

Abstract
Purpose.
Müller cells, a major type of glial cell found in the eye, are postulated to play an important role in many retinal diseases, including diabetic retinopathy (DR). Complement is an integral part of innate immunity, and the activation of complement has been associated with retinal diseases. However, the role of complement in the regulation of Müller cell function remains unclear. We were trying to address these issues in this study.
Methods.
Using primary human Müller cells and a spontaneously immortalized human Müller cell line, we examined the expression of complement receptor C5aR both at mRNA and protein levels. Regulation of C5aR expression on Müller cells by prostaglandin E2 and by hyperglycemia, both of which are integrally involved in DR, were studied. Significance of C5aR on Müller cells was also investigated by examining relevant cytokine productions and their impacts on retinal endothelial cell proliferation/permeability after ligating the receptor using its ligand, C5a.
Results.
C5aR is constitutively expressed in human Müller cells. Prostaglandin E2 and hyperglycemia individually and synergistically upregulate C5aR expression in Müller cells. Signaling through C5aR on Müller cells upregulates production of IL-6 and VEGF, which promotes the proliferation of human retinal endothelial cells and increases their permeability.
Conclusions.
These results indicate that complement can regulate Müller cells through C5aR, which may contribute to the pathogenesis of retinal diseases, including DR.
Keywords: diabetic retinopathy, retina, innate immunity
Human retinal Müller cells express C5aR. C5aR expression in Müller cells is upregulated by PGE2 and/or hyperglycemia. Signaling through C5aR stimulates Müller cells to produce IL-6 and VEGF, cytokines that are integrally involved in the pathogenesis of diabetic retinopathy.
Introduction
Diabetic retinopathy (DR) is one of the most common causes of blindness in developed countries.1 In the retinas of patients with DR, abnormal proliferation of retinal endothelial cells leads to neovascularization, a major feature of complications of this disease.2 It has been demonstrated that levels of inflammatory and angiogenesis factors, including prostaglandin E2 (PGE2), IL-6, and VEGF, are elevated either systemically or locally in the retinas of patients with DR, which could contribute to the pathogenesis of this disease.3–5 PGE2 is a small-molecule derivative of arachidonic acid, produced by cyclooxygenases.6 The receptors for PGE2, EP1 to EP4, are expressed in multiple types of cells,7 including Müller cells in the retina.8
Müller cells constitute a major glial cell population in the retina, and they are a major source of angiogenesis factors in the retina in many other ocular diseases. Previous studies have shown that advanced glycation end products and lipopolysaccharides stimulate Müller cells to produce IL-6 and VEGF, cytokines that potently promote endothelial cell proliferation9,10 and increase vascular permeability.
Complement, as an important part of the innate immune response, is integrally involved in the pathogenesis of many ocular disorders, including AMD,11,12 autoimmune uveitis,13 and DR.14 When complement is activated, the complement activation product C5a is released into the fluid phase, which regulates cellular activities, including the stimulation of IL-6 and VEGF production from a variety of cells that express its receptor, C5aR. C5aR is a G-protein–coupled receptor that was originally thought to be expressed only in myeloid-derived blood cells, but later studies found that C5aR is present in many nonmyeloid cells, including astrocytes,15 alveolar epithelial cells,16 and bronchial epithelial cells.17 In the retina, C5aR expression has been detected in RPE and in choroidal endothelial cells.18,19 Although complement activation products have been found in the retinas of DR patients and in animal models, whether retinal Müller cells express C5aR and whether complement regulates Müller cell activity remain unknown. We report results of our study using primary human retinal Müller cells and a spontaneously immortalized human Müller cell line. We studied the presence and potential role of C5aR in regulating Müller cells under normal and hyperglycemic culture conditions and examined the direct impact of C5aR signaling in Müller cells in modulating the proliferation and permeability of primary human retinal endothelial cells (hRECs).
Materials and Methods
Müller Cell Culture
The primary human retinal Müller cells were isolated and characterized as described before, with minor modifications.20 In brief, retinas were digested with trypsin (0.25%; Mediatech, Inc., Manassas, VA) for 1 hour at 37°C. Cells were filtered through a 40-μm mesh and cultured in Dulbecco's modified Eagle's medium (DMEM)/F12 containing 20% fetal bovine serum (FBS). Cultures were greater than 95% pure for Müller cells (vimentin+, CRALBP+, and GFAP−). Human Müller cells were used between passages three through six. The spontaneously immortalized human Müller cell line (MIO-M1 cells, obtained from G. Astrid Limb, PhD, University College London Institute of Ophthalmology, London, UK)21 were grown in DMEM containing a normal (5 mM) or a high (30 mM) concentration of glucose supplemented with 10% FBS and 1% Pen-Strep solution (Invitrogen, Carlsbad, CA). The high-glucose media was changed every day for 7 days before experiments.
RT-PCR Detection of C5aR in Müller Cells
Total RNA was isolated from the primary cells and MIO-M1 cells using TRIzol (Invitrogen). RNA was reverse transcribed into cDNA with a SuperScript First-Strand kit (Invitrogen). The C5aR transcripts were detected by PCR with primers located on different C5aR exons: P1, 5′-TCC TCG GGG AGC CCA GGA GAC-3′ and P2, 5′-CCA GCA GAA AGC GGT CGG CG-3′ using a PTC 200 thermal cycler (Bio-Rad, Hercules, CA) with the following amplification conditions: 94°C for 30 seconds, 58°C for 1 minute, and 72°C for 1 minute, 40 cycles. The same amount of total RNA that had not undergone reverse transcription was included in the PCR reactions as negative controls.
Quantitative RT-PCR Measurements of C5aR Transcription in Müller Cells
Total RNA was isolated using TRIzol after 0, 6, and 24 hours of stimulating the Müller cells with 1 μM PGE2. A 1.0-μg aliquot of RNA was treated with deoxyribonuclease I following the manufacturer's manual (Invitrogen), and reverse-transcribed into cDNA with random primers and superscript II reverse transcriptase using the manufacturer's protocol (Invitrogen). Quantitative PCR was performed using primers GGAGCCCAGGAGACCAGAAC and TATCCTTGTCATCATAGTGCCCATA on an Applied Biosystems 7300 Real-Time PCR System in duplicate with SYBR Green PCR master mix (Applied Biosystems, Carlsbad, CA) according to the manufacturer's instructions. PCR consisted of 40 cycles of 95°C for 15 seconds and 60°C for 60 seconds. Finally, the dissociation curve was generated to confirm a single product. The quantity was determined with a standard curve, and expression levels were normalized against the 18S RNA.
Flow Cytometry Analysis of C5aR on Müller Cells
The primary cells or MIO-M1 cells cultured in normal versus high-glucose media were subcultured in six-well plates (1 × 105/well). On the following day, some cells were left unstimulated and some were stimulated with 1 μM PGE2 for 24 hours. After this step, the cells were washed with 1 mL FACS buffer (PBS plus 5% bovine serum albumin), then stained with a monoclonal rabbit anti-human C5aR antibody (BD Biosciences, San Jose, CA) or the same concentration of rabbit IgG, followed by a second staining with FITC-conjugated goat anti-rabbit IgGs (5 μg/mL). After washing, cells were analyzed using an LSR II flow cytometer (BD Biosciences).
Confocal Analysis of C5aR on Müller Cells
The primary cells and MIO-M1 cells were first cultured in a chamber slide system (Nalge Nunc International Corp., Rochester, NY), then either left untreated or treated with 1 μM PGE2 for 24 hours. The slides were then washed with PBS and fixed with cold acetone for 10 minutes. After washing the slides again with PBS, the cells were blocked with 10 μg/mL goat IgG for 30 minutes at room temperature, then stained with either the monoclonal rabbit anti-human C5aR antibody (BD Biosciences) or the same concentration of rabbit IgG at 37°C for 30 minutes, followed by the second staining with FITC-conjugated goat anti-rabbit IgG antibody (5 μg/mL) at 37°C for 30 minutes. All cells were analyzed under a confocal microscope (Zeiss, Oberkochen, Germany) in the Imaging Core Facility of the Visual Sciences Research Center at Case Western Reserve University (Cleveland, OH).
Functional Assays of C5aR on Müller Cells
The function of C5aR on Müller cells was assessed by studying IL-6 and VEGF production from MIO-M1 cells after stimulation of the C5aR ligand C5a. In brief, MIO-M1 cells were cultured in normal (5 mM) or high-glucose (30 mM) media for 7 days with daily media change, then subcultured in wells of 24-well plates (3 × 104/well). On the following day, in some wells, 1 μM PGE2 was added into the media; other wells were left untreated. Twenty-four hours later, media with PGE2 was removed, and fresh media with 50 ng/mL, 100 ng/mL, or 200 ng/mL of C5a was added. As controls, 50 μM of C5aR antagonist (C5aRA: JPE-1375; custom synthesized by AnaSpec, Fremont, CA) was added into some of the wells. Culture supernatants were collected after 24 hours and 48 hours, and levels of IL-6 (BioLegend, San Diego, CA) and VEGF (Peprotech, Rocky Hill, NJ) were measured by ELISA using the respective kits and following manufacturers' protocols.
Human REC Proliferation Assays
Primary hRECs were isolated and characterized as reported before.22 These hRECs were cultured in DMEM/Ham's F12 mix supplemented with 10% FBS, 1% endothelial cell growth supplement (15 μg/mL; Sigma-Aldrich, St. Louis, MO), 1% insulin/transferrin, selenium (Sigma-Aldrich), 1% antibiotic/antimycotic, and 1% GlutaMax (Invitrogen). For proliferation assays, 5000 cells were seeded in each well of a 96-well plate. On the following day, cells were starved in medium without FBS for 4 hours, then cultured with 50% Müller cell–conditioned media for 72 hours. Cell proliferation was determined using the CellTiter-Glo Luminescent Cell Assay kit (Promega, Madison, WI) following the manufacturer's protocol, and normalized against the controls.
Human REC Permeability Assays
Human RECs (5 × 104) were seeded on each of the Transwell inserts (6.5-mm diameter, 3-μm pore size; Corning, Inc., Lowell, MA) with 0.3 mL media in the top insert and 0.7 mL media in the bottom wells. Media were changed every 2 days until REC monolayers were formed 7 days later as assessed by microscopy. Then 0.3 mL of different Müller cell–conditioned media or control media was added into the top wells for 24 hours at 37°C. The permeability of the REC monolayer was assessed by measuring the leakage of FITC-labeled dextran (Sigma-Aldrich) from the top inserts into the bottom wells at different time points. In brief, 100 μL FITC-labeled dextran (1 mg/mL) was added into each of the inserts with REC monolayers, then 30 minutes, 60 minutes, and 120 minutes later, 50-μL aliquots were removed from each of the bottom wells, and the levels of leaked FITC-labeled dextran were measured by a fluorescence reader (Molecular Devices, Sunnyvale, CA) using 490 nm and 530 nm as the excitation and emission wavelengths, respectively.
Results
Müller Cells Express C5aR
Detection of C5aR transcripts by RT-PCR in primary Müller cells using C5aR-specific primers that span different C5aR exons (to avoid false-positive results) demonstrated that C5aR transcripts were present in the primary Müller cells. RT-PCR of RNA purified from the spontaneously immortalized Müller cell line MIO-M1 revealed that C5aR was also expressed in the MIO-M1 cells (Fig. 1). These results suggest that C5aR is constitutively expressed in Müller cells.
Figure 1.
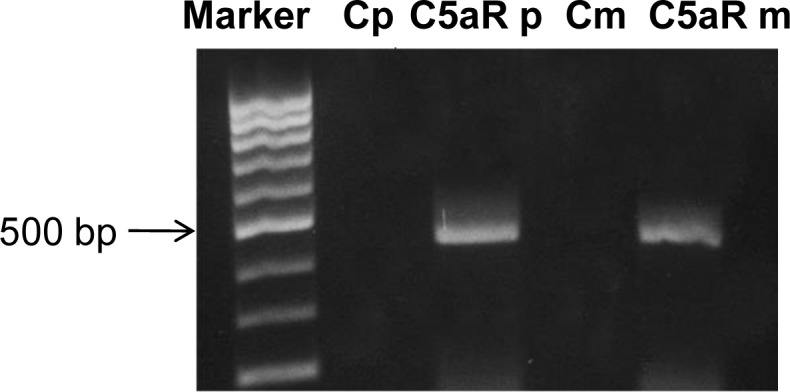
Detection of C5aR transcripts in both primary Müller cells and MIO-M1 cells. Total RNA isolated from primary Müller cells and MIO-M1 cells was reverse transcribed and used as a template to amplify a fragment of approximately 450 bp within the C5aR transcript spanning different exons. RNA that had not undergone reverse transcription was used as a negative control. Cp, primary cell control; Cm, MIO-M1 control. Marker: 100-bp DNA ladder.
To detect C5aR protein on the surface of Müller cells, we examined both the primary and the immortalized Müller cells by flow cytometry using an anti-C5aR monoclonal antibody (mAb) and its isotypes. These assays showed that under normal culture conditions, C5aR protein was barely detectable by flow cytometry on the surface of both types of Müller cells (Figs. 2A, 2C). In light of previous reports that PGE2 levels are elevated in the retina of DR patients23 and that PGE2 upregulates C5aR expression in human dendritic cells,24 we treated the MIO-M1 Müller cells with 1 μM PGE2 for 6 or 24 hours, then assessed C5aR transcript levels by qRT-PCR. These experiments showed that PGE2 upregulated C5aR transcription in Müller cells even at 6 hours after PGE2 stimulation (Fig. 2A). To verify the upregulation at protein levels, we again treated both the MIO-M1 and the primary Müller cells with 1 μM PGE2 for 24 hours, then assessed the cell surface C5aR protein levels again by flow cytometry. These experiments showed that C5aR protein levels were upregulated on both the primary and the immortalized Müller cells by PGE2 and now it was detectable by flow cytometry (Fig. 2B). Confocal microscopy analysis of PGE2-treated and untreated Müller cells further confirmed the flow cytometry results (Fig. 2C).
Figure 2.

C5aR expression in Müller cells is upregulated by PGE2 as examined by qRT-PCR (A), flow cytometry (B), and confocal microscopy (C). (A) Quantitative RT-PCR assessment of C5aR expression in Müller cells after PGE2 stimulation. Müller cells were treated with 1 μM PGE2, and total RNAs were isolated after 0, 6, and 24 hours of PGE2 stimulation. C5aR transcript levels were measured by qRT-PCR and normalized against 18S RNA levels. (B) Flow analysis of C5aR expression on surface of Müller cells. C5aR was barely detectable in both primary cells (upper panel) and MIO-M1 cells (lower panel) under normal culture conditions (without PGE2); C5aR expression was increased and detectable in both types of cells after incubation with 1 μM PGE2 for 24 hours (with PGE2). Dotted line, isotype control; solid line, anti-C5aR mAb staining. Representative results from three individual experiments. (C) Confocal analysis of C5aR expression on Müller cells, showing results comparable to those from flow cytometry analysis.
C5aR Signaling Upregulates IL-6 and VEGF Production From Müller Cells
To determine whether the C5aR detected on Müller cells might regulate cellular activities, we cultured MIO-M1 cells in the presence or absence of PGE2 for 24 hours. After this step, the cells were washed and incubated with different concentrations of C5a or the same concentrations of C5a together with 50 μM of C5aRA as controls. We collected the culture supernatants 24 hours and 48 hours after C5a stimulation to measure levels of IL-6 and VEGF using the respective ELISAs. These experiments showed that Müller cells constitutively produce IL-6 and VEGF. Without the pretreatment of PGE2, C5a stimulation barely changed the production of these cytokines. With the upregulation of C5aR by PGE2, C5a stimulated Müller cells to produce both IL-6 and VEGF in a dose-dependent manner compared with the PGE2 pretreated cells cultured in the absence of C5a. The upregulation of IL-6 and VEGF was the direct result of C5a-C5aR interactions in Müller cells, as indicated by the fact that including a C5aR antagonist in the culture totally abolished the effects of C5a on the upregulation of these cytokines (Fig. 3).
Figure 3.

C5a induces production of IL-6 (A) and VEGF (B) from Müller cells. MIO-M1 cells were treated with 1 μM PGE2 for 24 hours or left untreated. After washing, 0, 50, 100, and 200 ng/mL C5a were added, either in the absence or presence of 50 μM of C5aRA. Culture supernatants were collected 24 and 48 hours later, and levels of IL-6 and VEGF were measured by standard ELISA. Data are shown as mean ± SD, from representative results of four independent experiments. (*P < 0.05 compared with group without PGE2 and C5a treatment.)
Conditioned Media From C5a-Treated Müller Cells Promote hREC Proliferation
To determine whether C5aR in Müller cells could regulate hREC proliferation through modulating IL-6 and VEGF production, we added into hREC cultures the above-described Müller cell–conditioned media containing C5a/C5aR signaling-stimulated IL-6 and VEGF, then assessed the endothelial cell proliferation 72 hours later using a CellTiter-Glo Luminescent Cell Assay kit (Promega). These assays showed that in proportion to the levels of IL-6 and VEGF, as measured by ELISA as described above, the conditioned media from C5a-stimulated Müller cells augmented the proliferation of RECs in a dose-dependent manner compared with conditioned media from Müller cells without C5a stimulation (Fig. 4A). To determine whether the IL-6 and VEGF in the conditioned media were responsible for the elevated proliferation of hRECs, we repeated the experiment adding 5 μg/mL anti–IL-6 and anti-VEGF mAbs in the cultures and measured REC proliferation 72 hours later. These experiments showed that the augmentation of hREC proliferation by the conditioned media was reduced after IL-6 and VEGF were blocked by the neutralizing antibodies (Fig. 4B).
Figure 4.

Conditioned media from C5a-treated Müller cells promote human REC proliferation. Culture supernatants collected 48 hours after C5a stimulation from the above studies were added into hREC cultures (1:1 dilution), and REC proliferation was assessed 72 hours later using a CellTiter-Glo Luminescent Cell Viability Assay Kit (A). To verify the role of IL-6 and VEGF in the culture supernatant in promoting hREC proliferation, the experiments were repeated with IL-6 and VEGF neutralization mAbs (B). *P < 0.05.
Conditioned Media From C5a-Stimulated Müller Cells Increases hREC Monolayer Permeability
In addition to its ability to promote REC proliferation, previous studies have shown that VEGF compromises endothelial cell tight junctions and results in increased vascular permeability. It has also been demonstrated that Müller cells are the major source of VEGF in the retina, at least in a model of DR.25 We examined whether conditioned media from Müller cells stimulated with C5a could also regulate REC permeability. We first grew hRECs into a monolayer in a Transwell cell culture system, then replaced half of the REC media with conditioned media from Müller cells that either had or had not undergone C5a stimulation. Twenty-four hours later, we assessed the permeability of the hRECs by adding FITC-labeled dextran into the inserts and measuring levels of the FITC-dextran leaked into the lower chamber at different time points. These experiments demonstrated that, compared with normal media (which maintained REC monolayer integrity), conditioned media from unstimulated Müller cells increased FITC-dextran leakage (Fig. 5). Conditioned media from C5a-stimulated Müller cells further increased hREC permeability compared with conditioned media from unstimulated Müller cells (Fig. 5).
Figure 5.
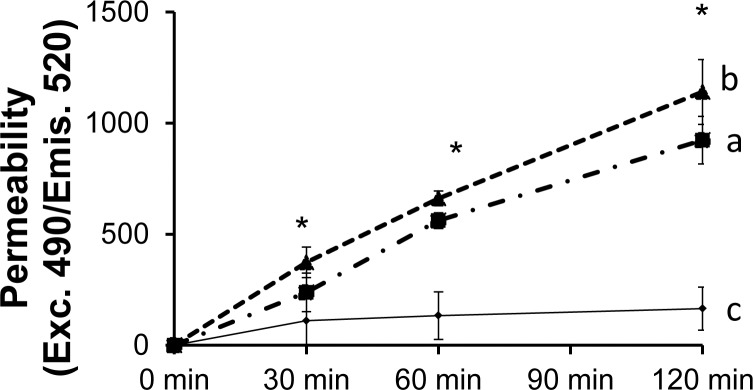
Conditioned media from C5a-treated Müller cells increase human REC monolayer permeability. Human RECs were seeded on a culture inset for 7 days to allow the formation of the REC monolayer. After this step, conditioned media from Müller cells cultured in the absence or presence of PGE2/C5a were added into the inserts at a 1:1 dilution. Then, FITC-labeled dextran was added into the inserts, and levels of leaked FITC-dextran in the bottom wells were measured at different time points by a fluorescence plate reader (Molecular Devices, Sunnyvale, CA) using 490 nm and 530 nm as the excitation and emission wavelengths, respectively. *P < 0.05.
Hyperglycemia Upregulates C5aR Expression on Müller Cells
The above studies indicate that PGE2 upregulates C5aR expression on Müller cells and that C5aR signaling augments IL-6 and VEGF production, which promotes REC proliferation and increases REC monolayer permeability, both of which are important in the pathogenesis of DR. Because patients and animals who develop DR have both hyperglycemia and increased PGE2 levels in the eye,23 to investigate the impacts of hyperglycemic conditions and PGE2 on C5aR expression in Müller cells, we cultured MIO-M1 Müller cells in media containing normal (5 mM) or high (30 mM) concentrations of glucose for 7 days with daily media change. On day 8, we left some cells untreated and treated other cells with PGE2 for 24 hours, following the protocols described above, then examined C5aR protein expression by flow cytometry. We found that the hyperglycemic state alone directly upregulated C5aR expression in Müller cells (Fig. 6A) and that PGE2 treatment of Müller cells cultured in media containing high-glucose media further upregulated C5aR expression (Fig. 6A). To verify the flow cytometry results with regard to C5aR functionality, we treated Müller cells cultured in normal or high-glucose media with or without PGE2 for 24 hours, then stimulated the cells with different concentrations of C5a following the protocols described above. After 48 hours, we compared IL-6 and VEGF levels in the culture supernatants by ELISA. These assays showed that in the absence of PGE2 treatment, Müller cells cultured under hyperglycemic conditions produced higher levels of IL-6 but not VEGF after C5a stimulation than cells cultured with media containing normal glucose (Fig. 6B). However, under both PGE2 stimulation and hyperglycemic state, Müller cells further increased the production of IL-6 and VEGF after C5aR activation by C5a.
Figure 6.

Hyperglycemia upregulates C5aR expression on Müller cells. (A) MIO-M1 cells were cultured in high-glucose (30 mM) media for 7 days with daily media change, then cells were cultured in the absence or presence of 1 μM PGE2 for another 24 hours, followed by flow cytometry analysis of C5aR expression on the surface of Müller cells. Dotted line, isotype control; solid line, anti-C5aR mAb staining. (B) MIO-M1 cells were cultured in normal (5 mM) or high-glucose (30 mM) media for 7 days with daily media change, then cultured in the absence or presence of 1 μM PGE2 for another 24 hours. Then different amounts of C5a, with or without 50 μM C5aRA, were added into each well. Forty-eight hours later, IL-6 and VEGF levels in the culture supernatants were measured by conventional ELISA. *P < 0.05.
Discussion
In this study, we demonstrated that Müller cells express C5aR by studying both the primary human retinal Müller cells and the spontaneously immortalized human retinal Müller cell line MIO-M1. We found that expression of C5aR was upregulated by PGE2 or hyperglycemia and that stimulating C5aR with its ligand C5a upregulated IL-6 and VEGF production, which enhanced human REC proliferation and increased REC monolayer permeability.
When complement is activated, C5a is released to the nearby environment and serves as a potent chemokine and activator for many types of cells that express its receptor, C5aR. In the retina, C5aR has been found in RPE cells and upregulates monocyte chemotactic protein-1, IL-1β, IL-6, and IL-8 after ligation with its ligand C5a.26,27 However, whether the retina Müller cells express C5aR was not clear. Results presented in this report demonstrate that primary Müller cells, as well as the immortalized Müller cell MIO-M1, constitutively express low levels of C5aR. Directly treating the Müller cells with the ligand C5a did not result in measurable IL-6 or VEGF upregulation, suggesting that complement, or at least C5a, does not have a significant effect on the production of these cytokines from Müller cells under normal conditions, potentially due to the extremely low levels of C5aR expressed. It also suggests that complement has to orchestrate with other pathological changes happening during the development of retinal disease (e.g., DR), to promote inflammation, neovascularization, and other complications.
PGE2 has been shown to upregulate C5aR expression in human monocyte-derived dendritic cells.28 It is also considered as a marker of inflammation, and its levels are elevated systemically in the plasma or locally in the retinas of patients or animals with DR,4,5,29 suggesting a possible role of PGE2 in the development of DR. We found that treating Müller cells with PGE2 significantly upregulated their C5aR expression as examined with qRT-PCR, flow cytometry, and confocal immunofluorescence analysis. The PGE2-stimulated C5aR on Müller cells was functional, as stimulation of these PGE2-treated Müller cells by C5a significantly upregulated their production of VEGF and IL-6, cytokines that are found critical for the development of DR by promoting inflammation and neovascularization.3,30 In addition, it appears that blocking the IL-6 and VEGF in the Müller cell–conditioned media only partially decreased hREC proliferation (Fig. 4B), suggesting that other factors might also be involved in the mechanism by which complement contributes to the development of DR These results provide a mechanistic link between PGE2, complement, and DR, suggesting that PGE2 might be involved in the development of DR by upregulating C5aR on Müller cells in the retina, which leads to augmented local production of IL-6 and VEGF after stimulation of the complement activation product C5a.
Although complement activation products have been detected in the retinas of DR patients or animal models of DR,31,32 and although C5a has been correlated with inflammatory cytokines (including VEGF) in the vitreous of DR patients,33 the potential mechanisms by which complement contributes to the development of DR remain elusive. C5a-stimulated VEGF production from RPE cells has been found important in a laser-induced retinal choroidal neovascularization model, in which genetic or pharmaceutical ablation of C5aR function in the retina reduced retinal VEGF levels and the severity of choroidal neovascularization.34 Elevated retinal VEGF levels have been found in DR patients, associated with complications including retina-blood barrier breakdown and neovascularization.35 Müller cells have been identified as a primary source of VEGF in several retinal diseases, including DR.36 In fact, by studying animals in which VEGF expression is selectively knocked out in Müller cells, studies by others have demonstrated the critical role of Müller cell–produced VEGF in inducing retinal neovascularization and in vascular leakage in an animal model of DR.37 In our study, the conditioned media containing elevated levels of VEGF enhanced hREC proliferation and increased REC monolayer permeability, suggesting that PGE2 and C5a could work together to stimulate local VEGF production from Müller cells to contribute to retinal neovascularization and vascular leakage.
Hyperglycemia is the main metabolic characteristic of diabetes mellitus, and it plays critical roles in the development of many diabetic complications. We found that hyperglycemia alone upregulated C5aR expression on Müller cells (Fig. 6A). Consequently, Müller cells cultured under hyperglycemic conditions produced higher levels of VEGF and IL-6 after C5a stimulation than cells cultured in normal media (Fig. 6B). These data, together with other results described in this report and previously published work, link hyperglycemic conditions with many hallmarks of DR patients, including retinal neovascularization, retinal vascular leakage, and elevated ocular levels of VEGF and IL-6. It is noteworthy that IL-6 may exert a protective role in the eye, despite its widely believed detrimental effects as a strong proinflammatory cytokine; previous studies have demonstrated that the addition of IL-6 protected Müller cells from hyperglycemia-induced injury.38
In summary, our studies found that C5aR is expressed in human retinal Müller cells. Müller cell–expressed C5aR is upregulated by PGE2 and by hyperglycemic conditions. Signaling through C5aR stimulates IL-6 and VEGF production from the Müller cells, which increases hREC proliferation and REC monolayer permeability. These results suggest that complement may regulate the production of inflammatory factors and angiogenesis factors from Müller cells through C5aR, which might contribute to the pathogenesis of many retinal disorders, especially DR.
Acknowledgments
The authors thank Astrid Limb from University Colleage London for providing the MIO-M1 cells, Lian-fu Wang at the Department of Surgery, Cleveland Clinic, for help on the qRT-PCR experiments, and Christine Kassuba from Cleveland Clinic for help with manuscript preparation.
Supported in part by American Diabetes Association Grant 1-13-BS-112-BR (FL) and EY019250 (CSS).
Disclosure: L. Cheng, None; H. Bu, None; J.-A.C. Portillo, None; Y. Li, None; C.S. Subauste, None; S.S. Huang, None; T.S. Kern, None; F. Lin, None
References
- 1. Moss SE, Klein R, Klein BEK. The 14-year incidence of visual loss in a diabetic population. Ophthalmology. 1998; 105: 998–1003 [DOI] [PubMed] [Google Scholar]
- 2. Davis MD. Diabetic retinopathy. A clinical overview. Diabetes Care. 1992; 15: 1844–1874 [DOI] [PubMed] [Google Scholar]
- 3. Koleva-Georgieva DN, Sivkova NP, Terzieva D. Serum inflammatory cytokines IL-1beta, IL-6, TNF-alpha and VEGF have influence on the development of diabetic retinopathy. Folia Medica (Plovdiv). 2011; 53: 44–50 [DOI] [PubMed] [Google Scholar]
- 4. Form DM, Auerbach R. PGE2 and angiogenesis. Proc Soc Exp Biol Med. 1983; 172: 214–218 [DOI] [PubMed] [Google Scholar]
- 5. Yanni SE, Barnett JM, Clark ML, Penn JS. The role of PGE2 receptor EP4 in pathologic ocular angiogenesis. Invest Ophthalmol Vis Sci. 2009; 50: 5479–5486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Phipps RP, Stein SH, Roper RL. A new view of prostaglandin E regulation of the immune response. Immunol Today. 1991; 12: 349–352 [DOI] [PubMed] [Google Scholar]
- 7. Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther. 2004; 103: 147–166 [DOI] [PubMed] [Google Scholar]
- 8. Yanni SE, Barnett JM, Clark ML, Penn JS. The role of PGE2 receptor EP4 in pathologic ocular angiogenesis. Invest Ophthalmol Vis Sci. 2009; 50: 5479–5486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tojo N, Kashiwagi Y, Nishitsuka K, et al. Interactions between vitreous-derived cells and vascular endothelial cells in vitreoretinal diseases. Acta Ophthalmol. 2010; 88: 564–570 [DOI] [PubMed] [Google Scholar]
- 10. Tojo N, Kashiwagi Y, Yamamoto S, Yamamoto T, Yamashita H. The in vitro response of human retinal endothelial cells to cytokines and other chemically active agents is altered by coculture with vitreous-derived hyalocytes. Acta Ophthalmol. 2010; 88: e66–e72 [DOI] [PubMed] [Google Scholar]
- 11. Bora PS, Sohn J-H, Cruz JMC, et al. Role of complement and complement membrane attack complex in laser-induced choroidal neovascularization. J Immunol. 2005; 174: 491–497 [DOI] [PubMed] [Google Scholar]
- 12. Edwards AO, Ritter R III, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005; 308: 421–424 [DOI] [PubMed] [Google Scholar]
- 13. Read RW, Szalaid AJ, Vogta SD, McGwine G, Barnumc SR. Genetic deficiency of C3 as well as CNS-targeted expression of the complement inhibitor sCrry ameliorates experimental autoimmune uveoretinitis. Exp Eye Res. 2006; 82: 389–394 [DOI] [PubMed] [Google Scholar]
- 14. Gerl VB, Bohl J, Pitz S, Stoffelns B, Pfeiffer N, Bhakdi S. Extensive deposits of complement C3d and C5b-9 in the choriocapillaris of eyes of patients with diabetic retinopathy. Invest Ophthalmol Vis Sci. 2002; 43: 1104–1108 [PubMed] [Google Scholar]
- 15. Gasque P, Singhrao SK, Neal JW, Götze O, Morgan BP. Expression of the receptor for complement C5a (CD88) is up-regulated on reactive astrocytes, microglia, and endothelial cells in the inflamed human central nervous system. Am J Pathol. 1997; 150: 31–41 [PMC free article] [PubMed] [Google Scholar]
- 16. Riedemann NC, Guo R-F, Sarma VJ, et al. Expression and function of the C5a receptor in rat alveolar epithelial cells. J Immunol. 2002; 168: 1919–1925 [DOI] [PubMed] [Google Scholar]
- 17. Drouin SM, Kildsgaard J, Haviland J, et al. Expression of the complement anaphylatoxin C3a and C5a receptors on bronchial epithelial and smooth muscle cells in models of sepsis and asthma. J Immunol. 2001; 166: 2025–2032 [DOI] [PubMed] [Google Scholar]
- 18. Yoshihiro F, Edward MM. C5a receptor-mediated production of IL-8 by the human retinal pigment epithelial cell line, ARPE-19. Curr Eye Res. 2001; 23: 320–325 [DOI] [PubMed] [Google Scholar]
- 19. Skeie JM, Fingert JH, Russell SR, Stone EM, Mullins RF. Complement component C5a activates ICAM-1 expression on human choroidal endothelial cells. Invest Ophthalmol Vis Sci. 2010; 51: 5336–5342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ikeda T, Puro DG. Nerve growth factor: a mitogenic signal for retinal Muller glial cells. Brain Res. 1994; 649: 260–264 [DOI] [PubMed] [Google Scholar]
- 21. Limb GA, Salt TE, Munro PM, Moss SE, Khaw PT. In vitro characterization of a spontaneously immortalized human Muller cell line (MIO-M1). Invest Ophthalmol Vis Sci. 2002; 43: 864–869 [PubMed] [Google Scholar]
- 22. Mizutani M, Kern TS, Lorenzi M. Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J Clin Invest. 1996; 97: 2883–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schoenberger SD, Kim SJ, Sheng J, Rezaei KA, Lalezary M, Cherney E. Increased prostaglandin E2 (PGE2) levels in proliferative diabetic retinopathy, and correlation with VEGF and inflammatory cytokines. Invest Ophthalmol Vis Sci. 2012; 53: 5906–5911 [DOI] [PubMed] [Google Scholar]
- 24. Weinmann O, Gutzmer R, Zwirner J, et al. Up-regulation of C5a receptor expression and function on human monocyte derived dendritic cells by prostaglandin E2. Immunology. 2003; 110: 458–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang J, Xu X, Elliott MH, Zhu M, Le YZ. Muller cell-derived VEGF is essential for diabetes-induced retinal inflammation and vascular leakage. Diabetes. 2010; 59: 2297–2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fukuoka Y, Medof EM. C5a receptor-mediated production of IL-8 by the human retinal pigment epithelial cell line, ARPE-19. Curr Eye Res. 2001; 23: 320–325 [DOI] [PubMed] [Google Scholar]
- 27. Fukuoka Y, Strainic M, Medof ME. Differential cytokine expression of human retinal pigment epithelial cells in response to stimulation by C5a. Clin Exp Immunol. 2003; 131: 248–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weinmann O, Gutzmer R, Zwirner J, et al. Up-regulation of C5a receptor expression and function on human monocyte derived dendritic cells by prostaglandin E2. Immunology. 2003; 110: 458–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Steinle JJ. Sympathetic neurotransmission modulates expression of inflammatory markers in the rat retina. Exp Eye Res. 2007; 84: 118–125 [DOI] [PubMed] [Google Scholar]
- 30. Myśliwiec M, Balcerska A, Zorena K, Myśliwska J, Lipowski P, Raczyńska K. The role of vascular endothelial growth factor, tumor necrosis factor alpha and interleukin-6 in pathogenesis of diabetic retinopathy. Diabetes Res Clin Pract. 2008; 79: 141–146 [DOI] [PubMed] [Google Scholar]
- 31. Gerl VB, Bohl J, Pitz S, Stoffelns B, Pfeiffer N, Bhakdi S. Extensive deposits of complement C3d and C5b-9 in the choriocapillaris of eyes of patients with diabetic retinopathy. Invest Ophthalmol Vis Sci. 2002; 43: 1104–1108 [PubMed] [Google Scholar]
- 32. Zhang J, Gerhardinger C, Lorenzi M. Early complement activation and decreased levels of glycosylphosphatidylinositol-anchored complement inhibitors in human and experimental diabetic retinopathy. Diabetes. 2002; 51: 3499–3504 [DOI] [PubMed] [Google Scholar]
- 33. Muramatsu D, Wakabayashi Y, Usui Y, Okunuki Y, Kezuka T, Goto H. Correlation of complement fragment C5a with inflammatory cytokines in the vitreous of patients with proliferative diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol. 2013; 251: 15–17 [DOI] [PubMed] [Google Scholar]
- 34. Nozaki M, Raisler BJ, Sakurai E, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci U S A. 2006; 103: 2328–2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Simo R, Carrasco E, Garcia-Ramirez M, Hernandez C. Angiogenic and antiangiogenic factors in proliferative diabetic retinopathy. Curr Diabetes Rev. 2006; 2: 71–98 [DOI] [PubMed] [Google Scholar]
- 36. Bai Y, Ma J-X, Guo J, et al. Müller cell-derived VEGF is a significant contributor to retinal neovascularization. J Pathol. 2009; 219: 446–454 [DOI] [PubMed] [Google Scholar]
- 37. Wang J, Xu X, Elliott MH, Zhu M, Le Y-Z. Müller cell-derived VEGF is essential for diabetes-induced retinal inflammation and vascular leakage. Diabetes. 2010; 59: 2297–2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yego EC, Vincent JA, Sarthy V, Busik JV, Mohr S. Differential regulation of high glucose-induced glyceraldehyde-3-phosphate dehydrogenase nuclear accumulation in Muller cells by IL-1beta and IL-6. Invest Ophthalmol Vis Sci. 2009; 50: 1920–1928 [DOI] [PubMed] [Google Scholar]