

Abstract
Purpose.
The inhibitory effects and mechanism(s) of type IV collagen α-6 chain–derived noncollagenous domain (α6[IV]NC1 or hexastatin) on elastin-derived peptide (EDP)–activated choroidal endothelial cell migration, kinase signaling, and membrane type 1 metalloproteinase (MT1-MMP) activation are explored.
Methods.
Mouse choroidal endothelial cells (MCECs) were incubated in media with soluble EDPs (kappa elastin, mouse elastin, and Val-Gly-Val-Ala-Pro-Gly [VGVAPG] hexapeptide) for different time intervals with or without α6(IV)NC1. The MCECs proliferation, migration, tube formation, MT1-MMP expression, and angiogenic signaling were analyzed in cells subjected to EDP and α6(IV)NC1 treatments. The MCECs also were subjected to EDPs, and specific inhibitors for evaluation of focal adhesion kinase (FAK) and protein kinase B (Akt) phosphorylation.
Results.
Kappa elastin, mouse elastin, and VGVAPG enhanced the migration, without affecting the proliferation of MCECs. The α6(IV)NC1 inhibited survival and EDP-activated migration of MCECs. The EDP-activated MCEC tube formation on matrigel also was inhibited by α6(IV)NC1. Further, EDP-activated MT1-MMP expression and FAK/phosphoinositide-3-kinase (PI-3K)/mammalian target of rapamycin (mToR)/Akt phosphorylation in MCECs, were reduced by α6(IV)NC1. The EDP-induced FAK and Akt phosphorylation was blocked by FAK- and Akt-specific inhibitors.
Conclusions.
The EDPs and α6(IV)NC1 are identified to exhibit opposing effects on MCEC angiogenic behavior and signaling. The α6(IV)NC1 inhibited cell survival, EDP-mediated migration, MT1-MMP expression and, FAK/PI-3K/mToR/Akt phosphorylation in MCECs. This work demonstrates α6(IV)NC1 as a prospective endogenous molecule for the treatment of diseases involving choroidal neovascularization in the eye.
Keywords: elastin-derived peptides, noncollagenous domains of type IV collagen, angiogenesis
Type IV collagen α-6 chain–derived noncollagenous domain (α6[IV]NC1) inhibited different elastin-derived peptides (EDPs), and promoted MCEC migration and tube formation. Further, α6(IV)NC1 inhibited EDP-mediated angiogenic activation by inhibiting MT1-MMP expression by inhibiting FAK/PI-3K/mToR/Akt signaling.
Introduction
Pathologic progression of choroidal neovascularization (CNV) in the wet form of age-related macular degeneration (AMD) includes the activation of angiogenic behavior in choroidal endothelial cells. This is promoted by different pathologic factors, including extracellular matrix (ECM) derivatives.1,2 Elastins and type IV collagen constitute major components of ECM in different tissues, such as skin, lungs, vascular basement membrane (VBM) of blood vessels, and ocular tissues.3,4 Degradation of elastin was identified as a pathologic factor leading to the progression of CNV, through substantial histologic, genetic, and clinical studies, with significant focus on elastin degradation products (EDPs).1,2 Histologic studies revealed conspicuous fragmentation and decrease in the thickness of Bruch's membrane–elastin layer in patients with AMD. Thus, damage in elastic Bruch's membrane was identified to promote intrusion of pathologic choriocapillaries in CNV.5–7 Elevation of serum EDP levels in patients with CNV further established the role of elastolysis in CNV of AMD.2 Such increased EDPs in CNV were generated through the elastolysis in skin, Bruch's membrane, and choriocapillaries. Thus, elastolysis in the extraocular tissues also affected CNV.2,8,9 The role of EDPs in promoting CNV has been demonstrated at cellular level through the elucidation of angiogenic effects activated by the EDPs on choroidal endothelial cells. These studies used soluble elastins generated through the elastolysis of bovine ligament elastins (kappa-elastin [κE]) and the synthetic elastin-derived bioactive peptide, Val-Gly-Val-Ala-Pro-Gly hexapeptide (VGVAPG [BP]).10
In contrast with the generation of angiogenic promoting EDPs through elastin metabolism, type IV collagen metabolism leads to the release of noncollagenous domains (NC1) with antiangiogenic properties on endothelial cells (ECs).11–18 Type IV collagen α-2 chain–derived noncollagenous domain (α2[IV]NC1) induced apoptotic activity in ECs and suppressed CNV.19 The NC1 domain of type IV collagen α-1 sub-chain suppressed angiogenic activation in retinal endothelial cells.20 Thus, contrasting roles of elastin and type IV collagen derivatives have been established in regulation of CNV. However, these earlier studies characterized the individual roles of either EDPs or type IV collagen NC1 domains on angiogenesis. Our study investigated the combined effects of different EDPs and α6(IV)NC1 in regulating angiogenic signaling in mouse choroidal endothelial cells (MCECs). Recombinant α6(IV)NC1 exhibited antiangiogenic properties in vitro and in vivo, though to our knowledge its angioinhibitory signaling mechanism is not known yet.21–23 In our study, the effects of α6(IV)NC1 on EDP-mediated angiogenic activities and signaling were evaluated systematically using MCECs. We showed that α6(IV)NC1 inhibits MCECs proliferation, and EDP mediated angiogenic signaling by negatively regulating focal adhesion kinase (FAK)/phosphoinositide-3-kinase (PI-3K)/mammalian target of rapamycin (mToR)/protein kinase B (Akt) phosphorylation. This resulted in inhibition of membrane type 1 metalloproteinase (MT1-MMP) expression, reducing migratory potential.
Materials and Methods
Mouse Choroidal Endothelial Cell Culture
The MCECs used in our study were a gift from Sheibani Nader (University of Wisconsin, Madison, WI). The MCECs were cultured in endothelial cell growth Medium (EGM-2; Lonza, Walkersville, MD) on 1% gelatin-coated plates and used for all in vitro studies.24
Recombinant Expression and Purification of α6(IV)NC1
Recombinant human α6(IV)NC1 was cloned, expressed, and purified from bacterial inclusion bodies by column chromatography using the L-arginine renaturation method.22 Endotoxin levels in the purified protein were estimated using the Limulus amoebocyte lysate assay kit (Lonza), following manufacturer's instructions.
Cell Viability or Proliferation Assay
The MCECs were cultured in endothelial basal medium EGM-2 (Lonza) medium and approximately 80% confluence cells were subjected to serum starvation for 8 hours. Serum-starved cells were trypsinized, resuspended in media with respective treatments, and distributed into 96-well plates at density of 5 × 104 cells per well. The endothelial cell basal medium (EBM)-2 without serum was used as negative control and EMB-2 with 2% fetal calf serum (FCS) was used as positive control. Treated wells contained cells resuspended in EBM-2 medium containing 2% FCS, added with one of the EDPs (bovine neck ligament soluble elastins/kappa-elastin [κE-2; μg/mL] or mouse lung elastin peptides/m-elastins [ME-2; μg/mL], or Bio active peptide-VGVAPG [BP-200; ng/mL]) from Elastin Products Company, Inc. (Owensville, MO) with or without 0.5 and 1.0 μM α6(IV)NC1.
Cells treated with 0.5 and 1.0 μM α6(IV)NC1 alone also were maintained to evaluate the individual effect of α6(IV)NC1 on MCECs. Three replicates were maintained for each treatment and experiments were repeated twice. Similar replicates of control and treated wells also were maintained by adding polymyxin-B (5 mg/mL) to the above-mentioned treatments, to evaluate the effects of endotoxin neutralization on cell proliferation. Cells were incubated for 48 hours in a 5% CO2 incubator at 37°C and proliferation was analyzed by using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide assay (MTT) reagent method.20
Migration Assay
Assessment of MCEC migration in the presence of EDPs, α6(IV)NC1, and their combinations was done using a 48-well Boyden chamber. The MCECs (1 × 104/30 μL) were resuspended in EBM-2 containing combinations of α6(IV)NC1 (0.5 and 1.0 μM) seeded into each upper well, separated by the 8 μ polycarbonate membrane from lower wells with either EBM-2 as negative control, and EBM-2 with EDPs (2 μg/mL κE/ME or 200 ng/mL VGVAPG, as positive controls). The number of cells migrating and attaching to the underside of the membrane was observed after staining with hematoxylin and eosin (H&E) through an Olympus CK2 light microscope (Olympus Corporation, Tokyo, Japan), as reported.22
Tube Formation Assay
Tube formation assay was done on the matrigel (BD Biosciences, Franklin Lakes, NJ) in 24-well plates. Briefly, matrigel was thawed overnight at 4°C, and 250 μL were added to each well and allowed to polymerize at 37°C, for approximately 30 minutes. The MCECs were suspended at a density of 5 × 104 cells/mL in EGM-2 (without antibiotics) containing different combinations of EDPs and α6(IV)NC1, and released onto the polymerized matrigel. Wells containing culture medium were used as controls, medium with only EDPs (2 μg/mL κE/ME or 200 ng/mL VGVAPG) were used as positive controls, and EDPs along with 0.5 or 1.0 μM purified α6(IV)NC1 were considered as treated wells. Tube formation was recorded after 48 hours of incubation at 37°C, using a Juli light microscope (Bulldog Bio, Inc., Portsmouth, NH).
Wound Healing Assay
Migration of MCECs into a scratch wound region in the presence and absence of EDPs and α6(IV)NC1 was evaluated using a 24-well culture plate assay. Briefly, MCECs were cultured to sub-confluence in 24-well culture plates in EGM-2 medium. In each well, the center of the cultured monolayer was scratched with a sterile tip, followed by washing with PBS. Cells were maintained in EBM-2 medium supplemented with 2% FCS to promote survival of MCECs, without proliferation as positive control. Cells maintained only in EBM-2 (controls), EMB2 supplemented with 2 μg/mL κE or ME, or 200 ng/mL VGVAPG, and in combinations of 2 μg/mL κE with 0.5 or 1.0 μM α6(IV)NC1, 2 μg/mL ME with 0.5 or 1.0 μM α6(IV)NC1, and 200 ng/mL BP with 0.5 or 1.0 μM α6(IV)NC1 were monitored at 0 to 48 hours. Images of cellular migration were captured at 48-hour fixed points under the Juli microscope. Quantitative analysis of the MCECs migration was assessed by measuring the width of wounds in each image, at three equidistant points using reference scale and Adobe Photoshop software (Adobe Systems, Mountain View, CA). Distance (in millimeters) migrated by the cells in 48 hours was obtained by subtracting the average width measured at approximately 48 hours from that at 0 hours, and expressed as mean ± SD.
Western Blot Analyses for Cell Signaling Experiments
The MCECs cultured to 80% confluence and serum-starved in 2% FCS medium for 8 hours were used to detect kinase activation. Serum-starved MCECs were treated with 2 μg/mL κE or ME and 200 ng/mL VGVAPG, with or without 0.5 or 1.0 μM α6(IV)NC1 for 2 hours at 37°C, in a 5% CO2 incubator. Cell extracts (30 μg/lane) from these cells were used for kinase detection after 12% SDS-PAGE separation and Western transfer using respective antibodies for detecting p-FAK, p-Akt, p-PI3K, p-mToR, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH), respectively (antibodies from Cell Signaling Technology, Beverly, MA). Enhancement of p-FAK and p-Akt in MCECs on EDP treatment was verified further by adding synthetic FAK and Akt phosphorylation inhibitors along with EDPs. Inhibitor of FAK phosphorylation, Y15 (500 nM; Sigma-Aldrich, St. Louis, MO), and Akt phosphorylation, GSK 690693 (1.0 μM; Tocris Bioscience, Bristol, UK) were added, along with κE and BP, and detection of p-FAK and p-Akt was done on the cell extracts after 2 hours of incubation, as described above. Serum-starved MCECs were incubated further for 24 hours in EMB-2 containing 2 μg/mL κE or ME and 200 ng/mL VGVAPG, with or without 0.5 or 1.0 μM α6(IV)NC1 at 37°C in a 5% CO2 incubator, and cell extracts (30 μg/lane) from these cells were used for Western blot analyses for detecting MT1-MMP and β-actin levels.
Results
Recombinant α6(IV)NC1 Inhibits MCECs Viability
Effect of 2% serum, EDPs, recombinant α6(IV)NC1, and polymyxin-B on MCECs survival/proliferation was evaluated, and cell density measured after 48 hours indicated that ECs viability was comparatively similar in treatments containing serum-free, 2% serum, EDPs, or polymyxin-B alone (Fig. 1). The MCEC survival was reduced with addition of 0.5 and 1.0 μM α6(IV)NC1 to the 2% serum medium or EDP-containing wells without polymyxin-B (Fig. 1A). Addition of polymyxin-B (5 mg/mL) along with α6(IV)NC1 or other factors did not affect the survival of MCECs considerably (Fig. 1B).
Figure 1.
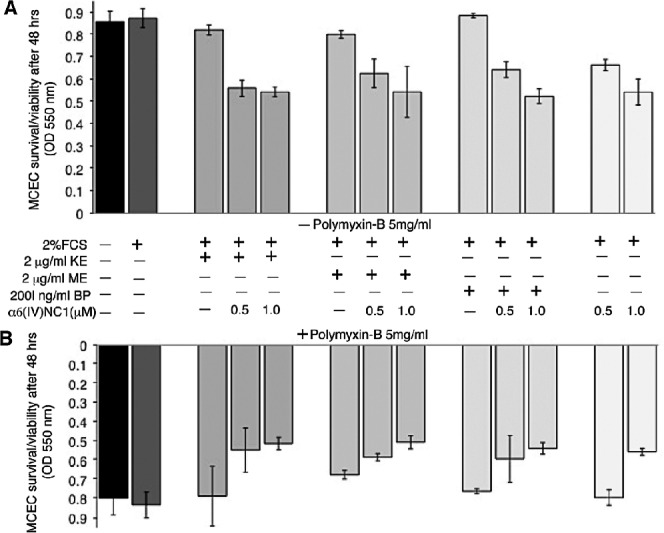
Inhibition of MCEC survival by α6(IV)NC1. (A) Upper graph showing survival of MCECs without polymyxin-B treatments. (B) Lower graph showing MCECs treated with polymyxin-B. Status bars indicate average cell survival of MCECs (mean ± SD of 3 replicates) after 48 hours of treatment. Presence (+) and absence (−) of factor in respective treatments are indicated.
α6(IV)NC1 Inhibits EDP-Promoted MCEC Migration
Previously, EDPs have been shown to promote choroidal endothelial cell (CEC) migration.10 Here, the effect of EDPs and α6(IV)NC1 on MCEC migration was evaluated using the Boyden chamber migration assay. In this experiment, the number of MCECs migrating towards lower wells was induced to varying degrees by treatment with EBM-2, κ-elastin (κE), mouse-elastin (ME), or VGVAPG (BP) (Fig. 2). Cellular migration towards these EDPs was drastically reduced when exposed to α6(IV)NC1 at 0.5 and 1.0 μM concentrations mixed along with the different EDPs (Figs. 2A, 2C, 2E). Different EDPs-mediated MCECs migrating was reduced by dose-dependent treatment of α6(IV)NC1. The inhibitory effect of α6(IV)NC1 on MCECs migration was lower for κE-induced cell migration compared to that of ME- or VGVAPG (BP)-induced migration (Figs. 2B, 2D, 2F).
Figure 2.

Inhibition of MCEC migration by α6(IV)NC1. Photographs showing MCECs from the underside of Boyden chamber membrane. Controls indicate cells migrating in EBM-2 (arrowheads). Relatively higher migration exhibited by MCECs towards wells containing (A) κE (upper right), (B) ME (upper right), and (E) VGVAPG (upper right) used as positive controls; and lowered migration exhibited by MCECs in treated wells containing 0.5 and 1.0 μM α6(IV)NC1 along with (A, C, E) EDPs (lower panels). Fields represent 5 of the replicates as observed at ×200 magnification with Olympus CK2 light microscope. Data graph representation of MCEC migration in presence or absence of EDPs and α6(IV)NC1 (B, D, F). Experiments were performed with three replicates and data in the graphs are represented as mean ± SD. Presence (+) and absence (−) of factor in respective treatments are indicated.
EDPs-Induced MCEC Wound Healing Response Is Inhibited by α6(IV)NC1
Scratch wound assays performed on MCECs also showed that α6(IV)NC1 reduced different EDPs-induced migration compared to controls (Figs. 3A–N). In contrast, migration of MCECs was reduced in the wells containing 1.0 μM α6(IV)NC1 along with 2% FCS, 2 μg/mL κE or ME, and 200 ng/mL of BP compared to controls κE or ME and BP (Figs. 3D–G versus Figs. 3H, 3L–N). Migratory capacity of MCECs was relatively higher in EDPs treatment compared to the control wells (Fig. 3O).
Figure 3.

The α6(IV)NC1 inhibits EDP-enhanced in vitro wound closure. (A–H) Wells showing migration of MCECs into artificial scratch wound made on the surface of culture wells at 48 hours. Migration of cells to words scratch wound was higher in 2 μg/mL κE (A, E), 2 μg/mL ME- (B, F), and 200 ng/mL BP- (C, G), 2% serum medium + 1.0 μM α6(IV)NC1 (D, H)–treated wells, and relatively lower in 1.0 μM α6(IV)NC1-treated wells (2 μg/mL κE + 1.0 μM α6[IV]NC1 [I, L], 2 μg/mL ME + 1.0 μM α6[IV]NC1 [J, M], and 200 ng/mL BP + 1.0 μM α6[IV]NC1 [K, N]), respectively (black bars in the scratch wound indicate 0.5 mm). Average gap covered by MCECs in 48 hours in each treatment is represented in graphic format (O). Status bars (O) indicate difference between initial and final gap measured at three equidistant points in each image, represented as mean ± SD. Presence (+) and absence (−) of factor in respective treatments are indicated.
EDPs Promoted MCEC Tube-Formation Inhibition by α6(IV)NC1
Based on the above results, it was presumed that α6(IV)NC1 also might inhibit tube formation by MCECs, which is an essential step in the angiogenic process. The in vitro tube formation assay performed on matrigel indicated inhibition of EDP-induced MCEC tube formation in a dose-dependent manner by α6(IV)NC1 (Fig. 4). Tubes formed by MCECs in the EGM-2 medium alone were relatively thin or exhibited incomplete partial networks (Figs. 4A, 4C, 4E, controls). Tubes formed by MCECs after 48 hours of incubation with 2.0 μg/mL κE or ME or 200 ng/mL of BP were relatively thick with complex network patterns (Figs. 4A, 4C, 4E; upper left panels). In contrast, fragmented tubes, cellular clumping, and detachment of MCECs were observed in wells containing 0.5 and 1.0 μM α6(IV)NC1 along with the EDPs (Figs. 4A, 4C, 4E; lower panels). The average number of tubes were lowest in the wells containing 1.0 μM α6(IV)NC1 along with 200 ng/mL of BP (Fig. 4F), and also decreased in wells containing α6(IV)NC1 along with κE or ME compared to those containing only EDPs (Figs. 4B, 4E).
Figure 4.
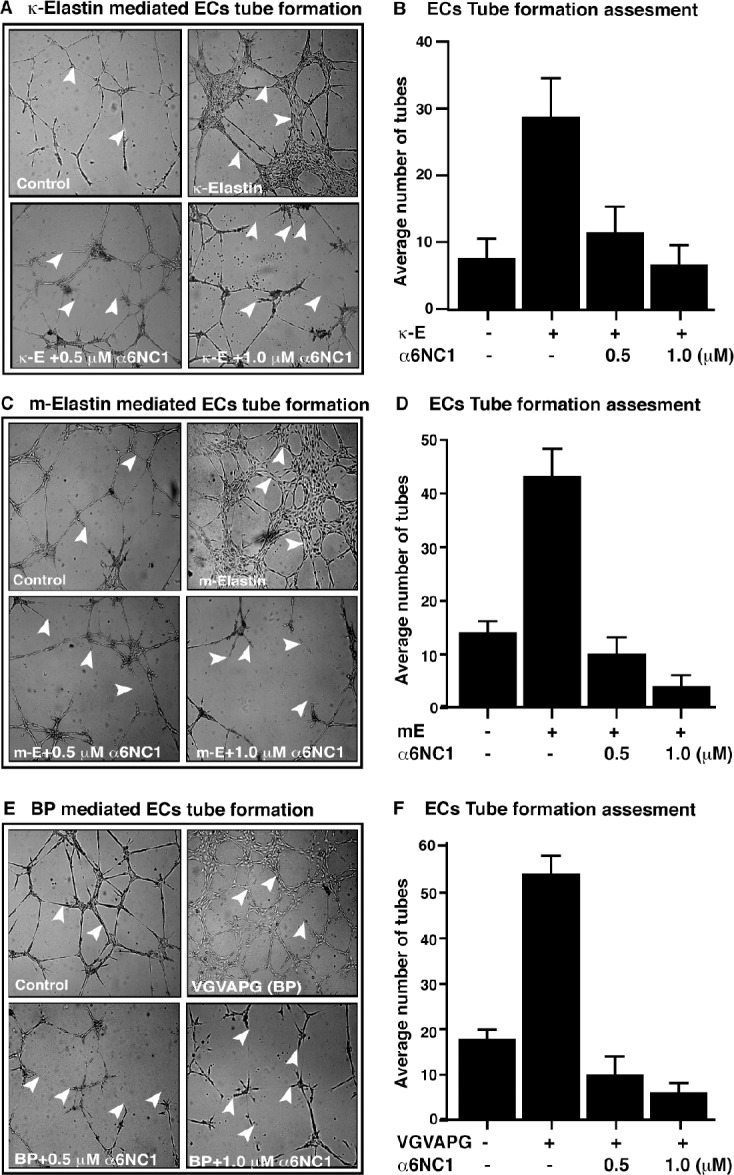
Inhibition of MCEC tube formation by α6(IV)NC1. Micrographs indicate incomplete and thin tubes in control wells containing only (A, C, E) EGM-2 (upper left panels), thick and higher number of tubes formed in presence of (A) κE (upper right), (C) ME (upper right), and (E) VGVAPG (upper right), respectively. Fragmented tubes and clumping of MCECs with incomplete tube formation evident in presence of 0.5 and 1.0 μM α6(IV)NC1 along with (A, C, E) EDPs (lower panels). Graphic representation of average number of tubes formed by MCECs in presence or absence of EDPs and α6(IV)NC1, represented in (B, D, F), respectively. Status bars indicate average of three measurements for each treatment represented as mean ± SD. Presence (+) and absence (−) of factor in respective treatments are indicated.
Regulation of EDP-Mediated MT1-MMP Expression by α6(IV)NC1
The EDPs are known to promote the expression of MT1-MMP and enhance choroidal EC migration.10 In tracing the mechanism(s) involved in inhibition of EDPs-promoted MCEC migration and tube formation upon incubation with α6(IV)NC1, we studied the effects of different EDPs and α6(IV)NC1 on MT1-MMP expression in MCECs. Levels of the pro-, processed forms of MT1-MMP in MCECs were enhanced by κE treatment compared to that of control cells. Further, incubation of cells with increasing concentrations of α6(IV)NC1, along with κE (2 μg/mL) reduced the levels of MT1-MMP, as observed by Western blot analyses (Fig. 5A, upper lane). Similar to the effect of κE on MT1-MMP levels in MCECs, VGVAPG and ME also enhanced MT1-MMP levels in MCECs at concentrations of 2 and 0.2 μg/mL, respectively, which were reduced by treatment of different doses of α6(IV)NC1 (Fig. 5B, upper lane).
Figure 5.
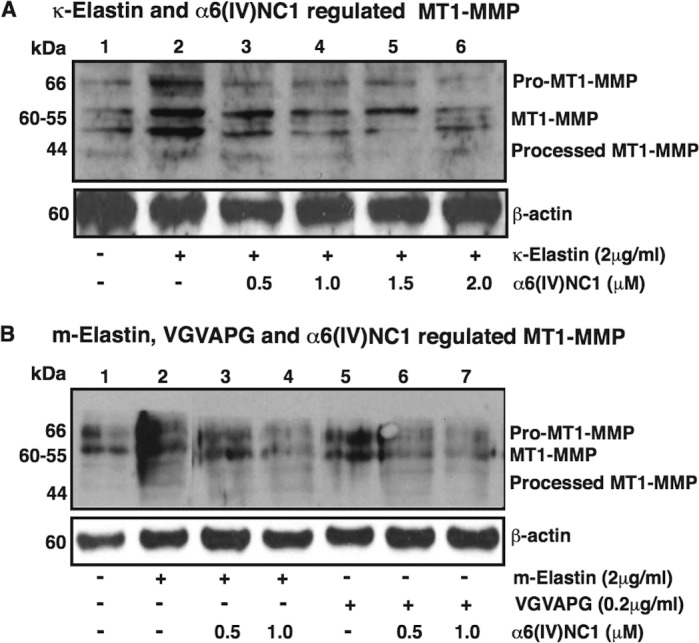
The α6(IV)NC1 inhibits EDP enhanced MT1-MMP levels in MCECs. Western blot analyses showing MT1-MMP levels in MCEC cell lysates treated in media containing combination of (A) κE and α6(IV)NC1 (upper lane), and (B) ME or BP and α6(IV)NC1 (upper lane), respectively. Presence (+) and absence (−) of factor in respective treatments are indicated. Lower Western blots in (A, B); β-actin is shown as loading control.
Inhibition of EDPs-Induced Phosphorylation of FAK, PI3K, Akt, and mToR by α6(IV)NC1
Regulation of angiogenic signaling response in MCECs by different EDPs and α6(IV)NC1 was studied through the evaluation of FAK, PI3K, Akt, and mToR phosphorylation. Phosphorylation of FAK, PI3K, Akt, and mToR was enhanced in MCECs treated with κE for 2 hours and decreased in a dose-dependent manner on treatment with increasing concentrations of α6(IV)NC1 under fixed κE treatment (Fig. 6A). Similar patterns of increased PI3K, Akt, and mToR phosphorylation on treatment with ME or BP, and decreased phosphorylation on α6(IV)NC1 treatment along with ME and BP also were observed (Fig. 6B).
Figure 6.
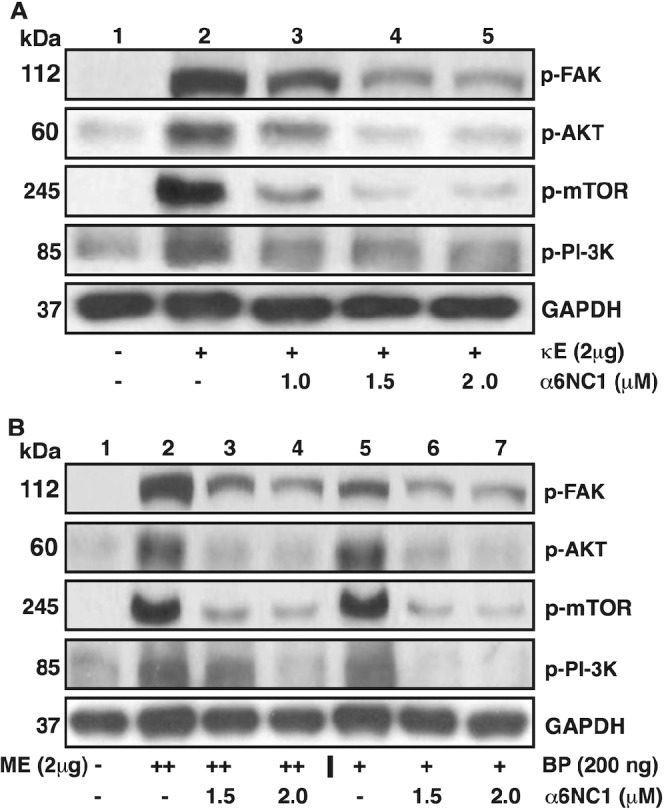
Inhibition of different bioactive peptides promoted kinase signaling in MCECs by α6(IV)NC1. (A) Inhibition of κ-E promoted kinase signaling in MCECs by α6(IV)NC1. Western blot analyses showing bands corresponding to p-FAK, p-Akt, p-mToR, and p-PI3K in MCEC lysates subjected to combinations of κE and α6(IV)NC1 treatments. Presence (+) and absence (−) of factor in respective treatments are indicated. (B) Inhibition of ME and BP promoted kinase signaling in MCECs by α6(IV)NC1. Western blot analyses showing bands corresponding to p-FAK, p-Akt, p-mToR, and p-PI3K in MCEC lysates subjected to combinations of ME, BP, and α6(IV)NC1 treatments. Presence (++/+) and absence (−) of factor in respective treatments are indicated. (A, B) GAPDH is shown as loading control.
Inhibition of EDP-Induced FAK and Akt Phosphorylation by Synthetic Inhibitors
To test further that the EDP mediated activation of FAK and Akt phosphorylation in MCECs, we examined the regulation of FAK and Akt activation in MCECs by the EDPs, in the presence of synthetic FAK- and Akt-specific inhibitors. Phosphorylation of FAK in MCECs treated with the 2 μg/mL κE or 200 ng/mL VGVAPG was reduced on simultaneous treatment for 2 hours with 500 nM p-FAK inhibitor, Y-15 (Fig. 7A). Similarly, enhanced phosphorylation of Akt by κE (2 μg/mL) or VGVAPG (200 ng/mL) also was reduced with the addition of p-Akt inhibitor, GSK 690693, at 1.0-μM concentration, along with EDPs (Fig. 7B).
Figure 7.
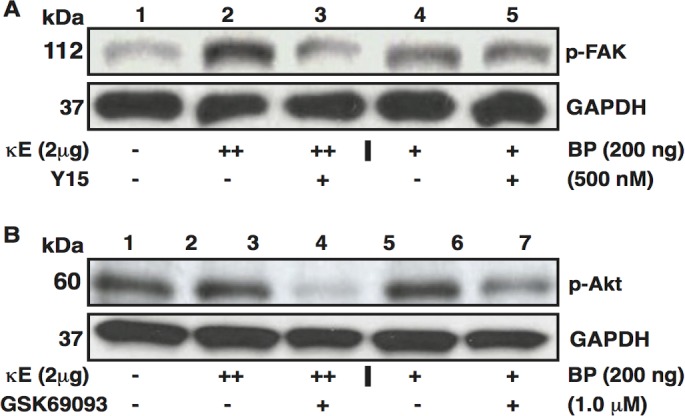
Inhibition of κE- and BP-enhanced kinase signaling in MCECs by synthetic FAK and Akt inhibitors. (A) Upper Western blots showing bands corresponding to p-FAK from MCEC lysates subjected to κE, BP, and Y15 treatments. (B) Upper Western blots showing bands corresponding to p-Akt from MCEC lysates subjected to κE, BP, and GSK69093 treatments. (A, B) Presence (++/+) or absence of factor in respective treatments is indicated. Lower Western blots in (A, B) show GAPDH as loading control.
Discussion
It has been shown previously that CNV in AMD is promoted by the EDPs released into vasculature through the abnormal metabolism of elastins, indicating a pathologic role of elastin metabolism in CNV.2,5 In contrast, degradation of type IV collagen leads to the release of NC1 domains with antiangiogenic properties, which inhibit pathologic angiogenesis.14,25–28 Our study was done to evaluate the net effects of α6(IV)NC1 domain and EDPs in regulation of MCECs angiogenic patterns in vitro. Angiogenic effects of different EDPs in CNV were studied previously using cultured CECs of monkey and human origin.9,10 The CECs of mouse origin were applied in our study for evaluating effects of EDPs and α6(IV)NC1, considering the significance of mouse as a model system for in vivo CNV studies.29
Another study reported that survival of endothelial cells is maintained normally in the presence of EDPs.10 In our study, addition of 2% serum or EDPs did not alter the proliferation of MCECs compared to serum-free medium. However, survival of MCECs was retained similarly in serum-free, 2% serum and EDP-treated wells. These results are in accordance with previous report indicating that EDPs do not alter the proliferation of endothelial cells in 48 hours compared to the controls containing Hank's balanced salt solution (HBSS).9 In our previous study, we identified that α6(IV)NC1 can inhibit proliferation of human umbilical vein endothelial cells (HUVECs) in vitro.22 In our study, α6(IV)NC1 (0.5 and 1.0 μM) applied in the media alone, or in combination with EDPs or 2% serum, inhibited MCEC survival. Addition of polymyxin-B alone or in combination with α6(IV)NC1 did not alter the proliferation significantly, showing that the antisurvival affect exhibited by α6(IV)NC1 was not due to the endotoxin present in this recombinant protein.
Chemotactic EDPs induce cellular migration for different cell types, including keratinocytes, fibroblasts, and choroidal endothelial cells.10,30,31 In our study, enhanced migration of MCECs by EDPs was inhibited by α6(IV)NC1. The positive effect of EDPs in enhancing CEC migration also was studied previously, where the migration was found to be higher for cells migrating towards fetal bovine serum (FBS) medium compared to EDPs.9 In our study, migration of MCECs was compared in the presence of EDPs and α6(IV)NC1, rather than FBS and EDPs. Here, we showed that α6(IV)NC1 inhibited EDP-induced MCEC migration in Boyden chamber and scratch wound assays. An earlier study with HUVECs showed that α6(IV)NC1 coated on well plates promoted the migration and adhesion of ECs.21 In our study, α6(IV)NC1 was presented as a soluble protein in the medium, rather than as a coating to the substrata, to evaluate its effects in comparison with the soluble EDPs. It may be presumed that soluble α6(IV)NC1 binds to the MCECs, and inhibits further adhesion and migration on the substrata as observed in the present migration experiments. Further, the reduction in migratory capability of MCECs also can be attributed to the inhibitory effect of α6(IV)NC1 on cell survival, as identified here in the cell survival experiment.
Endothelial cells exhibit sequential angiogenic responses on angiogenic activation, including migration and tube formation, leading to the initiation of neocapillary development in vivo.32 Earlier studies indicate that κE and VGVAPG (BP) promoted tube formation of CECs cultured on Matrigel.10 In accordance with the previous study, our results demonstrated that κE and VGVAPG (BP) enhanced tube formation in cultured MCECs. In addition, we showed that ME also exhibited a similar enhancing effect on MCEC tube formation. Addition of α6(IV)NC1 along with the three different EDPs reduced tube formation in MCECs cultured on matrigel with distorted and irregular tube development, which may pertain partially to the antisurvival effect exhibited by α6(IV)NC1. The tube formation that was exhibited by α6(IV)NC1 on MCECs is similar to that of other IV collagen NC1 domains on other human ECs.11,20,22
Vascular basement membranes (VBM) act as a scaffold for the attachment of ECs to the luminal surface of blood vessels.33,34 Therefore, degradation of the VBM scaffold is essential for EC migration during angiogenesis, which is facilitated by MT1-MMP on the EC surface.35,36 The role of MT1-MMP in EDP-induced CEC migration was established in an earlier study.10 In accordance with the observations of the previous study, in our study the levels of the pro- and active MT1-MMP were enhanced in the EDP-treated MCECs. Further, the levels of pro- and active MT1-MMP also reduced upon coincubation of MCECs with EDPs and α6(IV)NC1. These results demonstrated that the inhibition of EDP enhanced MT1-MMP expression by α6(IV)NC1. This can be a complementary regulatory mechanism through which α6(IV)NC1 inhibits MCEC migration and tube formation, in addition to the antisurvival effect shown by this NC1 domain, in the presence of EDPs. A similar effect of decrease in MT1-MMP expression levels was reported previously in human microvascular endothelial cells-1 (HMEC-1) subjected to NC1 domain derived from collagen XIX.37
Regulation of angiogenic responses in ECs by NC1 domains of type IV collagen and EDPs are facilitated through the binding of these angiogenic regulators to the cell surface receptors.17,21,28,38–40 Such binding leads to integrin receptor activation and downstream kinase activation promoting EC migration.15,41 Antiangiogenic effects of other NC1 domains of type IV collagen were shown earlier to be elicited through the inhibition of FAK-mediated kinase signaling.14 In this study, increased phosphorylation of FAK/PI-3K/Akt/mToR in EDP-treated MCECs suggested a role for this signaling cascade in angiogenic activation of MCECs by EDPs. Furthermore, the inhibition of EDP-induced FAK and Akt phosphorylation by their respective synthetic inhibitors (Y15 and GSK690693) supported a possible role for FAK and Akt in EDP- and α6(IV)NC1-regulated MCEC angiogenic responses.
Another recent study reported that EDPs enhance CNV through induction of collagen.42 However, our study was focused on the noncollagenous domain component of type IV collagen, and showed the combined effects of the type IV collagen-derived metabolite, α6(IV)NC1, and different elastin-derived metabolites in regulation of angiogenic behavior in CECs. Here, our findings demonstrated that α6(IV)NC1 inhibits proangiogenic signals and, thus, may have therapeutic potential for the treatment of diseases involving choroidal neovascularization, such as the wet form of AMD.
Acknowledgments
Supported by Flight Attendant Medical Research Institute (Miami, Florida) Grant FAMRI-062558, National Institutes of Health/National Cancer Institute (NIH/NCI; Bethesda, Maryland) Grant NIH/HCI-RO1CA143128, and Dobleman Head and Neck Cancer Institute (Omaha, Nebraska) Grant DHNCI-61905 (YS).
Disclosure: V. Gunda, None; R.K. Verma, None; Y.A. Sudhakar, None
References
- 1. Guymer RH, Bird AC, Hageman GS. Cytoarchitecture of choroidal capillary endothelial cells. Invest Ophthalmol Vis Sci. 2004; 45: 1660–1666 [DOI] [PubMed] [Google Scholar]
- 2. Sivaprasad S, Chong NV, Bailey TA. Serum elastin-derived peptides in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2005; 46: 3046–3051 [DOI] [PubMed] [Google Scholar]
- 3. Chen L, Miyamura N, Ninomiya Y, Handa JT. Distribution of the collagen IV isoforms in human Bruch's membrane. Br J Ophthalmol. 2003; 87: 212–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kielty CM, Sherratt MJ, Shuttleworth CA. Elastic fibres. J Cell Sci. 2002; 115: 2817–2828 [DOI] [PubMed] [Google Scholar]
- 5. Chong NH, Keonin J, Luthert PJ, et al. Decreased thickness and integrity of the macular elastic layer of Bruch's membrane correspond to the distribution of lesions associated with age-related macular degeneration. Am J Pathol. 2005; 166: 241–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Spraul CW, Grossniklaus HE. Characteristics of drusen and Bruch's membrane in postmortem eyes with age-related macular degeneration. Arch Ophthalmol. 1997; 115: 267–273 [DOI] [PubMed] [Google Scholar]
- 7. Spraul CW, Lang GE, Grossniklaus HE, Lang GK. Histologic and morphometric analysis of the choroid, Bruch's membrane, and retinal pigment epithelium in postmortem eyes with age-related macular degeneration and histologic examination of surgically excised choroidal neovascular membranes. Surv Ophthalmol. 1999; 44 (suppl 1): S10–S32 [DOI] [PubMed] [Google Scholar]
- 8. Blumenkranz MS, Russell SR, Robey MG, Kott-Blumenkranz R, Penneys N. Risk factors in age-related maculopathy complicated by choroidal neovascularization. Ophthalmology. 1986; 93: 552–558 [DOI] [PubMed] [Google Scholar]
- 9. Skeie JM, Mullins RF. Elastin-mediated choroidal endothelial cell migration: possible role in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2008; 49: 5574–5580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Robinet A, Fahem A, Cauchard JH, et al. Elastin-derived peptides enhance angiogenesis by promoting endothelial cell migration and tubulogenesis through upregulation of MT1-MMP. J Cell Sci. 2005; 118: 343–356 [DOI] [PubMed] [Google Scholar]
- 11. Sudhakar A, Sugimoto H, Yang C, Lively J, Zeisberg M, Kalluri R. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc Natl Acad Sci U S A. 2003; 100: 4766–4771 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 12. Nyberg P, Xie L, Kalluri R. Endogenous inhibitors of angiogenesis. Cancer Res. 2005; 65: 3967–3979 [DOI] [PubMed] [Google Scholar]
- 13. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011; 473: 298–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sudhakar A. The matrix reloaded: new insights from type IV collagen derived endogenous angiogenesis inhibitors and their mechanism of action. J Bioequiv Bioavailab. 2009; 1: 52–62 [Google Scholar]
- 15. Kalluri R. Discovery of type IV collagen noncollagenous domains as novel integrin ligands and endogenous inhibitors of angiogenesis. Cold Spring Harb Symp Quant Biol. 2002; 67: 255–266 [DOI] [PubMed] [Google Scholar]
- 16. Ingber D, Folkman J. Inhibition of angiogenesis through modulation of collagen metabolism. Lab Invest. 1988; 59: 44–51 [PubMed] [Google Scholar]
- 17. Sudhakar A, Nyberg P, Keshamouni VG, et al. Human alpha1 type IV collagen NC1 domain exhibits distinct antiangiogenic activity mediated by alpha1beta1 integrin. J Clin Invest. 2005; 115: 2801–2810 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18. Maeshima Y, Sudhakar A, Lively JC, et al. Tumstatin, an endothelial cell-specific inhibitor of protein synthesis. Science. 2002; 295: 140–143 [DOI] [PubMed] [Google Scholar]
- 19. Lima ESR, Kachi S, Akiyama H, et al. Recombinant noncollagenous domain of alpha2(IV) collagen causes involution of choroidal neovascularization by inducing apoptosis. J Cell Physiol. 2006; 208: 161–166 [DOI] [PubMed] [Google Scholar]
- 20. Boosani CS, Nalabothula N, Sheibani N, Sudhakar A. Inhibitory effects of arresten on bFGF-induced proliferation, migration, and matrix metalloproteinase-2 activation in mouse retinal endothelial cells. Curr Eye Res. 2010; 35: 45–55 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21. Petitclerc E, Boutaud A, Prestayko A, et al. New functions for noncollagenous domains of human collagen type IV. Novel integrin ligands inhibiting angiogenesis and tumor growth in vivo. J Biol Chem. 2000; 275: 8051–8061 [DOI] [PubMed] [Google Scholar]
- 22. Gunda V, Boosani CS, Verma RK, Guda C, Sudhakar YA. L-Arginine mediated renaturation enhances yield of human, a6 type IV collagen noncollagenous domain from bacterial inclusion bodies. Prot Pept Letts. 2012; 19: 1112–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mundel TM, Yliniemi AM, Maeshima Y, Sugimoto H, Kieran M, Kalluri R. Type IV collagen alpha6 chain-derived noncollagenous domain 1 (alpha6(IV)NC1) inhibits angiogenesis and tumor growth. Int J Cancer. 2008; 122: 1738–1744 [DOI] [PubMed] [Google Scholar]
- 24. Dutta D, Ray S, Home P, et al. Regulation of angiogenesis by histone chaperone HIRA-mediated incorporation of lysine 56-acetylated histone H3.3 at chromatin domains of endothelial genes. J Biol Chem. 2010; 285: 41567–41577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kalluri R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer. 2003; 3: 422–433 [DOI] [PubMed] [Google Scholar]
- 26. Rosenbloom J. Elastin: biosynthesis, structure, degradation and role in disease processes. Connect Tissue Res. 1982; 10: 73–91 [DOI] [PubMed] [Google Scholar]
- 27. Drozdz M, Kucharz E, Olczyk K. Elastin structure and metabolism under physiological and pathological conditions [in Polish]. Postepy Hig Med Dosw. 1976; 30: 709–734 [PubMed] [Google Scholar]
- 28. Gunda V, Sudhakar YA. Regulation of angiogenesis in choroidal neovascularization of age related macular degeneration by endogenous angioinhibitors. In: Rumelt S. ed Advances in Ophthalmology. Rijeka, Croatia: InTech; 2012: 409–423 [Google Scholar]
- 29. Wang S, Sorenson CM, Sheibani N. Lack of thrombospondin 1 and exacerbation of choroidal neovascularization. Arch Ophthalmol. 2012; 1; 130: 615–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Senior RM, Griffin GL, Mecham RP. Chemotactic responses of fibroblasts to tropoelastin and elastin-derived peptides. J Clin Invest. 1982; 70: 614–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fujimoto N, Tajima S, Ishibashi A. Elastin peptides induce migration and terminal differentiation of cultured keratinocytes via 67 kDa elastin receptor in vitro: 67 kDa elastin receptor is expressed in the keratinocytes eliminating elastic materials in elastosis perforans serpiginosa. J Invest Dermatol. 2000; 115: 633–639 [DOI] [PubMed] [Google Scholar]
- 32. Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000; 6: 389–395 [DOI] [PubMed] [Google Scholar]
- 33. Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006; 86: 279–367 [DOI] [PubMed] [Google Scholar]
- 34. Sottile J. Regulation of angiogenesis by extracellular matrix. Biochim Biophys Acta. 2004; 1654: 13–22 [DOI] [PubMed] [Google Scholar]
- 35. Davis GE, Senger DR. Endothelial extracellular matrix: biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ Res. 2005; 97: 1093–1107 [DOI] [PubMed] [Google Scholar]
- 36. Galvez BG, Matias-Roman S, Albar JP, Sanchez-Madrid F, Arroyo AG. Membrane type 1-matrix metalloproteinase is activated during migration of human endothelial cells and modulates endothelial motility and matrix remodeling. J Biol Chem. 2001; 276: 37491–37500 [DOI] [PubMed] [Google Scholar]
- 37. Ramont L, Brassart-Pasco S, Thevenard J, et al. The NC1 domain of type XIX collagen inhibits in vivo melanoma growth. Mol Cancer Ther. 2007; 6: 506–514 [DOI] [PubMed] [Google Scholar]
- 38. Mundel TM, Kalluri R. Type IV collagen-derived angiogenesis inhibitors. Microvasc Res. 2007; 74: 85–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hamano Y, Zeisberg M, Sugimoto H, et al. Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell. 2003; 3: 589–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maquart FX, Bellon G, Pasco S, Monboisse JC. Matrikines in the regulation of extracellular matrix degradation. Biochimie. 2005; 87: 353–360 [DOI] [PubMed] [Google Scholar]
- 41. Sudhakar A, Boosani CS. Signaling mechanisms of endogenous angiogenesis inhibitors derived from type IV collagen. Gene Regul Syst Bio. 2007; 1: 217–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Skeie JM, Hernandez J, Hinek A, et al. Molecular responses of choroidal endothelial cells to elastin derived peptides through the elastin-binding protein (GLB1). Matrix Biol. 2012; 31: 113–119 [DOI] [PMC free article] [PubMed] [Google Scholar]