

Abstract
Cellular senescence, an irreversible cell cycle arrest induced by a diversity of stimuli, has been considered as an innate tumor suppressing mechanism with implications and applications in cancer therapy. Using a targeted proteomics approach we show that fibroblasts induced into senescence by expression of oncogenic Ras exhibit a decrease of global acetylation on all core histones, consistent with formation of senescence-associated heterochromatic foci. We also detected clear increases in repressive markers (e.g., >50% elevation of H3K27me2/3) along with decreases in histone marks associated with increased transcriptional expression/elongation (e.g., H3K36me2/3). Despite the increases in repressive marks of chromatin, 179 loci (of 2206 total) were found to be upregulated by global quantitative proteomics. The changes in the cytosolic proteome indicated an upregulation of mitochondrial proteins and downregulation of proteins involved in glycolysis. These alterations in primary metabolism are opposite of the well-known Warburg effect observed in cancer cells. This study significantly improves our understanding of stress-induced senescence and provides a potential application for triggering it in anti-proliferative strategies that target the primary metabolism in cancer cells.
Keywords: histones, mass spectrometry, metabolism, oxidative phosphorylation, proteomics, senescence
INTRODUCTION
In normal tissues, proliferative balance is carefully maintained by the highly regulated process of cell division. Cancers arise as somatic cells escape the controls which normally restrain them against unregulated expansion in renewable tissues [1]. Evolutionally, several mechanisms have developed to prevent uncontrolled cell division. These mechanisms work coordinately in an extraordinarily effective way to suppress tumorigenesis. Given the trillions of cells in a multicellular organism that are constantly exposed to cancer-causing environmental insults, the development of cancer is relatively rare.
Cellular senescence is one of several tumor-suppressing mechanisms and refers to the result of a signal transduction program that leads to an irreversible arrest of the cell cycle [1]. The senescence phenomenon was first associated with aging as Hayflick and colleagues observed that normal human fibroblasts lost their proliferative capacity after a finite number of cell divisions in culture [2]. This phenomenon termed replicative senescence has been shown to be a result of telomere attrition [3]. Senescent cells display a characteristic phenotype, including enlarged cell size, flat vacuolated cell morphology, inability to synthesize DNA, formation of senescence-associated heterochromatin foci (SAHF) and expression of an endogenous β-galactosidase activity [4]. A similar phenotype can occur in “young” cells in response to multiple stimuli, including DNA damage, oncogenic signals and oxidative stress [5].
Oncogene activation acting to potentiate or overstimulate normal growth signaling is a hallmark of cancer [5]. However, in “normal” cells oncogene activation does not lead to transformation, but instead provokes cellular senescence. Expression of a variety of oncogenes, including HRAS [6], BRAF [7], E2F1 [8], and MYC [9], drives a burst of cell hyperproliferation followed by a proliferation slow down and eventually cell growth arrest. It has recently been observed that oncogene induced senescence (OIS) is associated with activation of the DNA damage response (DDR) [6, 10]. DNA damage signals contribute to activation of the p53 and pRB pathways, two key regulators of the senescence program, indicated by the fact that inactivation of either one usually abolishes cellular senescence in both mouse and human cells [11].
Cellular senescence serves as a tumor-suppressor in two ways to inhibit proliferation. First, cell growth arrest certainly blocks tumor growth. Second, proliferation arrest should prevent the acquisition of additional mutations that can drive oncogenic events such as increased metastatic potential. Even though some cancer cells can escape the senescence program, they still retain the capacity to undergo senescence [5]. From a potential therapeutic perspective, senescence initiated by genotoxic stress, including chemotherapeutic drugs, is able to contribute to tumor regression in vivo [12, 13]. Senescent cells are also able to secret immune regulators which can trigger clearance of senescent cells by the innate immune system [14]. Moreover, cellular senescence could serve as a backup program that inhibits growth when tumors have defects in apoptotic pathways or developed resistance to chemotherapeutic drug-induced apoptosis [4].
Though great progress towards understanding the senescence phenotype and its underlying molecular mechanisms has been made over the last decade, a global view at the systems level is still lacking. It has been shown that senescent cells display a distinct gene expression profile at the transcriptome level [15], with such “omic” studies mostly focused on replicative senescence [16, 17]. Moreover, transcriptome dynamics do not reflect direct changes at the proteome level [18, 19]. Hence, a mechanistic investigation of the proteome in oncogene induced senescent cells could provide information about differential regulation of responsive genes in the senescence program.
In this work, we applied quantitative mass spectrometry to study global histone modifications and protein expression changes in OIS cells. While core histones showed marks consistent with heterochromatin formation in bulk chromatin, we quantified nearly 200 proteins with elevated expression levels in senescent cells. These upregulated loci agreed with the view that senescent cells are arrested in the G1 phase of the cell cycle and cell growth is uncoupled from cell division. Upregulation of proteins involved in oxidative phosphorylation and downregulation of proteins involved in glycolysis indicated that the metabolic signature of oncogene induced senescence was opposite of the well-known Warburg effect in cancer cells. By changing protein expression and activity to mimic such a metabolic signature, control fibroblasts, were forced into senescence in elevated numbers. These findings further our understanding of the senescence program and its antagonistic role to the development of cancer.
MATERIALS & METHODS
Cell culture and gene transfer
Human diploid fibroblasts IMR-90 were purchased from ATCC and cultured in DMEM supplemented with 10% FBS and antibiotics. The retroviral vectors (pBabe-Puro and pBabe-Ras (H-RasV12)) were kind gifts from Dr. Scott Lowe at Cold Spring Harbor Laboratory. Retroviruses were packed using phoenix cells (ATCC) as described previously [20]. Pseudo-retrovirus used in SILAC experiments with VSV-g was packed and concentrated at the Vector Core facility at University of Michigan.
Intact histone profiling and peptide preparation
Whole core histones were purified from control cells and senescent cells, respectively, and analyzed by Top Down LC-MS as described previously [21]. For Bottom Up LC-MS of histone peptides, bulk histones from control and senescent cells were chemically derivatized by treatment with propionic anhydride and digested by trypsin as described before [22]. They were analyzed on a triple quadrupole mass spectrometer (vide infra).
SILAC labeling
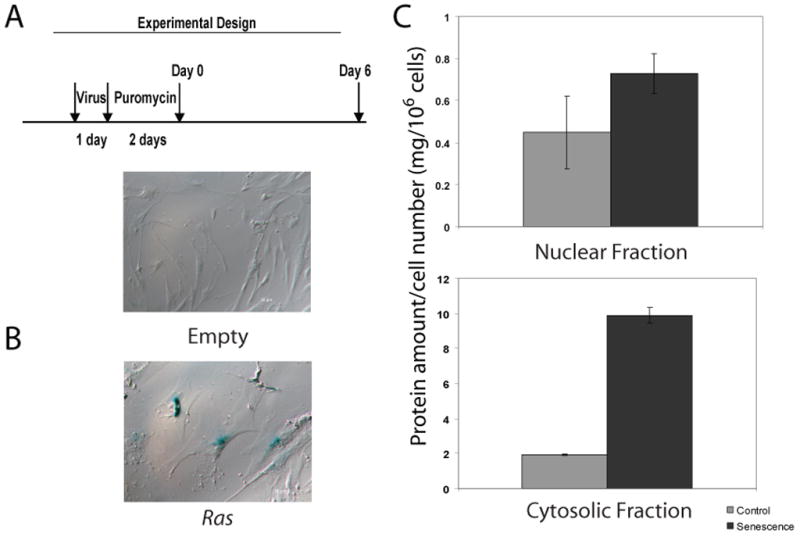
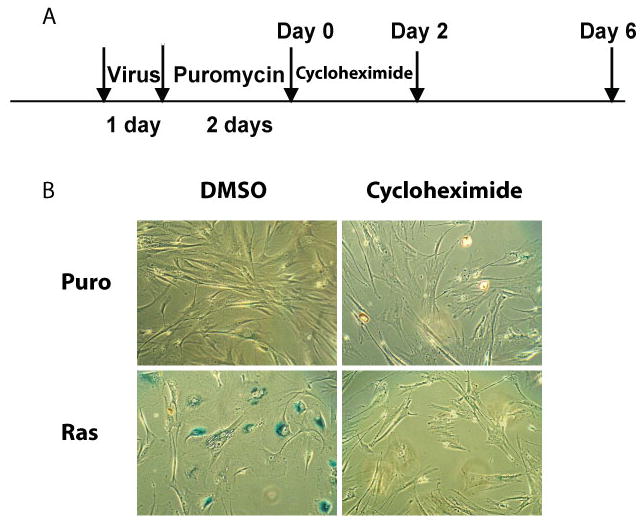
“Heavy” media was prepared in house with L-13C6, 15N4 –arginine, L-13C6, 15N2-lysine and dialyzed FBS. IMR-90 cells were cultured in heavy media for 3 days prior to retroviral infection. Concentrated retroviruses containing pBabe-Puro plasmid were diluted with heavy media in a 1:20 ratio to reduce light amino acid incorporation during virus infection. The infected population was selected in heavy media with 2 μg/mL puromycin for 2 days and cultured in fresh heavy media for another 6 days (Figure 1A).
Figure 1.
Protein harvest, fractionation and digestion
Control and senescent cells were trypsinized and counted with a hemocytometer. Cell pellets were washed by cold PBS 3 times and lysed in nuclear isolation buffer containing 15 mM Tris–hydrochloride; pH 7.5, 60 mM potassium chloride, 11 mM calcium chloride, 5 mM sodium chloride, 5 mM magnesium chloride, 250 mM sucrose, 1 mM DTT, 10 mM sodium butyrate, protease inhibitor cocktails (Sigma), phosphatase inhibitor cocktails (Sigma) and 0.3% NP-40. The cell lysate was centrifuged at 1000 × g for 5 minutes at 4° C. The supernatant was saved as the cytosolic fraction. The pellet (nuclear fraction) was dissolved in 50 mM Tris buffer with 4% SDS supplemented with 10 mM sodium butyrate, 1 mM DTT, phosphatase inhibitor cocktails and protease inhibitor cocktails. Protein concentration was determined by BCA assays (Fisher). Equal amounts of the cytosolic or nuclear proteins from control and senescent cells were mixed, respectively, resulting in 80–100 μg of total protein.
Protein mixtures were separated by 1D SDS-PAGE, using 1 mm NuPage Noves Bis-Tris 4%–20% gradient gels (Invitrogen) according to the instructions. The gel was stained with Coomassie blue using mass spectrometry compatible staining kit (Thermo Pierce) and destained in HPLC grade water. Each gel lane was cut into 14 slices and protein bands were excised and digested with trypsin as described previously [23]. Digested peptides were extracted from the gel pieces and dried down in a speed-vac prior to storage at −80°C.
Bottom Up Nanocapillary LC-MS/MS
Digested proteins were separated by online reverse-phase nanoscale capillary liquid chromatography (nanoLC) coupled with electrospray tandem mass spectrometry (MS/MS). The peptide mixtures were resuspended in 10 μL of buffer A (100% LC-MS water containing 0.2% formic acid) and loaded onto a trap column (150 μm internal diameter, in-house packed with C18 resin to a length of 3 cm) using an Eksigent 1D+ autosampler (Eksigent Technologies, Dublin, CA) at a high flow rate of 5 μL/min. Columns were in-house packed with 10μm Jupiter C18 (Phenomenex, Torrance, CA) material. Subsequently, the peptides were eluted off the trap column and further separated by a C18 analytical column (75 μm × 10 cm) with a nanospray emitter containing an integrated frit (PicoFrit, New Objective, Inc., Woburn, MA) with 5 μm C18 resin at a flow rate of 300 nL/min. A 120 min. gradient with buffer A (as above) and buffer B (5% water/95% acetonitrile, containing 0.2% formic acid) was used: buffer B concentration increased from 5% to 30% over 50 min. and from 30% to 45% from 50 to 70 min. Within the next 70 to 72 min., buffer B was ramped to 80% and remained at 80% from 72to 77 min. before declining to 5% by 79 min. The column was further washed with 2 gradients of 5% B to 80% B from 79 to 99 min. and then equilibrated at 0% B from 99 to 120 min.
Samples were analyzed on a 12 Tesla LTQ-FT Ultra (Thermo Fisher Scientific, San Jose, CA) instrument fitted with a digitally controlled nanospray ionization source (PicoView DPV-550, New Objective, Inc.). This FTMS instrument was operated in data dependent mode to automatically switch between MS and MS/MS acquisition. Survey scan spectra were acquired in the FT-ICR cell from m/z 400 to 1600 with resolution R=170,000 at m/z 400 and AGC set to 1,000,000 charges. The MS/MS spectra were acquired in the linear ion trap with the five most intense ions sequentially isolated and fragmented in the ion trap using CID with a target value of 30,000 and maximum fill time of 150 ms. Dynamic exclusion was set to 30 s with a repeat count of 2. An activation Q of 0.25 and activation time of 30 ms were applied to MS/MS acquisitions.
Top Down Nanocapillary LC-MS
Intact histone proteins were analyzed as described previously. Briefly, HPLC purified histones underwent RPLC-MS analysis. Buffer A is 95% water, 5% acetonitrile, and 0.2% formic acid while buffer B is 5% water, 95% acetonitrile, and 0.2% formic acid. The columns were in-house packed with 5 μm, 1000Å PLRP-S material (Agilent Technologies, Inc., Santa Clara, CA). The proteins were loading onto a trap column (150 μm internal diameter, with PLRP-S length of 3 cm) at a high flow rate of 3 μL/min. The proteins were eluted off of the trap column at a flow rate of 300 nL/min onto an analytical column (75 μm internal diameter, 10 cm PLRP-S length). The gradient consisted of buffer B being increased from 0% to 30% by 20 min., then from 30% to 60% from 20 to 100 min. Between LC-MS runs, the column was washed and equilibrated by three short 0% to 100% B gradients. The LC-MS experiments were performed on a 7 Tesla LTQ-FT mass spectrometer (Thermo Fisher Scientific, San Jose, CA). The LTQ-FT method performed a low resolution ion trap scan followed by a high resolution FTMS scan with resolution of 100,000 at m/z 400. Intact monoisotopic masses were calculated by the Xtract algorithm (Thermo Fisher Scientific, San Jose, CA).
Nano-LC triple quadrupole mass spectrometry
Histones from isolated nuclei were acid-extracted and derivatized with propionic anhydride both prior to and following trypsin digestion as previously described [25, 26]. Briefly, two rounds of propionylation are performed. The first round occurs before trypsin digestion to block the cleavage in unmodified lysine while the second round comes after trypsin digestion to derivative newly exposed N-termini. In addition to producing uniformity in peptide length, histone derivitization can also increase the hydrophobicity of the small, basic peptides enough to allow retention on reverse phase columns. The first round normalizes peptide length and both derivitization rounds serve to increase hydrophobicity of the peptides. The net effect of the two rounds of derivitization is that N-terminal, unmodified, and singly methylated lysines gain one propionyl group. However, no propionyl group is added to lysines with di- and tri-methylation. Propionyl derivatized histone peptides were resuspended in 20 μL water with 1% TFA. The nanoLC-MS system for peptide analysis consisted of a Dionex UltiMate 3000 coupled to a ThermoFisher Scientific TSQ Quantum triple quadrupole mass spectrometer. Buffer A was 100% LC-MS grade water with 0.1% formic acid and buffer B was 100% acetonitrile. The propionylated peptides were loaded onto a C18 trapping column (2 cm × 150 μm; Vydac 218MS C18 5μm beads) for 5 min. at a flow-rate of 5 μL/min. in 0.1% TFA loading buffer. The peptides were separated by a gradient from 2 to 35% buffer B from 5 to 31 min. The analytical column (10 cm × 75 μm) consisted of the same C18 material as the trapping column; both were packed in-house. The triple quadrupole settings were as follows: collision gas pressure of 1.5 mTorr; Q1 peak width of 0.7 (FWHM); cycle time of 3.5 s; skimmer offset of 10 V; electrospray voltage of 2.6 kV. Nanospray was from a New Objective emitter with 10 μm tip, attached to a custom-built stage.
Selected reaction monitoring (SRM) mass spectrometer transitions were developed and data were analyzed using Skyline software (v0.7; MacCoss Lab, University of Washington) [27]. Synthetic methylated and acetylated histone peptides (Anaspec) were employed in SRM-based assay development. The synthetic peptides were different singly modified forms of either the H3 1-21 aa peptide or the H3 21-44 aa peptide and were derivatized and trypsin digested as above. The different forms of the H3 1-21 aa peptide were unmodified, K4ac, K4me1, K4me2, K4me3, K9me1, K9me2, K9me3, K9ac, and K14ac. The forms for the H3 21-44 aa peptide were K27me1, K27me2, K27me3, K36me1, K36me2, and K36me3.
Each SRM transition has a predefined precursor mass as well as between one and three predefined fragmentation product ions (Supplementary Table 4). Briefly, method development consisted of surveying a wide range of predicted b and y ions for each peptide; where isobaric forms of the peptide existed (e.g. K27 and K36 methylation forms of the H3 27-40 aa peptide), transitions were selected that distinguished between the different forms. The most intense of these fragment ions were selected for collision energy optimization. The histone SRM analysis was performed in biological triplicate and technical duplicate.
Data analysis
SILAC-MS data were analyzed by MaxQuant software (v 1. 0. 13. 13) [28]. Proteins were identified by database searching using Mascot (Matrix Science Ltd., Beachwood, OH) against the human IPI database supplemented with frequently observed contaminants and the reverse sequences of each entry. Search parameters specified a MS deviation of 7 ppm and an MS/MS tolerance of 0.5 Da with up to three miscleavages allowed. Carbamidomethylation of cysteine was set as a fixed modification and oxidation of methionines and N-terminal protein acetylation were allowed as variable modifications. The false discovery rates (FDR) at the peptide and protein level were both set to 1%, and proteins identified with one unique peptide were manually validated. All the post-search bioinformatics analysis was performed with the lead proteins in a protein group. Data generated from triple quad mass spectrometry were processed using Skyline software [27], with Savitzky-Golay smoothing of peaks. Automatic peak assignment and retention times were verified manually. Total area of transitions corresponding to specific histone marks were employed to create the data of Figure 2B and Supplementary Figure 4.
Figure 2.
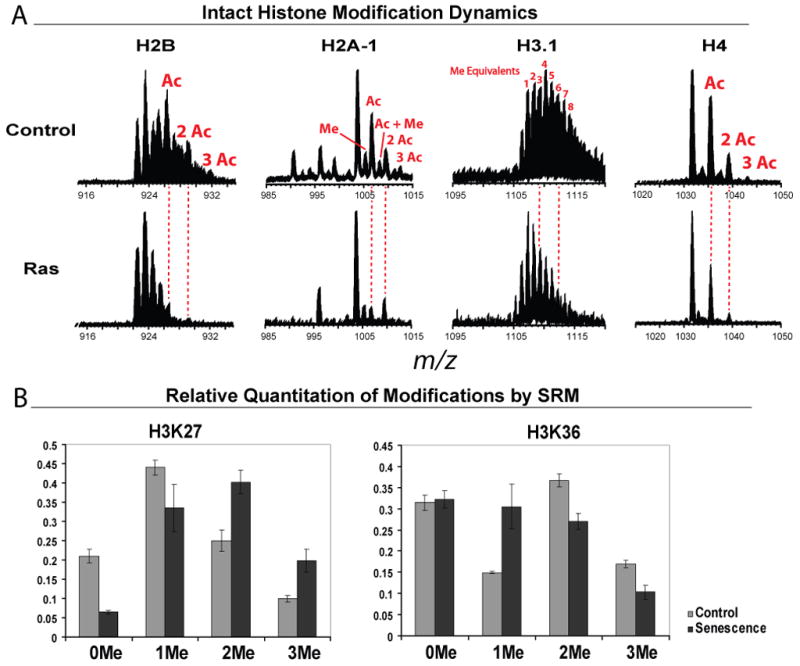
Intact histone MS data were analyzed by Xtract (Thermo Fisher, San Jose, CA). Manual Xtract analysis of the selected ion chromatograms of eluted protein species yielded monoisotopic masses. The masses were then matched to the Uniprot database masses to confirm identity. Acetylation forms (+42 Da for each acetylation) were also found using the manual Xtract data analysis approach.
SA-β-gal assays and SAHF images
SA-β-gal activity was detected as previously described [20]. DNA was visualized by DAPI staining (1 μg/mL).
Western blotting
Western blotting analysis was carried out using 20 μg of whole cell lysate. The membranes were probed with primary antibodies against p16 (1: 200, Santa Cruz), HDAC1 (1: 1000, Millipore), and HDAC2 (1: 1000, Millipore) in 0.05% Tween 20/PBS and then with an HRP-labeled secondary antibody (1:5000; Santa Cruz). Proteins were visualized by chemiluminescence with ECL Western blotting detection reagents (Thermo Pierce).
RESULTS
Reduction of global histone acetylation during oncogene induced senescence
In order to examine histone modifications in global chromatin, we analyzed intact histones from oncogenic Ras induced senescent cells (6 days after puromycin selection, Figure 1A) by LC-FTMS. The senescence phenotype was first confirmed by examining several well established senescence markers, including SAHF formation (>70% of cells), SA-β-gal assays and upregulation of protein p16INK4A (Figure 1B and Supplementary Figure 1). We observed a significant decrease of relative global histone acetylation levels from all core histone proteins in senescent cells compared to control cells (Figure 2A). Histone H2A, H2B and H4 showed decreased intensity of their acetylated forms in the spectra. Though H3 has a more complicated spectrum, interrogated in detail elsewhere [29, 30], we clearly observed the mass shift from higher m/z to lower m/z (base peak shifted from m/z 1110 to m/z 1107) indicative of deacetylation (Figure 1B).
To localize and quantify the modifications on specific lysine residues, we chemically derivatized bulk histones with propionic anhydride to produce tryptic peptides well retained on the HPLC column for analysis by LC-MS. Similarly, a peptide from the histone H4 tail also showed decreased acetylation levels by 50% of the mono-acetylated form in senescent cells (Supplementary Figure 2) but no significant changes for H4K20me1/2/3 were observed (data not shown) which is the major methylation site on histone H4 [31]. Next, we examined the protein expression level of selected histone deacetylases (HDACs) in senescent cells by western blotting. No upregulation of HDAC1 or HDAC2 expression was observed in senescent cells (Supplementary Figure 3) suggesting that global hypoacetylation in senescent cells might not result from changing the expression level of a single HDAC. This was also in agreement with our earlier reports showing that knocking down one or two HDACs did not change global histone acetylation levels in HeLa S3 cells [21].
Comprehensive quantitation of methylation pattern on histone H3 in senescent cells by SRM mass spectrometry
Using this strategy, we quantitatively analyzed methylation level changes in senescent cells on 4 different lysine residues (K9, K27, K36 and K79) on histone H3. The di- and tri-methylation of K27 and K36 have been associated with down- and upregulation of transcription, respectively, with the di- and tri-methylation of K27 having been shown to antagonize methylation of K36 [26, 32, 33]. Here, di- and tri-methylation on K27 significantly increased in senescent cells while the methylation pattern on K36 shifted from di- and tri-methylation to mono-methylation in senescent cells (Figure 2B). Methylation levels on histone H3 K79 increased slightly (Supplementary Figure 4A). While histone H3 K9 tri-methylation is associated with transcriptionally silent heterochromatin and has been shown to be enriched on SAHFs [34], the methylation pattern on the global chromatin did not change significantly as the di- and tri-methylation levels remained low (Supplementary Figure 4B).
Quantitative analysis of protein expression changes in senescent cells
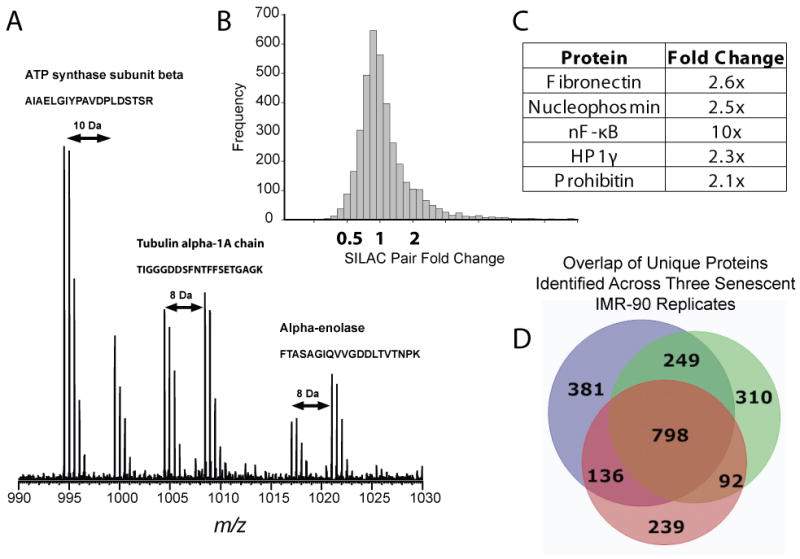
To characterize protein expression level changes in oncogene induced senescent cells, we SILAC-labeled IMR-90 cells transduced with pBabe-puro plasmids (control) and compared its proteome to that of oncogenic Ras induced senescent cells 6 days after puromycin selection. As primary fibroblasts have a slower growth rate when compared to cancer cell lines, we adapted the standard SILAC protocol to allow sufficient time for a full labeling of all proteins with heavy isotope versions of the amino acids (Supplementary Figure 5). Proteins from an aliquot of labeled cells were digested and analyzed by nano-LC-MS/MS and a labeling efficiency greater than 98% was observed (data not shown). We also observed no Arg-Pro conversion in these labeled cells [35]. Since senescent cells displayed a dramatic cell size enlargement, particularly in the cytoplasm, we measured the protein content from the cell nucleus and cytoplasm. The enlarged cytoplasm did translate into a ~four fold increase in amount of protein in the cell cytoplasm (Figure 1C). Hence, the same protein amounts were mixed during sample preparation instead of mixing the same number of cells in the SILAC experiments. Peptides resulting from trypsin digestion of the combined samples were then separated and detected by MS in form of peptide pairs, and their ratios reflect relative changes in protein abundance (Figure 3A). We used high resolution MS to identify 2206 proteins from three biological replicates at a protein false discovery rate of <1% (Supplementary Table 3). As shown by the Venn diagram in Figure 3D, a list of proteins was identified for each of the three biological replicates, with 60% of proteins identified at least in two biological replicates. For the 609 proteins present in all three biological replicates, the mean of the ratio between treated and control is 1.178 with a standard deviation of 0.457. We considered average changes in protein abundance greater than 1.916 as significant, with 1.916 being two standard deviations from the mean (Figure 3B).
Figure 3.
Proteins upregulated in oncogene induced senescent cells
In our data set, 179 proteins, ~9% of the identified proteome, showed significant increases of protein expression levels (Supplementary Table 1). In this list, several proteins have already been shown to play a role in cellular senescence from previous studies (Figure 3C). Fibronectin is one of the proteins shown to be upregulated in replicative senescent cells at both the mRNA and protein levels [36]. Nucleophosmin has been shown to be able to induce senescence in murine primary fibroblast cells by overexpression [37]. Also, some protein components of SAHFs, including HP1γ [15] and prohibitin [38] showed increased protein expression levels in our data set. NF-κB, a transcription factor with vital roles in cellular growth and inflammatory stimuli, is seen at much higher protein expression levels in senescent cells. NF-κB expression has been associated with oncogenic Ras expression in fibroblasts [39].
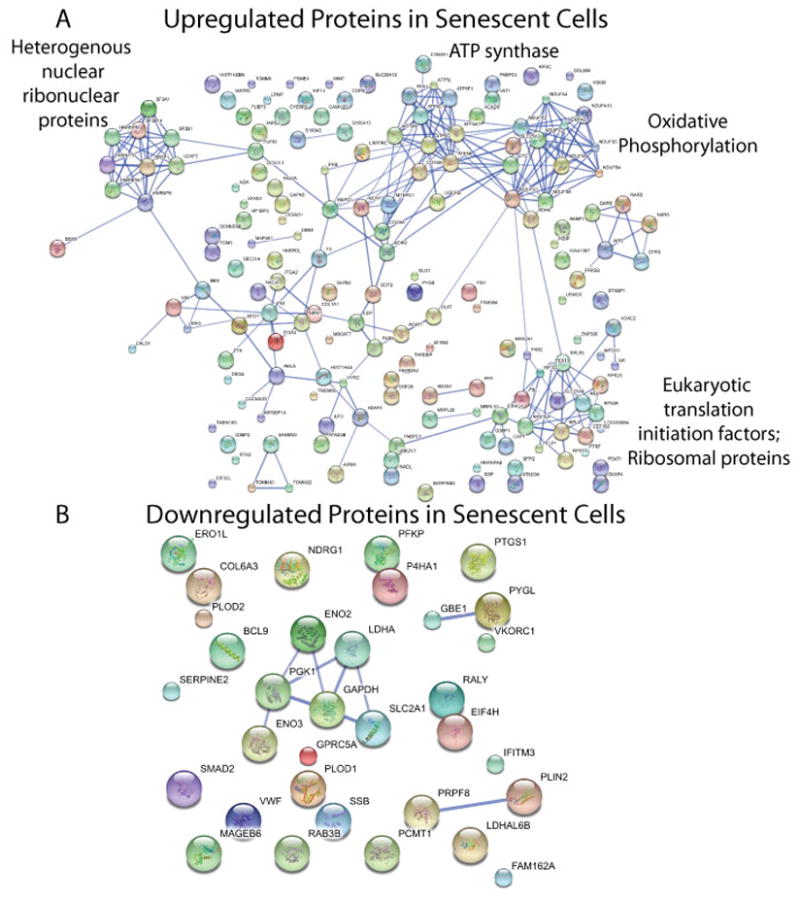
In order to visualize functional linkages of these upregulated proteins on a systems level, we applied the network analysis tool ‘STRING’ [40] which maps all of the upregulated proteins from our data set onto known protein-protein interactions. We retrieved all interactions using a cutoff of ≥0.7 for STRING’s “confidence score” (i.e., high confidence). In the resulting interaction network built from upregulated proteins, four clusters emerged (Figure 4A). The first cluster includes a large number of heterogeneous nuclear ribonucleoproteins (hnRNPs) and RNA splicing factors which function during transcription and mRNA processing. The second cluster includes eukaryotic translation initiation factors and ribosomal proteins. The other two clusters contain mitochondrial proteins involved in oxidative phosphorylation and ATP synthesis. In contrast, we randomly selected sets of ~200 proteins as null hypotheses and did not observe such noticeable aggregates of functional modules with the same analysis (Supplementary Figure 6).
Figure 4.
Among mitochondrial proteins involved in oxidative phosphorylation, five subunits of ATP synthase were found to be upregulated in at least two biological replicates. Additionally, four NADH dehydrogenase subunits and two cytochrome c oxidase subunits were found to be upregulated (Supplementary Figure 7). These upregulated mitochondrial proteins had an average ratio of 2.047 with a standard deviation of 0.058.
Protein synthesis is required for OIS
To study the function of the increased expression level of a group of functionally connected proteins in senescent cells, we examined the involvement of protein synthesis in senescence program. Senescent cells were treated with protein synthesis inhibitor, cycloheximide, at a final concentration of 1 μg/mL for 2 days after puromycin selection and incubated in fresh media for another 4 days. As shown in Figure 5, such treatment with cycloheximide abolished the senescence phenotype indicated by negative SA-β-Gal staining. This suggests that functional protein synthesis is required for developing the senescence phenotype.
Figure 5.
Proteins downregulated in oncogene induced senescent cells
Compared to upregulated proteins, a smaller number of proteins showed decreased protein expression level in senescent cells. A total of 31 proteins, about 1.5% of the identified proteome, showed significantly decreased protein abundance in senescent cells (Supplementary Table 2). The list of downregulated proteins was submitted to the STRING analysis and a group of proteins involved in glycolysis appeared as the most significant cluster in the network map (Figure 4B). Proteins in this category include Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), Enolase (ENO2, ENO3), Phosphoglycerate kinase 1 (PGK), and glucose transporter GLUT1 (SLC2A1) (Supplementary Table 2).
Global proteome changes indicate an altered metabolism in senescent cells
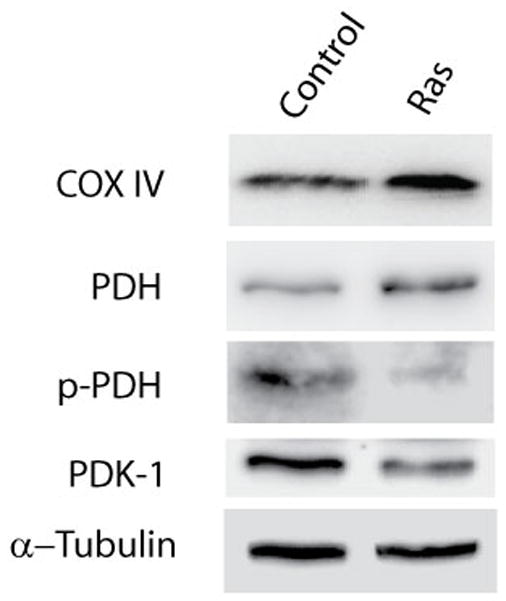
To verify our proteomic findings, we measured by Western blot the levels of proteins involved in glycolysis and oxidative phosphorylation. One of the important proteins connecting glycolysis and oxidative phosphorylation is pyruvate dehydrogenase (PDH) which catalyzes the conversion of pyruvate to acetyl-CoA in mitochondria. The activity of PDH is controlled by phosphorylation. When non-phosphorylated, PDH is active, whereas the phosphorylated form is inactive and results in the channeling of pyruvate to lactate. Therefore, we examined the activity of PDH in senescent cells as reported by its phosphorylation status. In senescent cells, the expression level of PDH is slightly increased while the phosphorylated form is significantly decreased (Figure 6). These data are consistent with PDH being more active in senescent cells, causing more pyruvate to flow into oxidative phosphorylation. This decreased phosphorylation of PDH was likely caused by the decreased expression levels of PDH kinases (e.g., PDK-1) in senescent cells. This was directly tested by immunoblot, which detected a decrease in PDK-1 level in senescent cells at day 6 (Figure 6). Also, one of the proteins of oxidative phosphorylation, COX IV, showed elevated protein abundance, further confirming the global view of metabolism provided by quantitative mass spectrometry data. Together these data suggest that oncogene induced senescent cells, while not dividing but growing, shifted their metabolism to favor oxidative phosphorylation to enhance ATP synthesis and biomass production.
Figure 6.
DISCUSSION
Previous functional proteomic studies have identified several proteins differentially regulated in replicative senescence from a set of different organisms [42–44]. Due to a much smaller data set and fewer proteins identified from these studies, they were primarily focused on the differential expression levels of single proteins. This study is the first large scale application of functional proteomics to oncogene induced senescence. Along with the comprehensive histone modification data, it provides a systems-level view to improve our understanding of cellular senescence and place its antagonistic role towards the progression of cancer in better context.
Profiling Histone Modifications in Global Chromatin
Our combined mass spectrometric analysis of histone modifications provides a comprehensive view of global histone modification patterns in cells undergoing accelerated senescence. Both hypoacetylation of the core histones and hypermethylation of H3K27 are features of senescent heterochromatin. Identified as a distinct type of facultative heterochromatin, SAHFs are contrary to the constitutive heterochromatin accumulated in senescent cells that is responsible for silencing E2F target genes [15]. At the >5% level in bulk chromatin, SAHFs contain several universal markers of heterochromatin but not the markers of condensed chromatin in mitotic and apoptotic cells, such as phosphoserine 10 or 28 of histone H3 and phosphoserine 14 of H2B [45, 46]. SAHFs are also characterized by their depletion of linker histone H1 and enrichment in two other proteins, the histone variant macroH2A and HMGA proteins [45, 47]. Other than the reported enrichment of histone modifications at local foci, our data indicate a global change at the histone acetylation level. Unlike local histone modifications, which reflect the chromatin status at specific regions of chromosomes (e.g., promoters, centromeres and telomeres), global histone modifications reflect the chromatin structure in bulk. Considering the evidence that single chromosomes condense into single SAHFs [34], it is possible that the deacetylation and increase of methylation on K27 might occur throughout the entire genome (except those regions driving OIS and requiring highly permissive euchromatin for transcription) and result in the global chromosome condensation seen as SAHFs by microscopy.
Histone acetylation is controlled by the balance between histone acetyltransferases (HATs) and HDACs. Due to redundancy and compensation between different members of the HAT and HDAC families, global histone acetylation might not be controlled by one or even a few HATs or HDACs [21]. Indeed, perturbation of chromatin modifications by either inhibition of HDAC or downregulation of HAT could induce a senescence phenotype [48, 49]. This suggests a possible link between the global chromatin structure and the senescence phenotype. With more information from genome wide approaches in the future, it will become clearer how local chromatin modifications and global chromatin structures contribute to the maintenance of the senescence state and how chromatin remodelers function to mediate cellular senescence.
Insights from Global Proteomics
Oncogene induced cellular senescence has been characterized as an irreversible cell cycle arrest at G1 phase of the cell cycle [20]. G1 is the major period of cell growth in the cell cycle and also when new cellular organelles are being synthesized. In G1, the cell requires both structural proteins and enzymes, resulting in a great amount of protein synthesis and high metabolic activity in the cell. Our quantitative proteomic data showing that proteins involved in transcript processing, translation and ATP synthesis were upregulated in oncogene induced senescent cells are consistent with these cells sharing some features with G1 cells. This also is consistent with cell growth being uncoupled with cell division.
Contrasting Senescence Metabolism to the Warburg Effect
Glycolysis is a universal metabolic pathway that converts glucose to pyruvate and releases free energy to produce ATP. In most mammalian cells, glucose is metabolized to carbon dioxide by oxidation of pyruvate in the tricarboxylic acid (TCA) cycle and following oxidative phosphorylation in the presence of oxygen. On the other hand, glycolic pyruvate is converted to lactic acid in the absence of oxygen. A very common characteristic of tumor cells is that the tumor cells exhibit increased glycolytic metabolism compared with normal cells, and this abnormal glycolytic metabolism has been discussed as a potential seventh hallmark of cancer beyond the six widely accepted hallmarks of cancer [51]. Also, inhibition of glycolytic enzymes has been shown to be able to induce senescence while overexpression of different glycolytic enzymes can bypass replicative and Ras induced senescence [52].
Our results for upregulated oxidative phosphorylation proteins in senescent cells suggest that the alterations in senescence cellular metabolism are opposite of those seen in cancer cells. We observed upregulation of mitochondrial proteins with an average ratio of 2.047 between treated and control when equal amounts of cytosolic proteins were used in the SILAC experiments. Considering ~4 fold increase of cytosolic protein in senescence cells, the average increase of mitochondrial proteins in each senescence cell was ~8 fold. Such an increase could be the result of greater mitochondrial density, more oxidative phosphorylation proteins per mitochondrion, or both. However, determination of the exact mechanism will require further investigation. In senescent cells, changes in protein expression level appear to cause a different metabolic profile which decreases the rate of glycolysis and channels more pyruvate to oxidative phosphorylation, opposite of that in cancer cells (Figure 7).
Figure 7.
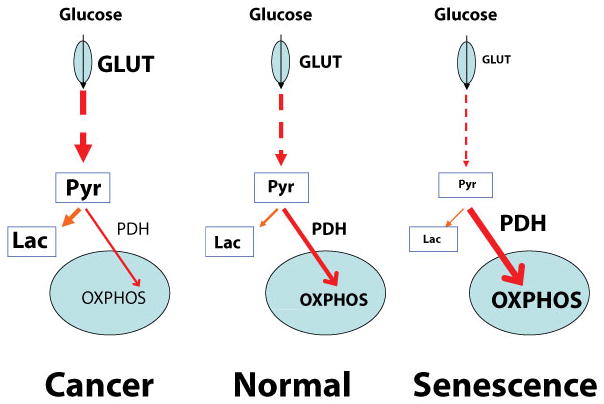
Compared to normal cells, cancer cells exhibit a signature metabolic shift, with a pronounced upregulation in glycolysis coupled with a decrease in oxidative phosphorylation regardless of the availability of oxygen to the cells [41]. This metabolic change occurs both in hypoxic and aerobic conditions and is referred to as the Warburg effect. In cancer biology, it is well established that PDK is the enzyme responsible for inhibiting PDH from pyruvate conversion [53]. Because of our observed shift in proteins involved in glycolysis and oxidative phosphorylation by global proteomics, we were interested in how the levels of PDK-1, the enzyme regulating conversion of cytoplasmic pyruvate into mitochondrial acetyl-CoA, changed in senescence. We observed by Western blot that PDK-1 was present at lower levels in senescence cells. Corroborating this observation was that of lower amounts of phosphorylated PDH, the target of PDK-1. The complete Western blot data presents additional evidence of an upregulation in oxidative phosphorylation, as more pyruvate will be converted to acetyl-CoA and available for use in the downstream mitochondrial metabolism. Our findings indicated a reduced level of glycolysis together with increased oxidative phosphorylation and led us to call this change in senescence metabolism an ‘anti-Warburg’ effect.
It has been demonstrated that in primary cells, increased Ras expression results in an increased level of intracellular reactive oxygen species (ROS) [50]. This ROS increase appears to be important for inducing and maintaining cellular senescence because as literature has shown treating the cells with hydrogen peroxide scavenging agents or culturing Ras expressing cells in low oxygen can block Ras-induced senescence [50]. Oxidative phosphorylation performs an essential part of metabolism with the transfer of electrons from electron donors to electron acceptors in order to release energy. During this process, reactive oxygen species such as hydrogen peroxide and superoxide are produced in mitochondria, which lead to propagation of free radicals that can damage cells. Our observation of upregulated proteins involved in oxidative phosphorylation suggests an explanation for the increased ROS levels seen in oncogene induced senescent cells. However, the precise mechanism for how oncogene expression causes upregulation of oxidative phosphorylation and the link between OIS and ROS will need to be fully elucidated.
Summary
The paradoxical nature of senescence is continued cell growth coupled with cell cycle arrest. The observation of SAHF formation in senescence has been known for some time but no proteomic study has surveyed the impact of the heterochromatin rearrangement on cellular protein levels. With a quantitative approach to both the changing protein levels and the fluctuating chromatin modification landscape brought through the induction of senescence, we have made an initial foray into broadly defining this complicated cellular decision making, which seems centered around an altered metabolism. By further studying the underlying mechanism controlling the molecular switches controlling metabolism, one could understand how cancer cells disrupt the normal limitations of metabolism control and hence bypass senescence. Restoration of innate growth restriction programs in cancer cells could prove beneficial for cancer therapy.
Supplementary Material
Acknowledgments
The authors are grateful to Dr. Scott Lowe for the pBabe-Puro and pBabe-Ras plasmids. The authors also thank all the members of the Kelleher group, Dr. Navdeep Chandel at Northwestern University for helpful discussions, and Brian Imai and Peter Yau from the mass spectrometry facility at University of Illinois at Urbana-Champaign for their help with data analysis. This work was supported by generous grants from the Packard Foundation and the National Institutes of Health (GM 067193) to N.L.K.
List of Abbreviations
- DDR
DNA damage response
- SAHF
senescence-associated heterochromatin foci
- OIS
oncogene induced senescence
- SILAC
stable isotope labeling by amino acids in cell culture
- SRM
selected reaction monitoring
- FDR
false discovery rate
- HDAC
histone deacetylase
- HAT
histone acetyltransferase
- ROS
reactive oxygen species
Footnotes
The authors declare no conflict of interest.
References
- 1.Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–315. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- 2.Hayflick L. LIMITED IN VITRO LIFETIME OF HUMAN DIPLOID CELL STRAINS. Experimental Cell Research. 1965;37:614. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 3.Wright WE, Shay JW. Telomere dynamics in cancer progression and prevention: fundamental differences in human and mouse telomere biology. Nature Medicine. 2000;6:849–851. doi: 10.1038/78592. [DOI] [PubMed] [Google Scholar]
- 4.Chiantore MV, Vannucchi S, Mangino G, Percario ZA, et al. Senescence and Cell Death Pathways and Their Role in Cancer Therapeutic Outcome. Current Medicinal Chemistry. 2009;16:287–300. doi: 10.2174/092986709787002691. [DOI] [PubMed] [Google Scholar]
- 5.Shay JW, Roninson IB. Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene. 2004;23:2919–2933. doi: 10.1038/sj.onc.1207518. [DOI] [PubMed] [Google Scholar]
- 6.Di Micco R, Fumagalli M, Cicalese A, Piccinin S, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 7.Michaloglou C, Vredeveld LCW, Soengas MS, Denoyelle C, et al. BRAF(E600)-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 8.Denchi EL, Attwooll C, Pasini D, Helin K. Deregulated E2F activity induces hyperplasia and senescence-like features in the mouse pituitary gland. Molecular and Cellular Biology. 2005;25:2660–2672. doi: 10.1128/MCB.25.7.2660-2672.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dominguez-Sola D, Ying CY, Grandori C, Ruggiero L, et al. Non-transcriptional control of DNA replication by c-Myc. Nature. 2007;448:445–U443. doi: 10.1038/nature05953. [DOI] [PubMed] [Google Scholar]
- 10.Bartkova J, Rezaei N, Liontos M, Karakaidos P, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 11.Campisi J, di Fagagna FD. Cellular senescence: when bad things happen to good cells. Nature Reviews Molecular Cell Biology. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 12.Schmitt CA, Fridman JS, Yang M, Lee S, et al. A senescence program controlled by p53 and p16(INK4a) contributes to the outcome of cancer therapy. Cell. 2002;109:335–346. doi: 10.1016/s0092-8674(02)00734-1. [DOI] [PubMed] [Google Scholar]
- 13.te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Research. 2002;62:1876–1883. [PubMed] [Google Scholar]
- 14.Xue W, Zender L, Miething C, Dickins RA, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Narita M, Nunez S, Heard E, Lin AW, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- 16.Shelton DN, Chang E, Whittier PS, Choi D, Funk WD. Microarray analysis of replicative senescence. Current Biology. 1999;9:939–945. doi: 10.1016/s0960-9822(99)80420-5. [DOI] [PubMed] [Google Scholar]
- 17.Yoon IK, Kim HK, Kim YK, Song IH, et al. Exploration of replicative senescence-associated genes in human dermal fibroblasts by cDNA microarray technology. Experimental Gerontology. 2004;39:1369–1378. doi: 10.1016/j.exger.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 18.Bonaldi T, Straub T, Cox J, Kumar C, et al. Combined use of RNAi and quantitative proteomics to study gene function in Drosophila. Mol Cell. 2008;31:762–772. doi: 10.1016/j.molcel.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 19.Graumann J, Hubner NC, Kim JB, Ko K, et al. Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC) and proteome quantitation of mouse embryonic stem cells to a depth of 5,111 proteins. Molecular & Cellular Proteomics. 2008;7:672–683. doi: 10.1074/mcp.M700460-MCP200. [DOI] [PubMed] [Google Scholar]
- 20.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16(INK4a) Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 21.Li MX, Jiang LH, Kelleher NL. Global histone profiling by LC-FTMS after inhibition and knockdown of deacetylases in human cells. Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences. 2009;877:3885–3892. doi: 10.1016/j.jchromb.2009.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plazas-Mayorca MD, Zee BM, Young NL, Fingerman IM, et al. One-Pot Shotgun Quantitative Mass Spectrometry Characterization of Histones. Journal of Proteome Research. 2009;8:5367–5374. doi: 10.1021/pr900777e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2006;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 24.Ong SE, Mittler G, Mann M. Identifying and quantifying in vivo methylation sites by heavy methyl SILAC. Nat Methods. 2004;1:119–126. doi: 10.1038/nmeth715. [DOI] [PubMed] [Google Scholar]
- 25.Garcia BA, Mollah S, Ueberheide BM, Busby SA, et al. Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat Protoc. 2007;2:933–938. doi: 10.1038/nprot.2007.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng Y, Sweet SM, Popovic R, Martinez-Garcia E, et al. Total kinetic analysis reveals how combinatorial methylation patterns are established on lysines 27 and 36 of histone H3. Proc Natl Acad Sci U S A. 2012;109:13549–13554. doi: 10.1073/pnas.1205707109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.MacLean B, Tomazela DM, Shulman N, Chambers M, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 29.Garcia BA, Pesavento JJ, Mizzen CA, Kelleher NL. Pervasive combinatorial modification of histone H3 in human cells. Nat Methods. 2007;4:487–489. doi: 10.1038/nmeth1052. [DOI] [PubMed] [Google Scholar]
- 30.Thomas CE, Kelleher NL, Mizzen CA. Mass spectrometric characterization of human histone H3: A bird’s eye view. Journal of Proteome Research. 2006;5:240–247. doi: 10.1021/pr050266a. [DOI] [PubMed] [Google Scholar]
- 31.Pesavento JJ, Mizzen CA, Kelleher NL. Quantitative analysis of modified proteins and their positional isomers by tandem mass spectrometry: Human histone H4. Analytical Chemistry. 2006;78:4271–4280. doi: 10.1021/ac0600050. [DOI] [PubMed] [Google Scholar]
- 32.Yuan W, Xu M, Huang C, Liu N, et al. H3K36 Methylation Antagonizes PRC2-mediated H3K27 Methylation. J Biol Chem. 2011;286:7983–7989. doi: 10.1074/jbc.M110.194027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmitges FW, Prusty AB, Faty M, Stutzer A, et al. Histone Methylation by PRC2 Is Inhibited by Active Chromatin Marks. Mol Cell. 2011;42:330–341. doi: 10.1016/j.molcel.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 34.Zhang RG, Chen W, Adams PD. Molecular dissection of formation of senescence-associated heterochromatin foci. Molecular and Cellular Biology. 2007;27:2343–2358. doi: 10.1128/MCB.02019-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ong SE, Kratchmarova I, Mann M. Properties of C-13-substituted arginine in stable isotope labeling by amino acids in cell culture (SILAC) Journal of Proteome Research. 2003;2:173–181. doi: 10.1021/pr0255708. [DOI] [PubMed] [Google Scholar]
- 36.Kumazaki T, Robetorye RS, Robetorye SC, Smith JR. FIBRONECTIN EXPRESSION INCREASES DURING INVITRO CELLULAR SENESCENCE - CORRELATION WITH INCREASED CELL AREA. Experimental Cell Research. 1991;195:13–19. doi: 10.1016/0014-4827(91)90494-f. [DOI] [PubMed] [Google Scholar]
- 37.Colombo E, Marine JC, Danovi D, Falini B, Pelicci PG. Nucleophosmin regulates the stability and transcriptional activity of p53. Nature Cell Biology. 2002;4:529–533. doi: 10.1038/ncb814. [DOI] [PubMed] [Google Scholar]
- 38.Rastogi S, Joshi B, Dasgupta P, Morris M, et al. Prohibitin facilitates cellular senescence by recruiting specific corepressors to inhibit E2F target genes. Molecular and Cellular Biology. 2006;26:4161–4171. doi: 10.1128/MCB.02142-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frost JA, Swantek JL, Stippec S, Yin MJ, et al. Stimulation of NF kappa B activity by multiple signaling pathways requires PAK1. J Biol Chem. 2000;275:19693–19699. doi: 10.1074/jbc.M909860199. [DOI] [PubMed] [Google Scholar]
- 40.Snel B, Lehmann G, Bork P, Huynen MA. STRING: a web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Research. 2000;28:3442–3444. doi: 10.1093/nar/28.18.3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (New York, NY) 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benvenuti S, Cramer R, Bruce J, Waterfield MD, Jat PS. Identification of novel candidates for replicative senescence by functional proteomics. Oncogene. 2002;21:4403–4413. doi: 10.1038/sj.onc.1205525. [DOI] [PubMed] [Google Scholar]
- 43.Benvenuti S, Cramer R, Quinn CC, Bruce J, et al. Differential proteome analysis of replicative senescence in rat embryo fibroblasts. Molecular & Cellular Proteomics. 2002;1:280–292. doi: 10.1074/mcp.m100028-mcp200. [DOI] [PubMed] [Google Scholar]
- 44.Dierick JF, Kalume DE, Wenders F, Salmon M, et al. Identification of 30 protein species involved in replicative senescence and stress-induced premature senescence. Febs Letters. 2002;531:499–504. doi: 10.1016/s0014-5793(02)03604-9. [DOI] [PubMed] [Google Scholar]
- 45.Funayama R, Saito M, Tanobe H, Ishikawa F. Loss of linker histone H1 in cellular senescence. Journal of Cell Biology. 2006;175:869–880. doi: 10.1083/jcb.200604005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peterson CL, Laniel MA. Histones and histone modifications. Current Biology. 2004;14:R546–R551. doi: 10.1016/j.cub.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 47.Zhang RG, Poustovoitov MV, Ye XF, Santos HA, et al. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Developmental Cell. 2005;8:19–30. doi: 10.1016/j.devcel.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 48.Bandyopadhyay D, Okan NA, Bales E, Nascimento L, et al. Down-regulation of p300/CBP histone acetyltransferase activates a senescence checkpoint in human melanocytes. Cancer Research. 2002;62:6231–6239. [PubMed] [Google Scholar]
- 49.Ogryzko VV, Hirai TH, Russanova VR, Barbie DA, Howard BH. Human fibroblast commitment to a senescence-like state in response to histone deacetylase inhibitors is cell cycle dependent. Molecular and Cellular Biology. 1996;16:5210–5218. doi: 10.1128/mcb.16.9.5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee AC, Fenster BE, Ito H, Takeda K, et al. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J Biol Chem. 1999;274:7936–7940. doi: 10.1074/jbc.274.12.7936. [DOI] [PubMed] [Google Scholar]
- 51.Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 52.Kondoh H, Lleonart ME, Gil J, Wang J, et al. Glycolytic enzymes can modulate cellular life span. Cancer Research. 2005;65:177–185. [PubMed] [Google Scholar]
- 53.Sutendra G, Michelakis ED. Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology. Frontiers in oncology. 2013;3:38. doi: 10.3389/fonc.2013.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.