

Abstract
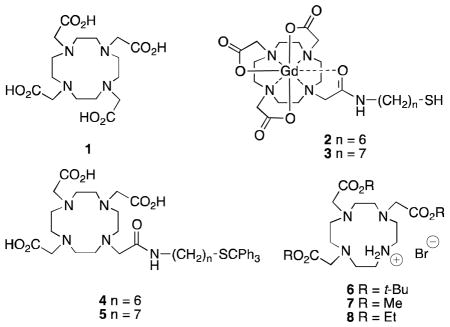
Alkylation of the hydrobromide salts of 1,4,7-tris(methoxycarbonylmethyl)-1,4,7,10-tetraazacyclododecane and 1,4,7-tris(ethoxycarbonylmethyl)-1,4,7,10-tetraazacyclododecane with appropriate α-bromoacetamides, followed by hydrolysis, provides convenient access to 10-(2-alkylamino-2-oxoethyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid derivatives that contain acid-sensitive functional groups. The utility of the method is demonstrated by improved syntheses of two known DOTA monoamides bearing acid-sensitive ω-tritylthio alkyl chains in much higher yields based on cyclen as the starting material.
Keywords: Cyclen, DOTA monoamide, DO3A monoamide, medical imaging, synthesis
INTRODUCTION
1,4,7,10-Tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA, 1) forms stable complexes with lanthanides and radiometals.1,2 As a result, DOTA-derived metal complexes are utilized as contrast agents in magnetic resonance imaging (MRI),3–5 in radiopharmaceuticals, and radionuclide imaging.6–8 In recent years, efforts to deliver biomolecules, such as proteins,9,10 peptides,11 and monoclonal antibodies,12,13 labeled with metal ions or radioactive metal ions to specific cellular targets for imaging and therapy have gained in importance. A most common approach to linking the metal ion to a biomolecule is by means of a modified DOTA chelate that has been rendered bifunctional.14 Many bifunctional derivatives of DOTA have been obtained by conversion of one of the pendant acid groups into an N-substituted acetamido derivative.15 The chelate strongly coordinates with the chosen metal ion, while a functional group incorporated in the nitrogen substituent of the acetamido group conjugates with the biomolecule. Several precursors to DOTA monoamides have been described,16–18 and some are commercially available. However, very little has been published relating to the synthesis of DOTA monoamides bearing acid-labile functional groups in the linker.
Previously we reported the synthesis of the gadolinium(III) DOTA monoamide complex 2 bearing a hexyl side chain that terminated with a thiol group.19 The complex 2 bound to human serum albumin (HSA) in a redox sensitive manner. More recently, we demonstrated the utility of 2 as an MRI biomarker of redox-active drugs in tumor-bearing mice.20 We also synthesized Gd-DOTA monoamide complex 3 bearing an ω-thioheptyl side chain that showed higher binding affinity for HSA as compared to 2.21 DOTA monoamides 4 and 5, intermediates in the published syntheses19,21 of complexes 2 and 3, respectively, were prepared by mono-activation of 1 with isobutyl chloroformate, followed by reaction with 6-tritylthiohexylamine and 7-tritylthioheptylamine, respectively, in good yields based on the amine starting materials. However, based on DOTA, the yields of 4 and 5 were 27% and 23%, respectively.22 Assuming a yield of 82% in the conversion of cyclen to DOTA,23 the yields of 4 and 5 based on cyclen were 22% and 19%, respectively. Existing procedures for the synthesis of DOTA monoamides that employ an excess of DOTA to avoid mixtures of products are not optimal, given the high cost of DOTA.24 Other shortcomings of some published methods are the use of high dilution conditions and long reaction times. These issues limit large-scale preparations of DOTA monoamides. We describe in this article an efficient and scalable synthesis that is amenable to preparations of a series of DOTA monoamides, including compounds with acid-labile functionality in the linker, such as 4 and 5.
RESULTS AND DISCUSSION
Of the methods reported for the preparation of DOTA monoamides,25 two are well suited for large-scale synthesis. The first involves monoalkylation of 1,4,7,10-tetraazacyclododecane (cyclen, 9) with an α-bromoacetamide, followed by incorporation of three acetate ester arms (methyl, ethyl, or tert-butyl) and their subsequent hydrolysis or cleavage to afford the DOTA monoamide. The second method involves the preparation of a DO3A tris-ester, such as the hydrobromides 6–8, followed by neutralization, alkylation with an α-bromoacetamide, and subsequent hydrolysis or cleavage to afford the DOTA monoamide. In either method, one can choose methyl, ethyl, or tert-butyl ester groups, depending on the acid- or base-sensitivity of moieties on the acetamide arm. In the first method, an excess of cyclen is generally used to promote monoalkylation. This method can work well for the synthesis of a single DOTA monoamide, but for the synthesis of a series of DOTA monoamides, will involve the preparation and characterization of many more intermediates than will the second method. Barge, et al.,26 prepared a set of DOTA monoamide derivatives from 6 by the second method. For our work, the necessary acidolysis of the t-butyl ester moieties undermines the application of this synthetic method. However, the use of 7 or 8 in place of 6 would substitute base hydrolysis for acidolysis and permit preparations of DOTA monoamides such as 4 and 5 possessing acid-labile functionality in the linker, possibly on large scales.
While commercially available 6 has been used in the synthesis of many bifunctional chelates,27,28 the related methyl and ethyl esters 729–32 and 833–36 have found limited use. Few methods for the synthesis of 7 and 8 have been published. Ester 7 was obtained as a free base in 54% yield by alkylation of 9 with four equivalents of methyl bromoacetate in refluxing methanol in the presence of triethylamine, followed by preparative TLC purification.29 Alternatively, 7 was obtained as a triflate salt by alkylation of N-formylcyclen37 with 3.3 equivalents of methyl bromoacetate, followed by removal of the formyl group.30 Ester 8 was reportedly isolated in 72% yield after reaction of cyclen with 2.24 equivalents of ethyl bromoacetate in dichloromethane without an additional base.38 More recently, Natrajan, et al.,39 prepared 8 in 63% yield as a hydrobromide salt by reaction of cyclen with 3.3 equivalents of ethyl bromoacetate in acetonitrile in the presence of sodium bicarbonate.
We described a synthesis of 6 as a hydrobromide salt in 79% yield by reaction of 9 with 3.3 equivalents of t-butyl bromoacetate and sodium acetate in dimethylacetamide at −20 °C.40 The work-up procedure involved adding the reaction mixture to water and isolation of the hydrobromide salt by addition of potassium bicarbonate. Following this procedure, 9 was allowed to react with three equivalents of methyl bromoacetate and sodium acetate in dimethylacetamide at −20 °C. However, the hydrobromide salt 7 could not be isolated from the aqueous solution upon addition of potassium bicarbonate, presumably because of its greater solubility in water. To avoid a water work-up, DMF was used as solvent instead of dimethylacetamide.
In a typical procedure, three equivalents of methyl bromoacetate in DMF were added to a stirred suspension of one equivalent of 9 and 3.3 equivalents of sodium acetate in DMF at −20 °C (Scheme 1). After stirring for six hours at room temperature, the reaction mixture was diluted with DCM and the salts removed by filtration. The filtrate was evaporated under reduced pressure and the crude oil was purified on a flash silica gel column eluted with dichloromethane and dichloromethane/5% methanol. The hydrobromide salt 7 was obtained as tan colored oil, which solidified to a white solid upon scratching in the presence of ether at −20 °C. The yield of 7 was 78%.
Scheme 1.

Synthesis of 7 and 8.
Recrystallization of 7 from toluene produced a shiny amorphous solid. However, slow evaporation of a toluene solution produced crystals that were suitable for X-ray crystallographic analysis. The hydrobromide salt 7 crystallized in space group P21/c with one molecule in the asymmetric unit and four molecules in the unit cell (Figure 1).41
Figure 1.

The structure of hydrobromide salt 7. A hydrogen bond interaction is present between N4-H and Br with a donor-acceptor distance of 3.277(4) Å. Displacement ellipsoids are at the 50% probability level.
Following a similar protocol, the hydrobromide salt 8 was synthesized in 77% yield by reaction of 9 with three equivalents of ethyl bromoacetate. With 7 and 8 in hand, we set about preparing 4 and 5 by an alternate synthetic route as described below.
Treatment of 1019 and 1121 with bromoacetyl bromide in DCM in the presence of 1N NaOH gave 12 and 13 in 92% and 94% yields, respectively (Scheme 2).35 While it was previously reported that alkylations of the hydrobromide salt of 6 gave poor yields, even in presence of K2CO3,26 we found that treatment of the hydrobromide salt 7 with K2CO3 in acetonitrile for one hour at 70 °C prior to the addition of bromoacetamide 12, followed by reaction at this temperature for one hour, produced 14 in 84% yield (Scheme 2). Reactions of 7 with 13, 8 with 12, and 8 with 13 under similar conditions gave 15, 16, and 17 in excellent yields. Finally, hydrolysis of esters 14 and 16 in refluxing 1N ethanolic KOH for 30 minutes gave DOTA monoamide 4 in 84% and 94% yields, respectively, while hydrolysis of esters 15 and 17 under these conditions gave the DOTA monoamide 5 in 84% and 81% yields, respectively.
Scheme 2.

Synthesis of DOTA monoamides 4 and 5
CONCLUSIONS
Efficient gram-scale syntheses of the hydrobromide salts 7 and 8 have been developed, and the procedures seem amenable to further scale–up. The first X-ray crystal structure of hydrobromide salt 7 has been obtained. Neutralization of the hydrobromide salts 7 and 8 with K2CO3 prior to addition of α-bromoacetamides 12 and 13 produced alkylation products 14–17 in excellent yields. Saponification of 14–17 gave 4 and 5 in high yields. Overall, the three-step reaction sequences starting from cyclen (9) produced DOTA monoamides 4 and 5 in 57–69% yields based on cyclen, about three-fold higher yields than the literature syntheses of these same compounds.19,21 Thus, hydrobromide salts 7 and 8 can be used as alternatives to 6 in the preparation of sets of DOTA monoamides, avoiding the synthesis and characterization of intermediates that would result from monoalkylation of 9 followed by incorporation of three acetate arms, or of single DOTA monoamide targets on large scales, and will be especially useful in cases where the DOTA monoamide derivatives contain acid sensitive moieties.
EXPERIMENTAL
[1,4,7-Tris(methoxycarbonylmethyl)]1,4,7,10-tetraazacyclododecane Hydrobromide (7)
To a suspension of cyclen (1.00 g, 5.81 mmol) and sodium acetate (1.57 g, 19.17 mmol) in DMF (8 mL) at −20 °C was added a solution of methyl bromoacetate (2.66 g, 1.70 mL, 17.43 mmol) in DMF (4 mL) dropwise over 10 min. The temperature was maintained at −20 °C during the addition, after which the reaction mixture was allowed to come to room temperature. After 6 h of vigorous stirring, the reaction mixture was diluted with CH2Cl2 (50 mL) and the salts removed by filtration. The filtrate was evaporated under reduced pressure and the resulting oil was loaded onto a flash silica gel column (100 g). Elution with CH2Cl2 (200 mL), followed by CH2Cl2/MeOH (95:5), gave a tan oil. Scratching of the oil under ether at −20 °C gave 7 (2.12 g, 4.53 mmol, 78%) as a white solid, mp 113–114 °C, Rf 0.42 (CH2Cl2/MeOH 5:1). 1H NMR (500 MHz, CDCl3) δ 2.85 (4, m), 2.91 (8, m), 3.10 (4, m), 3.42 (2, s), 3.48 (4, s), 3.69 (9, s), 9.97 (1, br s); 13C NMR (125 MHz, CDCl3) δ 47.3, 48.3, 49.3, 51.4, 51.6, 57.0, 170.6, 171.4. Anal. Calcd for C17H33BrN4O6: C, 43.50; H, 7.09; N, 11.94. Found: C, 43.25; H, 7.05; N, 12.01.
2-Bromo-N-(6-trityl-thiohexyl)acetamide (12)
To a well stirred, ice-cold mixture of amine 10 (5.34 g, 14 mmol) in CH2Cl2 (45 mL) and 1 N NaOH (45 mL, 45 mmol) was added bromoacetyl bromide (5.75 g, 2.5 mL, 28 mmol) dropwise. After 45 min, water (25 mL) was added, the layers were separated, and the aqueous layer extracted with CH2Cl2 (3 × 25 mL). The combined organic fractions were dried (MgSO4), filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel eluted with hexanes/EtOAc (6:4) to afford 12 (6.36 g, 12.8 mmol, 92%) as a white solid, mp 78–79 °C, Rf 0.58 (EtOAc/hexanes 1:1). 1H NMR (500 MHz, CDCl3) δ 1.16–1.22 (2, m), 1.23–1.29 (2, m), 1.38 (2, quintet, J = 7.5 Hz), 1.48 (2, quintet, J = 7.5 Hz), 2.13 (2, t, J = 7.5 Hz), 3.19–3.24 (2, m), 3.85 (2, s), 6.41 (1, s), 7.17–7.23 (3, m), 7.25–7.29 (6, m), 7.38–7.41 (6, m); 13C NMR (125 MHz, CDCl3) δ 26.3, 28.4, 28.5, 29.0, 29.3, 31.8, 40.1, 66.4, 126.5, 127.7, 129.5, 144.9, 165.0. Anal. Calcd for C27H30BrNOS: C, 65.31; H, 6.09; N, 2.82; S, 6.46. Found: C, 64.93; H, 6.48; N, 2.52; S, 6.84.
[1,4,7-Tris(methoxycarbonylmethyl)-10-[N-(6-trityl-thiohexyl)carbamoyl]-1,4,7,10-tetraazacyclo-dodecane (14)
To a stirred solution of 7 (0.94 g, 2 mmol) in dry CH3CN (20 mL) was added anhydrous K2CO3 (1.10 g, 8 mmol) and the mixture heated at 70 °C. After 1 h, heating was discontinued, bromoacetamide 12 (1.04 g, 2.1 mmol) was added, heating was resumed, and the reaction mixture stirred for an additional 1 h at 70 °C. The reaction mixture was cooled, filtered, and the filtrate evaporated to dryness under reduced pressure. The residue was purified by flash column chromatography on silica gel (100 g). Elution with MeOH/CH2Cl2 (98:2, 200 mL) followed by MeOH/CH2Cl2 (95:5) gave 14 (1.39 g, 1.74 mmol, 87%) as a white foam, Rf 0.52 (CH2Cl2/MeOH 5:1). 1H NMR (500 MHz, CDCl3) δ 1.14-1-26 (4, m), 1.34–1.40 (2, quintet, J = 7.5 Hz), 1.46–1.52 (2, quintet, J = 7.5 Hz), 2.15 (2, t, J = 7.5 Hz), 2.20–3.30 (24, m), 3.39 (2, s), 3.70 (6, s), 3.72 (3, m), 7.18–7.21 (3, m), 7.26–7.29 (6, m), 7.38–7.40 (6, m), 8.52 (1, m); 13C NMR (125 MHz, CDCl3) δ 26.6, 28.5, 28.6, 29.2, 31.9, 39.1, 52.1, 53.3, 55.0, 56.5, 66.2, 126.3, 127.6, 129.4, 144.9, 171.6, 173.4. HRMS (ESI+, m/z) calculated for C44H62N5O7S 804.43645, found 804.43563.
[1,4,7-Tris(carboxymethyl)-10-[N-(6-trityl-thiohexyl)carbamoyl]-1,4,7,10-tetraazacyclododecane (4)
The triester monoamide 14 (0.80 g, 1 mmol) was added to 1N KOH solution in EtOH/H2O (95:5, 20 mL) and the mixture heated to reflux. After 0.5 h, the solvent was removed under reduced pressure and the residue loaded onto a flash silica gel column. Elution with CH2Cl2/MeOH/aq. NH4OH (5:3:0.3) followed by removal of solvents gave a viscous oil which was dissolved in a minimal amount of water and freeze-dried. By this procedure, 4 (0.64 g, 0.84 mmol, 84%) was obtained as a white fluffy solid. The 1H NMR, 13C NMR, and mass spectra were in agreement with published procedure.19
Supplementary Material
Acknowledgments
This work was supported by grants R01-CA118359, P01-CA017094, and P30-CA023074 from the National Institutes of Health.
Footnotes
Supplementary materials are available for this article. Go to the publisher’s online edition of Synthetic Communications® for the following free supplemental resource(s): Full experimental and spectral details.
Supporting Information: Complete experimental details, 1H and 13C NMR spectra of compounds 7, 8, 12–17, and an X-ray crystallographic report for compound 7 can be found via the “Supplementary Content” section of this article’s webpage.
REFERENCES AND NOTES
- 1.Lauffer RB. Chem Rev. 1987;87:901–927. [Google Scholar]
- 2.Volkert WA, Hoffman TJ. Chem Rev. 1999;99:2269–2292. doi: 10.1021/cr9804386. [DOI] [PubMed] [Google Scholar]
- 3.Caravan P. Chem Soc Rev. 2006;35:512. doi: 10.1039/b510982p. [DOI] [PubMed] [Google Scholar]
- 4.Aime S, Botta M, Terreno E. Adv Inorg Chem. 2005;57:173. [Google Scholar]
- 5.Merbach AE, Tóth É, editors. The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging. Wiley; Chichester: 2001. [Google Scholar]
- 6.Sarko D, Eisenhut M, Haberkorn U, Mier W. Current Med Chem. 2012;19:2667–2688. doi: 10.2174/092986712800609751. [DOI] [PubMed] [Google Scholar]
- 7.Fowler JC, Solanki CA, Barber RW, Ballinger JR, Peters AM. Acta Oncologica. 2007;46:105–110. doi: 10.1080/02841860600635854. [DOI] [PubMed] [Google Scholar]
- 8.Al-Nahhas A, Win Z, Szyszko T, Singh A, Nanni C, Fanti S, Rubello D. Anticancer Res. 2007;27:4087–4094. [PubMed] [Google Scholar]
- 9.Hao G, Fukumura T, Nakao R, Suzuki H, Szelecsényi F, KovácsSuzuki K. Appl Radiat Isotop. 2009;67:511–515. doi: 10.1016/j.apradiso.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 10.Li C, Winnard PT, Jr, Takagi T, Artemov D, Bhujwalla ZM. J Am Chem Soc. 2006;128:15072–15073. doi: 10.1021/ja066199i. [DOI] [PubMed] [Google Scholar]
- 11.Lee S, Xie J, Chen X. Chem Rev. 2010;110:3087–3111. doi: 10.1021/cr900361p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lewis MR, Kao JY, Anderson AJ, Shively JE, Raubitschek AA. Bioconjugate Chem. 2001;12:320–324. doi: 10.1021/bc0000886. [DOI] [PubMed] [Google Scholar]
- 13.Li L, Tsai SW, Anderson AL, Keire DA, Raubitschek AA, Shively JE. Bioconjugate Chem. 2002;13:110–115. doi: 10.1021/bc015535b. [DOI] [PubMed] [Google Scholar]
- 14.De León-Rodriguez LM, Kovacs Z. Bioconjugate Chem. 2008;19:391–402. doi: 10.1021/bc700328s. [DOI] [PubMed] [Google Scholar]
- 15.There is some ambiguity in the literature as to whether compounds such as4and5should be termed “DOTA monoamides” or “DO3A monoamides”. CAS names such compounds as 10-(2-alkylamino-2-oxoethyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid derivatives, which is inconsistent with both of these common usages. We employ “DOTA monoamide” in this article since this usage is consistent with use of the term “DOTA tetraamide” to indicate all four acetate arms of the DOTA are present as amides.
- 16.Heppeler A, Froidevaux S, Macke HR, Jermann E, Behe M, Powell P, Hennig M. Chem Eur J. 1999;5:1974–1981. [Google Scholar]
- 17.Anelli PL, Lattuada L, Gabellini M, Recanati P. Bioconjugate Chem. 2001;12:1081–1084. doi: 10.1021/bc010046x. [DOI] [PubMed] [Google Scholar]
- 18.Li C, Winnard P, Bhujwalla ZM. Tetrahedron Lett. 2009;50:2929–2931. doi: 10.1016/j.tetlet.2009.03.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raghunand N, Jagadish B, Trouard TP, Galons JP, Gillies RJ, Mash EA. Magn Reson Med. 2006;55:1272–1280. doi: 10.1002/mrm.20904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guntle GP, Jagadish B, Mash EA, Powis G, Dorr RT, Raghunand N. Trans Oncol. 2012;5:190–199. doi: 10.1593/tlo.11322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jagadish B, Guntle GP, Zhao D, Gokhale V, Ozumerzifon TJ, Ahad AM, Mash EA, Raghunand N. J Med Chem. 2012;55:10378–10386. doi: 10.1021/jm300736f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.A structurally related DOTA monoamide bearing a β-tritylthioethyl group was prepared by reacting DOTA with β-tritylthioethylamine in 23% yield. See Wängler C, Wängler B, Eisenhut M, Haberkorn U, Mier W. Bioorg Med Chem. 2008;16:2606–2616. doi: 10.1016/j.bmc.2007.11.044.
- 23.Desreux JF. Inorg Chem. 1980;19:1319–1324. [Google Scholar]
- 24.List price for 25 grams of DOTA is $2470 from Macrocyclics, Dallas, TX.
- 25.Frullano L, Caravan P. Curr Org Synth. 2011;8:535–565. doi: 10.2174/157017911796117250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barge A, Tei L, Upadhyaya D, Fedeli F, Beltrami L, Stefania R, Aime S, Cravotto G. Org Biomol Chem. 2008;6:1176–1184. doi: 10.1039/b715844k. [DOI] [PubMed] [Google Scholar]
- 27.Suchy M, Hudson RHE. Eur J Org Chem. 2008;29:4847–4865. [Google Scholar]
- 28.Lattuada L, Barge A, Cravotto G, Giovenzana GB, Tei L. Chem Soc Rev. 2011;40:3019–3049. doi: 10.1039/c0cs00199f. [DOI] [PubMed] [Google Scholar]
- 29.Kline SJ, Betebenner DA, Johnson DK. Bioconjugate Chem. 1991;2:26–31. doi: 10.1021/bc00007a005. [DOI] [PubMed] [Google Scholar]
- 30.Huang Z, Sengar RS, Nigam A, Abadjian MC, Potter DM, Grotjahn DB, Wiener EC. Invest Radiol. 2010;45:641–654. doi: 10.1097/RLI.0b013e3181ee6e06. [DOI] [PubMed] [Google Scholar]
- 31.Giardiello M, Botta M, Lowe MPJ. Incl Phenom Macrocycl Chem. 2011;71:435–444. [Google Scholar]
- 32.Ansari MH, Ahmad M, Dicke KA. Bioorg Med Chem Lett. 1993;3:1071–1072. [Google Scholar]
- 33.Chauvin T, Torres S, Rosetto R, Kotek J, Badet B, Durand P, Tóth É. Chem Eur J. 2012;18:1408–1418. doi: 10.1002/chem.201101779. [DOI] [PubMed] [Google Scholar]
- 34.Kiviniemi A, Makela J, Makila J, Saanikoji T, Liljenback H, Poijarvi-Virta P, Lonnberg H, Laitala-Leinonen T, Roivainen A, Virta P. Biconjugate Chem. 2012;23:1981–1988. doi: 10.1021/bc300253t. [DOI] [PubMed] [Google Scholar]
- 35.Othman M, Desmaele D, Couvreur P, Elst LV, Laurent S, Muller RN, Bourgaux C, Morvan E, Pouget T, Lepetre-Mouelhi S, Durand P, Gref R. Org Bimol Chem. 2011;9:4367–4386. doi: 10.1039/c1ob00015b. [DOI] [PubMed] [Google Scholar]
- 36.Battistini E, Gianolio E, Gref R, Couvreur P, Fuzerova S, Othman M, Aime S, Badet B, Durand P. Chem Eur J. 2008;14:4551–4561. doi: 10.1002/chem.200701587. [DOI] [PubMed] [Google Scholar]
- 37.Dischino DD, Delaney EJ, Emswiler JE, Gaughan GT, Prasad JS, Srivastava SK, Tweedle MF. Inorg Chem. 1991;30:1265–1269. [Google Scholar]
- 38.Mishra AK, Draillard K, Faivre-Chauvet A, Gestin JF, Curtet C, Chatal JF. New J Chem. 2001;25:336–339. [Google Scholar]
- 39.Natrajan LS, Villaraza AJL, Kenwright AM, Faulkner S. Chem Commun. 2009:6020–6022. doi: 10.1039/b913702e. [DOI] [PubMed] [Google Scholar]
- 40.Jagadish B, Bricket-Albrecht GL, Nichol GS, Mash EA, Raghunand N. Tetrahedron Lett. 2011;52:2058–2061. doi: 10.1016/j.tetlet.2010.10.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crystal data for hydrobromide salt7: C17H33N4O6Br, Mr = 469.38 g mol−1; colorless block 0.46 × 0.17 × 0.14 mm3; T = 100.31 K. MoKα, radiation (0.71073 Å); space group P21/c, unit cell parameters a = 6.6414(12) Å, b = 6.6414(12) Å, c = 26.354(5) Å, V = 2445.6(7) Å3; 24,826 measured reflections., 8025 unique reflections; final R1 = 0.0383 (F2 > 2σ), wR2 = 0.0822 (all data).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.