

The detailed characterization of endogenous proteins and use of non-natural amino acid engineering allows the identification and structural and functional analysis of a post-translational modification in regulating ligand binding and enzyme activity.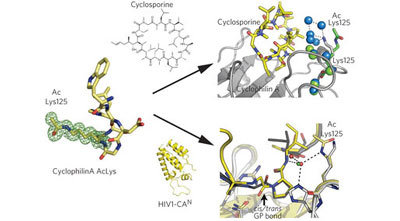
Supplementary information
The online version of this article (doi:10.1038/nchembio.342) contains supplementary material, which is available to authorized users.
Subject terms: HIV infections, Immunosuppression, Acetylation, Structural biology
Abstract
Cyclophilin A (CypA) is a ubiquitous cis–trans prolyl isomerase with key roles in immunity and viral infection. CypA suppresses T-cell activation through cyclosporine complexation and is required for effective HIV-1 replication in host cells. We show that CypA is acetylated in diverse human cell lines and use a synthetically evolved acetyllysyl-tRNA synthetase/tRNACUA pair to produce recombinant acetylated CypA in Escherichia coli. We determined atomic-resolution structures of acetylated CypA and its complexes with cyclosporine and HIV-1 capsid. Acetylation markedly inhibited CypA catalysis of cis to trans isomerization and stabilized cis rather than trans forms of the HIV-1 capsid. Furthermore, CypA acetylation antagonized the immunosuppressive effects of cyclosporine by inhibiting the sequential steps of cyclosporine binding and calcineurin inhibition. Our results reveal that acetylation regulates key functions of CypA in immunity and viral infection and provide a general set of mechanisms by which acetylation modulates interactions to regulate cell function.
Supplementary information
The online version of this article (doi:10.1038/nchembio.342) contains supplementary material, which is available to authorized users.
Main
Cyclophilins, also known as immunophilins, are peptidyl-prolyl cis–trans isomerases found in all kingdoms of life and all tissues. Cyclophilin A (CypA), the archetypal cyclophilin, is central to protein folding1, signal transduction2, trafficking3, receptor assembly4, cell cycle regulation5 and stress response and has an expanding catalog of roles6. Two of CypA's most important roles in human health are in controlling immunosuppression and viral infection. CypA is the target of the widely used immunosuppressive cyclosporine (1). The complex formed by CypA and cyclosporine binds to and allosterically inhibits calcineurin, a protein phosphatase, leading to suppression of the T-cell mediated immune response by means of an IL-2–mediated downregulation of T-cell activation7. CypA is also an essential host protein in the efficient replication of several viruses, including vesicular stomatitis virus5, severe acute respiratory syndrome (SARS) virus6, hepatitis C virus (HCV)8, vaccinia virus and HIV type 1 (refs. 9,10). During HIV-1 infection, CypA interacts with the capsid protein gag and is packaged into budding HIV-1 virions9,10. Inhibition of CypA by cyclosporine substantially decreases viral titer in human cells11. The importance of CypA in lentiviral replication is underscored by the independent evolution in two different primate species of antiviral restriction factors that use a retrotransposed copy of CypA to provide viral targeting12. Despite the importance of CypA in immunosuppression, viral infection and other key cellular processes, the molecular mechanisms by which the varied and crucial functions of CypA are regulated remains unclear.
Acetylation of the ϵ-amine of specific lysine residues in proteins is a reversible post-translational modification with diverse roles and a functional importance that rivals that of phosphorylation13. Acetylation is mediated by acetyl-CoA–dependent histone acetyltransferases14 and reversed by zinc-dependent histone deacetylases or NAD dependent sirtuins15. Acetylated targets can be specifically recognized by bromodomain-containing proteins16. Recent mass spectrometry and immunofluorescence studies demonstrate that hundreds of nonhistone proteins are acetylated in mammalian cells17; however, the molecular mechanisms by which acetylation may control protein function and effect cellular regulation are largely unknown.
A recent proteomics screen isolated a peptide whose sequence matched CypA but which contained an Nϵ-acetyl-L-lysine (acetyllysine, 2) in place of Lys125 (ref. 17). However, because there are many CypA gene fusions in the genome, the origin of this peptide was ambiguous. Here we show that the free enzyme form of CypA is acetylated in human cells. We produced homogeneously and site-specifically acetylated recombinant CypA using an acetyllysyl-tRNA synthetase/tRNACUA pair that co-translationally directs the incorporation of acetyllysine in response to an amber codon18 placed in a CypA gene. This approach allowed us to perform structural and biophysical measurements on acetylated CypA for the first time. These results reveal how acetylation modulates key functions of CypA, including suppressing its catalytic activity, cyclosporine binding and calcineurin inhibition and altering its recognition of the HIV-1 capsid. Furthermore, the molecular details of these effects establish a general set of mechanisms by which acetylation can regulate protein activity.
Results
CypA is acetylated in human cells
To investigate whether CypA is acetylated in human cells, we immunoprecipitated endogenous CypA from HeLa and Jurkat T cells using a CypA-specific antibody. Detection by western blot using anti-CypA demonstrated that endogenous CypA was present in the cell extract (Fig. 1a; see Supplementary Fig. 1 for full-length gels and blots). Detection using an antibody to acetyllysine showed that this endogenous CypA was acetylated in both HeLa and Jurkat T cells (Fig. 1b). The anti-acetyllysine did not detect the unacetylated control (Fig. 1b). These data demonstrate that CypA is acetylated in human cells—including T cells, in which CypA mediates its effects on immunosuppression and HIV-1 infection.
Figure 1. Homogenous and site-specific incorporation of acetyllysine into CypA.
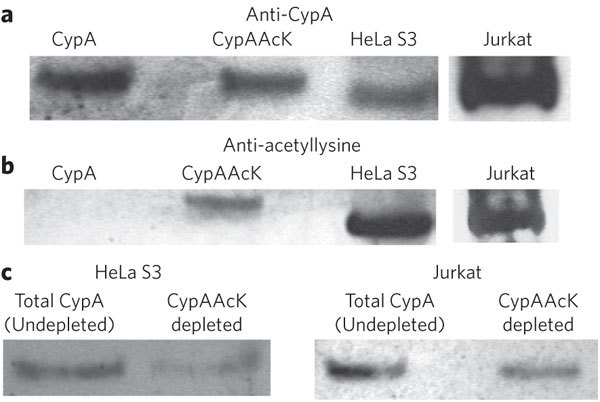
(a,b) CypA can be immunoprecipitated from HeLa and Jurkat T cells (a) and the acetylated fraction detected by anti-acetyllysine in both cell types (b). Recombinant CypA and CypAAcK represent controls. (c) Quantification of acetylated CypA in HeLa and Jurkat cells using an immunodepletion approach. Endogenous CypA from cell lysate was depleted with anti-acetyllysine and compared to undepleted material by western blot with anti-CypA. A substantial proportion of CypA is acetylated in both HeLa and Jurkat cells. See Supplementary Figure 1 for full-length gels and blots.
To determine the proportion of endogenous CypA that was acetylated in each cell type, we performed an immunodepletion experiment. Acetylated CypA was depleted from lysate using anti-acetyllysine bound to protein A–Sepharose. We compared the depleted material, by western blot with anti-CypA, to undepleted material processed in the absence of the anti-acetyllysine (Fig. 1c). To obtain a quantitative estimate of the proportion of acetylated CypA, we performed a standardization experiment with varying ratios of acetylated:nonacetylated recombinant protein (Supplementary Fig. 2). Using this standardization, we estimated that 50% ± 5% (s.d.) of CypA was acetylated in HeLa cells and 38% ± 3% in Jurkat T cells. These results indicated that a substantial proportion of CypA was acetylated in both HeLa and Jurkat T cells. Although we expect the proportion of acetylated protein to be regulated in response to stimuli, these results demonstrate that the effects of acetylation on CypA are relevant to, and important for, understanding CypA-mediated processes.
Expression of homogenously Lys125-acetylated CypA
To determine the consequences of Lys125 acetylation on CypA structure and function, we needed a way to synthesize large amounts of site-specifically, homogeneously acetylated protein. We produced recombinant acetylated CypA (CypAAcK) from E. coli using an evolved orthogonal aminoacyl-tRNA synthetase/tRNACUA pair that cotranslationally directs the incorporation of exogenously added acetyllysine into recombinant proteins in response to amber codons placed in the genes encoding them18,19. The CypA gene bearing an amber codon in place of the Lys125 codon was expressed from a T7 promoter in cells bearing acetyllysyl-tRNA synthetase and the corresponding amber suppressor tRNA. The cells were supplemented with 10 mM acetyllysine and 20 mM nicotinamide and protein expression induced at log phase. Recombinant CypAAcK was purified at >10 mg per liter of culture in the presence of 20 mM nicotinamide to inhibit cobB, an NAD-dependent deacetylase present in E. coli. The presence of the acetyl group was confirmed by anti-acetyllysine western blot (Supplementary Fig. 3a), and the quantitative incorporation of acetyllysine was further demonstrated by ESI-MS (Supplementary Fig. 3b).
Atomic structure of acetylated CypA
Recombinant CypA is most stable in basic buffers between pH 8.0 and 8.5; however, the acetylated CypA rapidly precipitates under these conditions. By screening a range of pH and salt conditions using absorbance at 320 nm as a measure of aggregation, we found that the stability of acetylated CypA increased markedly in acidic buffers (pH 5.4–6.4). Under mildly acidic conditions, acetylated CypA was still soluble at concentrations around 3 mM. This suggested that Lys125 had a prominent role in surface electrostatics and that its acetylation had a substantial impact on CypA behavior. To assess the effect of acetylation on the physicochemistry of CypA, we determined the native crystal structure. Acetylated CypA crystallized in two different forms, one in an orthorhombic space group and one in a trigonal space group. We determined the structure of both forms to assess crystal packing effects. Crystal contacts are provided in Supplementary Table 1. The structures were refined to 1.2 Å and 1.4 Å, respectively, with two copies in the asymmetric unit in the case of the trigonal crystal and a single copy in the case of the orthorhombic crystal. These are, to our knowledge, the first structures containing a specific, biologically relevant post-translational lysine acetylation. In both CypA structures there is clear density for the acetyllysine side chain, as evidenced by the omit maps (Fig. 2a,b). This further demonstrates the homogeneity of acetyllysine incorporation and shows that the acetyllysine has a well-defined conformation. Finally, the two structures and the two chains from the trigonal crystal superpose closely, with only minimal differences in the extreme N-terminal residues (Fig. 2c).
Figure 2. Structure of acetylated CypA.
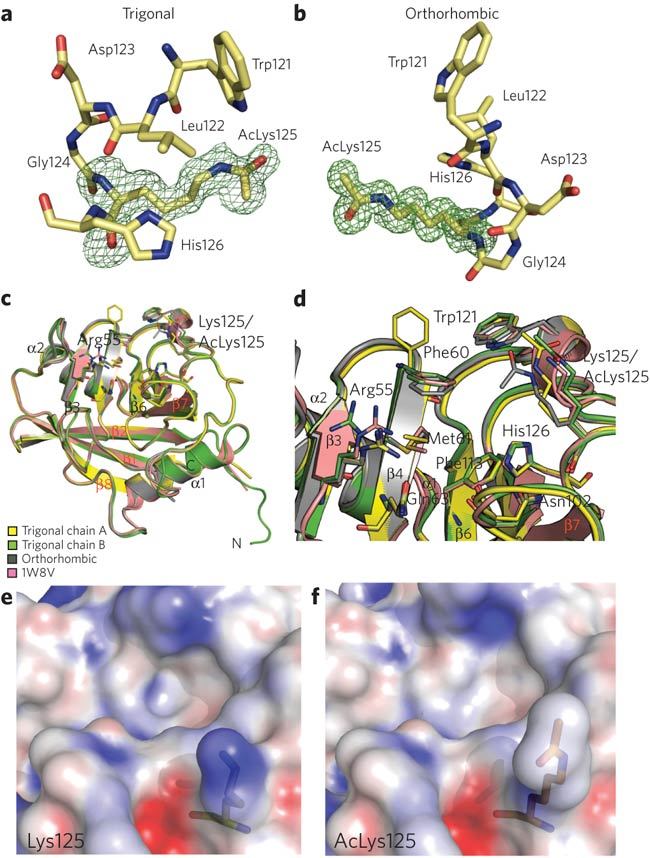
(a,b) Omit maps of the acetyllysine density for the trigonal (a) and orthorhombic structures (b) contoured at 1σ. (c,d) Comparison of unacetylated (PDB 1W8V) and acetylated CypA structures. There are no significant conformational changes associated with incorporation of the acetyl group. (e,f) Electrostatic surface potential of the active site around Lys125. Blue represents a positive charge and red negative. Scaled from –20 to 20 kT e−1.
The secondary structure of the acetylated CypA superposes closely with the known structure of native cyclophilin, showing that there are no large conformational changes induced by acetylation (Fig. 2c,d). Comparison of the active sites reveals differences in only three residues: Phe60, Arg55 and Lys125. The Phe60 side chain of trigonal chain A is rotated 90° compared to the other structures. This is due to crystal contacts between Phe60 and its symmetry-related copy. Phe60, together with Met61 and Phe113, form a hydrophobic core at the base of the substrate binding pocket. Arg55 is a key catalytic residue, facilitating catalysis through stabilization of the proline in the transition state20. Differences in the side chain orientation of Arg55 in the acetylated and unacetylated structures are likely to result from the inherent flexibility required in this region to accommodate diverse CypA substrates. Acetylation of CypA does not therefore seem to distort its conformation or substantially rearrange the active site. However, acetylation does markedly affect its surface electrostatics. Calculation of the surface electrostatic potential using the Adaptive Poisson-Boltzmann Solver (APBS) shows that Lys125 provides the strongest patch of positive charge in the active site (Fig. 2e,f) and that this charge is largely neutralized by the acetyl group. Such a marked shift in electrostatics correlates with our observation that acetylation switches CypA from requiring a basic to an acidic environment.
Acetylation inhibits cyclosporine binding
The formation of the CypA–cyclosporine complex is a prerequisite for realizing the immunosuppressive effects of cyclosporine. To investigate the effect of CypA acetylation on cyclosporine binding, we compared the steady-state affinity of cyclosporine to acetylated and unacetylated CypA by following the fluorescence enhancement of the single CypA tryptophan residue that occurs upon complex formation. Using this method, we measured a CypA–cyclosporine dissociation constant of 1 ± 1.2 nM, which is within the previously reported range (1–205 nM)21. Measurements of acetylated CypA binding to cyclosporine, however, showed that acetylation reduced the affinity of CypA for cyclosporine 20-fold. to 19 ± 2 nM (Supplementary Fig. 4a,b). We confirmed the effect of acetylation on cyclosporine binding by enzyme inhibition. Cyclosporine gave an inhibition constant of 1.3 ± 0.2 nM for CypA and an inhibition constant of 27 ± 5.6 nM for acetylated CypA (Supplementary Fig. 5).
To determine the molecular basis of the affinity change, we solved the structure of cyclosporine in complex with acetylated CypA. The complex crystallized in a hexagonal space group with five molecules in the asymmetric unit. The acetylated complex superposed closely with the previously solved unacetylated structure; however, comparison of the electrostatics showed that the changes in charge mediated by the acetyl group directly altered the environment of the bound substrate (Fig. 3a,b). A principal effect of this change was to disrupt the water network between CypA and cyclosporine. In the unacetylated complex, Lys125 mediates a shell of waters at the interface between CypA and cyclosporine (Fig. 3c). Acetylation of Lys125 releases the network of five water molecules ordered by the ϵ-amino group of this lysine (Fig. 3c). This solvent remodeling may explain the effect of acetylation on the thermodynamics of cyclosporine binding and illustrates how acetylation may control protein–small molecule interactions without eliciting rearrangement of structure or inducing allosteric changes.
Figure 3. Structural comparison of the unacetylated and acetylated CypA–cyclosporine complexes.
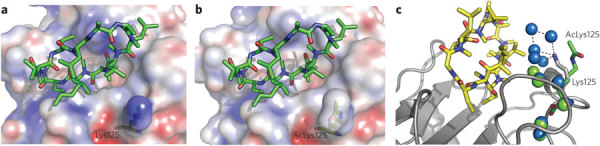
(a,b) The binding site of the unacetylated complex (a) and acetylated complex (b) from the superposed structures, with the cyclosporine in green and CypA as a molecular surface upon which the electrostatic surface potential has been mapped. Acetylation alters the active site charge. (c) The role of water molecules at the CypA–cyclosporine interface. Cyclosporine is in yellow, CypA and Lys125 in gray and the acetyllysine side chain in green. Waters belonging to the unacetylated complex are in, blue; waters belonging to the acetylated complex are in green. Acetylation results in loss of much of the water network at the interface. The previously solved structure PDB 2CPL38 was used in the comparison.
Acetylation modulates calcineurin inhibition
The CypA–cyclosporine complex is a potent inhibitor of the phosphatase calcineurin, and the immunosuppressive effects of cyclosporine are a direct result of this inhibition. To examine the effect of acetylation on the ability of the CypA–cyclosporine complex to inhibit calcineurin, we assayed the calcineurin-mediated release of phosphate from the RII-phosphopeptide22 as a function of concentration of CypA–cyclosporine. When cyclosporine was in excess and equal amounts of CypA–cyclosporine were formed with the acetylated and unacetylated cyclophilin, we found an inhibition constant (Ki) for the unacetylated complex of 497 nM, which is similar to the inhibition constants previously measured for this interaction22. Acetylation reduced inhibition of calcineurin by twofold, to 972 nM (Supplementary Fig. 6). The potency of cyclosporine immunosuppression is determined by the affinity of cyclosporine for CypA and the efficiency with which the CypA–cyclosporine complex inhibits calcineurin. As we found that acetylated CypA bound cyclosporine 20-fold more weakly than the unacetylated CypA, and as the acetylated CypA–cyclosporine complex mediates a twofold decrease in calcineurin inhibition, the total effect of acetylation on CypA-mediated immunosuppression may be multiplicatively larger than either of these individual effects. However, the potent immunosuppression mediated by cyclosporine in humans suggests that the acetylation levels in T cells in vivo or acetylation itself is not sufficient to completely abolish calcineurin inhibition in these cells.
Acetylation inhibits CypA catalysis
To determine how acetylation alters the enzymatic activity of CypA, we used a modified form of the linked chymotrypsin assay23 to measure the kcat/KM of cis to trans isomerization. In this assay, cleavage of a nitroanilide peptide substrate by chymotrypsin was detected by measuring the absorbance of liberated 4-nitroaniline. The peptide is also a substrate for CypA and contains a proline residue that at equilibrium is approximately 90% in trans and 10% in cis. The trans form of the peptide is readily cleaved by chymotrypsin, whereas the cis form can only be cleaved after its isomerization by CypA. Previous studies have used steady-state methods to determine the kcat/KM of CypA isomerization; however, this results in loss of data during mixing of the reaction components. Moreover, this approach cannot differentiate the two kinetic events and therefore does not take into account any contribution of the fast cleavage of trans peptide to the apparent rate of isomerization. We used a stopped-flow spectrophotometer, which allows pre–steady state measurement (owing to rapid mixing, <1 ms) and the resolution of separate kinetic events. Using the stopped-flow method, we observed two discrete phases in 4-nitroaniline production, corresponding to a fast cleavage step and a slower isomerization step (Fig. 4a). The molecular processes underpinning the fast and slow steps are confirmed by their relative amplitudes, which correspond closely to the expected cis–trans distribution of the substrate such that the slower, isomerization step represents ∼10% of the total amplitude change. By fitting each experiment to a double exponential, we obtained an accurate measurement of the apparent rate of cis to trans isomerization at different CypA concentrations. We then calculated the kcat/KM by fitting the increase in apparent rate (kobs) with increasing CypA concentrations to kobs = (kcat/KM)[CypA]. Comparison of CypA to acetylated CypA showed that acetylation decreased kcat/KM by 35-fold from 1 × 107 M−1 s−1 to 3 × 105 M−1 s−1 (Fig. 4b).
Figure 4. Acetylation decreases CypA-catalyzed cis to trans isomerization.
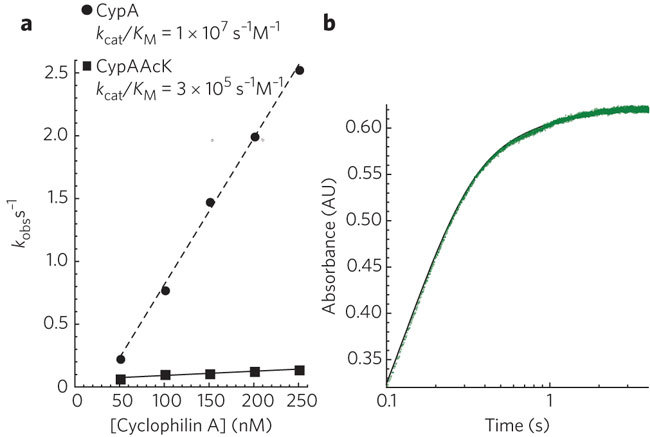
(a) Cleavage of a nitroanilide peptide substrate for both chymotrypsin and CypA results in two kinetic events corresponding to cleavage of trans substrate and cis–trans isomerization. (b) These events are fit to a double exponential to determine the apparent relaxation rate kobs. Plotting either CypA or acetylated CypA concentration against kobs and fitting to kobs = (kcat/KM)[CypA] gives kcat/KM.
Acetylation alters HIV-1 capsid interaction
CypA is an important host cofactor in HIV-1 replication. Inhibition of the CypA–HIV-1 capsid interaction by cyclosporine or mutation of the CypA-binding loop reduces infectivity10,11,24,25. NMR measurements demonstrate that the Gly89–Pro90 peptide bond in HIV-1 capsid is isomerized by CypA, and it has been proposed that CypA may catalyze the disassembly of viral capsid26. To investigate how acetylation affects the thermodynamics of CypA–capsid interaction, we performed isothermal titration calorimetry (ITC). We found that the acetylated and unacetylated forms of CypA bound HIV-1 capsid with similar affinities. However, acetylation altered the thermodynamics of interaction (Table 1). The interaction of unacetylated CypA and HIV-1-CAN (N-terminal capsid domain) was mainly enthalpy driven (−7.4 kcal mol−1); the entropic contribution was only marginal (0.6 kcal mol−1). Acetylation substantially reduced enthalpic favorability (by 2.8 kcal mol−1), but an increase in entropy (by 1.8 kcal mol−1) compensated for this.
Table 1.
Effect of acetylation on CypA interaction with HIV-1 capsid
| Protein | KD (μM) | ΔH (kcal mol−1) | TΔS (kcal mol−1) | N |
|---|---|---|---|---|
| CypAWT | 7.5 ± 0.8 | –7.4 ± 0.2 | 0.6 | 0.9 ± 0.01 |
| CypAAcK | 6.1 ± 0.9 | –4.6 ± 0.1 | 2.4 | 0.9 ± 0.02 |
| CypA-K125Q | 10 ± 0.7 | –6.1 ± 0.1 | 0.6 | 0.8 + 0.01 |
Isothermal titration calorimetry was used to determine the thermodynamics of interaction of HIV-1 N-terminal capsid domain with unacetylated CypA, acetylated CypA and acetylation-mimic mutant K125Q. Acetylation caused no significant change to the affinity (KD) but altered the entropy (TΔS) and enthalpy (ΔH) components, indicating that it altered the mechanism of binding. The K125Q data indicate that glutamine was not a good mimic of acetyllysine. N is the stoichiometry.
The replacement of lysine by glutamine has often been used to mimic acetyllysine and the effects of acetylation. We used ITC characterization of capsid binding to determine how effective glutamine is as a substitute. The affinity of the glutamine mutant K125Q was lower than that of the acetylated CypA (Table 1). Furthermore the thermodynamics of K125Q binding to capsid was similar to that of unacetylated CypA. This indicated that glutamine is not a good mimic of acetyllysine in this case.
Because enthalpy–entropy compensation can indicate a change in binding mechanism, we investigated the effects of CypA acetylation on capsid recognition by determining the structure of acetylated CypA:HIV-1 M group CAN at a resolution of 1.95 Å. We compared this structure to a previously solved unacetylated complex27 to reveal the molecular differences in the complexes that result from acetylation of CypA. The acetylated and unacetylated CypA:HIV1-CAN complexes superposed closely but with minor changes in the orientation of the capsid scaffold with respect to the CypA-binding-loop (Fig. 5a). The main difference between the two structures was that whereas both copies of the capsid in the unacetylated structure were bound with their Gly89–Pro90 peptide bond in a trans conformation, both copies in the acetylated structure were bound with this bond in a cis conformation (Fig. 5b,c). This change in HIV-1 capsid binding from trans to cis is a direct result of acetylation of Lys125. In the unacetylated complex, the trans Gly89 peptide oxygen is orientated toward the positive electrostatic patch provided by Lys125 and His126; in the acetylated complex, the oxygen is orientated away.
Figure 5. Structural comparison of unacetylated and acetylated CypA–HIV-1 complexes.
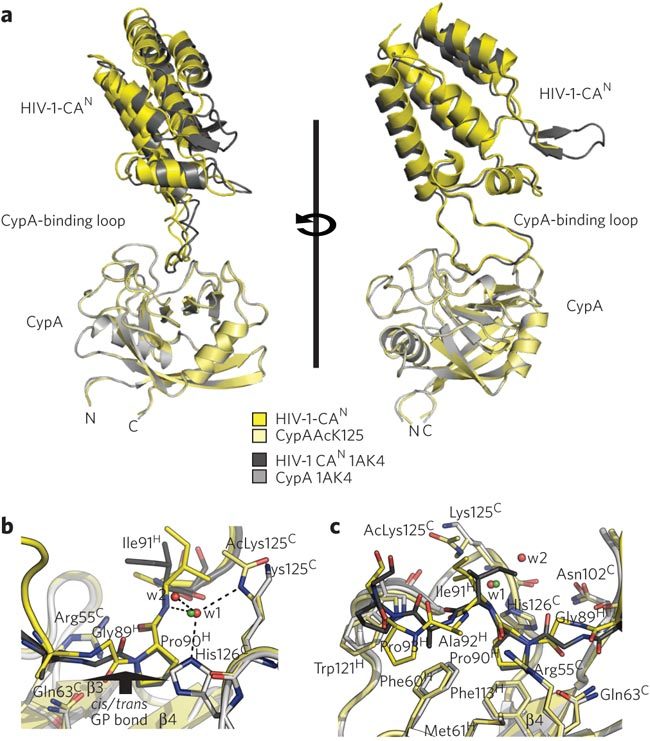
(a) Ribbon representation of the superposed complexes in two views. (b,c) Interaction between CypA active site residues and the capsid, showing the isomerized Gly89–Pro90 cis-trans peptidyl-prolyl bond. b and c show different parts of the HIV-Cyp interaction site. Acetylation alters both selectivity for this bond and the conformation of capsid bound by CypA. The acetylated complex is in yellow; the unacetylated complex is in gray. Red spheres represent waters from the acetylated structure; green spheres are from the unacetylated structure. Water w1 is conserved in the two complexes. Superscript H and C indicate HIV residues and Cyp residues, respectively.
This altered substrate binding correlates with our solution data on the effect of acetylation on CypA catalysis. Enzymatic characterization showed that acetylation inhibited isomerization and reduced kcat/KM by 35-fold (Fig. 4). Altered HIV-1 binding—namely, the switch from trans to cis proline in the HIV-1 complex—is consistent with this reduced kcat/KM, which is a measure of cis to trans isomerization. In addition to switching cis/trans preference through charge neutralization, the acetyllysine also stabilized the cis conformation of HIV-1 capsid by making new hydrogen bond and hydrophobic interactions. The methyl group of the acetyl moiety formed a new hydrophobic interaction with an alternative side chain conformer of Ile91. This interaction resulted in main-chain changes that were propagated to C-terminal residues Ala92 and Pro93, altering their interactions in turn. In the unacetylated complex, Pro93 forms a hydrophobic stacking interaction with CypA residue Trp121. The rearrangement in the position of Pro93 caused by acetylation led to an alternative stacking interaction with the aromatic ring of conserved CypA residue Phe60. Finally, the acetyllysine mediated a new water network involving the side chain of highly conserved His126, the main-chain amide of Ile91 and two water molecules (Fig. 5b). Acetylated CypA therefore stabilizes the HIV-1 capsid in its cis form by engaging residues C-terminal to the isomerized Pro90 in a different conformation. This correlates with the proposed catalytic pathway for nitroanilide peptide substrates, in which the position of residues C-terminal to the isomerized proline alters according to whether the peptide binds in cis or trans28.
Discussion
We have shown that CypA is substantially post-translationally acetylated in human cells and that this acetylation modulates key CypA functions, including cyclosporine binding, calcineurin inhibition and HIV-1 capsid interaction. We have solved to our knowledge the first high-resolution crystal structures of an acetylated protein, both in its free form and in complex with small molecules and protein ligands. These structures and accompanying biochemical and biophysical studies reveal that CypA acetylation antagonizes sequential binding and catalytic activities in the pathway that regulates the immunosuppressive effects of cyclosporine, decreases the catalytic efficiency of CypA cis to trans isomerization 35-fold and switches the binding of CypA from trans to cis HIV-1 capsid.
Acetylation represents a previously unexamined aspect of CypA biology. Identifying the acetyltransferases and deacetylases that modulate CypA acetylation levels will be important in exploring how CypA acetylation modulates function in response to stimuli such as immune stimulation. CypA is widely expressed, yet different tissues show different CypA phenotypes. For instance, cyclosporine and its derivatives are potent inhibitors and widely used immunosuppressives, but their off-target effects are limited. A study on calcineurin inhibition by cyclosporine in different tissues found wide variation in potency that was not always explicable by variation in CypA levels29. Furthermore, HIV-1 interaction with CypA in different cell lines and different species gives rise to diverse and unpredictable phenotypes30,31. Differences in acetylation levels in different tissues may underpin some of this variation.
In binding to HIV-1 capsid, as in binding to cyclosporine, acetylation mediates its effects through changes in both the charge and water network in the active site. However, in the case of HIV-1 binding (and in contrast to cyclosporine binding), acetylation also mediates changes in the orientation of key interaction residues, generating new interactions and abolishing interactions present in the unacetylated complex. Disassembly of the viral capsid is an essential step in HIV-1 infection and may be catalyzed by CypA isomerization of the Gly89–Pro90 peptide bond26. By reducing CypA activity, as indicated both by the enzyme kinetics and the altered substrate binding in the complexed structure, acetylation may interfere with this process and reduce viral infectivity. In addition to HIV-1, HCV has also been shown to require CypA for efficient replication8. As CypA inhibitors represent potential anti-HCV drugs32, it will be interesting to see whether acetylation affects the role of CypA in HCV replication. Finally, CypA is part of a larger CypA family, including proteins such as CypB that are almost identical in sequence and yet have discrete specificities33. Post-translational modification of CypA but not CypB may help differentiate their function. Acetylation may inhibit binding to some substrates but allow CypA to isomerize new ones.
A persistent challenge in studying the role of acetylation has been in the preparation of homogeneously and site-specifically acetylated proteins. Histone proteins have been acetylated using semisynthetic methods34, but these methods are generally limited to the termini of proteins and yield small amounts of material, making biophysical studies challenging. Enzymatic modification with histone acetyltransferases is generally nonquantitative35 and leads to heterogeneity in the site and extent of acetylation. These methods of acetylation lead to inhomogeneous samples that are unsuitable for quantitative biophysical studies or structural biology. Our experiments demonstrate that homogeneously and site-specifically acetylated proteins that are suitable for structural studies and quantitative measurements can be made in excellent yield using an acetyllysyl-tRNA synthetase/tRNACUA pair18. This approach will make it possible to further explore the role of acetylation in controlling organism function.
Whereas acetylation is often considered to simply neutralize electrostatic interactions, our data reveal that the modification can regulate protein function by at least four general mechanisms: solvent remodeling, electrostatic quenching, hydrophobic interactions and surface complementarity. Because many acetylation sites have now been identified, it will be interesting to investigate how the molecular mechanisms we have uncovered are manipulated and augmented by cells to control organism function and possibly to combat viral infection.
Methods
Recombinant proteins.
All CypA proteins were expressed in E. coli C41 and purified through their N-terminal His6 tag, followed by size exclusion chromatography. Acetyl-CypA (CypAAcK) was expressed from a pCDFDuet-vector containing coding regions for the Methanosarcna barkeri MS tRNACUA (MbtRNACUA) and the CypA-Lys125amber, respectively. The incorporation of pyrrolysine in M. barkeri MS is directed by a pyrrolysyl-tRNA synthetase (MbPylRS) and its cognate amber suppressor, MbtRNACUA, in response to an amber codon. For further details see Supplementary Methods.
Isothermal titration calorimetry.
The interaction of CypA, CypAAcK and CypA-K125Q with HIV1-CAN capsids was measured by ITC based on ref. 36 using a MicroCal VP-ITC 200 microcalorimeter. For details see Supplementary Methods.
Linked chymotrypsin assay.
A modified form of the linked chymotrypsin assay23 was used to allow pre–steady state measurement of CypA-catalyzed cis to trans isomerization. Experiments were carried out at 5 °C using a Hi-Tech KinetAsyst Stopped-Flow System in a 1:1 mixing mode and with the photomultiplier in line with the long dimension of the cell. Briefly, an equal volume of 100 μM N-succinyl-Ala-Ala-Pro-Phe-p-nitroanilide peptide (Sigma) in one syringe was rapidly mixed with a solution in a second syringe containing 5 mg ml−1 bovine α-chymotrypsin (Sigma) and concentrations of CypA increasing from 0 to 500 nM. Biphasic liberation of nitroanilide was observed and fit to a double exponential, where the first, fast step, with greater amplitude, corresponds to cleavage of trans peptide by chymotrypsin and the second, slower step, with smaller amplitude, corresponds to cis to trans isomerization by CypA. The apparent relaxation rate for isomerization was determined on an average of at least three independent measurements. The results of experiments at different CypA concentrations were then used to determine kcat/KM as described in the main text. For inhibition experiments see Supplementary Methods.
Fluorescence equilibrium titration measurements.
All experiments were performed using a Perkin Elmer LS55 fluorimeter at 20 °C in buffer A. The interaction of cyclosporine and CypA was followed by measuring the intrinsic tryptophan fluorescence of W121 (λexcitation = 280 nm, λemission = 338 nm). Cyclosporine was added stepwise (final concentrations, 1–240 nM) to 10–50 nM CypA or CypAAcK, rapidly mixed and the fluorescence intensity measured. The final intensity at each titration point was calculated from the average of 20 measurements at equilibrium. The relative fluorescence-intensity was plotted as a function of cyclosporine concentration and the obtained curve was analyzed by fitting a quadratic function to the data. For data analysis, we used Grafit 3.0–5.0. Final KD values were calculated from the mean of at least two independent titration experiments.
Crystallization and structure determination.
For details of crystallization conditions see Supplementary Methods. All crystals were obtained using sitting drop for initial screens and the hanging drop/vapor diffusion method for optimization. Crystals were flash frozen in liquid nitrogen using the cryoprotectants described above. Data collection was performed at the European Synchrotron Radiation Facility in Grenoble, France at beamline ID29. For data analysis see Supplementary Methods and Supplementary Table 2.
Cell culture, immunoprecipitation and western blotting.
Suspension HeLa S3 cells were grown in DMEM supplemented with 10% (v/v) FCS and 100 U ml−1 penicillin, and 100 μg ml−1 streptomycin in 2-liter cell culture flasks (density, 0.5 × 106 cells ml−1; viability, >90%). Lysis was performed in lysis buffer (0.3% (v/v) Triton X-100, 20 mM nicotinamide, 1 μM trichostatin A, 50 μM sirtinol in PBS) for 1 h on ice. Preclearing was done by adding 100 μl of protein A–Sepharose slurry (GE Healthcare) to 1 ml of cell lysate (30 min, 4 °C). Immunoprecipitation was done by addition of 1.5 μg ml−1 of the polyclonal rabbit anti-CypA (Santa Cruz Biotechnology; 1 h, 4 °C). After addition of 100 μl of prepared protein A–Sepharose, the antibody–protein complexes were bound to the beads for 4 h. The beads were washed three times with lysis buffer and 50 μl of twofold SDS running buffer was added. Western blotting was done using a standard protocol. Monoclonal anti-acetyllysine (1:2,000, Biomol) and polyclonal anti-CypA (1:500, Santa Cruz Biotechnology) were used. We used an electrochemiluminescence detection system (GE Healthcare) with horseradish peroxidase–coupled secondary antibody.
Immunodepletion experiments.
HeLa S3 cells and Jurkat T-cells were grown in DMEM or RPMI medium, respectively. We collected 500 ml of cells with a titer of 1 × 106 cells ml−1, washed them with PBS and resuspended them in lysis buffer (0.3% (v/v) Triton X-100, 1 mM trichostatin A, 50 mM sirtinol, 20 mM nicotinamide, protease inhibitors). Lysate was precleared as described above before addition of 1.5 μg ml−1 anti-CypA (Biomol) for immunoprecipitation and incubation for 1 h at 4 °C. Then 100 μl protein A–Sepharose beads were added and incubated overnight at 4 °C. After washing the beads three times with lysis buffer, CypA was eluted from the Sepharose and split into two samples. One sample was incubated with 15 μg ml−1 anti-acetyllysine and the other with the same volume of PBS (1 h, 4 °C). Protein A–Sepharose (50 μl) was added and incubated over night at 4 °C. Depleted supernatant was then loaded onto SDS-PAGE for western analysis using anti-CypA. S.d. values were calculated from at least three independent measurements. See Supplementary Figure 2 for standardization details.
Calcineurin-phosphatase assay.
Calcineurin activity was determined using a colorimetric assay (Biomol), in which released phosphate from an RII-phosphopeptide (DLDVPIPGRFDRRVpSVAAE, where pS is phosphoserine) is detected upon complex formation with malachite green by means of its absorbance at 620 nm (ref. 37). For full details see Supplementary Methods.
Compounds.
Sirtinol (purity ≥95%, HPLC), trichostatin A (5 mM in DMSO), nicotinamide (purity ≥99.5%, HPLC) and cyclosporine (purity ≥98.5%, TLC) were purchased from Sigma. Acetyllysine was from Bachem (purity ≥99%, TLC).
Statistical data analysis.
Where multiple experiments were performed, the mean value has been quoted, followed by the s.d. as calculated using the standard equation s.d. = [∑(X − M)2/(n − 1)]1/2, where X is the individual score, M is the mean and n is the sample size.
Accession codes.
Protein Data Bank: Coordinates for orthogonal free acetyl-CypA, trigonal free acetyl-CypA, cyclosporine complex and HIV-1 N-terminal capsid complex have been deposited with accession codes 2X25, 2X2A, 2X2C and 2X2D, respectively.
Additional information
Supplementary information and chemical compound information is available online at http://www.nature.com/naturechemicalbiology/. Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/. Correspondence and requests for materials should be addressed to J.W.C. or L.C.J.
Supplementary information
Supplementary Methods, Supplementary Results, Supplementary Figures 1–6, Supplementary Table 2 (PDF 17803 kb)
Supplementary Table 1 (PDF 492 kb)
Acknowledgements
This work was supported by the UK Medical Research Council.
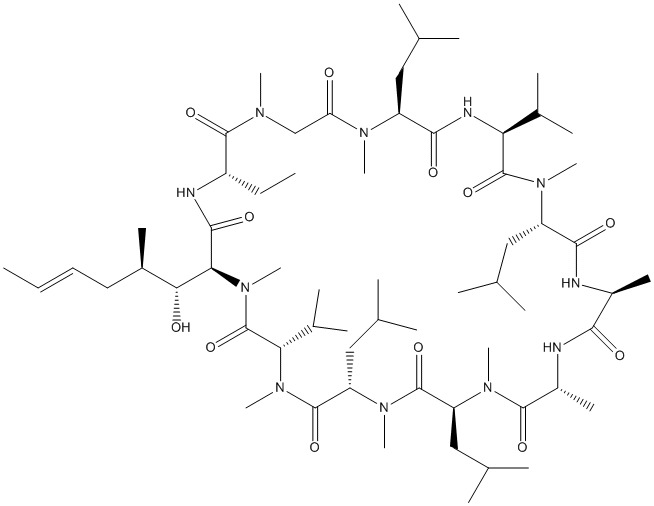
Cyclosporine
- PubChemID
90744369
- InChIKey
PMATZTZNYRCHOR-CGLBZJNRSA-N
- Synonyms
-
Cyclosporine
Ciclosporin
Neoral
Cyclosporine (USP)
Antibiotic S 7481F1
BMT-ABA-SAR-MLE-VAL-MLE-ALA-ALA-MLE-MLE-MVA
S-Neoral
Cipol N
HSDB 6881
S 7481F1
DRG 0275
Optimmune
(3S,6S,9S,12R,15S,18S,21S,24S,30S,33S)-30-Ethyl-33-[(1R,2R,4E)-1-hydroxy-2-methylhex-4-en-1-yl]-6,9,18,24-tetraisobutyl-3,21-diisopropyl-1,4,7,10,12,15,19,25,28-nonamethyl-1,4,7,10,13,16,19,22,25,28,31-undecaazacyclotritriacontane-2,5,8,11,14,17,20,23,26,29,32-undecone
1,4,7,10,13,16,19,22,25,28,31-undecaazacyclotritriacontane-2,5,8,11,14,17,20,23,26,29,32-undecone, 30-ethyl-33-[(1R,2R,4E)-1-hydroxy-2-methyl-4-hexenyl]-1,4,7,10,12,15,19,25,28-nonamethyl-3,21-bis(1-m
ethylethyl)-6,9,18,24-tetrakis(2-methylpropyl)-, (3S,6S,9S,12R,15S,18S,21S,24S,30S,33S)-
(3S,6S,9S,12R,15S,18S,21S,24S,30S,33S)-30-ethyl-33-[(1R,2R,4E)-1-hydroxy-2-methylhex-4-en-1-yl]-1,4,7,10,12,15,19,25,28-nonamethyl-3,21-bis(1-methylethyl)-6,9,18,24-tetrakis(2-methylpropyl)-1,4,7,10,13,16,19,22,25,28,31-undecaazacyclotritriacontane-2,5,8,11,14,17,20,23,26,29,32-undecone
1,4,7,10,13,16,19,22,25,28,31-undecaazacyclotritriacontane-2,5,8,11,14,17,20,23,26,29,32-undecone, 30-ethyl-33-[(1R,2R,4E)-1-hydroxy-2-methyl-4-hexenyl]-1,4,7,10,12,15,19,25,28-nonamethyl-3,21-bis(1-methylethyl)-6,9,18,24-tetrakis(2-methylpropyl)-, (3S,6S,9S,12R,15S,18S,21S,24S,30S,33S)-
(3S,6S,9S,12R,15S,18S,21S,24S,30S,33S)-30-Ethyl-33-[(1R,2R,4E)-1-hydroxy-2-methylhex-4-en-1-yl]-1,4,7,10,12,15,19,25,28-nonamethyl-3,21-bis(1-methylethyl)-6,9,18,24-tetrakis(2-methylpropyl)-1,4,7,10,13,16,19,22,25,28,31-undecaazacyclotritriacontan-2,5,8,11,14,17,20,23,26,29,32-undecon
(3S,6S,9S,12R,15S,18S,21S,24S,30S,33S)-30-Ethyl-33-[(1R,2R,4E)-1-hydroxy-2-methyl-4-hexen-1-yl]-6,9,18,24-tetraisobutyl-3,21-diisopropyl-1,4,7,10,12,15,19,25,28-nonamethyl-1,4,7,10,13,16,19,22,25,28,31-undecaazacyclotritriacontane-2,5,8,11,14,17,20,23,26,29,32-undecone
1,4,7,10,13,16,19,22,25,28,31-undecaazacyclotritriacontane-2,5,8,11,14,17,20,23,26,29,32-undecone, 30-ethyl-33-[(1R,2R,4E)-1-hydroxy-2-methyl-4-hexen-1-yl]-1,4,7,10,12,15,19,25,28-nonamethyl-3,21-bis(1-methylethyl)-6,9,18,24-tetrakis(2-methylpropyl)-, (3S,6S,9S,12R,15S,18S,21S,24S,30S,33S)-
(3S,6S,9S,12R,15S,18S,21S,24S,30S,33S)-30-ethyl-33-[(1R,2R,4E)-1-hydroxy-2-methylhex-4-en-1-yl]-1,4,7,10,12,15,19,25,28-nonamethyl-6,9,18,24-tetrakis(2-methylpropyl)-3,21-di(propan-2-yl)-1,4,7,10,13,16,19,22,25,28,31-undecaazacyclotritriacontane-2,5,8,11,14,17,20,23,26,29,32-undecone
1H-imidazole-1-carboxamide, N-propyl-N-[2-(2,4,6-trichlorophenoxy)ethyl]-, compd. with (3S,6S,9S,12R,15S,18S,21S,24S,30S,33S)-30-ethyl-33-[(1R,2R,4E)-1-hydroxy-2-methyl-4-hexen-1-yl]-1,4,7,10,12,15,19,25,28-nonamethyl-3,21-bis(1-methylethyl)-6,9,18,24-tetrakis(2-methylpropyl)-1,4,7,10,13,16,19,22,25,28,31-undecaazacyclotritriacontane-2,5,8,11,14,17,20,23,26,29,32-undecone (1:1)
N-propyl-N-[2-(2,4,6-trichlorophenoxy)ethyl]-1H-imidazole-1-carboxamide - (3S,6S,9S,12R,15S,18S,21S,24S,30S,33S)-30-ethyl-33-[(1R,2R,4E)-1-hydroxy-2-methylhex-4-en-1-yl]-1,4,7,10,12,15,19,25,28-nonamethyl-6,9,18,24-tetrakis(2-methylpropyl)-3,21-di(propan-2-yl)-1,4,7,10,13,16,19,22,25,28,31-undecaazacyclotritriacontane-2,5,8,11,14,17,20,23,26,29,32-undecone (1:1)
- MDL Molfile
- 41589_2010_BFnchembio342_MOESM1_ESM.mol (7.6KB, mol)
- ChemDraw
- 41589_2010_BFnchembio342_MOESM2_ESM.cdx (9.7KB, cdx)
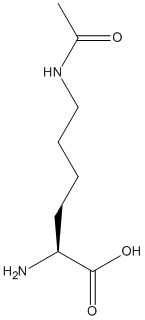
Nϵ-Acetyl-L-lysine
- PubChemID
90744368
- InChIKey
DTERQYGMUDWYAZ-ZETCQYMHSA-N
- Synonyms
-
692-04-6
Ne-Acetyl-L-lysine
N-6-Acetyl-L-lysine
- MDL Molfile
- 41589_2010_BFnchembio342_MOESM3_ESM.mol (1.2KB, mol)
- ChemDraw
- 41589_2010_BFnchembio342_MOESM4_ESM.cdx (2.1KB, cdx)
Author Contributions
M.L., H.N., J.W.C. and L.C.J. designed experiments and analyzed data.
Accession codes
Primary accessions
Protein Data Bank
Referenced accessions
Protein Data Bank
Competing interests
The authors declare no competing financial interests.
Contributor Information
Jason W Chin, Email: chin@mrc-lmb.cam.ac.uk.
Leo C James, Email: lcj@mrc-lmb.cam.ac.uk.
References
- 1.Ou WB, Luo W, Park YD, Zhou HM. Chaperone-like activity of peptidyl-prolyl cis-trans isomerase during creatine kinase refolding. Protein Sci. 2001;10:2346–2353. doi: 10.1110/ps.23301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Min L, Fulton DB, Andreotti AH. A case study of proline isomerization in cell signaling. Front. Biosci. 2005;10:385–397. doi: 10.2741/1536. [DOI] [PubMed] [Google Scholar]
- 3.Uittenbogaard A, Ying Y, Smart EJ. Characterization of a cytosolic heat-shock protein-caveolin chaperone complex. Involvement in cholesterol trafficking. J. Biol. Chem. 1998;273:6525–6532. doi: 10.1074/jbc.273.11.6525. [DOI] [PubMed] [Google Scholar]
- 4.Helekar SA, Char D, Neff S, Patrick J. Prolyl isomerase requirement for the expression of functional homo-oligomeric ligand-gated ion channels. Neuron. 1994;12:179–189. doi: 10.1016/0896-6273(94)90162-7. [DOI] [PubMed] [Google Scholar]
- 5.Nahreini P, et al. Effects of altered cyclophilin A expression on growth and differentiation of human and mouse neuronal cells. Cell. Mol. Neurobiol. 2001;21:65–79. doi: 10.1023/A:1007173329237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Decker ED, Zhang Y, Cocklin RR, Witzmann FA, Wang M. Proteomic analysis of differential protein expression induced by ultraviolet light radiation in HeLa cells. Proteomics. 2003;3:2019–2027. doi: 10.1002/pmic.200300473. [DOI] [PubMed] [Google Scholar]
- 7.Roehrl MH, et al. Selective inhibition of calcineurin-NFAT signaling by blocking protein-protein interaction with small organic molecules. Proc. Natl. Acad. Sci. USA. 2004;101:7554–7559. doi: 10.1073/pnas.0401835101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chatterji U, et al. The isomerase active site of cyclophilin A is critical for hepatitis C virus replication. J. Biol. Chem. 2009;284:16998–17005. doi: 10.1074/jbc.M109.007625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franke EK, Yuan HE, Luban J. Specific incorporation of cyclophilin A into HIV-1 virions. Nature. 1994;372:359–362. doi: 10.1038/372359a0. [DOI] [PubMed] [Google Scholar]
- 10.Thali M, et al. Functional association of cyclophilin A with HIV-1 virions. Nature. 1994;372:363–365. doi: 10.1038/372363a0. [DOI] [PubMed] [Google Scholar]
- 11.Sokolskaja E, Sayah DM, Luban J. Target cell cyclophilin A modulates human immunodeficiency virus type 1 infectivity. J. Virol. 2004;78:12800–12808. doi: 10.1128/JVI.78.23.12800-12808.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Price AJ, et al. Active site remodeling switches HIV specificity of antiretroviral TRIMCyp. Nat. Struct. Mol. Biol. 2009;16:1036–1042. doi: 10.1038/nsmb.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kouzarides T. Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 2000;19:1176–1179. doi: 10.1093/emboj/19.6.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marmorstein R, Roth SY. Histone acetyltransferases: function, structure, and catalysis. Curr. Opin. Genet. Dev. 2001;11:155–161. doi: 10.1016/S0959-437X(00)00173-8. [DOI] [PubMed] [Google Scholar]
- 15.Denu JM. The Sir 2 family of protein deacetylases. Curr. Opin. Chem. Biol. 2005;9:431–440. doi: 10.1016/j.cbpa.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 16.Mujtaba S, Zeng L, Zhou MM. Structure and acetyl-lysine recognition of the bromodomain. Oncogene. 2007;26:5521–5527. doi: 10.1038/sj.onc.1210618. [DOI] [PubMed] [Google Scholar]
- 17.Kim SC, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell. 2006;23:607–618. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 18.Neumann H, Peak-Chew SY, Chin JW. Genetically encoding Nϵ-acetyllysine in recombinant proteins. Nat. Chem. Biol. 2008;4:232–234. doi: 10.1038/nchembio.73. [DOI] [PubMed] [Google Scholar]
- 19.Neumann H, et al. A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol. Cell. 2009;36:153–163. doi: 10.1016/j.molcel.2009.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Howard BR, Vajdos FF, Li S, Sundquist WI, Hill CP. Structural insights into the catalytic mechanism of cyclophilin A. Nat. Struct. Biol. 2003;10:475–481. doi: 10.1038/nsb927. [DOI] [PubMed] [Google Scholar]
- 21.Fanghanel J, Fischer G. Thermodynamic characterization of the interaction of human cyclophilin 18 with cyclosporin A. Biophys. Chem. 2003;100:351–366. doi: 10.1016/S0301-4622(02)00292-2. [DOI] [PubMed] [Google Scholar]
- 22.Etzkorn FA, Chang ZY, Stolz LA, Walsh CT. Cyclophilin residues that affect noncompetitive inhibition of the protein serine phosphatase activity of calcineurin by the cyclophilin.cyclosporin A complex. Biochemistry. 1994;33:2380–2388. doi: 10.1021/bi00175a005. [DOI] [PubMed] [Google Scholar]
- 23.Kofron JL, Kuzmic P, Kishore V, Colon-Bonilla E, Rich DH. Determination of kinetic constants for peptidyl prolyl cis-trans isomerases by an improved spectrophotometric assay. Biochemistry. 1991;30:6127–6134. doi: 10.1021/bi00239a007. [DOI] [PubMed] [Google Scholar]
- 24.Braaten D, Franke EK, Luban J. Cyclophilin A is required for the replication of group M human immunodeficiency virus type 1 (HIV-1) and simian immunodeficiency virus SIV(CPZ)GAB but not group O HIV-1 or other primate immunodeficiency viruses. J. Virol. 1996;70:4220–4227. doi: 10.1128/jvi.70.7.4220-4227.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Towers GJ, et al. Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat. Med. 2003;9:1138–1143. doi: 10.1038/nm910. [DOI] [PubMed] [Google Scholar]
- 26.Bosco DA, Eisenmesser EZ, Pochapsky S, Sundquist WI, Kern D. Catalysis of cis/trans isomerization in native HIV-1 capsid by human cyclophilin A. Proc. Natl. Acad. Sci. USA. 2002;99:5247–5252. doi: 10.1073/pnas.082100499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gamble TR, et al. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell. 1996;87:1285–1294. doi: 10.1016/S0092-8674(00)81823-1. [DOI] [PubMed] [Google Scholar]
- 28.Eisenmesser EZ, Bosco DA, Akke M, Kern D. Enzyme dynamics during catalysis. Science. 2002;295:1520–1523. doi: 10.1126/science.1066176. [DOI] [PubMed] [Google Scholar]
- 29.Kung L, et al. Tissue distribution of calcineurin and its sensitivity to inhibition by cyclosporine. Am. J. Transplant. 2001;1:325–333. doi: 10.1034/j.1600-6143.2001.10407.x. [DOI] [PubMed] [Google Scholar]
- 30.Hatziioannou T, Perez-Caballero D, Cowan S, Bieniasz PD. Cyclophilin interactions with incoming human immunodeficiency virus type 1 capsids with opposing effects on infectivity in human cells. J. Virol. 2005;79:176–183. doi: 10.1128/JVI.79.1.176-183.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ylinen LMJ, et al. Cyclophilin A levels dictate infection efficiency of Human Immunodeficiency Virus Type 1 capsid escape mutants A92E and G94D. J. Virol. 2009;83:2044–2047. doi: 10.1128/JVI.01876-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watashi K, Shimotohno K. Chemical genetics approach to hepatitis C virus replication: cyclophilin as a target for anti-hepatitis C virus strategy. Rev. Med. Virol. 2007;17:245–252. doi: 10.1002/rmv.534. [DOI] [PubMed] [Google Scholar]
- 33.Watashi K, et al. Cyclophilin B is a functional regulator of hepatitis C virus RNA polymerase. Mol. Cell. 2005;19:111–122. doi: 10.1016/j.molcel.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 34.Shogren-Knaak M, et al. Histone H4–K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 35.Robinson PJ, et al. 30 nm chromatin fibre decompaction requires both H4–K16 acetylation and linker histone eviction. J. Mol. Biol. 2008;381:816–825. doi: 10.1016/j.jmb.2008.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiseman T, Williston S, Brandts JF, Lin LN. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem. 1989;179:131–137. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]
- 37.Enz A, Shapiro G, Chappuis A, Dattler A. Nonradioactive assay for protein phosphatase 2B (calcineurin) activity using a partial sequence of the subunit of cAMP-dependent protein kinase as substrate. Anal. Biochem. 1994;216:147–153. doi: 10.1006/abio.1994.1018. [DOI] [PubMed] [Google Scholar]
- 38.Ke H. Similarities and differences between human cyclophilin A and other beta-barrel structures. Structural refinement at 1.63 A resolution. J. Mol. Biol. 1992;228:539–550. doi: 10.1016/0022-2836(92)90841-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods, Supplementary Results, Supplementary Figures 1–6, Supplementary Table 2 (PDF 17803 kb)
Supplementary Table 1 (PDF 492 kb)