

Abstract
Adoptive immunotherapy with ex vivo expanded T cells is a promising approach to prevent or treat leukemia. Myeloid leukemias express tumor-associated antigens (TAA) that induce antigen-specific cytotoxic T lymphocyte (CTL) responses in healthy individuals. We explored the feasibility of generating TAA-specific CTLs from stem cell donors of patients with myeloid leukemia to enhance the graft-versus-leukemia effect after stem cell transplantation. CTL lines were manufactured from peripheral blood of 10 healthy donors by stimulation with 15mer peptide libraries of five TAA (proteinase 3 (Pr3), preferentially expressed antigen in melanoma, Wilms tumor gene 1 (WT1), human neutrophil elastase (NE) and melanoma-associated antigen A3) known to be expressed in myeloid leukemias. All CTL lines responded to the mix of five TAA and were multi-specific as assessed by interferon-γ enzyme-linked immunospot. Although donors showed individual patterns of antigen recognition, all responded comparably to the TAAmix. Immunogenic peptides of WT1, Pr3 or NE could be identified by epitope mapping in all donor CTL lines. In vitro experiments showed recognition of partially human leukocyte antigen (HLA)-matched myeloid leukemia blasts. These findings support the development of a single clinical grade multi-tumor antigen-specific T-cell product from the stem cell source, capable of broad reactivity against myeloid malignancies for use in donor-recipient pairs without limitation to a certain HLA-type.
Keywords: immunotherapy, cytotoxic T cells, tumor-associated antigens, myeloid leukemia, stem cell transplantation, graft-versus-leukemia effect
Introduction
The cure of myeloid leukemia after allogeneic stem cell transplantation (SCT) is mediated by donor-derived natural killer cells and by T cells, which recognize antigens on leukemia cells. These antigens fall into two categories: minor histocompatibility antigens expressed by leukemia progenitors and tumor-associated antigens (TAA) representing diverse proteins overexpressed by the leukemia.1 The graft-versus-leukemia (GVL) effect has been shown in autologous2 and allogeneic transplant settings. Induction of high rates of sustained remissions are observed in ‘good risk’ patients transplanted in remission early in the course of disease, but relapse rates of up to 60% can be anticipated in patients transplanted in relapse or those in second or subsequent remission.3 Hence, there is a need to enhance the GVL effect by additional immunotherapeutic approaches especially in those high-risk patients. In the first months following SCT, in a milieu of cytokine-driven T-cell proliferation, donor-derived T cells, which recognize tumor antigens can expand and have an important role for the GVL effect.4 The early post-transplant period thus presents a favorable opportunity for in vivo expansion of adoptively transferred TAA-specific T cells.5 We therefore explored the possibility of generating TAA-specific cytotoxic T lymphocytes (CTLs) from the donor for infusion into the recipient after SCT to enhance the GVL effect.
A number of TAA are known to be widely expressed by myeloid leukemias. For inducing TAA-specific T-cell expansion, we selected five TAA: Wilms tumor gene 1 (WT1), proteinase 3 (Pr3), human neutrophil elastase (NE), melanoma-associated antigen A3 (MAGE-A3) and preferentially expressed antigen in melanoma (PRAME), based on their known antigenicity and in some instances, association with induction of immune responses corresponding with clinical efficacy. The WT1 protein has been the TAA most extensively characterized. A series of MHC class I and II epitopes have been described to be immunogenic,6,7 and peptide vaccines have been successfully used to generate WT1-specific T cells in healthy individuals.8,9 Such T-cell responses were associated with disease control or remission in several vaccine studies, and WT1-specific T cells increase after SCT in patients with hematological malignancies and are associated with sustained disease remission.10 Pr3 is overexpressed in AML, and T cells recognizing the human leukocyte antigen-A2 (HLA-A2)-restricted peptide PR1 have been found after SCT and in patients with a variety of myeloid malignancies.11 Furthermore, a PR1 vaccine has been shown to induce remission in some patients relapsing after SCT.12 We found that the PR1 epitope sequence is also present in the closely related protein NE, which is overexpressed in AML. NE-specific CD4+ and CD8+ T-cell responses can be induced in healthy donors and are detectable after SCT, suggesting that NE contains a variety of potential immunogenic peptides.13,14 Similarly, T cells recognizing PRAME occur in post SCT patients and can be detected in healthy subjects.15 MAGE antigens are expressed by a wide variety of malignant cells and are also overexpressed in myeloid malignancies.16
Although the identification of specific HLA-restricted peptide epitopes is clearly important in defining immunogenic regions in the parent protein, current knowledge extends to only a handful of well-characterized peptide sequences, most of which are restricted to HLA-A2.7,17 A strategy targeting a small number of single TAA peptides could not have universal application. Furthermore, although a number of immunodominant peptides that induce CD8+ CTL responses have been described for TAA,7,18 the use of single peptides to generate CTLs would limit the approach to recipients of a relevant HLA-type and would lose the potential help and additional cytotoxicity from CD4+ T cells, which have been shown to be important for GVL reactivity.19,20 To overcome these constraints, we developed an approach to generate multi-TAA-specific CD4+ and CD8+ CTLs using peptide libraries of 15mer peptides overlapping by 11 amino acids spanning the whole amino acid sequence of a target antigen. Here, we show that it is possible to generate a clinical grade donor-derived CTL product to prevent or treat relapse of myeloid leukemia after allogeneic SCT.
Materials and Methods
Samples and cell lines
Healthy donor peripheral blood was obtained from the Department of Transfusion Medicine, NIH, Bethesda, MD, USA. Cord blood units were obtained from the MD Anderson cord blood bank. Peripheral blood mononuclear cells (PBMC) were isolated by density gradient centrifugation and cryopreserved. PBMC were stimulated with the mitogen phyto-hemagglutinine-P (PHA-P, 5 μg/ml) (Sigma, St Louis, MO, USA) in the presence of interleukin 2 (IL2) to promote blast formation (PHA-blast).
Generation of dendritic cells
Monocyte-derived dendritic cells (DC) were generated from CD14+ cells selected according to the instructions of the manufacturer (Miltenyi, Bergisch Gladbach, Germany), cultured in Cellgro DC media (Cellgenix, Freiburg, Germany) in the presence of IL4 (1000 U/ml) and granulocyte-macrophage colony-stimulating factor (800 U/ml) (R&D, Minneapolis, MN, USA). On day 5, immature DC were matured with a cytokine cocktail consisting of IL4, granulocyte-macrophage colony-stimulating factor, IL6 (10 ng/ml), tumor necrosis factor (TNF)α (10 ng/ml), IL1β (10 ng/ml)(R&D) and prostaglandin E2 (1 μg/ml)(Sigma), and harvested after 48 h.
Generation of T-cell lines
Antigen-specific T-cell lines were generated by stimulation of PBMC with peptide-pulsed DC at an effector-to-target ratio of 10:1 in RPMI medium 1640 supplemented with 45% Clicks media (Irvine Scientific, Santa Ana, CA, USA), 5% human AB-serum and 2 mM Glutamax. For initial stimulation, a cytokine-mix containing IL7 (10 ng/ml), IL12 (10 ng/ml), IL15 (5 ng/ml) and IL6 (100 ng/ml) was added, for the second stimulation IL7 and IL2 (50 U/ml) (all Peprotech, Rocky Hill, NJ, USA) for the further maintenance of CTLs.21 Restimulation of CTLs was carried out weekly with peptide-pulsed DC at an effector-to-target ratio of 20:1.
Peptides
Peptide libraries for WT1, NE and Pr3 of 15mer peptides overlapping by 11 amino acids spanning the whole protein were obtained from Biosynthesis (Lewisville, TX, USA) and Princeton Biomolecules (Langhorne, CA, USA). Peptide pools were designed to represent each peptide in two pools, as previously described for cytomegalovirus pp65.22 A total pool containing all peptides was reconstituted accordingly. Pepmixes for MAGE-A3, PRAME, cytomegalovirus pp65 and IE-1 were purchased from JPT (Berlin, Germany).
IFNγ enzyme-linked immunospot assay
Peptide recognition was tested in an interferon-γ (IFNγ) enzyme-linked immunospot (ELISpot) assay as previously published.7 Briefly, 105 CTLs were plated, stimulated with peptide and incubated for 16h at 37 °C. Plates were developed and spots counted with a Zeiss Elispot-Reader.
Intracellular cytokine detection and CD107a assay
CTLs were stimulated for 6h with pepmix or single peptide under costimulation with anti-CD28 and CD49d antibodies (1 μg/ml, BD Biosciences, San Jose, CA, USA) in presence of Brefeldin A (Golgiplug, BD Biosciences) and Monensin (GolgiStop, BD Biosciences). For simultaneous assessment of cytokine release and CD107a-expression, anti-CD107a antibody (BD Biosciences) was added during the incubation. Cells were stained with extracellular antibodies against CD3, CD4 and CD8 (BD Biosciences) and Vivid viability dye (Invitrogen, Grand Island, NY, USA) to exclude dead cells. Intracellular staining of IFNγ, TNFα, IL2, IL4, IL10, MIP1β, CD154 (BD Biosciences) of permeabilized cells (Cytofix/Cytoperm, BD Biosciences) was analyzed on a BD Fortessa Flow Cytometer using an automated plate reader. Controls labeled with appropriate isotype-matched antibodies were included in each experiment. Data were analyzed using FlowJo Flow Cytometry software (Treestar, Ashland, OR, USA).
Cytotoxicity assay
Target cells were fluorescence labeled with carboxyfluorescein succinimi-dyl ester (CFSE, Invitrogen), peptide-pulsed and incubated with CTLs at different effector-to-target ratios for 6–8h. Ethidium homodimer (Invitrogen) was added after incubation to stain dead cells. Samples were acquired on a BD Fortessa Flow Cytometer. The number of live target cells was determined by gating on carboxyfluorescein succinimidyl ester-positive, ethidium homodimer-negative cells, and used to calculate cytotolytic activity as follows: Lysis (%) = 100—((live target cells sample/live target cells control) × 100).
Coculture with primary blasts samples
Recognition of primary blast samples was tested in coculture with partially HLA-matched AML blast samples in presence of IL2 (25 U/ml). Remaining leukemia cells were quantified by costaining with anti-CD33 and CD34 antibodies, and samples were acquired on a BD Biosciences FACS Calibur Flow Cytometer.
Colony forming unit assay
Colony forming unit (CFU) assays were performed to show effective blast elimination of progenitor cells. CTLs were preincubated for 6 h with partially HLA-matched AML blasts in Iscove's Modified Dulbecco's Media medium (Invitrogen) with 10% fetal bovine serum and plated in methocult methylcellulose media (Stem cell Technologies, Vancouver, Canada). Colonies were counted on day 14 and relative inhibition of colony formation calculated in comparison with unspecific control CTLs as follows: Relative inhibition (%) = 100—((CFU specific CTLs/CFU unspecific CTLs) × 100).
Results
Multi-tumor antigen-specific CTLs can be generated from stem cell donors
To evaluate whether CTLs could be expanded from the peripheral blood of potential stem cell donors, healthy donor PBMC were primed with autologous DC pulsed with 5 TAAs in the presence of IL7, IL15, IL12 and IL6.21 After three rounds of stimulation, CTLs expanded from 10 donors showed a mean expansion of 12.5-fold (range 4–20) (Figure 1a). These CTLs were predominantly CD3+ (mean 97.9%) with varying content of CD4+ and CD8+ T cells (mean 42.2%, range 9.5–89.5% and mean 43.9%, range 8.4–84.4%, respectively). CTLs contained few natural killer cells (mean 0.6%) and negligible B-cells (mean 0.04%) (Figure 1b). Analysis of CTLs showed a predominance of CD45RO+/CD62L−/CCR7− T cells in accordance with an effector memory phenotype (Figure 1c). To confirm the multi-TAA-specific activity of the CTLs, the cultures were evaluated for IFNγ-secretion in response to peptide libraries for each antigen in ELISpot. All 10 donors showed recognition of the TAAmix consisting of five TAA: WT1, Pr3, NE, MAGE-A3 and PRAME. Multi-TAA-specific CTLs, which were ex vivo expanded using DC pulsed with the TAAmix, recognized a mean of 2.6 (range 1–4) antigens, versus a mean of 3.1 (range 1–5) in CTLs stimulated with each antigen individually in separate cultures (Figure 1d). Antigen recognition in IFNγ-ELISpot showed highest spot counts for NE (mean 596 spot-forming units (SFU)/105 cells; range 3–1404) followed by MAGE-A3 (mean 278; range 0–735). Against TAAmix a mean of 455 SFU/105 cells (range 78–969) was achieved (Figure 1e). Consistent with the ELISpot results, highest cytolytic activity was observed against NE (mean 29.1%, range 0–83% at an effector-to-target ratio of 40:1); lysis of TAAmix-pulsed target cells was in a similar range (mean 32.3%, range 0–72%) (Figure 1f). Further, simultaneous lysis of up to three antigens was observed in a cytotoxicity assay using peptide-pulsed autologous PHA-blasts as target cells. Two representative examples are shown in Figure 2.
Figure 1.

Multi-antigen-specific CTL lines are reproducibly expanded from adult donors and CB samples. (a) Fold expansion of TAAmix-specific CTLs generated from 10 healthy individuals by repeated stimulation of PBMC with pepmix-pulsed DC (mean ± s.d.). (b) Phenotype of TAAmix-specific CTL lines after three stimulations (% of lymphocyte gate, and mean) showing balanced predominance of CD4+ and CD8+ T cells. (c) T-cell subsets with predominance of effector memory phenotype (% of CD3+ cells, and mean). (d) Recognition of individual target antigens in IFNγ-ELISpot assay by CTLs generated by TAAmix (left) versus single antigen (SA) stimulation (right). Percentage of CTL lines recognizing from one to five antigens are shown. (e) Mean spot counts ± s.e. in response to target antigen stimulation by IFNγ-ELISpot assay of TAAmix CTLs. (f) mean cytolytic activity ± s.e. of TAAmix CTL lines (n = 8) against peptide-pulsed autologous PHA-blasts (PHAB) in a carboxyfluorescein succinimidyl ester (CFSE)-based cytotoxicity assay at an effector-to-target ratio of 40:1. (g) Phenotype of six cord blood-derived CTL lines after three restimulations (% of lymphocyte gate, and mean). (h) Phenotype of T-cell subsets (% of CD3+ cells, and mean) of these cord blood-derived CTL lines. (i) Mean of INFγ spot counts ± s.e. in response to target antigen stimulation by ELISpot assay of six cord blood TAAmix CTL lines showing recognition of all antigens.
Figure 2.

Functional assays in TAAmix-specific CTL lines in two donors. (a) Recognition of TAAmix and SAs by IFNγ-ELISpot (mean spot count ± s.d.) and (b) cytolytic activity (mean ± s.d.) in a CFSE-based cytotoxicity assay against peptide-pulsed target cells correlating with these results, with no lysis of unpulsed PHAB (dotted line). (c) Intracellular cytokine detection of peptide stimulated CTLs in the presence of CD28/CD49d, Brefeldin A and Monensin showing secretion of multiple cytokines, (IFNγ, TNFα, IL2) and CD154 and CD107a-expression as a marker of activation and degranulation, respectively, in both CD4+ and CD8+ T-cell populations. (d) Recognition of TAAmix and SAs in IFNγ-ELISpot assay (mean spot count ± s.d.) by TAAmix CTLs from a representative donor. (e) Specific lysis (mean ± s.d.) of peptide-pulsed autologous PHAB in a CFSE-based cytotoxicity assay. (f) Cytokine secretion by CD4+ and CD8+ T cells.
Another critical source of stem cells for allogeneic transplant is umbilical cord blood (CB), therefore we also explored the feasibility of generating multi-antigen-specific CTLs from CB samples (n = 6). CB mononuclear cells were stimulated with peptide-pulsed autologous DC, according to the protocol described for PBMC. After three stimulations, phenotyping of CB-derived TAAmix-specific CTLs revealed a similar phenotype as PBMC-derived CTLs with a mean CD3+ content of 89.3% (range 79.3–97.2%) and mean 49.4% CD4+ (range 18.6–73.9%) and mean 43.1% CD8+ (range 4.1–78.9%) (Figure 1g), and the majority of the product comprising CD3+/CD45RO+/CCR7−/CD62L− effector memory T cells (mean 54.4%, range 39.2–69.4%) (Figure 1h). In IFNγ-ELISpot assays these CTLs showed reactivity against all antigens tested; for TAAmix mean spot count was 224 (range 174-270 SFU/105 cells), for WT1 119 (range 51–182), NE 102 (range 41–168), Pr3 125 (66–179), MAGE-A3 125 (range 47–213) and PRAME 143 (range 50–180)(background mean 66, range 15–193) (Figure 1i). Unlike PBMC-derived CTLs, there was no predominance of T-cells directed towards any one antigen and responses were generally lower than those observed in PBMC-derived CTLs. However, CB-derived CTLs recognized the tumor antigens in a more even distribution with a mean of 4.3 antigens recognized by each CB-derived CTL line (range 3–5) compared with PBMC-derived CTLs (mean 2.6).
Comparison of TAAmix and single antigen recognition in individual donors
To evaluate the efficacy of CTL generation and the breadth of the responses to the TAAmix, CTLs were incubated separately with the TAAmix and individual antigens in IFNγ-ELISpot and cytotoxicity assays. Of the 10 lines evaluated, all demonstrated IFNγ-secretion in response to TAAmix and 6 of 8 lines tested showed cytolytic activity against TAAmix-pulsed autologous target cells. Up to five antigens were recognized in IFNγ-ELISpot and up to three antigens simultaneously in cytotoxicity assays. Stimulation with the TAAmix did not seem to influence or inhibit responses to any of the antigens tested. The CTL lines were shown to be multifunctional, secreting several cytokines, which has been shown to be important for in vivo antitumor activity, comprising both CD4+ and CD8+ T-cell responses.
As shown in representative donors two (Figures 2a–c) and five (Figures 2d–f), recognition of TAAmix and individual antigens in IFNγ-ELISpot (Figures 2a and d) correlated with the cytolytic activity of these CTLs against targets pulsed with TAAmix and the same antigens in a carboxyfluorescein succinimidyl ester-based cytotoxicity assay (Figures 2b and e). Further, intracellular cytokine detection after stimulation with TAAmix or the individual antigens showed production of multiple cytokines in both the CD4+ and CD8+ populations, with an increased predominance of Th1 cytokines secreted by the CD8+ populations, as well as expression of the degranulation marker CD107a. In both donors responses for IFNγ and CD107a were ∼10-fold higher in the CD8+ populations compared with the CD4+ populations (Figures 2c and f).
Antileukemic activity against partially HLA-matched AML blasts
To evaluate the antileukemic activity of TAAmix-specific CTLs in vitro, CTLs were cocultured with primary leukemia blast samples matched in at least one HLA-antigen (range1–3), including pairs, which were matched solely at HLA class II alleles. Where available, AML blast samples were evaluated for expression of MAGE-A3 and PRAME. Low expression was detectable by immunohistochemistry (data not shown). Figure 3 shows the results of five CTL lines tested against a panel of five primary AML blast samples. Specific leukemia recognition and elimination occurred even with single HLA class I or II allele matched targets. As control for nonspecific lysis or allogeneic reactivity, CTL lines with irrelevant specificity (viral antigens) generated from the same donor were used in all experiments. All donor-derived TAAmix-specific CTLs showed leukemia-specific killing, as well as CTL activation as determined by CFU assay, IFNγ-ELISpot and intracellular cytokine detection.
Figure 3.
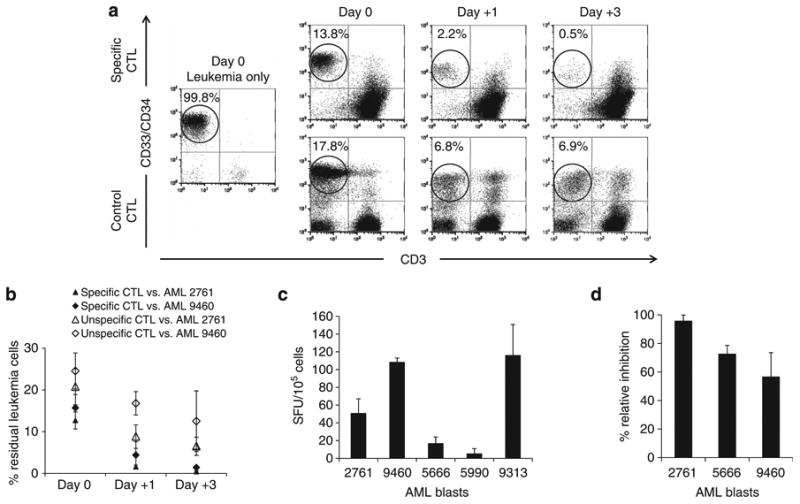
Recognition of primary AML blast cells by TAAmix CTL lines. (a) Coculture of TAAmix CTLs with partially HLA-matched AML blast sample no. 2761. Leukemia blasts were quantified by anti-CD33/CD34 costaining and CTLs by CD3 staining. Leukemia blasts were eliminated over time when cocultured with TAAmix CTLs, but remained at higher levels in culture when incubated with control CTLs (virus-specific CTLs) from the same donor. Analysis on day 0 and after 1 and 3 days of coculture. (b) Summary of coculture experiments of TAAmix CTLs (black symbols) generated from five donors with partially HLA-matched AML blast samples no. 2761 and no. 9460 and nonspecific control CTL lines derived from the same donors (open symbols); % remaining leukemia blasts ± s.d. on day 0, and after 1 and 3 days of coculture. (c) Production of IFNγ by TAAmix CTLs coincubated with partially HLA-matched AML blasts at a 1:1 ratio in ELISpot (mean spot counts ± s.e. of five CTL lines). (d) Relative inhibition of AML blast colony formation in a CFU assay (calculated in comparison to nonspecific CTLs generated from the same donor; mean ± s.d. of 5 CTL lines).
A representative example of a coculture experiment with AML blast sample no. 2761 is shown in Figure 3a. A 3 day coculture with specific CTLs eliminated leukemia blasts (0.5%), compared with 6% blasts persisting in cocultures with control CTLs. Further demonstration of leukemia elimination by the expanded TAA-specific CTLs, is shown in the Supplementary Figure, which demonstrates reduction in absolute blast counts in the coculture.
The summary of coculture experiments of five healthy donor TAAmix-specific CTL lines with AML no. 2761 and no. 9460 blasts shows a mean of 0.4 and 1.4% residual leukemia cells, compared with 6.5 and 12.5% in the controls (Figure 3b). Analysis of IFNγ and IL4-secretion in the coculture supernatants by ELISA showed secretion of both Th1 and Th2 cytokines with a predominance of IFNγ (data not shown). TAAmix-specific CTLs were also incubated with AML blast samples in short-term coculture. IFNγ-ELISpot and specific IFNγ-release were observed in response to HLA-matched blasts (Figure 3c). Finally, in CFU assays, inhibition of colony formation occurred when TAAmix-specific CTLs were incubated with leukemia blasts. The relative inhibition of colony growth ranged from 56.8–95.9% in the AML samples evaluable for analysis (Figure 3d).
Identification of immunogenic peptides for WT1, NE and Pr3
To extend the known repertoire of HLA-A2-restricted WT1 and Pr3 antigens currently used in vaccine trials we sought peptide epitopes restricted by other HLA-molecules. In our cohort of donors, the strongest responses were seen against NE, which has not been extensively characterized, but is generally expressed in myeloid leukemia. We therefore selected NE, WT1 and Pr3 for further epitope mapping. At least one immunogenic peptide was found for each of the CTL lines tested (n = 10), irrespective of the donor HLA-type, and up to five epitopes were found in one individual. We identified one novel and six previously described epitopes for WT1 (Table 1), six epitopes for PR3 including two previously described (Table 2) and seven new epitopes for NE (Table 3). Within the previously described epitopes, recognition was restricted by different HLA-alleles in all but two cases for Pr3.
Table 1. WT1 peptides identified by epitope mapping.
| Donor ID | WT1 | Peptide sequence | HLA restriction | AA # | HLA-A | HLA-B | HLA-Cw | HLA-DRB | HLA-DQB | Reference and HLA-restriction |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | No. 60 | TWNQMNLGATLKGVA | HLA-DR | 237–251 | 0201 | 15/39 | 03/12 | 0403/0701 | 0202/03 | Oka et al.42 A*0201 Doubrovina et al.43 HLA-DRB1*0402, 1104, A*0203, *2402, *0101, *0201 |
| 3 | No. 1 | MGSDVRDLNALLPAV | Class I | 1–15 | 0201/26 | 27/39 | 07/12 | 1501/1601 | 0502/0602 | Knights et al.44 HLA-DRB1*0401 |
| 4 | No. 97/98 | CKTCQRKFSRSDHLKTHTR | HLA-DP | 385–402 | 0201/11 | 44/49 | 02/07 | 0101/07 | 0303/0501 | Doubrovina et al.43 HLA-A*0201 |
| 7 | No. 52 | PTDSCTGSQALLLRT | HLA-DP | 205–219 | 0201 | 44 | 04/05 | 03/0401 | 0201/03 | Doubrovina et al.43 HLA-A*0101, B*3802, *4402 |
| No. 58 | MTSQLECMTWNQMNL | Not identified | 229–243 | Doubrovina et al.43 HLA-A*0201 | ||||||
| No. 91 | ERRFSRSDQLKRHQR | HLA-DR | 361–375 | |||||||
| 10 | No. 60 | TWNQMNLGATLKGVA | HLA-DR | 237–251 | 0301/30 | 15/42 | 03/17 | 0401/0901 | 02/03 | See above |
Abbreviation: AA, amino acids, HLA, human leukocyte antigen.
Table 2. Proteinase 3 (Pr3) peptides identified by epitope mapping.
| Donor ID | Pr3 | Peptide sequence | HLA restriction | AA no. | HLA-A | HLA-B | HLA-Cw | HLA-DRB | HLA-DQB | Reference and HLA-restriction |
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | No. 9/10 | EAQPHSRPYMASLQMRGNP | Class I | 33–51 | 0201/01 | 08/44 | 05/07 | 0301/0401 | 0201/0301 | |
| 3 | No. 16 | IHPSFVLTAPHCLRD | Not identified | 61–75 | 0201/26 | 27/39 | 07/12 | 1501/1601 | 0502/0602 | |
| 9 | No. 15 | GGTLIHPSFVLTAPH | HLA-DR | 57–71 | 03/11 | 07/56 | 01/07 | 0101/0701 | 02/0501 | |
| 10 | No. 25 | QQHFSVAQVFLNNYD | Not identified | 97–111 | 0301/30 | 15/42 | 03/17 | 0401/0901 | 02/03 | |
| No. 29 | ENKLNDILLIQLSSP | Not identified | 113–127 | |||||||
| No. 57 | ATRLFPDFFTRVALY | Not identified | 225–239 | Lacey et al.45 HLA-A*0201 | ||||||
| NO. 61 | DWIRSTLRRVEAKGR | Not identified | 241–255 | Piesche et al.46 HLA-DR |
Abbreviation: AA, amino acids, HLA, human leukocyte antigen.
Table 3. Neutrophil elastase (NE) peptides identified by epitope mapping.
| Donor ID | NE | Peptide sequence | HLA restriction | AA no. | HLA-A | HLA-B | HLA-Cw | HLA-DRB | HLA-DQB |
|---|---|---|---|---|---|---|---|---|---|
| 1 | No. 2 | RRLACLFLACVLPAL | Class I | 5–19 | 0201 | 15/39 | 03/12 | 0403/0701 | 0202/03 |
| 2 | No. 2 | RRLACLFLACVLPAL | Class I | 5–19 | 0201/01 | 08/44 | 05/07 | 0301/0401 | 0201/0301 |
| 3 | No. 2 | RRLACLFLACVLPAL | Class I | 5–19 | 0201/26 | 27/39 | 07/12 | 1501/1601 | 0502/0602 |
| No. 9 | GRRARPHAWPFMVSL | Class I | 33–47 | ||||||
| No. 16 | APNFVMSAAHCVANV | HLA-DR | 61–75 | ||||||
| 4 | No. 5 | PALLLGGTALASEIV | Class I | 17–31 | |||||
| 5 | No. 2 | RRLACLFLACVLPAL | Class I | 5–19 | 01/0201 | 08/44 | 05/07 | 0301/0401 | 02/0301 |
| No. 8 | EIVGGRRARPHAWPF | Class I | 29–43 | ||||||
| 6 | No. 2 | RRLACLFLACVLPAL | Class I | 5–19 | 0201 | 15/57 | 01/06 | 07/1501 | 0303/0602 |
| No. 30 | DIVILQLNGSATINA | HLA-DR | 117–131 | ||||||
| 7 | No. 2 | RRLACLFLACVLPAL | Class I | 5–19 | 0201 | 44 | 04/05 | 03/0401 | 0201/03 |
| No. 5 | PALLLGGTALASEIV | Class I | 17–31 | ||||||
| 9 | No. 59 | APVAQFVNWIDSIIQ | Class I | 223–247 | 3/11 | 07/56 | 01/07 | 0101/0701 | 02/0501 |
| No. 9-11 | GRRARPHAWPFMVSLQLRGGHFC | HLA-DR | 33–51 |
Abbreviation: AA, amino acids, HLA, human leukocyte antigen.
Several CTL lines recognized a WT1 epitope lying within WT1 peptide no. 60 (TWNQMNLGATLKGVA). As shown for a representative donor, epitope mapping resulted in recognition of pools F and U that share WT1 peptide no. 60 (Figures 4a and b). However, the cytolytic response to targets pulsed with WT1 peptide no. 60 was relatively low (Figure 4c), consistent with the predominantly CD4+-restricted antigen recognition determined by antibody blocking experiments in IFNγ-ELISpot (Figure 4d). As shown in Table 1, the CTL lines recognizing WT1 no. 60 shared HLA-DRβ1*04. Further, blocking with anti-HLA-DR antibody led to a reduction of cytokine secretion close to background levels (Figure 4d). The HLA-DR-restriction could further be confirmed by incubating CTLs with partially HLA-matched antigen-presenting cells loaded with WT1 no. 60 presenting the peptide in the context of HLA-DRβ1*04, but with the expression of different alleles as shown in comparison to autologous antigen-presenting cells (Figure 4e). As it is common for MHC class II-restricted antigens, promiscuity for several HLA-DRβ1*04 alleles was observed with WT1 no. 60 recognition in the context of HLA-DRβ1*0403, *0404, *0406, *0407 and *0411, but not HLA-DRβ1*04-negative antigen-presenting cells (Figure 4e). Further, these T-cell lines recognized primary myeloid leukemia blasts matched at HLA-DRβ1*04 but not HLA-mismatched blasts in IFNγ-ELISpot (Figure 4f) and by intracellular cytokine detection (Figure 4g). Stimulation with WT1 no. 60 peptide as well as HLA-DRβ1*04+ AML and CML blast samples induced cytokine secretion predominantly in the CD4+ population, but only background levels were observed in response to the peptide control and healthy donor PBMC (Figure 4g).
Figure 4.

Characterization of a MHC class II-restricted WT1 epitope. (a) Epitope mapping for WT1 by testing recognition of peptide pools (A–U). Each pool contained 10 or 11 15mer peptides spanning the entire WT1 protein. Recognition of pools F and U indicating recognition of WT1 peptide no. 60, confirmed in (b) by testing WT1 no. 60 and flanking peptides. (c) Specific cytotolytic activity against WT1 no. 60-pulsed autologous PHAB (black line) compared with unpulsed PHAB (dotted line) in a CFSE-based cytotoxicity assay (mean ± s.d.). (d) Recognition of WT1 no. 60 was blocked by preincubation of peptide-pulsed target cells with anti-HLA class II antibody, in particular anti-HLA-DR, indicating a CD4+-restricted recognition of this peptide. (e) Recognition of WT1 no. 60 in IFNγ-ELISpot presented by a variety of HLA-DRβ1*04-positive and -negative antigen-presenting cells (APCs) compared with autologous APCs showing promiscuity in the recognition of WT1 no. 60 for several HLA-DRβ1*04 alleles (mean spot counts ± s.d.). (f) To test for recognition of primary leukemia blasts, CTLs were stimulated with HLA-DRβ1*04-positive and -negative myeloid blast cells and healthy donor PBMC in IFNγ-ELISpot assay (mean ± s.d.) and (g) intracellular cytokine detection; the predominant response was seen mainly in the CD4+ compartment.
An example of a newly identified NE epitope APVAQFVNWID-SIIQ (NE no. 59) is shown in Figure 5. Figure 5a illustrates that the CTLs showed reactivity against pools C and P in IFNγ-ELISpot, and according to the matrix (Figure 5b) this suggests that the immunogenic epitope lay within NE peptide no. 59. This was further confirmed by testing reactivity against the 15mer peptide NE no. 59 in IFNγ-ELISpot and cytotoxicity assay (Figures 5c and d). HLA-restriction of peptide no. 59 recognition was then evaluated by blocking antigen reactivity on peptide-pulsed autologous PHAB in IFNγ-ELISpot with anti-HLA class I and II blocking antibodies. As shown in Figure 5e, IFNγ-secretion was reduced to levels close to background when the T cells were preincubated with anti-HLA class I antibody, indicating that NE no. 59 recognition is mediated by CD8+ T cells, which is consistent with the Th1 cytokine prolife (IFNγ, TNFα and IL2-secretion) and activation (CD154) and degranulation marker (CD107a) expression of this CTL line (Figure 5f).
Figure 5.

Identification of a novel MHC class I-restricted NE peptide. (a) Epitope mapping for NE with the complete peptide library and minipools (A–P) by INFγ-ELISpot. (b) Peptide matrix for NE—each pool contains 8 15mer peptides. (c) Recognition of pools C and P, corresponding with recognition of peptide no. 59 in INFγ-ELISpot (mean ± s.d.). (d) Cytolytic activity of TAAmix CTLs against PHAB pulsed with NE no. 59 antigen or NE total pool (control: unpulsed PHAB, mean ± s.d.). (e) Blocking experiments with anti-HLA class I and II blocking antibodies using autologous peptide-pulsed PHAB as targets (mean ± s.d.). IFNγ-secretion was reduced to near background (unpulsed autologous PHAB) by preincubation with anti-HLA class I antibody, indicating MHC class I-restriction. (f) Intracellular cytokine staining of TAAmix CTLs stimulated with NE no. 59 showing predominant response in the CD8+ population.
Discussion
Our results show that it is possible to rapidly and reproducibly expand multi-TAA-specific CTLs ex vivo by peptide stimulation from healthy donors regardless of their HLA-type. These CTL lines comprised a mix of CD4+ and CD8+ cells and showed broad reactivity against multiple target antigens accompanied by the multifunctional cytokine profile associated with effective antitumor activity in vivo. Furthermore, we showed recognition and killing of partially HLA-matched primary myeloid leukemia blasts by TAA-specific CTLs.
In patients immunocompromised after SCT, the infusion of ex vivo generated antiviral CTLs can decrease viral load resulting in durable clinical improvement.23–25 Unselected donor lymphocytes can also exert antileukemic activity in patients relapsing with leukemia after SCT.26,27 Furthermore, antigen-specific CTLs prophylactically administered to patients with CML after SCT can induce molecular remission without the induction of graft-versus-host disease.28 These studies provide a rationale for developing TAA-specific CTLs to prevent or treat leukemic relapse after transplantation.
The strategy we used to generate multi-specific CTLs is practicable using lymphocytes from healthy stem cell donors as well as from cord blood donations. The approach has clinical applicability—from a small starting volume of blood, cell numbers expanded over 10-fold, making it feasible to manufacture lymphocyte donations of 107 cells to deliver TAA-specific CTLs in the range of 105 CTL/kg—a dose that has been shown to be clinically effective with virus-specific CTLs after SCT.25,29 In scaling up this approach to a clinical grade product, a further log expansion is achievable if gas-permeable culture flasks are used.30 Given that around 107 lymphocytes can be obtained from the 20% fraction of a CB unit, it should also be possible to expand sufficient CTLs from this stem cell source. Scale-up under good manufacturing practice conditions should not present novel regulatory complications because the in vitro manipulations use clinical grade or FDA-approved reagents that have largely been validated in the development of both multivirus-and multi-tumor-specific CTLs under similar conditions.21,31–33 The expanded TAA-specific CTLs had an effector memory phenotype and included CD4+ and CD8+ T cells, features which are associated with prolonged persistence after infusion of virus-specific CTLs generated in a similar manner.25,34
Generation of antigen-specific CTLs from CB units indicates that specific CTLs can also be derived from unprimed naïve T cells, an approach previously described in adult donor-derived naïve T-cells targeting WT1.35 It is likely that our multi-TAA-specific CTLs were also predominantly derived from naïve T cells, and therefore killing of AML blasts observed with adult-derived CTLs would also occur in CB-derived CTLs, rich in naïve T cells. However, as we already observed with virus-specific CTLs, TAA-specific CB CTLs show different HLA-restricted epitope specificity compared with adult donor CTLs.31 Further studies will therefore be needed to define the TAA-specific cytotoxicity of CB CTLs using panels of AML blasts of the relevant HLA-type.
Additionally, a general concern with this strategy is the potential for tumor immune escape from downregulation or loss of the antigen being targeted.36 In an effort to overcome tumor immune escape, we generated CTLs recognizing five TAA. Given the absence of outcompetition by a single TAA using this approach it should be possible to further enlarge the TAAmix, should this be necessary to reduce the risk of tumor escape. This approach can be extended to cover a broader spectrum of antigens that are recognized simultaneously within one CTL line and comprising a mix of CD4+ and CD8+ T cells.
We evaluated the antileukemic efficacy of multi-TAA-specific CTLs by a variety of parameters including cytokine release, CD107a-expression and cytotoxicity to peptide-pulsed target cells and primary leukemias. The CTLs demonstrated multi-cytokine profiles characteristic of effective CTLs with coexpression of IFNγ, TNFα and IL2, and strong CD107a-expression indicating degranu-lation upon antigen encounter. Cytotoxicity was elicited against bulk leukemias as well as by the inhibition of colony formation. It should be noted, however, that we did not directly test CTLs against defined leukemia progenitors. Direct proof of a durable antileukemic effect of these CTLs would require the demonstration of their ability to prevent leukemia engraftment in a humanized mouse leukemia model.37
The target antigens selected were based on available data, indicating a therapeutic potential of CTLs recognizing these proteins. Currently there is insufficient data to determine the optimum array of TAA for targeting the majority of AML. For this reason we focused on TAA known to be widely expressed in myeloid leukemia. Optimally, TAA should only be expressed by tumor cells and only in organs that cannot be targeted by T cells (for example, the testis), but not in healthy tissue. It will therefore be necessary to identify off-target expression of TAA in other tissues, before incorporating them into a clinical grade CTL product and monitoring for unwanted side-effects and graft-versus-host disease. Another source of off-target cytotoxicity is alloreactivity from the CTL lines against minor histocompatibility antigens presented by non-leukemic tissues. However, using virus-specific CTLs from the same donor as a control, we were able to show that only the TAA-specific CTLs responded to the leukemia in an HLA-restricted fashion (Figure 3). This excluded the role of an alloantigen-driven cytotoxic response to the leukemia.
In our experiments, the combination of five different antigens in the TAAmix did not lead to the inhibition of any single antigen response. Comparable results have been reported for the generation of multivirus-specific CTLs with a similar protocol.32 As antigenic competition does not appear to be a limiting factor for CTL generation, other TAA such as Aurora kinase,38 BMI-139 and RHAMM40 could also be incorporated to broaden the antigen repertoire and increase the spectrum of possible target cells or tumor entities.
A limitation of adoptive T-cell therapy is the possibility that TAA are present in the malignant cells, but not sufficiently over-expressed to be recognized by CTLs. The coadministration of demethylating agents such as 5-azacytidine might be helpful in upregulating weak TAA, and rendering the leukemia target more susceptible to CTL-induced cytotoxicity.16 Further studies on TAA expression in AML, including TAA expression by leukemia stem cells would be required to refine the repertoire of TAA used in the TAAmix.
Most of the antigens used for CTL generation are expressed in a wide range of hematological malignancies, including myelodys-plastic syndrome, advanced CML and acute lymphoblastic leukemia, as well as solid tumors. Currently, a similar strategy using patient-derived TAA-specific CTLs targeting multiple lymphoma antigens21 is being evaluated clinically (NCT01333046) outside of the context of allogeneic hematopoietic stem cell transplantation, demonstrating that this approach is feasible in the autologous setting. Therefore, by including the additional TAAs being targeted in this study, multi-TAA-specific CTLs could find application in the treatment of various diseases and within one T-cell product that might facilitate the generation of a universal antitumor T-cell therapy.
By testing TAA-specific CTLs against peptide pools, we were able to characterize a number of epitopes, and we could use this strategy to identify critical peptides inducing both CD8+ and CD4+ responses. In some cases the responses were class II-mediated in a region where HLA class I-restricted epitopes have been previously described. The possibility of class I and II response-inducing epitopes coexisting within the same region of the target antigen may improve immune responses in vivo when using this sequence for CTL generation or vaccination, inducing a combined CD4+ and CD8+ response, associated with enhanced functionality, increased TAA-CTL frequencies and prolonged persistence in vivo.6 Second generation peptide libraries could therefore be envisaged, where TAA pepmixes are spiked with a number of more relevant peptides tailored for an array of common HLA-antigens. Ultimately, as the number of identified individual epitopes is expanded, a smaller but focused pepmix of antigens could be used to maximize the induction of T-cell responses. Furthermore, the infusion of in vitro generated TAA-specific CTLs lends itself to strategies where infused T cells could be further boosted and expanded by vaccination with whole TAA pepmixes or a selection of predefined epitopes, resulting in longer T-cell persistence in vivo.
Although the majority of individuals tested were HLA-A2+, we did not find reactivity against previously described HLA-A2-restricted epitopes, indicating that other epitopes could be more immunogenic and effective in targeting AML. This is in contrast to observations in EBV-associated Hodgkin's lymphoma where HLA-A2 has been shown to be protective and associated with strong responses against epitopes presented by this HLA-allele.41
In conclusion, these data show that multi-TAA-specific CTLs can be generated from stem cell donors to prevent relapse of myeloid leukemia after SCT. Clinical scale-up of this technique and production of a good manufacturing practice-grade cell product should present no major obstacle. In designing a pilot clinical trial, the 3 week production process would permit the delivery of a T-cell infusion as early as the third week after SCT. A safety study with stopping rules for unacceptable graft-versus-host disease and off-target effects would be necessary. However, early infusion of CTLs could be beneficial, targeting the leukemia at a minimal residual disease state achieved by the leukemia suppressive conditioning regimen, and taking advantage of the homeostatic drive of lymphoproliferation.4 Successful application of this approach in AML SCT should encourage the use of multi-tumor antigen-specific T-cell infusions as a strategy to enhance the graft-versus-tumor effect after SCT.
Supplementary Material
Acknowledgments
This work was supported in parts by NIH grant 1P01CA148600-01 and CPRIT Grant RP100484. CMB was also supported by the career development award from the Leukemia Lymphoma Society and an award from the Gillson Longenbaugh Foundation.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
References
- 1.Barrett AJ. Understanding and harnessing the graft-versus-leukaemia effect. Br J Haematol. 2008;142:877–888. doi: 10.1111/j.1365-2141.2008.07260.x. [DOI] [PubMed] [Google Scholar]
- 2.Montagna D, Maccario R, Locatelli F, Montini E, Pagani S, Bonetti F, et al. Emergence of antitumor cytolytic T cells is associated with maintenance of hemato-logic remission in children with acute myeloid leukemia. Blood. 2006;108:3843–3850. doi: 10.1182/blood-2006-05-021535. [DOI] [PubMed] [Google Scholar]
- 3.Burnett AK, Knapper S. Acute Myeloid Leukemia. In: Treleaven J, Barrett AJ, editors. Haematopoietic Stem Cell Transplantation in Clinical Practice. Elsevier; 2009. [Google Scholar]
- 4.Porter DL, June CH. T-cell reconstitution and expansion after hematopoietic stem cell transplantation: T' it up! Bone Marrow Transplant. 2005;35:935–942. doi: 10.1038/sj.bmt.1704953. [DOI] [PubMed] [Google Scholar]
- 5.Rezvani K, Barrett AJ. Characterizing and optimizing immune responses to leukaemia antigens after allogeneic stem cell transplantation. Best Pract Res Clin Haematol. 2008;21:437–453. doi: 10.1016/j.beha.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fujiki F, Oka Y, Tsuboi A, Kawakami M, Kawakatsu M, Nakajima H, et al. Identification and characterization of a WT1 (Wilms Tumor Gene) protein-derived HLA-DRB1*0405-restricted 16-mer helper peptide that promotes the induction and activation of WT1-specific cytotoxic T lymphocytes. J Immunother. 2007;30:282–293. doi: 10.1097/01.cji.0000211337.91513.94. [DOI] [PubMed] [Google Scholar]
- 7.Weber G, Karbach J, Kuci S, Kreyenberg H, Willasch A, Koscielniak E, et al. WT1 peptide-specific T cells generated from peripheral blood of healthy donors: possible implications for adoptive immunotherapy after allogeneic stem cell transplantation. Leukemia. 2009;23:1634–1642. doi: 10.1038/leu.2009.70. [DOI] [PubMed] [Google Scholar]
- 8.Maslak PG, Dao T, Krug LM, Chanel S, Korontsvit T, Zakhaleva V, et al. Vaccination with synthetic analog peptides derived from WT1 oncoprotein induces T-cell responses in patients with complete remission from acute myeloid leukemia. Blood. 2010;116:171–179. doi: 10.1182/blood-2009-10-250993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rezvani K, Yong AS, Mielke S, Savani BN, Musse L, Superata J, et al. Leukemia-associated antigen-specific T-cell responses following combined PR1 and WT1 peptide vaccination in patients with myeloid malignancies. Blood. 2008;111:236–242. doi: 10.1182/blood-2007-08-108241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rezvani K, Yong AS, Savani BN, Mielke S, Keyvanfar K, Gostick E, et al. Graft-versus-leukemia effects associated with detectable Wilms tumor-1 specific T lymphocytes after allogeneic stem-cell transplantation for acute lymphoblastic leukemia. Blood. 2007;110:1924–1932. doi: 10.1182/blood-2007-03-076844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rezvani K, Price DA, Brenchley JM, Kilical Y, Gostick E, Sconocchia G, et al. Transfer of PR1-specific T-cell clones from donor to recipient by stem cell transplantation and association with GvL activity. Cytotherapy. 2007;9:245–251. doi: 10.1080/14653240701218524. [DOI] [PubMed] [Google Scholar]
- 12.Qazilbash M, Wieder E, Rios R, Lu S, Kant S, Giralt S, et al. Vaccination with the PR1 leukemia-associated antigen can induce complete remission in patients with myeloid leukemia. ASH Annu Meet Abstr. 2004;104:259. [Google Scholar]
- 13.Fujiwara H, El OF, Grube M, Price DA, Rezvani K, Gostick E, et al. Identification and in vitro expansion of CD4+ and CD8+ T cells specific for human neutrophil elastase. Blood. 2004;103:3076–3083. doi: 10.1182/blood-2003-07-2424. [DOI] [PubMed] [Google Scholar]
- 14.Rezvani K, Yong AS, Mielke S, Savani BN, Jafarpour B, Eniafe R, et al. Lymphode-pletion is permissive to the development of spontaneous T-cell responses to the self-antigen PR1 early after allogeneic stem cell transplantation and in patients with acute myeloid leukemia undergoing WT1 peptide vaccination following chemotherapy. Cancer Immunol Immunother. 2012;61:1125–1136. doi: 10.1007/s00262-011-1187-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rezvani K, Yong AS, Tawab A, Jafarpour B, Eniafe R, Mielke S, et al. Ex-vivo characterization of polyclonal memory CD8+ T-cell responses to PRAME-specific peptides in patients with acute lymphoblastic leukemia and acute and chronic myeloid leukemia. Blood. 2009;113:2245–2255. doi: 10.1182/blood-2008-03-144071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goodyear O, Agathanggelou A, Novitzky-Basso I, Siddique S, McSkeane T, Ryan G, et al. Induction of a CD8+ T-cell response to the MAGE cancer testis antigen by combined treatment with azacitidine and sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood. 2010;116:1908–1918. doi: 10.1182/blood-2009-11-249474. [DOI] [PubMed] [Google Scholar]
- 17.Rezvani K, Brenchley JM, Price DA, Kilical Y, Gostick E, Sewell AK, et al. T-cell responses directed against multiple HLA-A*0201-restricted epitopes derived from Wilms' tumor 1 protein in patients with leukemia and healthy donors: identification, quantification, and characterization. Clin Cancer Res. 2005;11(Pt 1):8799–8807. doi: 10.1158/1078-0432.CCR-05-1314. [DOI] [PubMed] [Google Scholar]
- 18.Schmitt M, Casalegno-Garduno R, Xu X, Schmitt A. Peptide vaccines for patients with acute myeloid leukemia. Expert Rev Vaccines. 2009;8:1415–1425. doi: 10.1586/erv.09.90. [DOI] [PubMed] [Google Scholar]
- 19.Bleakley M, Riddell SR. Exploiting T cells specific for human minor histocompatibility antigens for therapy of leukemia. Immunol Cell Biol. 2011;89:396–407. doi: 10.1038/icb.2010.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang YZ, Barrett J. The allogeneic CD4+ T-cell-mediated graft-versus-leukemia effect. Leuk Lymphoma. 1997;28:33–42. doi: 10.3109/10428199709058328. [DOI] [PubMed] [Google Scholar]
- 21.Gerdemann U, Katari U, Christin AS, Cruz CR, Tripic T, Rousseau A, et al. Cytotoxic T lymphocytes simultaneously targeting multiple tumor-associated antigens to treat EBV negative lymphoma. Mol Ther. 2011;19:2258–2268. doi: 10.1038/mt.2011.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoffmeister B, Kiecker F, Tesfa L, Volk HD, Picker LJ, Kern F. Mapping T cell epitopes by flow cytometry. Methods. 2003;29:270–281. doi: 10.1016/s1046-2023(02)00349-3. [DOI] [PubMed] [Google Scholar]
- 23.Feuchtinger T, Matthes-Martin S, Richard C, Lion T, Fuhrer M, Hamprecht K, et al. Safe adoptive transfer of virus-specific T-cell immunity for the treatment of systemic adenovirus infection after allogeneic stem cell transplantation. Br J Haematol. 2006;134:64–76. doi: 10.1111/j.1365-2141.2006.06108.x. [DOI] [PubMed] [Google Scholar]
- 24.Feuchtinger T, Opherk K, Bethge WA, Topp MS, Schuster FR, Weissinger EM, et al. Adoptive transfer of pp65-specific T cells for the treatment of chemorefractory cytomegalovirus disease or reactivation after haploidentical and matched unrelated stem cell transplantation. Blood. 2010;116:4360–4367. doi: 10.1182/blood-2010-01-262089. [DOI] [PubMed] [Google Scholar]
- 25.Leen AM, Myers GD, Sili U, Huls MH, Weiss H, Leung KS, et al. Monoculture-derived T lymphocytes specific for multiple viruses expand and produce clinically relevant effects in immunocompromised individuals. Nat Med. 2006;12:1160–1166. doi: 10.1038/nm1475. [DOI] [PubMed] [Google Scholar]
- 26.Kolb HJ, Schattenberg A, Goldman JM, Hertenstein B, Jacobsen N, Arcese W, et al. Graft-versus-leukemia effect of donor lymphocyte transfusions in marrow grafted patients. Blood. 1995;86:2041–2050. [PubMed] [Google Scholar]
- 27.Kolb HJ, Schmid C, Barrett AJ, Schendel DJ. Graft-versus-leukemia reactions in allogeneic chimeras. Blood. 2004;103:767–776. doi: 10.1182/blood-2003-02-0342. [DOI] [PubMed] [Google Scholar]
- 28.Bornhauser M, Thiede C, Platzbecker U, Kiani A, Oelschlaegel U, Babatz J, et al. Prophylactic transfer of BCR-ABL-, PR1-, and WT1-reactive donor T cells after T cell-depleted allogeneic hematopoietic cell transplantation in patients with chronic myeloid leukemia. Blood. 2011;117:7174–7184. doi: 10.1182/blood-2010-09-308569. [DOI] [PubMed] [Google Scholar]
- 29.Leen AM, Christin A, Myers GD, Liu H, Cruz CR, Hanley PJ, et al. Cytotoxic T lymphocyte therapy with donor T cells prevents and treats adenovirus and Epstein-Barr virus infections after haploidentical and matched unrelated stem cell transplantation. Blood. 2009;114:4283–4292. doi: 10.1182/blood-2009-07-232454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vera JF, Brenner LJ, Gerdemann U, Ngo MC, Sili U, Liu H, et al. Accelerated production of antigen-specific T cells for preclinical and clinical applications using gas-permeable rapid expansion cultureware (G-Rex) J Immunother. 2010;33:305–315. doi: 10.1097/CJI.0b013e3181c0c3cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanley PJ, Cruz CR, Savoldo B, Leen AM, Stanojevic M, Khalil M, et al. Functionally active virus-specific T cells that target CMV, adenovirus, and EBV can be expanded from naive T-cell populations in cord blood and will target a range of viral epitopes. Blood. 2009;114:1958–1967. doi: 10.1182/blood-2009-03-213256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gerdemann U, Keirnan JM, Katari UL, Yanagisawa R, Christin AS, Huye LE, et al. Rapidly generated multivirus-specific cytotoxic T lymphocytes for the prophylaxis and treatment of viral infections. Mol Ther. 2012;20:1622–1632. doi: 10.1038/mt.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bollard CM, Gottschalk S, Helen HM, Leen AM, Gee AP, Rooney CM. Good manufacturing practice-grade cytotoxic T lymphocytes specific for latent membrane proteins (LMP)-1 and LMP2 for patients with Epstein-Barr virus-associated lymphoma. Cytotherapy. 2011;13:518–522. doi: 10.3109/14653249.2011.561983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bollard CM, Gottschalk S, Leen AM, Weiss H, Straathof KC, Carrum G, et al. Complete responses of relapsed lymphoma following genetic modification of tumor-antigen presenting cells and T-lymphocyte transfer. Blood. 2007;110:2838–2845. doi: 10.1182/blood-2007-05-091280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wolfl M, Kuball J, Ho WY, Nguyen H, Manley TJ, Bleakley M, et al. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood. 2007;110:201–210. doi: 10.1182/blood-2006-11-056168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gottschalk S, Ng CY, Perez M, Smith CA, Sample C, Brenner MK, et al. An Epstein-Barr virus deletion mutant associated with fatal lymphoproliferative disease unresponsive to therapy with virus-specific CTLs. Blood. 2001;97:835–843. doi: 10.1182/blood.v97.4.835. [DOI] [PubMed] [Google Scholar]
- 37.Ma Q, Wang C, Jones D, Quintanilla KE, Li D, Wang Y, et al. Adoptive transfer of PR1 cytotoxic T lymphocytes associated with reduced leukemia burden in a mouse acute myeloid leukemia xenograft model. Cytotherapy. 2010;12:1056–1062. doi: 10.3109/14653249.2010.506506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ochi T, Fujiwara H, Suemori K, Azuma T, Yakushijin Y, Hato T, et al. Aurora-A kinase: a novel target of cellular immunotherapy for leukemia. Blood. 2009;113:66–74. doi: 10.1182/blood-2008-06-164889. [DOI] [PubMed] [Google Scholar]
- 39.Yong AS, Stephens N, Weber G, Li Y, Savani BN, Eniafe R, et al. Improved outcome following allogeneic stem cell transplantation in chronic myeloid leukemia is associated with higher expression of BMI-1 and immune responses to BMI-1 protein. Leukemia. 2011;25:629–637. doi: 10.1038/leu.2010.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmitt M, Schmitt A, Rojewski MT, Chen J, Giannopoulos K, Fei F, et al. RHAMM-R3 peptide vaccination in patients with acute myeloid leukemia, myelodysplastic syndrome, and multiple myeloma elicits immunologic and clinical responses. Blood. 2008;111:1357–1365. doi: 10.1182/blood-2007-07-099366. [DOI] [PubMed] [Google Scholar]
- 41.Niens M, van den BA, Diepstra A, Nolte IM, van der SG, Gallagher A, et al. The human leukocyte antigen class I region is associated with EBV-positive Hodgkin's lymphoma: HLA-A and HLA complex group 9 are putative candidate genes. Cancer Epidemiol Biomarkers Prev. 2006;15:2280–2284. doi: 10.1158/1055-9965.EPI-06-0476. [DOI] [PubMed] [Google Scholar]
- 42.Oka Y, Elisseeva OA, Tsuboi A, Ogawa H, Tamaki H, Li H, et al. Human cytotoxic T-lymphocyte responses specific for peptides of the wild-type Wilms' tumor gene (WT1) product. Immunogenetics. 2000;51:99–107. doi: 10.1007/s002510050018. [DOI] [PubMed] [Google Scholar]
- 43.Doubrovina E, Carpenter T, Pankov D, Selvakumar A, Hasan A, O'Reilly RJ. Mapping of novel peptides of WT-1 and presenting HLA alleles that induce epitope-specific HLA-restricted T cells with cytotoxic activity against WT-1(+) leukemias. Blood. 2012;120:1633–1646. doi: 10.1182/blood-2011-11-394619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knights AJ, Zaniou A, Rees RC, Pawelec G, Muller L. Prediction of an HLA-DR-binding peptide derived from Wilms' tumour 1 protein and demonstration of in vitro immunogenicity of WT1(124-138)-pulsed dendritic cells generated according to an optimised protocol. Cancer Immunol Immunother. 2002;51:271–281. doi: 10.1007/s00262-002-0278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lacey SF, La RC, Kaltcheva T, Srivastava T, Seidel A, Zhou W, et al. Characterization of immunologic properties of a second HLA-A2 epitope from a granule protease in CML patients and HLA-A2 transgenic mice. Blood. 2011;118:2159–2169. doi: 10.1182/blood-2011-04-349951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Piesche M, Hildebrandt Y, Chapuy B, Wulf GG, Trumper L, Schroers R. Characterization of HLA-DR-restricted T-cell epitopes derived from human proteinase 3. Vaccine. 2009;27:4718–4723. doi: 10.1016/j.vaccine.2009.05.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.