiophene 5,5-dioxide (9)
The typical procedure for bromination was followed, starting with 24 (1.82 g, 6.95 mmol) and compound 9 (2.1 g, 89%) was obtained as a pale yellow solid. 1H NMR (DMSO-d6, 400 MHz) δ 9.10 (s, 1H), 8.44-8.47 (m, 3H), 8.33 (d, J=8.0 Hz, 1H), 8.11 (dd, J=8.0, 4.0 Hz, 1H).
3-(1,4-Diazabicyclo[3.2.2]nonan-4-yl)-6-nitrodibenzo[b,d]thiophene 5,5-dioxide (10)
The typical procedure for Buchwald-Hartwig cross-coupling reaction was followed starting with 8 (0.129 g, 0.38 mmol). Note: the reaction mixture was heated at 105 °C for 48 h. The title compound 10 was obtained as a reddish solid (80 mg, 55% yield). 1H NMR (DMSO-d6, 400 MHz) δ 8.40 (d, J=4.0 Hz, 1H), 8.15 (d, J=8.0 Hz, 1H), 7.97 (d, J=8.0 Hz, 1H), 7.94 (d, J=8.0 Hz, 1H), 7.26 (d, J=4.0 Hz, 1H), 7.15 (d, J=4.0 Hz, 1H), 4.21 (s, 1H), 3.71 (m, 2H), 3.00-2.85 (m, 6H), 2.00 (s, 2H), 1.71 (m, 2H); HRMS calculated for C19H20N3O4S ([M+H]) 386.1169; found, 386.1150; Elemental Analysis for C19H19N3O4S•H2O, Calcd: C, 56.56; H, 5.25; N, 10.42; Found: C, 56.65; H, 4.99; N, 10.50.
7-(1, 4-Diazabicyclo[3.2.2]nonan-4-yl)-2-nitrodibenzo[b,d]thiophene 5,5-dioxide (11)
The typical procedure for Buchwald-Hartwig cross-coupling reaction was followed, starting with 9 (1.83 g, 5.38 mmol). The title compound 11 was obtained as a reddish solid (0.836 g, 61% yield). 1H NMR (DMSO-d6, 400 MHz) δ 8.77 (s, 1H), 8.20-8.12 (m, 3H), 7.32 (d, J=4.0 Hz, 1H), 7.16 (dd, J=8.0, 4.0 Hz, 1H), 4.23 (s, 1H), 3.72 (m, 2H), 3.00-2.88 (m, 6H), 2.00 (br s, 2H), 1.74-1.69 (m, 2H); HRMS calculated for C19H20N3O4S ([M+H]) 386.1169; found, 386.1152. Elemental Analysis for C19H19N3O4S • 1.25 H2O, Calcd.: C, 55.94; H, 5.31; N, 10.30; found: C, 55.98; H, 5.17; N, 10.15.
6-Amino-3-(1,4-diazabicyclo[3.2.2]nonan-4-yl)-dibenzo[b,d]thiophene 5,5-dioxide (12)
The typical procedure for reduction of nitro derivatives was followed starting with 10 (0.34 g, 0.88 mmol) and compound 12 (146 mg, 46%) was obtained as a yellow solid. 1H NMR (DMSO-d6, 400 MHz) δ 7.73 (d, J=12.0 Hz, 1H), 7.25 (t, J=8.0 Hz, 1H), 7.11 (br s, 1H), 7.05 (d, J=8.0 Hz, 1H), 6.97 (d, J=4.0 Hz, 1H), 6.62 (d, J=8.0 Hz, 1H), 5.87 (br s, 2 H), 4.17 (s, 1H), 3.66 (m, 2H), 2.98-2.91 (m, 6H), 2.01 (s, 2H), 1.72 (m, 2H).
2-Amino-7-(1,4-diazabicyclo[3.2.2]nonan-4-yl)dibenzo[b,d]thiophene 5,5-dioxide (13)
The typical procedure for reduction of nitro derivatives was followed starting with 11 (0.68 g, 1.76 mmol) and compound 13 was obtained as a yellow solid (585 mg, 93%). 1H NMR (DMSO-d6, 400 MHz) δ 7.67 (d, J=12 Hz 1H), 7.43 (d, J=12 Hz 1H), 7.27 (s, 1H), 7.14 (d, J=8.0, 4.0 Hz, 1H), 6.91 (s, 1H), 6.53 (d, J=8.0, 4.0 Hz, 1H), 6.17 (s, 2H), 4.40 (s, 1H), 3.87 (br s, 3H), 3.07 (m, 1H), 2.15 (br s, 3H), 2.02 (br s, 3H), 1.20 (m, 2H).
3-(1,4-Diazabicyclo[3.2.2]nonan-4-yl)-6-iododibenzo[b,d]thiophene 5,5-dioxide (14)
Compound 12 (143 mg, 0.4 mmol) was dissolved in a mixture of 4 N H2SO4 (0.8 mL) and CH3CN (1 mL), and the solution was cooled to −5 °C. Sodium nitrite (55 mg, 0.8 mmol) dissolved in H2O (0.5 mL) was added dropwise at the same temperature. After stirring for 60 min a solution of diazonium salt was formed. To a mixture of CuI (268 mg, 1.4 mmol) and saturated KI solution (2.5 mL) at 70 °C was added above prepared solution of diazonium salt dropwise over 10 min and further stirred at 70 °C for 30 min. The reaction mixture was cooled and 28% ammonia solution was added (2 mL). The aqueous suspension was repeatedly extracted with CHCl3 and the combined organic layers were washed with brine (10 mL), dried (Na2SO4) and concentrated in vacuo. The crude product was purified by flash chromatography on silica gel (CHCl3/i-PrOH/Et3N 10:1:0.1 to 3:1:0.2) to give 14 (28 mg, 15%). 1H NMR (DMSO-d6, 400 MHz) δ 7.96 (d, J=8.0 Hz, 1H), 7.92 (d, J=4.0 Hz, 1H), 7.81 (d, J=8.0 Hz, 1H), 7.38 (t, J=8.0 Hz, 1H), 7.32 (d, J=4.0 Hz, 1H), 7.18 (d, J=8.0 Hz, 1H), 4.34 (s, 1H), 3.82 (m, 2H), 3.22-3.18 (m, 6H), 2.09 (m, 2H), 1.87 (m, 2H); Elemental Analysis for C19H19IN2O2S •2.5 H2O, Calcd: C, 44.63; H, 4.73; N, 5.48; Found: C, 44.88; H, 4.41; N, 5.48.
7-(1,4-Diazabicyclo[3.2.2]nonan-4-yl)-2-iododibenzo[b,d]thiophene 5,5-dioxide (15)
Compound 13 (285 mg, 0.8 mmol) was dissolved in a mixture of 4 N H2SO4 (1.5 mL) and CH3CN (2 mL), and the solution was cooled to –5 °C. NaNO2 (110 mg, 1.6 mmol, 2 equiv) dissolved in H2O (1 mL) was added dropwise at the same temperature. After stirring for 60 min a solution of diazonium salt was formed. To a mixture of CuI (536 mg, 2.8 mmol, 3.5 eqiv.) and saturated KI solution (2.5 mL) at 70 °C was added above prepared solution of diazonium salt dropwise over 10 min and further stirred at 70 °C for 30 min. The reaction mixture was cooled and sat. NH4OH was added (4 mL). The aqueous suspension was repeatedly extracted with CHCl3 and the combined organic layers were washed with brine (10 mL), dried (Na2SO4) and concentrated in vacuo. The crude product was purified by flash chromatography on silica gel (CHCl3/i-PrOH/Et3N 10:1:0.1 to 3:1:0.2) to give 15 (75 mg, 20%). 1H NMR (DMSO-d6, 400 MHz) δ 8.40 (d, J=4.0 Hz, 1H), 7.93 (d, J=8.0 Hz, 1H), 7.78 (dd, J=8.0, 1.8 Hz, 1H), 7.60 (d, J=12.0 Hz, 1H), 7.25 (d, J=1.8 Hz, 1H), 7.11 (dd, J=8.0, 4.0 Hz, 1H), 4.20 (s, 1H), 3.70 (m, 2H), 2.98-2.88 (m, 6H), 2.00 (s, 2H), 1.72 (m, 2H); HRMS calculated for C19H20IN2O2S ([M+H]) 467.0285; found, 467.0306; Elemental Analysis for C19H21IN2O3S, Calcd: C, 47.12; H, 4.37; N, 5.78; Found: C, 47.24; H, 4.53; N, 5.87.
(5-Bromo-2-nitrophenyl)(2-fluorophenyl)sulfane (18)
Cesium carbonate (4.3 g, 13.2 mmol) was added to a solution of 4-bromo-2-fluoro-nitrobenzene 16 (2.42, 11 mmol, Aldrich) and 2-fluorobenzene thiol 17 (1.4 g, 11 mmol, Aldrich) in DMF (60 mL) and the mixture was stirred for 5 h at room temperature. Water (200 mL) and ethyl acetate (100 mL) were added, the organic layer was separated and washed sequentially with water (100 mL) then brine (100 mL). The organic phase was separated, dried and concentrated to yield a yellow solid that was purified by silica gel chromatography (Hexanes/EtOAc 8:1 to 3:1) to give 18 (2.88 g, 80%). 1H NMR (CDCl3, 400 MHz) δ 8.17 (d, J=8.0 Hz, 1H), 7.68-7.60 (m, 2H), 7.41-7.29 (m, 3H), 6.95 (s, 1H).
4-Bromo-2-((2-fluorophenyl)thio)aniline (19)
The typical procedure for reduction of nitro derivatives was followed, starting with 18 (3.2 g, 9.75 mmol) and the title compound 19 was obtained as a brown solid (2.46 g, 85%). 1H NMR (CDCl3, 400 MHz) δ 7.59 (d, J=4.0 Hz, 1H), 7.33 (dd, J=8.0, 4.0 Hz, 1H), 7.21-7.15 (m, 1 H), 7.10-7.00 (m, 2H), 6.92-6.87 (m, 1H), 6.69 (d, J=8.0 Hz, 1H), 4.37 (br s, 2H).
3-Bromo-6-fluorodibenzo[b,d]thiophene (20)
Compound 19 (1.18 g, 3.96 mmol) was dissolved in 37% HCl (11 mL) and the solution was cooled below 5 °C. To this reaction mixture, sodium nitrite (408 mg, 5.93 mmol) was added slowly at temperature below 5 °C. After addition, the reaction was stirred for 30 min below 5 °C. Then sodium tetrafluoroborate (865 mg, 7.92 mmol) was added and the reaction mixture was stirred for another 30 min at temperature below 5 °C. This reaction solution was then added to the stirred solution of copper (I) oxide (1.14 mg, 7.92 mmol) in 0.1 N sulfuric acid (390 mL) at 35–40 °C. The reaction mixture was stirred for 15–30 min. Ethyl acetate was added to the reaction mixture and the mixture was filtered to remove inorganic compound. The filtrate was then extracted with ethyl acetate (3 × 120 mL). The organic extract was washed with water followed by brine and then concentrated under vacuum. The residue was purified by silica gel chromatography (hexanes) to give 20 (600 mg, 54%). 1H NMR (CDCl3, 400 MHz) δ 8.04 (d, J=4.0 Hz, 1H), 8.02 (d, J=8.0 Hz, 1H), 7.93 (d, J=8.0 Hz, 1H), 7.62 (dd, J=8.0, 4.0 Hz, 1H), 7.47 (ddd, J=12.0, 8.0, 4.0 Hz, 1H), 7.22 (t, J=8.0 Hz, 1H).
3-Nitrodibenzo[b,d]thiophene 5,5-dioxide (23)
Dibenzo[b,d]thiophene 5,5-dioxide 21 (10 g, 46 mmol, Aldrich) was slowly added to a stirred mixture of glacial acetic acid (34 mL) and sulfuric acid (96%, 34 mL). The slurry was stirred and red fuming nitric acid (36 mL) was added dropwise over a period of 90 min at temperature −5°C – 5°C. The slurry was stirred for another 30 min and poured over ice. The precipitate was filtered, rinsed with water and dried at room temperature. The crude product was recrystallized with acetonitrile to give 23 as yellow crystals (8.7 g, 72%).1H NMR (DMSO-d6, 400 MHz) δ 8.84 (d, J=8.0 Hz, 1H), 8.65 (dd, J=8.0, 2.0 Hz, 1H), 8.50 (d, J=8.0, Hz, 1H), 8.39 (d, J=8.0, Hz, 1H), 8.13 (d, J=8.0, Hz, 1H), 7.93 (t, J=8.0, Hz, 1H), 7.81 (t, J=8.0, Hz, 1H).
2-Nitro-dibenzo[b,d]thiophene 5,5-dioxide (24)
The typical procedure for oxidation of 1,4-dibenzothiophene was followed starting with 22 (489 mg, 2.13 mmol, Oakwood Chemical) and the title compound 24 (510 mg, 90%) was obtained as white crystals. 1H NMR (CDCl3, 400 MHz) δ 8.66 (d, J=4 Hz 1H), 8.43 (dd, J=8, 4 Hz, 1H), 8.04 (d, J=8 Hz, 1H), 7.96 (d, J=8 Hz, 1H), 7.92 (d, J=8 Hz, 1H), 7.79 (t, J=8 Hz, 1H), 7.69 (t, J=8.0 Hz, 1H).
3-Bromo-7-nitrodibenzo[b,d]thiophene 5,5-dioxide (25)
The typical procedure for bromination was followed starting with 23 (2.59 g, 9.9 mmol) and brown solid was obtained and recrystallized with benzene to yield 25 as a yellow solid (1.73 g, 51%). 1H NMR (DMSO-d6, 400 MHz) δ 8.89 (d, J=4.0 Hz, 1H), 8.67 (dd, J=8.0, 3.0 Hz, 1H), 8.52-8.49 (m, 2H), 8.35 (d, J=8.0, Hz, 1H), 8.15 (dd, J=8.0, 2.0 Hz, 1H).
7-Bromodibenzo[b,d]thiophen-3-amine 5,5-dioxide (26)
A solution of stannous chloride dihydrate (12.4 g, 56 mmol) in 37% hydrochloric acid (21 mL) was added to a mixture of 25 (1.7 g, 5 mmol) in glacial acetic acid (50 mL). The reaction mixture was stirred at 100 °C for 60 min and cooled down to 5 °C. The precipitate was filtered off, rinsed with water on the filter and dispersed in water. The dispersion was made basic (pH=10) by addition of an excess of 1M sodium hydroxide and stirred for 3 h. The precipitate was filtered off, rinsed with water and dried overnight on the filter to yield 26 (0.7 g, 45%) as a pale white solid. 1H NMR (DMSO-d6, 400 MHz) δ 8.12 (s, 1H), 7.87-7.77 (m, 3H), 6.95 (s, 1H), 6.87 (dd, J=8, 4 Hz, 1H), 6.20 (br s, 2H).
2-Amino-7-bromodibenzo[b,d]thiophene 5,5-dioxide (27)
The typical procedure for reduction of nitro derivatives was followed staring with 9 (0.60 g, 1.76 mmol). The title compound 27 (496 mg, 91%) was obtained as a white solid. 1H NMR (DMSO-d6, 400 MHz) δ 8.16 (d, J=4 Hz, 1H), 7.93(d, J=8.0 Hz, 1H), 7.87 (d, J=12 Hz, 1H), 7.56 (d, J=8 Hz, 1H), 7.08 (s, 1H), 6.71 (d, J=8 Hz, 1H), 6.36 (br s, 2H).
4-Fluorodibenzo[b,d]thiophene 5,5-dioxide (29)
The typical procedure for oxidation of 1,4-dibenzothiophene was followed starting with 4-fluorodibenzo[b,d]thiophene 2845 (1.62 g, 8 mmol). The title compound 29 (1.8 g, 96%) was obtained as white crystals. 1H NMR (CDCl3, 400 MHz) δ 7.85 (d, J=8.0 Hz, 1H), 7.82 (d, J=8.0 Hz, 1H), 7.71-7.57 (m, 4H), 7.20 (t, J=8.0 Hz, 1H).
4-Nitrodibenzo[b,d]thiophene 5,5-dioxide (31)
The typical procedure for oxidation of 1,4-dibenzothiophene was followed starting with 3046 (1.08 g, 4.71 mmol). The final compound 31 (1.1g, 90%) was obtained as pale yellow crystals. 1H NMR (DMSO-d6, 400 MHz) δ 8.69 (d, J=8.0 Hz, 1H), 8.42 (d, J=8.0, Hz, 1H), 8.33 (d, J=8.0, Hz, 1H), 8.11 (t, J=8.0 Hz, 1H), 8.07 (d, J=8.0, Hz, 1H), 7.88 (t, J=8.0 Hz, 1H), 7.77 (t, J=8.0 Hz, 1H). HRMS calculated for C12H7NNaO4S ([M+Na]) 283.9988; found, 283.9994.
Radiosynthesis of [18F]7a and [18F]7c
The same radiolabeling method was used for both radioligands [18F]7a and [18F]7c. A solution of the [18F]fluoride, 15–20 mg of Kryptofix 222®, and 1–2 mg of K2C2O4 in 1 mL of 50% aqueous acetonitrile was added to a reaction vessel of a GE MicroLab box. The mixture was heated at 120–135 °C under a stream of argon, while water was evaporated azeotropically after the additions of 2 mL of CH3CN. A solution of the corresponding nitro precursor (10 or 11) (2 mg) in anhydrous DMSO (0.8 mL) was added to the reaction vessel and heated at 160 °C for 12 min. The reaction mixture was cooled, diluted with 0.7 mL of water, injected onto the reverse-phase semi-preparative HPLC column (Table 6). The radioactive peak was collected in 50 mL of HPLC water. The water solution was transferred through an activated Waters C-18 Oasis HLB light solid-phase extraction (SPE) cartridge. After washing the SPE with 10 mL saline, the product was eluted with a mixture of 1 mL of ethanol and 0.04 mL 1N HCl through a 0.2 µM sterile filter into a sterile, pyrogen-free multi-dose vial and 10 mL of 0.9% saline and 0.05 mL sterile 8.4% solution sodium bicarbonate was added through the same filter. The final products [18F]7a or [18F]7c were then analyzed by analytical HPLC (Table 6) using a UV detector at 340 nm to determine the radiochemical purity and specific radioactivity at the time synthesis ended. The total synthesis time including QC was 70–80 min.
In vitro binding assay
α7-nAChR assay with rat brain membranes
The assay was done commercially by Caliper PerkinElmer (Hanover, MD). In brief, rat cortical membranes were incubated with [125I]α-Bungarotoxin (KD = 0.7 nM) at a concentration of 0.1 nM in a buffer consisting of 50 mM Tris, 120 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 0.003 mM atropine sulfate at pH 7.4 for 150 min at 0 °C.63 The binding was terminated by rapid vacuum filtration of the assay contents onto GF/C filters presoaked in PEI. Radioactivity trapped onto the filters was assessed using a gamma-counter. Nonspecific binding was defined as that remaining in the presence of 1 µM α-Bungarotoxin. The assays were done two times independently, each in duplicate, at multiple concentrations of the test compounds. Binding assay results were analyzed using a one-site competition model, and IC50 curves were generated based on a sigmoidal dose response with variable slope. The Ki values were calculated using the Cheng-Prusoff equation. Methyllycaconitine (MLA) was used as a reference compound in all assays.
HEK 293 Cell culture and stable transfections (heteromeric nAChR)
HEK 293 cells (ATCC CRL 1573) were maintained at 37 °C with 5% CO2 in a humidified incubator. Growth medium for the HEK 293 cells was the minimum essential medium supplemented with 10% fetal bovine serum, 100 units/mL penicillin G, and 100 µg/mL streptomycin. Transfections of these cells, and selection and establishment of stable cell lines were carried out as described previously.52, 55
Membrane homogenate preparation (heteromeric nAChR)
Membrane homogenates for ligand binding assays were made as described previously.52, 55 Briefly, cultured cells at >90% confluency were removed from the culture flask (80 cm2) with a disposable cell scraper and placed in 10 mL of 50 mM Tris.HCl buffer (pH 7.4, 4 °C). The cell suspension was centrifuged at 1,000 × g for 5 min and the pellet was collected. The cell pellet was then homogenized in 10 mL buffer with a polytron homogenizer for 20 s and centrifuged at 35,000 g for 10 min at 4 °C. Membrane pellets were resuspended in fresh buffer.
Binding to heteromeric nAChR
subunit combinations, which represent possible heteromeric nAChRs, was measured with 0.5 nM [3H]epibatidine in HEK cells expressing these subunits (KD = 0.021 nM (α2β2-nAChR), KD = 0.084 nM (α2β4-nAChR), KD = 0.034 nM (α3β2-nAChR), KD = 0.29 nM (α3β4-nAChR), KD = 0.046 nM (α4β2-nAChR), KD = 0.094 nM (α4β4-nAChR)).55 Aliquots of the membrane homogenates containing 30 to 200 µg protein were used for the binding assays, which were carried out in a final volume of 100 µL in borosilicate glass tubes. After incubation at 24 °C for 2 h, the samples were collected with a cell harvester (Brandel M-48) onto Whatman GF/C filters pre-wet with 0.5% polyethylenimine. After harvesting the samples, the filters were washed three times with 5 mL of 50 mM Tris.HCl buffer, and then counted in a liquid scintillation counter. Nonspecific binding was measured in samples incubated in parallel containing 300 µM nicotine for [3H]epibatidine binding. Specific binding was defined as the difference between total binding and nonspecific binding. Data from these competition binding assays were analyzed using Prism 5 (GraphPad Software, San Diego, CA).
5-HT3(h) binding assay
The assay was done commercially by Caliper PerkinElmer (Hanover, MD) using recombinant HEK293 cells and 0.35 nM [3H]GR65630 (KD = 0.5 nM).
Biodistribution studies in CD-1 mice
Baseline study
Male, CD-1 mice weighing 25–30 g from Charles River Laboratories, (Wilmington, MA) were used for biodistribution studies. The animals were sacrificed by cervical dislocation at various times following injection of [18F]7a or [18F]7c (70 µCi, specific radioactivity 8000–12000 mCi/µmol, in 0.2 mL saline) into a lateral tail vein; three animals per time point. The brains were rapidly removed and dissected on ice. The brain regions of interest were weighed and their radioactivity content was determined in an automated γ-counter with a counting error below 3%. Aliquots of the injectate were prepared as standards and their radioactivity content was counted along with the tissue samples. The percent of injected dose per gram of tissue (%ID/g tissue) was calculated. All experimental protocols were approved by the Animal Care and Use Committee of the Johns Hopkins Medical Institutions.
Self-blockade of [18F] 7a binding with 7a
In vivo saturation blockade studies were done by i.v. co-administration of the radiotracer [18F]7a (70 µCi, SA = 9200 mCi/µmol, 0.2 mL) with various doses of “cold” 7a per animal (0 µg (vehicle); 0.0048 µg; 7.2 µg). Compound 7a was dissolved in saline at pH = 5.5. Ninety min after administration of the tracer and blocker, brain tissues were harvested, and their regional radioactivity content was determined. The self-blockade of [18F]7c with 7c was done similarly.
Blockade of [18F] 7a binding with 1
In vivo α7-nAChR receptor blocking studies were done by intravenous (i.v.) co-administration of the radiotracer [18F]7a (70 µCi, SA = 7900 mCi/µmol, 0.2 mL) with various doses of 1 (0 µg (vehicle); 0.02 mg/kg; 0.2 mg/kg, 1 mg/kg and 3 mg/kg). Three animals per dose were used. 1 was dissolved in a vehicle (saline:alcohol (9:1) at pH=5.5). Ninety min after administration of the tracer, brain tissues were harvested, and their regional radioactivity content was determined. The dose-dependent blockade study of [18F]7c with 5 was done the same way.
Blockade of [18F] 7a with nicotine and cytisine
In vivo CB1 receptor blocking studies were carried out by subcutaneous (s.c.) administration of (−)nicotine tartrate (5 mg/kg) or cytisine (1 mg/kg) followed by i.v. injection of the radiotracer [18F]7a (70 µCi, specific radioactivity ~14000 mCi/µmol, 0.2 mL) 5 min thereafter. The drugs were dissolved in saline and administered in a volume of 0.1 mL. Control animals were injected with 0.1 mL of saline. Ninety min after administration of the tracer, brain tissues were harvested, and their radioactivity content was determined.
Blockade of [18F] 7a with non-α7-nAChR drugs
In vivo receptor blocking studies were performed by administration of six drugs (Table 5), followed by i.v. injection of the radiotracer [18F]7a (70 µCi, specific radioactivity ~14000 mCi/µmol, 0.2 mL). The drugs (2 mg/kg, s.c.) were dissolved in a vehicle (saline:DMSO 5:1) and administered in a volume of 0.1 mL. Control animals were injected with 0.1 mL of the vehicle solution. Ninety min after administration of the tracer, brain tissues were harvested, and their radioactivity content was determined.
Scheme 5.

Reagents and conditions: (a) 30% H2O2, acetic acid, 60 °C, 24 h; (b) NBS, H2SO4, 24 h.
Acknowledgements
We thank Dr. Richard Wahl for fruitful discussions. We thank Dr. Hiroto Kuwabara for reading the manuscript and for his useful comments. We thank Dr. Yuchuan Wang for useful comments and references. We are grateful to Paige Finley and Gilbert Green for their valuable help with animal experiments. We are thankful to Judy W. Buchanan for editorial help. We very much thank Drs. Peter Brust (Helmholtz-Zentrum Dresden-Rossendorf Institute of Radiopharmacy) and Dan Peters (DanPET AB) for the samples of compounds 2 and 3 and Dr. Martin Pomper for the sample of 4 for our inhibition binding assay studies.
This research was supported by NIH Grants MH079017 and AG037298 (AGH) and, in part, by the Division of Nuclear Medicine of The Johns Hopkins University School of Medicine.
List of non-standard abbreviations and acronyms
- BINAP
2,2'-bis(diphenylphosphino)-1,1'-binaphthyl
- Bmax
receptor density in tissue
- BPND
binding potential
- EtOAc
ethylacetate
- EtOH
ethanol
- Et2O
diethylether
- Et3N
triethylamine
- KD
dissociation constant of the radioligand-receptor complex
- nAChR
nicotinic acetylcholine receptor
- i-PrOH
isopropyl alcohol
References
- 1.Gotti C, Clementi F. Neuronal nicotinic receptors: from structure to pathology. Prog Neurobiol. 2004;74:363–396. doi: 10.1016/j.pneurobio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 2.Lukas RJ, Changeux JP, Le Novere N, Albuquerque EX, Balfour DJ, Berg DK, Bertrand D, Chiappinelli VA, Clarke PB, Collins AC, Dani JA, Grady SR, Kellar KJ, Lindstrom JM, Marks MJ, Quik M, Taylor PW, Wonnacott S. International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev. 1999;51:397–401. [PubMed] [Google Scholar]
- 3.Dani JA, Bertrand D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol. 2007;47:699–729. doi: 10.1146/annurev.pharmtox.47.120505.105214. [DOI] [PubMed] [Google Scholar]
- 4.Philip NS, Carpenter LL, Tyrka AR, Price LH. Nicotinic acetylcholine receptors and depression: a review of the preclinical and clinical literature. Psychopharmacology (Berl) 2010;212:1–12. doi: 10.1007/s00213-010-1932-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ishikawa M, Hashimoto K. alpha7 nicotinic acetylcholine receptor as a potential therapeutic target for schizophrenia. Curr Pharm Des. 2011;17:121–129. doi: 10.2174/138161211795049561. [DOI] [PubMed] [Google Scholar]
- 6.Parri HR, Hernandez CM, Dineley KT. Research update: Alpha7 nicotinic acetylcholine receptor mechanisms in Alzheimer's disease. Biochem Pharmacol. 2011;82:931–942. doi: 10.1016/j.bcp.2011.06.039. [DOI] [PubMed] [Google Scholar]
- 7.Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woodruff-Pak DS, Gould TJ. Neuronal nicotinic acetylcholine receptors: involvement in Alzheimer's disease and schizophrenia. Behav Cogn Neurosci Rev. 2002;1:5–20. doi: 10.1177/1534582302001001002. [DOI] [PubMed] [Google Scholar]
- 9.D'Hoedt D, Bertrand D. Nicotinic acetylcholine receptors: an overview on drug discovery. Expert Opin Ther Targets. 2009;13:395–411. doi: 10.1517/14728220902841045. [DOI] [PubMed] [Google Scholar]
- 10.Hoffmeister PG, Donat CK, Schuhmann MU, Voigt C, Walter B, Nieber K, Meixensberger J, Bauer R, Brust P. Traumatic brain injury elicits similar alterations in alpha7 nicotinic receptor density in two different experimental models. Neuromolecular Med. 2011;13:44–53. doi: 10.1007/s12017-010-8136-4. [DOI] [PubMed] [Google Scholar]
- 11.Verbois SL, Sullivan PG, Scheff SW, Pauly JR. Traumatic brain injury reduces hippocampal alpha7 nicotinic cholinergic receptor binding. J Neurotrauma. 2000;17:1001–1011. doi: 10.1089/neu.2000.17.1001. [DOI] [PubMed] [Google Scholar]
- 12.Verbois SL, Scheff SW, Pauly JR. Time-dependent changes in rat brain cholinergic receptor expression after experimental brain injury. J Neurotrauma. 2002;19:1569–1585. doi: 10.1089/089771502762300238. [DOI] [PubMed] [Google Scholar]
- 13.Olincy A, Harris JG, Johnson LL, Pender V, Kongs S, Allensworth D, Ellis J, Zerbe GO, Leonard S, Stevens KE, Stevens JO, Martin L, Adler LE, Soti F, Kem WR, Freedman R. Proof-of-concept trial of an alpha7 nicotinic agonist in schizophrenia. Arch Gen Psychiatry. 2006;63:630–638. doi: 10.1001/archpsyc.63.6.630. [DOI] [PubMed] [Google Scholar]
- 14.Thomsen MS, Hansen HH, Timmerman DB, Mikkelsen JD. Cognitive improvement by activation of alpha7 nicotinic acetylcholine receptors: from animal models to human pathophysiology. Curr Pharm Des. 2010;16:323–343. doi: 10.2174/138161210790170094. [DOI] [PubMed] [Google Scholar]
- 15.Mazurov AA, Speake JD, Yohannes D. Discovery and development of alpha7 nicotinic acetylcholine receptor modulators. J Med Chem. 2011;54:7943–7961. doi: 10.1021/jm2007672. [DOI] [PubMed] [Google Scholar]
- 16.Taly A, Charon S. alpha7 nicotinic acetylcholine receptors: a therapeutic target in the structure era. Curr Drug Targets. 2012;13:695–706. doi: 10.2174/138945012800398919. [DOI] [PubMed] [Google Scholar]
- 17.Wallace TL, Bertrand D. Alpha7 neuronal nicotinic receptors as a drug target in schizophrenia. Expert Opin Ther Targets. 2013;17:139–155. doi: 10.1517/14728222.2013.736498. [DOI] [PubMed] [Google Scholar]
- 18.Pomper MG, Phillips E, Fan H, McCarthy DJ, Keith RA, Gordon JC, Scheffel U, Dannals RF, Musachio JL. Synthesis and biodistribution of radiolabeled alpha 7 nicotinic acetylcholine receptor ligands. J Nucl Med. 2005;46:326–334. [PubMed] [Google Scholar]
- 19.Hashimoto K, Nishiyama S, Ohba H, Matsuo M, Kobashi T, Takahagi M, Iyo M, Kitashoji T, Tsukada H. [11C]CHIBA-1001 as a novel PET ligand for alpha7 nicotinic receptors in the brain: a PET study in conscious monkeys. PLoS One. 2008;3:e3231. doi: 10.1371/journal.pone.0003231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ogawa M, Nishiyama S, Tsukada H, Hatano K, Fuchigami T, Yamaguchi H, Matsushima Y, Ito K, Magata Y. Synthesis and evaluation of new imaging agent for central nicotinic acetylcholine receptor alpha7 subtype. Nucl Med Biol. 2010;37:347–355. doi: 10.1016/j.nucmedbio.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 21.Dolle F, Valette H, Hinnen F, Vaufrey F, Demphel S, Coulon C, Ottaviani M, Bottlaender M, Crouzel C. Synthesis and preliminary evaluation of a carbon-11-labelled agonist of the a7 nicotinic acetylcholine receptor. J Label Compd Radiopharm. 2001;44:785–795. [Google Scholar]
- 22.Toyohara J, Ishiwata K, Sakata M, Wu J, Nishiyama S, Tsukada H, Hashimoto K. In vivo evaluation of alpha7 nicotinic acetylcholine receptor agonists [11C]A-582941 and [11C]A-844606 in mice and conscious monkeys. PLoS One. 2010;5:e8961. doi: 10.1371/journal.pone.0008961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horti AG, Ravert HT, Gao Y, Holt DP, Bunnelle WH, Schrimpf MR, Li T, Ji J, Valentine H, Scheffel U, Kuwabara H, Wong DF, Dannals RF. Synthesis and evaluation of new radioligands [(11)C]A-833834 and [(11)C]A-752274 for positron-emission tomography of alpha7-nicotinic acetylcholine receptors. Nucl Med Biol. 2013;40:395–402. doi: 10.1016/j.nucmedbio.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao Y, Ravert HT, Valentine H, Scheffel U, Finley P, Wong DF, Dannals RF, Horti AG. 5-(5-(6-[(11)C]methyl-3,6-diazabicyclo[3.2.0]heptan-3-yl)pyridin-2-yl)-1H-indole as a potential PET radioligand for imaging cerebral alpha7-nAChR in mice. Bioorg Med Chem. 2012;20:3698–3702. doi: 10.1016/j.bmc.2012.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Toyohara J, Sakata M, Wu J, Ishikawa M, Oda K, Ishii K, Iyo M, Hashimoto K, Ishiwata K. Preclinical and the first clinical studies on [11C]CHIBA-1001 for mapping alpha7 nicotinic receptors by positron emission tomography. Ann Nucl Med. 2009;23:301–309. doi: 10.1007/s12149-009-0240-x. [DOI] [PubMed] [Google Scholar]
- 26.Ettrup A, Mikkelsen JD, Lehel S, Madsen J, Nielsen EO, Palner M, Timmermann DB, Peters D, Knudsen GM. 11C-NS14492 as a novel PET radioligand for imaging cerebral alpha7 nicotinic acetylcholine receptors: in vivo evaluation and drug occupancy measurements. J Nucl Med. 2011;52:1449–1456. doi: 10.2967/jnumed.111.088815. [DOI] [PubMed] [Google Scholar]
- 27.Ravert HT, Dorff P, Foss CA, Mease RC, Fan H, Holmquist CR, Phillips E, McCarthy DJ, Heys JR, Holt DP, Wang Y, Endres CJ, Dannals RF, Pomper MG. Radiochemical synthesis and in vivo evaluation of [18F]AZ11637326: An agonist probe for the alpha7 nicotinic acetylcholine receptor. Nucl Med Biol. 2013;40:731–739. doi: 10.1016/j.nucmedbio.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 28.Rotering S, Scheunemann M, Fischer S, Hiller A, Peters D, Deuther-Conrad W, Brust P. Radiosynthesis and first evaluation in mice of [(18)F]NS14490 for molecular imaging of alpha7 nicotinic acetylcholine receptors. Bioorg Med Chem. 2013;21:2635–2642. doi: 10.1016/j.bmc.2013.02.018. [DOI] [PubMed] [Google Scholar]
- 29.Deuther-Conrad W, Fischer S, Hiller A, Becker G, Cumming P, Xiong G, Funke U, Sabri O, Peters D, Brust P. Assessment of alpha7 nicotinic acetylcholine receptor availability in juvenile pig brain with [18F]NS10743. Eur J Nucl Med Mol Imaging. 2011;38:1541–1549. doi: 10.1007/s00259-011-1808-y. [DOI] [PubMed] [Google Scholar]
- 30.Horti AG, Villemagne VL. The quest for Eldorado: development of radioligands for in vivo imaging of nicotinic acetylcholine receptors in human brain. Curr Pharm Design. 2006;12:3877–3900. doi: 10.2174/138161206778559605. [DOI] [PubMed] [Google Scholar]
- 31.Toyohara J, Wu J, Hashimoto K. Recent development of radioligands for imaging alpha7 nicotinic acetylcholine receptors in the brain. Curr Top Med Chem. 2010;10:1544–1557. doi: 10.2174/156802610793176828. [DOI] [PubMed] [Google Scholar]
- 32.Brust P, Peters D, Deuther-Conrad W. Development of Radioligands for the Imaging of alpha7 Nicotinic Acetylcholine Receptors with Positron Emission Tomography. Curr Drug Targets. 2012;13:594–601. doi: 10.2174/138945012800398955. [DOI] [PubMed] [Google Scholar]
- 33.Marutle A, Zhang X, Court J, Piggott M, Johnson M, Perry R, Perry E, Nordberg A. Laminar distribution of nicotinic receptor subtypes in cortical regions in schizophrenia. J Chem Neuroanat. 2001;22:115–126. doi: 10.1016/s0891-0618(01)00117-x. [DOI] [PubMed] [Google Scholar]
- 34.Kulak JM, Schneider JS. Differences in alpha7 nicotinic acetylcholine receptor binding in motor symptomatic and asymptomatic MPTP-treated monkeys. Brain Res. 2004;999:193–202. doi: 10.1016/j.brainres.2003.10.062. [DOI] [PubMed] [Google Scholar]
- 35.Kulak JM, Carroll FI, Schneider JS. [125I]Iodomethyllycaconitine binds to alpha7 nicotinic acetylcholine receptors in monkey brain. Eur J Neurosci. 2006;23:2604–2610. doi: 10.1111/j.1460-9568.2006.04804.x. [DOI] [PubMed] [Google Scholar]
- 36.Zhang L, Villalobos A, Beck EM, Bocan T, Chappie TA, Chen L, Grimwood S, Heck SD, Helal CJ, Hou X, Humphrey JM, Lu J, Skaddan MB, McCarthy TJ, Verhoest PR, Wager TT, Zasadny K. Design and Selection Parameters to Accelerate the Discovery of Novel Central Nervous System Positron Emission Tomography (PET) Ligands and Their Application in the Development of a Novel Phosphodiesterase 2A PET Ligand. J Med Chem. 2013;56:4568–4579. doi: 10.1021/jm400312y. [DOI] [PubMed] [Google Scholar]
- 37.Eckelman WC, Reba RC, Gibson RE. Receptor-binding radiotracers: A class of potential radiopharmaceuticals. J Nucl Med. 1979;20:350–357. [PubMed] [Google Scholar]
- 38.Tanibuchi Y, Wu J, Toyohara J, Fujita Y, Iyo M, Hashimoto K. Characterization of [(3)H]CHIBA-1001 binding to alpha7 nicotinic acetylcholine receptors in the brain from rat, monkey, and human. Brain Res. 2010;1348:200–208. doi: 10.1016/j.brainres.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 39.Ding M, Ghanekar S, Elmore CS, Zysk JR, Werkheiser JL, Lee CM, Liu J, Chhajlani V, Maier DL. [3H]Chiba-1001 (methyl-SSR180711) has low in vitro binding affinity and poor in vivo selectivity to nicotinic alpha-7 receptor in rodent brain. Synapse. 2012;66:315–322. doi: 10.1002/syn.21513. [DOI] [PubMed] [Google Scholar]
- 40.Maier DL, Hill G, Ding M, Tuke D, Einstein E, Gurley D, Gordon JC, Bock MJ, Smith JS, Bialecki R, Eisman M, Elmore CS, Werkheiser JL. Pre-clinical validation of a novel alpha-7 nicotinic receptor radiotracer, [3H]AZ11637326: target localization, biodistribution and ligand occupancy in the rat brain. Neuropharmacology. 2011;61:161–171. doi: 10.1016/j.neuropharm.2011.03.024. [DOI] [PubMed] [Google Scholar]
- 41.Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN, Holden J, Houle S, Huang SC, Ichise M, Iida H, Ito H, Kimura Y, Koeppe RA, Knudsen GM, Knuuti J, Lammertsma AA, Laruelle M, Logan J, Maguire RP, Mintun MA, Morris ED, Parsey R, Price JC, Slifstein M, Sossi V, Suhara T, Votaw JR, Wong DF, Carson RE. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab. 2007;27:1533–1539. doi: 10.1038/sj.jcbfm.9600493. [DOI] [PubMed] [Google Scholar]
- 42.Tichauer KM, Samkoe KS, Sexton KJ, Hextrum SK, Yang HH, Klubben WS, Gunn JR, Hasan T, Pogue BW. In Vivo Quantification of Tumor Receptor Binding Potential with Dual-Reporter Molecular Imaging. Mol Imaging Biol. 2011;14:584–592. doi: 10.1007/s11307-011-0534-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schrimpf MR, Sippy KB, Briggs CA, Anderson DJ, Li T, Ji J, Frost JM, Surowy CS, Bunnelle WH, Gopalakrishnan M, Meyer MD. SAR of alpha7 nicotinic receptor agonists derived from tilorone: exploration of a novel nicotinic pharmacophore. Bioorg Med Chem Lett. 2012;22:1633–1638. doi: 10.1016/j.bmcl.2011.12.126. [DOI] [PubMed] [Google Scholar]
- 44.Muller K, Faeh C, Diederich F. Fluorine in pharmaceuticals: looking beyond intuition. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 45.Nag M, Jenks WS. Photochemistry of substituted dibenzothiophene oxides: the effect of trapping groups. J Org Chem. 2005;70:3458–3463. doi: 10.1021/jo047995e. [DOI] [PubMed] [Google Scholar]
- 46.Manna S, Maity S, Rana S, Agasti S, Maiti D. ipso-Nitration of arylboronic acids with bismuth nitrate and perdisulfate. Org Lett. 2012;14:1736–1739. doi: 10.1021/ol300325t. [DOI] [PubMed] [Google Scholar]
- 47.Smith MB, March J. March's advanced organic chemistry. Hoboken, New Jersey: John Wiley & Sons, Inc.; 2007. pp. 853–869. [Google Scholar]
- 48.Kubinyi H. The quantitative analysis of structure-activity relationships. In: Wolff ME, editor. Burger's Medicinal Chemistry and Drug Discovery. New York: John Wiley & Sons; 1995. pp. 497–571. [Google Scholar]
- 49.Miller PW, Long NJ, Vilar R, Gee AD. Synthesis of 11C, 18F, 15O, and 13N radiolabels for positron emission tomography. Angew Chem Int Ed Engl. 2008;47:8998–9033. doi: 10.1002/anie.200800222. [DOI] [PubMed] [Google Scholar]
- 50.Hudlicky M, Pavlath AE. Chemistry of organic fluorine compounds II. A critical review. Washington, DC: Americal Chemical Society; 1995. pp. 286–289. [Google Scholar]
- 51.Clark JH, Wails D. Fluorodenitration of activated diphenyl sulphones using tetramethylammonium fluoride. J Fluor Chem. 1995;70:201–205. [Google Scholar]
- 52.Xiao Y, Abdrakhmanova GR, Baydyuk M, Hernandez S, Kellar KJ. Rat neuronal nicotinic acetylcholine receptors containing alpha7 subunit: pharmacological properties of ligand binding and function. Acta Pharmacol Sin. 2009;30:842–850. doi: 10.1038/aps.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anderson DJ, Bunnelle W, Surber B, Du J, Surowy C, Tribollet E, Marguerat A, Bertrand D, Gopalakrishnan M. [3H]A-585539 [(1S,4S)-2,2-dimethyl-5-(6-phenylpyridazin-3-yl)-5-aza-2-azoniabicyclo[2.2.1]hept ane], a novel high-affinity alpha7 neuronal nicotinic receptor agonist: radioligand binding characterization to rat and human brain. J Pharmacol Exp Ther. 2008;324:179–187. doi: 10.1124/jpet.107.130062. [DOI] [PubMed] [Google Scholar]
- 54.Navarro HA, Zhong D, Abrahm P, Xu H, Carroll FI. Synthesis and pharmacological characterization of ([125I]iodomethyllycaconitine ([125I]iodo-MLA). A new ligand for the a7 nicotinic acetylcholine receptor. J Med Chem. 2000;43:142–145. doi: 10.1021/jm990544f. [DOI] [PubMed] [Google Scholar]
- 55.Xiao Y, Kellar K. The comparative pharmacology and up-regulation of rat neuronal nicotinic receptor subtype binding sites stably expressed in transfected mammalian cells. J Pharmacol Exp Ther. 2004;310:98–107. doi: 10.1124/jpet.104.066787. [DOI] [PubMed] [Google Scholar]
- 56.Clarke PB, Schwartz RD, Paul SM, Pert CB, Pert A. Nicotinic binding in rat brain: autoradiographic comparison of [3H]acetylcholine, [3H]nicotine, and [125I]-alpha-bungarotoxin. J Neurosci. 1985;5:1307–1315. doi: 10.1523/JNEUROSCI.05-05-01307.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Whiteaker P, Davies AR, Marks MJ, Blagbrough IS, Potter BV, Wolstenholme AJ, Collins AC, Wonnacott S. An autoradiographic study of the distribution of binding sites for the novel alpha7-selective nicotinic radioligand [3H]-methyllycaconitine in the mouse brain. Eur J Neurosci. 1999;11:2689–2696. doi: 10.1046/j.1460-9568.1999.00685.x. [DOI] [PubMed] [Google Scholar]
- 58.Biton B, Bergis OE, Galli F, Nedelec A, Lochead AW, Jegham S, Godet D, Lanneau C, Santamaria R, Chesney F, Leonardon J, Granger P, Debono MW, Bohme GA, Sgard F, Besnard F, Graham D, Coste A, Oblin A, Curet O, Vige X, Voltz C, Rouquier L, Souilhac J, Santucci V, Gueudet C, Francon D, Steinberg R, Griebel G, Oury-Donat F, George P, Avenet P, Scatton B. SSR180711, a novel selective alpha7 nicotinic receptor partial agonist: (1) binding and functional profile. Neuropsychopharmacology. 2007;32:1–16. doi: 10.1038/sj.npp.1301189. [DOI] [PubMed] [Google Scholar]
- 59.Hashimoto K, Ishima T, Fujita Y, Matsuo M, Kobashi T, Takahagi M, Tsukada H, Iyo M. Phencyclidine-induced cognitive deficits in mice are improved by subsequent subchronic administration of the novel selective alpha7 nicotinic receptor agonist SSR180711. Biol Psychiatry. 2008;63:92–97. doi: 10.1016/j.biopsych.2007.04.034. [DOI] [PubMed] [Google Scholar]
- 60.Pichat P, Bergis OE, Terranova JP, Urani A, Duarte C, Santucci V, Gueudet C, Voltz C, Steinberg R, Stemmelin J, Oury-Donat F, Avenet P, Griebel G, Scatton B. SSR180711, a novel selective alpha7 nicotinic receptor partial agonist: (II) efficacy in experimental models predictive of activity against cognitive symptoms of schizophrenia. Neuropsychopharmacology. 2007;32:17–34. doi: 10.1038/sj.npp.1301188. [DOI] [PubMed] [Google Scholar]
- 61.Rollema H, Shrikhande A, Ward KM, Tingley FD, 3rd, Coe JW, O'Neill BT, Tseng E, Wang EQ, Mather RJ, Hurst RS, Williams KE, de Vries M, Cremers T, Bertrand S, Bertrand D. Pre-clinical properties of the alpha4beta2 nicotinic acetylcholine receptor partial agonists varenicline, cytisine and dianicline translate to clinical efficacy for nicotine dependence. Br J Pharmacol. 2010;160:334–345. doi: 10.1111/j.1476-5381.2010.00682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mintun MA, Raichle ME, Kilbourn MR, Wooten GF, Welch MJ. A quantitative model for the in vivo assessment of drug binding sites with positron emission tomography. Ann Neurol. 1984;15:217–227. doi: 10.1002/ana.410150302. [DOI] [PubMed] [Google Scholar]
- 63.Meyer EM, Kuryatov A, Gerzanich V, Lindstrom J, Papke RL. Analysis of 3-(4-hydroxy, 2-Methoxybenzylidene)anabaseine selectivity and activity at human and rat alpha-7 nicotinic receptors. J Pharmacol Exp Ther. 1998;287:918–925. [PubMed] [Google Scholar]