

Abstract
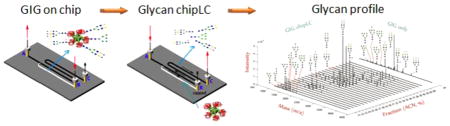
Glycosylation is one of the most common protein modifications and profoundly regulates many biological processes. Aberrant glycosylation is reported to associate with diseases such as cancers, human immunodeficiency virus, and immune disorders. It is considerably important to study protein glycosylation and the associated glycans for diagnostics and disease prognostics. Unlike other protein modifications, glycans attached to proteins are enormously complex. Therefore, the comprehensive analysis of glycans from biological or clinical samples is an unmet technical challenge. Development of the high-throughput method will facilitate the glycomics analysis. In this study, we developed a novel method for the high-throughput analysis of N-glycans from glycoproteins using glycoprotein immobilization for glycan extraction (GIG) coupled with liquid chromatography (LC) in an integrated microfluidic platform (chipLC). The separated glycans were then analyzed by mass spectrometry. Briefly, proteins were first immobilized on a solid support. Glycans on immobilized glycoproteins were modified on solid phase to increase the detection and structure analysis. N-Glycans were then enzymatically released and subsequentially separated by porous graphitized carbon particles packed in the same device. By applying the GIG-chipLC for glycomic analysis of human sera, we identified N-glycans with 148 distinct N-glycan masses. The platform was used to analyze N-glycans from mouse heart tissue and serum. The extracted N-glycans from tissues indicated that unique unsialylated N-glycans were detected in tissues that were missing from the proximal or distal serum, whereas common N-glycans from tissues and serum have mature and sialylated structures. The GIG-chipLC provides a simple and robust platform for glycomic analysis of complex biological and clinical samples.
Protein glycosylation is one of the most common and diverse protein modifications in which complex glycans are attached to glycoproteins. It is estimated that over 70% of all human proteins are glycosylated.1 Glycan biosynthesis relies on a great number of highly competitive processes involving glycosyltransferases and glycosidases, substrate availability, and the expression and structure of the glycosylated proteins and glycosylation sites. Therefore, protein glycosylation greatly depends on its biochemical environment.2 Aberrant glycosylation is likely associated with the occurrence of diseases such as cancers,3 inflammation,4 human immunodeficiency virus,5 and atherosclerosis,6,7 and thus, glycomics analysis could contribute to the discovery of novel disease biomarkers or therapeutics. In addition, glycans affect protein stability, binding, and immunogenicity; they play critical roles in developing glycoprotein therapeutics such as monoclonal antibodies. However, compared to genomics and proteomics, analytical techniques for glycomics lag far behind. The development of robust methods will facilitate the effective glycomics analysis.
Rapid isolation and separation of glycans from complex biological samples is crucial for glycomics analysis in order to analyze glycans by different instruments such as fluorescence spectroscopy and mass spectrometry (MS). While lectins can enrich glycans by affinity interactions, each lectin may be only effective on a certain type of glycans so that it lacks capability for global glycan enrichment and subsequent glycan profiling.8 Chromatographic methods such as size exclusion and hydrophilic columns are commonly used for global glycan enrichment and separation.9 However, sample loss is inevitable due to the physical methods. Solid-phase glycomics analysis via chemoselective approaches recently became popular for glycan isolation.10 For example, the hydrazide-functionalized beads react to aldehyde groups of reducing ends of glycans, and other nonconjugated molecules are removed for purification before release of the glycans from beads via hydrazone hydrolysis.10–12
The separation of glycans can be implemented in a number of chromatographic methods, such as size-exclusion chromatography, 13 high-performance anion-exchange chromatography, 14 capillary electrophoresis,15 hydrophilic interaction liquid chromatography,16 and reverse-phase liquid chromatography (RPLC).17,18 The choice of an optimal separation method generally depends on the glycans of interest to be analyzed. As a mature technique, RPLC is an effective method for the separation of glycans according to their structural elements.19 The retention of glycans is attributed to the creation of a solute-sized cavity in the stationary phase.20 One of the most common sorbents includes porous graphitized carbons (PGC) because of its remarkable selectivity for isomers and increased retention particularly for charged glycans.21–26 The strong absorption of polar glycans on the PGC compared to other sorbents allows for the analysis of both native and modified glycans.27–29
Microfluidics has been applied for glycan analysis, with miniaturized devices offering a number of advantages over traditional lab practices such as high integration, high throughput, versatile interfacing, low consumption on samples and reagents, and fast analysis.30,31 Microfluidics has thus attracted a lot of attention upon being proposed in the early 1990s32,33 and since then has been successfully demonstrated for analysis of amino acids,34 DNA,35 metabolites,36,37 proteins,38,39 and peptides.40,41 Native or derivatized glycans have been analyzed by chip-based liquid chromatography42,43 or hydrophilic interaction chromatography or porous graphite carbon column such as Agilent ChipCube LC-MS.18 In the latter system, glycans are separated by chip packed with PGC particles and directly introduced into an MS by electrospray ionization. Liquid chromatography (LC) in an integrated microfluidic platform (chipLC) can separate glycan isomers containing the same masses that MS analysis alone fails to distinguish.
Matrix-assisted laser desorption/ionization (MALDI) has been one of the popular methods for a snapshot of the mass profiles along with glycan structures.44,45 However, the sialylated glycans are fragile and have in-source decay during MALDI ionization without modification. To increase the detectability and stabilize sialylated glycans, they are often modified before MALDI-MS analysis. Recently, we reported the development of a robust method on the basis of a chemoselective technique for sialylated glycan modification and enrichment, termed as glycoprotein immobilization for glycan extraction (GIG).46,47 Proteins are covalently immobilized onto the solid support, and glycans from immobilized glycoproteins are released enzymatically or chemically for glycomic analysis. The immobilization of proteins on solid-phase also facilitates derivatization of glycans.42,43 The technique has been applied for the analysis of human serum with excellent coverage of N-glycans. 46 Over 66 N-glycans were detected in 0.2 μL of human serum proteins by MALDI-MS without separation. However, it is estimated that over 350 N-glycans are expected in human serum.48 It is thus possible to further improve glycan coverage by separation of glycans extracted from GIG.
In this study, glycans were first isolated from glycoproteins by a chemoenzymatic method and subsequently cleaned and separated by graphitized carbon chromatography in the integrated ChipLC system for glycan profiling using MALDIMS. Briefly, proteins were first conjugated to aldehyde beads packed in a microchip, and sialylated glycans were labeled on the beads. N-Glycans were released from beads by PNGase F and separated on the porous graphitized carbon particles (PGC). Finally, glycans were analyzed by MALDI-MS.
MATERIALS AND METHODS
Materials
AminoLink resin was from Pierce (Thermo Fisher Scientific Inc.; Rockford, IL); peptide-N-glycosidase F (PNGase F), denaturing buffer, and G7 were from New England BioLabs (Ipswich, MA). p-Toluidine, ribonuclease B (RNase B) from bovine pancreas, 2,5-dihydroxybenzoic acid (DHB), N,N-dimethylaniline (DMA), and carbon (mesoporous; particle size, 45 ± 5 μm; pore size, 100 ± 10 Å) were purchased from Sigma-Aldrich (St. Louis, MO). Mouse heart and serum were provided by Dr. Xingde Li from Biomedical Engineering Department, Johns Hopkins University. Pooled human sera were collected from healthy men with the approval of the Institutional Review Board of Johns Hopkins University. All other chemicals were purchased from Sigma-Aldrich unless specified. Microchip design and fabrication were described in the Supporting Information materials.
GIG Glycan Extraction
The detailed procedure of GIG was described in our previous work.46 Briefly, proteins (20 μL of RNase B at 10 μg/μL; 20 μL of human serum; or 400 μg of mouse serum or tissue proteins) were denatured in a total of 200 μL of binding buffer (40 mM sodium citrate and 20 mM sodium carbonate, pH 10) at 100 °C for 10 min. AminoLink beads packed in microchip were washed with 400 μL of binding buffer (pH 10) at a flow rate of 20 μL/min (note: the flow rate remains the same unless specifically mentioned) using a syringe pump (Pump 11 Elite Infusion/Withdrawal Programmable Dual Syringe; Harvard Apparatus; Holliston, MA). Protein solution was then infused into the channel from B to A and incubated for 2 h to conjugate proteins to AminoLink beads. The conjugated proteins were reduced by NaCNBH3 for 2 h by infusing 50 mM NaCNBH3 in 1× PBS from B to A. The free aldehyde groups on the beads were further blocked by 1 M Tris-HCl in the presence of 50 mM NaCNBH3. To prepare the solution for sialic acid modification, p-toluidine (47 mg) was dissolved in 367 μL of DI and 33 μL of HCl (36–38%). Then, EDC (40 μL) and HCl (25 μL) were added to 400 μL of p-toluidine solution, and final pH was adjusted to 4–6 by addition of either EDC (increase pH) or HCl (decrease pH). The p-toluidine solution (460 μL) was infused into the channel from B to A and incubated for 4 h while the needle C was capped. After washing the channel from B to A with 10% formic acid, 10% acetonitrile, 1 M sodium chloride and DI in sequence, PNGase F (4 μL) was injected in channel B after mixing with 4 μL of G7 buffer (New England Biolabs) in 32 μL of DI water and incubated for 2 h at 37 °C. All needles were capped to avoid buffer evaporation during incubation.
ChipLC Using Porous Graphitized Carbon
To condition porous graphitized carbons (PGC), the port B was capped while 200 μL of 80% acetonitrile (0.1% FA) was flushed through channel A to C; 400 μL of 0.1% FA was then injected through the same channel (Figure 1). This step was performed before proteins were immobilized to the beads described above. Following the glycan release from solid support, washing solution (400 μL, 0.1% formic acid (FA)) was injected from needle B while C was capped. Glycans were enriched at the intersection between channel A–B and A–C for further separation. The needle B was then capped while the needle A was connected to C for collecting elution. The eluate consisted of 0.1% FA in a variety of solutions consisting of acetonitrile and HPLC grade water, starting from 0% acetonitrile up to 80%. The details of acetonitrile concentration used are given in the results.
Figure 1.

GIG-chipLC for glycan analysis. The schematic diagram of the GIG-chipLC for glycan capture and separation and the location of A, B, and C for reservoirs and needle insertion. Two constrained channels (I and II) were constructed to block Aminolink beads and porous graphitized carbon particles (i); capture of proteins by infusing the proteins into the Aminolink bead-packed channel (ii); separation of released glycans in porous graphitized carbon particles (iii).
MALDI-TOF MS Analysis
Glycan fractions eluted from microchip were analyzed by an AXIMA Resonance mass spectrometer (Shimadzu; Columbia, MD). The MALDI matrix was prepared by mixing 4 μL of DMA in 200 μL of DHB (100 μg/μL in 50% acetonitrile, 0.1 mM NaCl) to increase the ionization of glycans. This matrix can form uniform crystals and improve glycan signal by enhancement of laser power absorption and ionization efficiency. The laser power was set to 100 for 2 shots each in 100 locations per spot. The assignment of glycan structures is described in our previous works.46
RESULTS AND DISCUSSION
Glycan isolation, modification, and separation have been widely achieved by chromatographic approaches in which glycans are first deglycosylated from glycoproteins by enzymatic digestion, separated from proteins and purified by columns, and derivatized by permethylation, reducing end labeling, or sialic acid modification before MS. These glycomic analysis procedures are time-consuming and could cause sample loss during the multistep sample handling.
In our newly developed approach for glycan identification, different components for glycan isolation, derivatization, and separation are integrated on a microchip. The component for glycoprotein capture and modifications (A–B) is virtually connected to the second component for glycan enrichment and separation (A–C). The interface between A–B and A–C (Figure 1i) is created by using a constrained channel I, which serves as a weir structure during bead packing. AminoLink beads are packed in the channel between B and the interface. They are firmly constrained in this channel since AminoLink beads are approximately 125 μm in diameter, whereas the diameter of the constrained channel I is 50 μm. Similarly, PGC (45 ± 5 μm) are fixed in the separation channel via prepacked AminoLink beads in the constrained channel II (Figure 1ii). The flow direction is turned on or off by capping the appropriate needle port (A, B, or C). With C capped, proteins are injected from B and conjugate on the AminoLink beads. Modifications on sialic acids are also conducted through infusion of p-toluidine solution from B. N-Glycans are released by PNGase F and eluted to separation component with port A off (Figure 1iii). The N-glycans are enriched at the interface in the separation channel by washing solution (0.1% FA), while salts and PNGase F buffer are removed. By capping the port B as shown in Figure 1ii, N-glycan fractions are collected through the port C without need of external valves.
As a proof-of-concept, we used standard glycoprotein, RNase B, for on-chip protein capture, glycan enzymatic release, and purification using PGC before detection by MALDI-MS. To demonstrate the performance of GIG on chip, 100 μL of beads (AminoLink Resin) was packed in microchip. Then, RNase B (200 μg) was conjugated to beads, and released N-glycans were purified and eluted from PGC using 40 μL of 80% acetonitrile in the presence of 0.1% FA. We observed all five N-glycans extracted from RNase B (Supporting Information Figure 1). Five N-glycans, Man5 to Man9, were observed according to their accurate masses.49,50 Consistent with previous reports, Man5 was the most dominant oligomannose in RNase B.51,52 These results indicate that GIG protein capture and glycan release and purification on the microchip is feasible for N-glycan analysis.
After glycans were captured and released using GIG in the first component of the chip, the second component packed with PGC could further improve the performance of glycan analysis by LC separation. We thus apply GIG-chipLC to extract N-glycans from complex samples, mouse blood serum (MBS), and mouse heart tissue (MT). To stabilize sialic acids and improve their hydrophobicity for MALDI-MS detection, p-toluidine was used to modify sialylated glycans on solid phase before glycan release.
To validate the method of sialic acid modification, we used a sialylglycopeptide (SGP) (Fushimi Pharmaceutical Co., Ltd.; Marugame, Kagawa, Japan) which contains six amino acids (Lys–Val–Ala–Asn–Lys–Thr).46 SGP (100 μg) was immobilized on beads as described in the Materials and Methods. p-Toluidine (400 μL) in the presence of EDC (N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide; Sigma) at pH 4–6 was added to the immobilized SGP. Carbodiimide coupling was terminated by replacing reaction buffer with 400 μL of 1% formic acid. We stopped the reaction at 0, 10, 30, 60, 120, and 240 min in triplicate. The internal standard DP7 (1 μL/1 μM; Sigma) was spiked to normalize the intensity. The reaction efficiency of sialic acid is given in Table 1. From specification of SGP (Fushimi), a negligible amount of glycans at 1663.2 (Da, [H]+) and 1977.3 (Da, [2Na]+) was observed. Without modification (0 min), 46% of glycan at 1663.2 and 27% of glycan at 1977.3 were observed in MALDI-MS; while only 25% of biantennary sialic acid (2290.2 Da, [3Na]+) was detected. The former two glycans are detected in MALDI due to the loss of two and one sialic acid. By continuous reaction, these two glycans gradually decreased to below 5% while the biantennary sialic acid (2424.3 Da) increased up to 93%. Some of the remained amount of glycan (~4%) at 1663.2 might come from the original sample. We did not observe one or two native sialic acids (peaks at 2290.2 and 2357.3 Da), indicating the complete labeling of biantennary sialic acids (2290.2 Da). Thus, carbodiimide coupling on solid phase is an effective method for sialylated glycan modification.
Table 1.
Reaction Efficiency of Sialic Acid by p-Toluidine Carbodiimide Coupling in the Presence of EDC at pH 4–6a
| Glycan | Glycan abundance after sialic acid modification by Carbodiimide coupling (normalized by DP7) (%)
|
Remarks | |||||
|---|---|---|---|---|---|---|---|
| 0 min | 10 min | 30 min | 60 min | 120 min | 240 min | ||
| 1663.2 |
46.5 ± 6.5 | 25.5 ± 2.5 | 13.5 ± 6.3 | 8.0 ± 3.0 | 7.5 ± 3.2 | 4.1 ± 0.9 | Loss two sialic acids |
| 1977.3 |
27.3 ± 3.1 | 17.02 ± 1.6 | 5.8 ± 1.5 | 2.2 ± 0.4 | 1.1 ± 0.5 | 0.24 ± 0.1 | Loss one sialic acid, without modification |
| 2290.2 |
25.58 ± 1.38 | 14.9 ± 4.0 | 4.6 ± 0.8 | 1.9 ± 0.3 | 1.1 ± 0.3 | 0.71 ± 0.3 | Two sialic acids, without modification |
| 2357.2 |
0.40 ± 0.01 | 27.7 ± 4.4 | 27.7 ± 2.9 | 18.0 ± 2.9 | 14.8 ± 1.9 | 1.78 ± 0.3 | Two sialic acids, one of them modified by p-toluidine |
| 2424.3 |
0.27 ± 0.09 | 14.8 ± 3.8 | 49.3 ± 4.6 | 69.8 ± 9.7 | 75.4 ± 5.9 | 93.2 ± 3.4 | Both sialic acids modified by p-toluidine |
100 μg of SGP is immobilized to 100 μL of Aminolink beads for carbodiimide coupling. An internal standard DP7 (1 μL, 1μM in DI) is spiked to normalize the intensity. blue ■: N-acetylglucosamine; green ●: mannose; yellow ●: galactose; pink ◆: N-acetylneuraminic acid; pink and gray ⬣: p-toluidine.
We then applied this method for mouse tissue and serum analysis. To demonstrate the reproducibility of chipLC, triplicate experiments were performed on both serum and tissue by glycan extraction, labeling, purification, and MS analysis. Tissue was first sonicated for 30 s in RIPA buffer at an interval of 30 s on ice for 3 min.46 The loading for each sample was 400 μg, and sialylated glycans were modified before release of N-glycans (in triplicates; three 400 μg of MT and three 400 μg of MBS; all from the same mouse). Glycans were analyzed by MALDI-MS without/with chipLC separation. When the released glycans were analyzed without chipLC separation, the PGC component was used as glycan purification cartridge and the glycans were eluted from the column with 80% acetonitrile in one fraction as described for RNase B analysis. On the basis of the MS analysis of the released glycans from mouse serum and heart tissue, several interesting results were observed. First, oligomannoses and complex N-glycans were detected in both serum and heart tissue. Second, tissue contained a higher level of oligomannoses than those from mouse blood serum (Figure 2A,B). Third, the level of sialylated glycans was higher in mouse blood serum compared to those from heart tissue (Figure 2A,B). Some of those abundant sialylated glycans have been previously reported in mouse serum,53 such as bi- or triantennary Nen5Gc glycans. Overall, without LC separation, glycans were detected in both MT and MBS. However, tissue-or serum-specific glycans, likely presented in low abundance and not detected in these spectra, might be detectable when the glycans were separated prior to MS analysis.
Figure 2.

N-Glycans from mouse serum or heart tissue by GIG without chipLC. (A) MS spectra of mouse glycans from blood serum (MBS) (ii) and mouse heart tissue (MT) (i) by GIG and MS. The number representation of several N-glycans is given in Supporting Information Table 1, where Man5–Man9 are oligomannoses. (B) Relative abundance of oligomannoses and sialylated N-glycans from mouse serum and heart tissue. Each sample is performed in triplicate.
To determine whether LC separation allowed one to detect specific glycans from heart tissue or blood serum, the extracted N-glycans from MBS and MT described above were separated by chipLC using stepwise acetonitrile elution (1% step from 0 to 80%) with 40 μL/fraction (flow rate: 10 μL/min). Each fraction was analyzed using MALDI-MS. We observed 65 distinct N-glycan masses commonly detected in both MBS and MT, including most fucosylated and sialylated glycans. The complete glycan list is given in Supporting Information Table 1. By GIG-chipLC, we detected 96 N-glycan masses in MBS and 72 N-glycan masses in MT. Seven N-glycan masses were only detected in MT, while 31 N-glycan masses were only detected from MBS. These results show that the microchip with integrated GIG and ChipLC facilitates the identification of tissue-specific glycans that might be relevant to the physiological or pathological status of the tissue. Several studies have focused on the identification of disease-associated glycan changes in blood serum due to its easy accessibility and noninvasive diagnosis;3,54–57 however, blood serum likely contains a large number of N-glycans, which are secreted from different organs. Alternatively, it might be more informative to study glycans from specific tissue of interest for organ-specific glycan changes. Therefore, specific glycan analysis of disease affected tissues could be an effective means for identification of abnormal glycans associated with a specific disease.
The N-glycans eluted in different percentages of acetonitrile by chipLC fraction were compared with the N-glycans without chipLC separation in a pseudo-3D plot (Figure 3; peak number corresponds to the glycan listed in Supporting Information Table 1). The high-abundance N-glycans, which were detected by MALDI-MS without chipLC fraction, was also prominent after chipLC separation, including oligomannoses and bi- and triantennary sialic acids (Figure 3). Oligomannoses were dominant N-glycans in mouse heart tissue (Figure 3i), while the sialylated glycans were mostly abundant in mouse blood serum (Figure 3ii). Remarkably, chipLC was reasonably reproducible on glycan fractionation (Supporting Information Figure 2). For example, Man5–Man9 glycans with masses lower than 2000 Da were eluted in a fraction of 21% acetonitrile across duplicated chipLC, while biantennary sialylated glycan (71) was eluted in 30% acetonitrile fraction and triantennary sialylated glycan (98) was detected at 37% acetonitrile fraction. Additionally, the reproducible tests using MBS (400 μg) showed (Supporting Information Table 3) the majority of N-glycans elutes in the same corresponding fraction, although the intensity of each N-glycans is varied after GIG-chipLC separation, ranging from 50 to 100%. In order to provide semiquantitative information, further improvement is required. The N-glycans extracted from GIG can be ideally modified using isobaric Aldehyde Reactive Tags (iARTs)58 and further separated using chipLC for accurate quantitation. Alternatively, use of electrospray ionization could improve quantification reproducibility without matrix effect as in MALDI.59
Figure 3.

Profiling of N-glycans from mouse serum or heart tissue by GIG-chipLC. Mouse blood serum (MBS) (ii) and mouse heart tissue (MT) (i) were analyzed by GIG-chipLC and MS. Oligomannoses were present in two fractions with acetonitrile concentrations at 21% and 22%; sialylated glycans were detected in eluents from 25% to 28% and 30% to 35%; triantennary and quadantennary sialylated glycans were detected in 40% and above.
The reproducible glycan elution in specific acetonitrile fraction from chipLC also indicated that PGC might be able to separate glycan isomers with the same mass (Figure 3). For example, oligomannoses (5, 10, 23, 46, 56) were coeluted in 21% acetonitrile; we also observed that a high intensity peak (5) was also eluted in 22% of acetonitrile, as well as a small amount of peak 10 and 23 in the same fraction (22%). These results could not have resulted from the incomplete elution of Man5–Man7 glycans by 21% acetonitrile since no glycans were detected at the end of 40 μL/fraction elution (flow rate: 10 μL/min). Similarly, biantennary sialic acid (71) was observed from fraction 30% to 34% and 37% but not between 34% and 36% (Figure 3ii). These results might suggest that isomers of these glycans were probably eluted in different fractions. According to a detailed study on oligomannose structures by tandem MS analysis, Man5, Man6, and Man7 have several isomers.52,60 For example, Man5 from RNase B has potentially four isomers, including one triantennary and three biantennary structures. It is possible that different isomers are eluted in 21% or 22% of acetonitrile. Similarly, Man6 isomers are eluted in 21% and 22% fractions. Additional studies would be needed to determine the identities of different oligomannose isomers after glycan permethylation, since PGC has good selective interactions with methyl substituted groups to further increase glycan retention. Identification of isomers by chipLC with permethylated glycans will further delineate glycan structures. Investigation on oligomannose isomers could be very useful to understanding the changes of oligomannoses in diseases such as the alterations of gp120 glycosylation in HIV.61
Glycomic analysis of complex biological samples by GIG and MALDI-MS showed increased glycan coverage by detecting low abundant glycans.46 When GIG was applied to the analysis of N-glycans from human serum, we detected 65 N-glycan masses without glycan fractionation (Figure 4i and Supporting Information Table 2). Over 40% of the detected 65 N-glycan masses were sialylated in which 13 N-glycans were also fucosylated. When GIG-chipLC was used to analyze N-glycans isolated from human serum with chipLC fraction, the number of N-glycan masses detected by MALDI-MS increased to 148 (Figure 4ii and Supporting Information Table 2). We detected 79 sialylated glycan masses and an additional 41 fucosylated glycan masses. The increased number of N-glycans observed after chipLC separation could be attributed to reduced dynamic range of glycan in each fraction. As a result, low-abundance glycans were effectively eluted in a specific fraction by PGC and detected by MALDI-MS without the ion competition of other high abundant glycan ions.
Figure 4.
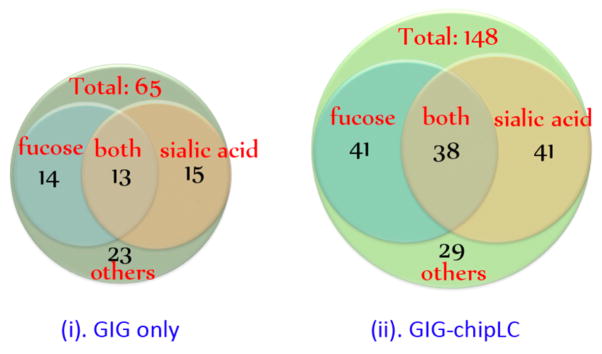
N-Glycan coverage from human serum. A total of 65 N-glycan masses were detected from human serum by GIG without LC fractionation (i); a total of 148 N-glycan masses were detected from human serum with GIG-chipLC (ii). The extracted N-glycans from human serum using GIG was directly analyzed by MALDI-MS.
The number of N-glycan structures could be much larger than the 148 N-glycan mass peaks detected from serum (Figure 4ii). Each N-glycan mass can potentially correspond to a number of isomers, sometimes over a dozen for a single mass.54 Importantly, each isomer may have its own physical and biological properties. Different isomers can further be distinguished by glycan permethylation and tandem MS. Permethylation of glycans can be implemented in the microfluidic system by packing sodium hydroxide particles in a microchip platform. Capillary permethylation has been one of the emerging technologies for glycan derivatization.62 The extracted glycans from GIG can be infused into a sodium hydroxide-packed microchip for glycan permethylation, followed by chipLC profiling. This innovation platform could be beneficial for glycomics analysis by providing linkage information corresponding to assigned glycan structures.
The use of integrated microchip for glycan isolation and separation described in this study demonstrates several advantages over traditional chromatography. In addition to advantages of microchip for solid-phase glycan extraction, the mesoporous PGC used for glycan separation has remarkably high surface area. The PGC particle size is around 45 μm in our study. The surface area in a single separation channel was estimated as follows: The channel cross section is 800 μm × 800 μm and length is 20 mm. Thus, the total separation channel volume equals 0.0128 cm3; the density of carbon particles is 0.5 cm3/g. Thus, a total of 25.6 mg of particles can be packed in the separation channel; the estimated surface area of packed carbon particles is 6.4 m2 based on its specific surface area. The commercial PGC, e.g., Hypercarb made by Thermo Scientific,63 has an average pore size of 250 Å and the specific surface area of 120 m2/g, that is 3.1 m2 for the same volume of particles. Subsequently, longer commercial columns are needed to achieve the same surface area of the fabricated microchip, resulting in increased back-pressure during separation. In addition, our microchip is low-cost, and separation particles can be repacked without sacrificing separation performance.
CONCLUSIONS
The glycans attached to glycoproteins are enormously complex due to their nontemplate biosynthesis. This complexity makes the comprehensive analysis of glycans from biological or clinical samples an unmet technical challenge. Development of high-throughput methods is indispensable for facilitating the glycomic analysis. We developed an integrated method which consists of high-throughput glycoprotein immobilization for glycan extraction (GIG) and porous graphitized carbon liquid chromatography (PGC-LC) for on-chip sialylated N-glycan modification, extraction, and separation. ChipLC by PGC minimizes dynamic range of glycan in fractions, resulting in detection of low-abundance glycans. In addition to an increase in the coverage of detected N-glycans by GIG-chipLC, it can facilitate MALDI-based analysis of N-glycans without the need for permethylation. Glycan isomers could also be separated by chipLC.
The developed GIG-chipLC can be used to analyze glycans from tissue and sera samples, thus providing a reliable tool for glycomic analysis. The reproducible performance in terms of glycan elution fraction (Supporting Information Table 3 and Supporting Information Figure 2) and ability to detect unique glycans from tissue samples could provide a powerful means for discovery of abnormal glycans associated with disease states.
Supplementary Material
Acknowledgments
The authors thank Dr. Lori Sokoll for providing human sera and acknowledge instrument support from Shimadzu Scientific Instruments. This work was supported by National Institutes of Health, National Cancer Institute, the Early Detection Research Network (EDRN, U01CA152813), the Clinical Proteomic Tumor Analysis Consortium (CPTAC, U24CA160036), National Institutes of Health, National Heart Lung and Blood Institute Programs of Excellence in Glycosicences (PEG, P01HL107153), and Johns Hopkins Proteomics Center (N01-HV-00240).
Footnotes
The authors declare no competing financial interest.
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Apweiler R, Hermjakob H, Sharon N. Biochim Biophys. 1999;1473:4–8. doi: 10.1016/s0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- 2.Bertozzi CR, Sasisekharan R. In: Essentials of Glycobiology. 2. Varki A, Cumings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2009. p. 784. [PubMed] [Google Scholar]
- 3.Kyselova Z, Mechref Y, Al Bataineh MM, Dobrolecki LE, Hickey RJ, Vinson J, Sweeney CJ, Novotny MV. J Proteome Res. 2007;6:1822–1832. doi: 10.1021/pr060664t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kyriakis JM, Avruch J. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 5.Montefiori DC, Robinson WE, Mitchell WM. Proc Natl Acad Sci. 1988;85:9248–9252. doi: 10.1073/pnas.85.23.9248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hansson GK. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 7.Lakatta EG. Circulation. 2003;107:490–497. doi: 10.1161/01.cir.0000048894.99865.02. [DOI] [PubMed] [Google Scholar]
- 8.Qiu R, Regnier FE. Anal Chem. 2005;77:2802–2809. doi: 10.1021/ac048751x. [DOI] [PubMed] [Google Scholar]
- 9.Qin H, Zhao L, Li R, Wu R, Zou H. Anal Chem. 2011;83:7721–7728. doi: 10.1021/ac201198q. [DOI] [PubMed] [Google Scholar]
- 10.Yang S, Zhang H. Proteomics Clin Appl. 2012;6:596–608. doi: 10.1002/prca.201200045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miura Y, Hato M, Shinohara Y, Kuramoto H, Furukawa J, Kurogochi M, Shimaoka H, Tada M, Nakanishi K, Ozaki M. Mol Cell Proteomics. 2008;7:370–377. doi: 10.1074/mcp.M700377-MCP200. [DOI] [PubMed] [Google Scholar]
- 12.Yang SJ, Zhang H. Anal Chem. 2012;84:2232–2238. doi: 10.1021/ac202769k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Packer NH, Lawson MA, Jardine DR, Redmond JW. Glycoconjugate J. 1998;15:737–747. doi: 10.1023/a:1006983125913. [DOI] [PubMed] [Google Scholar]
- 14.Lee Y. J Chromatogr, A. 1996;720:137–149. [Google Scholar]
- 15.Okafo G, Burrow L, Carr SA, Roberts GD, Johnson W, Camilleri P. Anal Chem. 1996;68:4424–4430. doi: 10.1021/ac960721+. [DOI] [PubMed] [Google Scholar]
- 16.Alpert AJ. J Chromatogr, A. 1990;499:177–196. doi: 10.1016/s0021-9673(00)96972-3. [DOI] [PubMed] [Google Scholar]
- 17.Bigge J, Patel T, Bruce J, Goulding P, Charles S, Parekh R. Anal Biochem. 1995;230:229–238. doi: 10.1006/abio.1995.1468. [DOI] [PubMed] [Google Scholar]
- 18.Alley WR, Jr, Madera M, Mechref Y, Novotny MV. Anal Chem. 2010;82:5095–5106. doi: 10.1021/ac100131e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen X, Flynn GC. Anal Biochem. 2007;370:147–161. doi: 10.1016/j.ab.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 20.Sentell KB, Dorsey JG. Anal Chem. 1989;61:930–934. doi: 10.1021/ac00184a003. [DOI] [PubMed] [Google Scholar]
- 21.Lipniunas PH, Neville DCA, Trimble RB, Townsend RR. Anal Biochem. 1996;243:203–209. doi: 10.1006/abio.1996.0507. [DOI] [PubMed] [Google Scholar]
- 22.Itoh S, Kawasaki N, Ohta M, Hyuga M, Hyuga S, Hayakawa T. J Chromatogr, A. 2002;968:89–100. doi: 10.1016/s0021-9673(02)00951-2. [DOI] [PubMed] [Google Scholar]
- 23.Morelle W, Michalski JC. Nat Protoc. 2007;2:1585–1602. doi: 10.1038/nprot.2007.227. [DOI] [PubMed] [Google Scholar]
- 24.Pabst M, Altmann F. Anal Chem. 2008;80:7534–7542. doi: 10.1021/ac801024r. [DOI] [PubMed] [Google Scholar]
- 25.Karlsson H, Halim A, Teneberg S. Glycobiology. 2010;20:1103–1116. doi: 10.1093/glycob/cwq070. [DOI] [PubMed] [Google Scholar]
- 26.Ruhaak LR, Deelder AM, Wuhrer M. Anal Bioanal Chem. 2009;394:163–174. doi: 10.1007/s00216-009-2664-5. [DOI] [PubMed] [Google Scholar]
- 27.Akira M, Toyoda H, Imanari T. Anal Sci. 1992;8:793–797. [Google Scholar]
- 28.Pabst M, Altmann F. Proteomics. 2011;11:631–643. doi: 10.1002/pmic.201000517. [DOI] [PubMed] [Google Scholar]
- 29.Ruhaak LR, Deelder AM, Wuhrer M. Anal Bioanal Chem. 2009;394:163–174. doi: 10.1007/s00216-009-2664-5. [DOI] [PubMed] [Google Scholar]
- 30.Jacobson SC, Hergenroder R, Koutny LB, Ramsey JM. Anal Chem. 1994;66:1114–1118. [Google Scholar]
- 31.Whitesides GM. Nature. 2006;442:368–373. doi: 10.1038/nature05058. [DOI] [PubMed] [Google Scholar]
- 32.Manz A, Graber N, Widmer H. Sens Actuators, B: Chem. 1990;1:244–248. [Google Scholar]
- 33.Harrison DJ, Fluri K, Seiler K, Fan Z, Effenhauser CS, Manz A. Science. 1993;261:895–895. doi: 10.1126/science.261.5123.895. [DOI] [PubMed] [Google Scholar]
- 34.Skelley AM, Scherer JR, Aubrey AD, Grover WH, Ivester RHC, Ehrenfreund P, Grunthaner FJ, Bada JL, Mathies RA. Proc Natl Acad Sci USA. 2005;102:1041–1046. doi: 10.1073/pnas.0406798102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woolley AT, Hadley D, Landre P, deMello AJ, Mathies RA, Northrup MA. Anal Chem. 1996;68:4081–4086. doi: 10.1021/ac960718q. [DOI] [PubMed] [Google Scholar]
- 36.Terabe S, Markuszewski MJ, Inoue N, Otsuka K, Nishioka T. Pure App Chem. 2001;73:1563–1572. [Google Scholar]
- 37.Ramautar R, Somsen GW, de Jong GJ. Electrophoresis. 2008;30:276–291. doi: 10.1002/elps.200800512. [DOI] [PubMed] [Google Scholar]
- 38.Yang S, Liu J, Lee CS, DeVoe DL. Lab Chip. 2008;9:592–599. doi: 10.1039/b805541f. [DOI] [PubMed] [Google Scholar]
- 39.Liu J, Yang S, Lee CS, DeVoe DL. Electrophoresis. 2008;29:2241–2250. doi: 10.1002/elps.200700608. [DOI] [PubMed] [Google Scholar]
- 40.Reichmuth DS, Shepodd TJ, Kirby BJ. Anal Chem. 2005;77:2997–3000. doi: 10.1021/ac048358r. [DOI] [PubMed] [Google Scholar]
- 41.Gao J, Xu J, Locascio LE, Lee CS. Anal Chem. 2001;73:2648–2655. doi: 10.1021/ac001126h. [DOI] [PubMed] [Google Scholar]
- 42.Ninonuevo M, An H, Yin H, Killeen K, Grimm R, Ward R, German B, Lebrilla C. Electrophoresis. 2005;26:3641–3649. doi: 10.1002/elps.200500246. [DOI] [PubMed] [Google Scholar]
- 43.Bynum MA, Yin H, Felts K, Lee YM, Monell CR, Killeen K. Anal Chem. 2009;81:8818–8825. doi: 10.1021/ac901326u. [DOI] [PubMed] [Google Scholar]
- 44.Raman R, Raguram S, Venkataraman G, Paulson JC, Sasisekharan R. Nat Methods. 2005;2:817–824. doi: 10.1038/nmeth807. [DOI] [PubMed] [Google Scholar]
- 45.Harvey DJ. Expert Rev Proteomics. 2005;2:87–101. doi: 10.1586/14789450.2.1.87. [DOI] [PubMed] [Google Scholar]
- 46.Yang S, Yan L, Shah P, Zhang H. Anal Chem. 2013;85:5555–5561. doi: 10.1021/ac400761e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shah P, Yang S, Sun SS, Aiyata P, Yarema K, Zhang H. Anal Chem. 2013;85:3606–3613. doi: 10.1021/ac3033867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aldredge D, An HJ, Tang N, Waddell K, Lebrilla CB. J Proteome Res. 2012;11:1958–1968. doi: 10.1021/pr2011439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berman E, Walters DE, Allerhand A. J Biol Chem. 1981;256:3853–3857. [PubMed] [Google Scholar]
- 50.Blixt O, Head S, Mondala T, Scanlan C, Huflejt ME, Alvarez R, Bryan MC, Fazio F, Calarese D, Stevens J. Proc Natl Acad Sci USA. 2004;101:17033–17038. doi: 10.1073/pnas.0407902101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mitra N, Sinha S, Ramya TN, Surolia A. Trends Biochem Sci. 2006;31:156–163. doi: 10.1016/j.tibs.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 52.Prien JM, Ashline DJ, Lapadula AJ, Zhang H, Reinhold VN. J Am Soc Mass Spectrom. 2009;20:539–556. doi: 10.1016/j.jasms.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kurogochi M, Matsushista T, Amano M, Furukawa J, Shinohara Y, Aoshima M, Nishimura SI. Mol Cell Proteomics. 2010;9:2354–2368. doi: 10.1074/mcp.M110.000430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kirmiz C, Li B, An HJ, Clowers BH, Chew HK, Lam KS, Ferrige A, Alecio R, Borowsky AD, Sulaimon S. Mol Cell Proteomics. 2007;6:43–55. doi: 10.1074/mcp.M600171-MCP200. [DOI] [PubMed] [Google Scholar]
- 55.Hamid UMA, Royle L, Saldova R, Radcliffe CM, Harvey DJ, Storr SJ, Pardo M, Antrobus R, Chapman CJ, Zitzmann N. Glycobiology. 2008;18:1105–1118. doi: 10.1093/glycob/cwn095. [DOI] [PubMed] [Google Scholar]
- 56.de Leoz MLA, Young LJT, An HJ, Kronewitter SR, Kim J, Miyamoto S, Borowsky AD, Chew HK, Lebrilla CB. Mol Cell Proteomics. 2011;10:1–9. doi: 10.1074/mcp.M110.002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Callewaert N, Van Vlierberghe H, Van Hecke A, Laroy W, Delanghe J, Contreras R. Nat Med. 2004;10:429–434. doi: 10.1038/nm1006. [DOI] [PubMed] [Google Scholar]
- 58.Yang S, Yuan W, Yang W, Zhou J, Harlan R, Edwards J, Li S, Zhang H. Anal Chem. 2013;85:8188–8195. doi: 10.1021/ac401226d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wada Y, Azadi P, Costello CE, Dell A, Dwek RA, Geyer H, Geyer R, Kakehi K, Karlsson NG, Kato K. Glycobiology. 2007;17:411–422. doi: 10.1093/glycob/cwl086. [DOI] [PubMed] [Google Scholar]
- 60.Pabst M, Grass J, Toegel S, Liebminger E, Strasser R, Altmann F. Glycobiology. 2012;22:389–399. doi: 10.1093/glycob/cwr138. [DOI] [PubMed] [Google Scholar]
- 61.Sanders RW, Venturi M, Schiffner L, Kalyanaraman R, Katinger H, Lloyd KO, Kwong PD, Moore JP. J Virol. 2002;76:7293–7305. doi: 10.1128/JVI.76.14.7293-7305.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kang P, Mechref Y, Klouckova I, Novotny MV. Rapid Commun Mass Spectrom. 2005;19:3421–3428. doi: 10.1002/rcm.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thermo Electron Corporation. Hypercarb HPLC Columns Technical Guide. http://www.interscience.nl/promotiesites/hypersil/topics/promotiesites/hypersil/nieuws/hypercarb_technical.pdf.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.