

Abstract
Tumor-associated macrophages are one of the major constituents of tumor stroma in many solid tumors and there is compelling preclinical and clinical evidence that macrophages promote cancer initiation and malignant progression. Therefore, these cells represent potential targets for therapeutic benefit. In this review, we will summarize macrophage phenotypic heterogeneity, the current understanding of how tumors take advantage of macrophage plasticity to generate immunosuppression, and how manipulation of specific macrophage populations can be used for therapeutic purposes through translational approaches.
Keywords: cancer, CCR2, CSF1R, macrophage, therapeutic
It has been well known for more than two centuries that dysplastic and malignant tissues are extensively infiltrated with leukocytes [1]. However, the specific role of individual leukocytic infiltrates in individual tumors remains less clear. Although clear experimental and epidemiological evidence supports the conclusion that tumors arise at sites of inflammation, there is also significant data to support a model in which the immune system simultaneously plays a role in destroying neoplastic cells and, thus, restrains tumor onset and progression [2,3]. Adding to this debate, the prognostic significance of individual tumor-infiltrating immune cell populations varies widely between different tumor types and/or even the markers used to define these immune cell infiltrates. This diversity of immunologic responses to malignancies makes the targeting of the immune system as part of anticancer therapies quite challenging. Nonetheless, tumor-associated macrophages (TAMs) are often a major constituent of tumor stroma in many solid tumors and are being actively pursued as a mediator of anticancer therapies. In this article, we will review the diversity of macrophage responses present in tumors and how these diverse populations might impact therapeutic targeting of these leukocytes in malignant disease.
Macrophages are an essential component of the innate immune system in humans and are a major constituent of normal tissues. Macrophages play a central role in tissue repair and remodeling under homeostasis and stress response, and are the first line of defense against pathogens. These cells originate from bone marrow precursors and extravasate into normal tissues, where they acquire distinct morphological and functional properties directed by the local tissue and immunological microenvironment. The hallmark feature of mononuclear cells is their subset heterogeneity and plasticity, which enables these cells to change their phenotype and functions in response to different innate and adaptive immune signals [4-6].
Cancer shares many features with chronic nonhealing wounds and is an extremely deregulated tissue in which several genetic and epigenetic changes regulate cell proliferation, survival and differentiation, and become initiators of tumor development [6]. However, these events do not occur in isolation; rather, they take place in the context of a diverse organ-specific tumor stroma. The tumor microenvironment encompasses a wide variety of cells including malignant and nonmalignant populations including stromal cells and leukocyte infiltrate. TAMs comprise the dominant portion of the leukocyte population. TAMs attempt to restore the normal function of damaged tissue, but their interaction with the neoplastic cells in the tumor microenvironment changes their properties, which results in immunosuppression and promotion of tumor growth [7]. These macrophages promote tumor growth, proliferation, vascularity, invasion, metastasis and chemotherapeutic resistance, and these features are linked to treatment resistance. The interaction of macrophages with tumor cells regulates cancer-related inflammation and the prevalence of these cells within tumors has been linked with worse overall prognosis emphasizing the importance of the molecular mechanisms of these interactions and properties of TAMs, so that they can be efficiently targeted.
Understanding macrophage heterogeneity and the molecular mechanisms by which malignant cells derail antitumor immune responses in favor of immune programs that facilitate tumor progression will allow for the identification of pharmacological targets that can be manipulated in order to achieve therapeutic benefit. In this article, we discuss the different types of macrophages and their properties within the tumor; we also discuss how the characterization and manipulation of specific macrophage populations might be used for therapeutic purposes.
Origin & subsets
Monocytes originate from progenitors in the bone marrow and traffic via the bloodstream to peripheral tissues. During both homeostasis and inflammation, circulating monocytes leave the bloodstream and migrate into tissues where, following conditioning by local growth factors, proinflammatory cytokines and microbial products, they differentiate into macrophage or dendritic cell populations [4]. Once recruited from the bloodstream these monocytes undergo tissue-specific functional and morphological adaptation. For example, under homeostatic conditions, monocytes can differentiate into alveolar macrophages (in the lung), osteoclasts (in bone marrow), microglial cells (in the CNS), histiocytes (in connective tissue) and Kupffer cells (in the liver) [8]. Thus, under homeostatic conditions tissue-specific microenvironments dictate the functional activities and differentiation programs of macrophages. This tissue specificity of macrophage differentiation is likely a critical regulator of macrophage phenotype during malignancy.
In addition to the phenotypic and functional heterogeneity of tissue resident macrophages under homeostatic conditions, circulating monocytes themselves also exhibit significant subtype heterogeneity. These subsets include both inflammatory monocytes (IMs) and resident monocytes (RMs) that can be identified by their cell surface markers [9]. In mice, both subsets express CD11b and the CSF1 receptor (CSF1R). IMs can be further identified by high expression of Ly6C, whereas RMs express low levels of Ly6C. In humans, IMs can be identified as CD14high/CD16− and RMs are CD14+/CD16+ [10]. In addition to phenotypic markers, these monocyte subsets express different repertoires of cytokine receptors. IMs express high levels of CC-chemokine receptor 2 (CCR2) but low levels of CX3C-chemokine receptor 1 (CX3CR1low). By contrast, RMs express low to negligible levels of CCR2 but high levels of CX3CR1. These differential receptor repertoires lead to distinct recruitment profiles likely key to the functional differences in response to various stimuli. Under homeostatic conditions, RMs ‘patrol’ the luminal side of vasculature to respond rapidly to danger signals, and play critical roles in viral responses. RMs can also promote tissue remodeling and repair via myofibroblast accumulation, angiogenesis and collagen deposition [11], and may be critical for resolution of inflammation. On the other hand, IMs are thought to promote inflammation typically expressing higher levels of TNF-α and IL-1β in response to stimuli. IMs are also critical for antimicrobial responses. These phenotypic and functional differences can influence tissue responses under nonmalignant conditions, as well as malignant conditions. Expansion of the circulating IM population is typical of many cancers [12,13], suggesting that this subset is dynamically regulated during tumor progression.
Tumor types produce a spectrum of chemokines and cytokines that attract monocytes. These can include CSF1, CCL2, CCL3, CCL4, CCL5, CCL8, SDF1, VEGF, MIP-1 and MIF. All of these cytokines have the ability to enhance monocyte and macrophage recruitment in various tumor models, and individual cancer types typically express them differentially. For example, luminal breast cancer cells often produce high levels of CSF1 [14], while CSF1 is not as frequently overexpressed by pancreatic cancer cells. By contrast, both of these tumor types frequently have high levels of CCL2 expression. Interestingly, a high level of production of these cytokines often correlates with poor overall survival [15-17]. For example, the expression of CSF1 in breast, endometrial, hepatocellular and colorectal cancer correlates with worse prognosis. As such, targeting these chemokines, or their receptors, is actively being pursued as one way to manipulate macrophage responses. While recruitment of monocytes and their development into TAMs is a thoroughly studied phenomenon, it may not be the only source of macrophages in tumors.
In addition to monocytes and macrophages derived from bone marrow precursors, new data suggest that some subsets of tissue-resident macrophages are also derived from progenitors in the placenta. Yolk sac macrophages develop independently of hematopoietic stem cells and do not require Myb, the transcription factor that regulates all hematopoietic stem cells. Yolk sac-derived macrophages can persist in the adult and become sources for some subsets of Kupffer cells and microglia [18]. This cellular subset can be sustained throughout life by local proliferation rather than recruitment and occurs in macrophages as diverse as alveolar macrophages, splenic white pulp and metallophilic macrophages [19], and Kupffer cells. Additionally in mice, granulocyte macrophage progenitors in the spleen can become a reservoir for monocyte mobilization independent of the bone marrow during tumorigenesis [20]. However, the role of these macrophage subsets in malignancy is not fully known and so targeting them for clinical benefit is yet to be determined.
Diversity of TAM responses
Once recruited to tumors, macrophages exhibit heterogeneous responses that lead to both pro- and antitumor properties of macrophages. The cellular functions are often dependent on the specific tissue microenvironment. One concept that has been widely used to try to explain the phenotypic heterogeneity is macrophage polarization. Macrophage responses have been traditionally classified into two major subtypes, M1 and M2, and can be viewed as two extremes on a linear scale. According to this classification, the M1 subtype includes classically activated/antitumor macrophages and the M2 are alternatively activated/protumor macrophages. This classification, although widely used to describe pro- and anti-tumor macrophages, does not always work well for tumor-related macrophages. Owing to the variations in their functions, we use the terms pro- and anti-tumor macrophages in addition to M1 and M2, in this article.
Classically activated macrophage responses are triggered by Toll-like receptor (TLR) agonists or by cell-mediated immune responses such as IFN-γ, TNF-α and GM-CSF. IFN-γ produced by adaptive or innate immune cells, such as NK cells, primes macrophages for enhanced tumoricidal capacity by secreting high levels of proinflammatory cytokines, such as superoxide anions and oxygen and nitrogen radicals, to increase their killing ability. NK cells can only sustain a transient population of protumor macrophages so an adaptive immune response is required for their constant maintenance. Classically activated macrophages also produce proinflammatory cytokines such as IL-1, IL-6 and IL-2, which are important in host defense. These antitumor macrophages induce the development of Th17 cells that produce IL-17, which causes neutrophil recruitment to the tumor. In addition to proinflammatory activity, in some instances, TAMs can play critical roles in antigen presentation and sustaining Th1 and cytotoxic T-cell responses through the production of IL-12. Unfortunately, in most clinically apparent tumors, there is little evidence of a large population of TAMs with M1 programming. However, therapeutics, which can enhance these functional activities of macrophages, are a promising treatment strategy (discussed later).
TAMs are also characterized as having activity similar to alternative activation. Traditional classification of alternative activation is subdivided into macrophages responding to either the Th2 type cytokines IL-4, IL-13 (M1a) and IL-10 (M1b), or TLR stimuli plus interaction with immune complexes (M1c) [5,21]. While in tumors, M2-like protumor activity can be driven by a large variety of stimuli, which include IL-4, IL-13, IL-1, glucocorticoids, TGF-β, Wnt-5a, IL-10 or hypoxia [22]. Existing macrophages in the tumor can also modify their phenotype by the inhibition of NF-κB signaling, which downregulates inflammatory genes, polarizing them towards a protumor phenotype. In addition to more typical polarizing stimuli, hyperactivation of recruitment or maturation pathways can also enhance protumor activity of macrophages; for example, CSF1 and CCL2, produced by breast cancer, specifically promote the protumor phenotype of macrophages [14]. In response to all these stimuli, protumor macrophages generally have high levels of expression of scavenger, mannose and galactose receptors; their metabolism is shifted to ornithine and polyamines [23], and they express lower levels of costimulatory molecules such as CD80 and CD86.
Unlike classically activated macrophages, most TAMs appear to promote immunosuppression. This can be mediated by expression of CCL17, CCL22 and CCL24, which play a significant role in recruitment of Tregs [24]. TAMs also often express PDL1 and PDL2, which can induce T-cell unresponsiveness. Thus, TAMs suppress the activity of antigen presentation and T-cell responses in tumors [25].
Interestingly, these pro- versus anti-tumor phenotypes appear to be readily reprogrammable. This is an important consideration for therapeutic intervention. It has been observed that macrophage phenotypes and functions change during tumor progression. For example, macrophages in early neoplastic tissue often play proinflammatory roles and support immune surveillance which may restrain tumor development. Notably, in the long term this chronic inflammatory program may be mutagenic [26]. By contrast, in advanced malignancy, the microenvironment is changed and macrophages can promote angiogenesis, enhance tumor cell dissemination and suppress antitumor immunity [27,28]. Thus the outcome of pharmacologic targeting of macrophage activities may depend on the disease stage, that is, premalignant, malignant or metastatic.
Subsets of macrophages with different phenotype can often also exist within the same tumor. Thus pro- versus anti-tumor phenotypes of macrophages may be influenced by regional effects, such as the predominance of stromal or tumors cells or local hypoxia. The impact of these heterogeneous populations can be observed in human clinical samples. For example, in non-small-cell lung cancer the stromal macrophages correlate with poor clinical outcome, whereas macrophages infiltrating tumor cell nests correlate with good clinical outcome and increased T-cell responses [29-31].
Macrophages promote tumor progression
In mouse models, the tumor-promoting properties of TAMs have been well studied. These properties have been extensively reviewed elsewhere [7,32-34]. Therefore, here we will only briefly highlight some of these properties that may impact the therapeutic targeting/reprogramming of these cells. TAMs have been shown to be capable of enhancing angiogenic, invasive and immunosuppressive programs in tumors (Figure 1). TAMs can enhance angiogenesis by the production of various chemokines including IL-8, MIF, VEGF, TNF-α and thymidine phosphorylase. These chemokines have been shown to promote tumor vascularity in breast, ovarian, endometrial and CNS malignancies. Tumor cell migration and invasion are also enhanced by protumor macrophages. Tumor invasion is facilitated by upregulation of proteolytic enzymes that mediate basement membrane breakdown. MMPs produced by protumor macrophages also play an important role in tumor invasion [35-37]. Furthermore, upon recruitment to tumors, macrophages increase the production of cathepsin-B expression, a cysteine-type lysosomal protease which plays an important role in tumor growth and lung metastasis [38]. Other than the well accepted role of protumor macrophages in promoting metastasis, resident macrophages in the liver have also been studied for their effects on establishment of metastasis. It was previously thought that liver macrophages or Kupffer cells were protective and destroyed circulating tumor cells because their depletion lead to increased tumor growth [39]. The latest evidence actually demonstrates the opposite. Recent data have demonstrated that Kupffer cells provide essential mitogens in hepatocellular carcinoma through an NF-κB-dependent signaling mechanism because its ablation reduces tumor burden [40].
Figure 1. Properties of pro- and anti-tumor-associated macrophages.
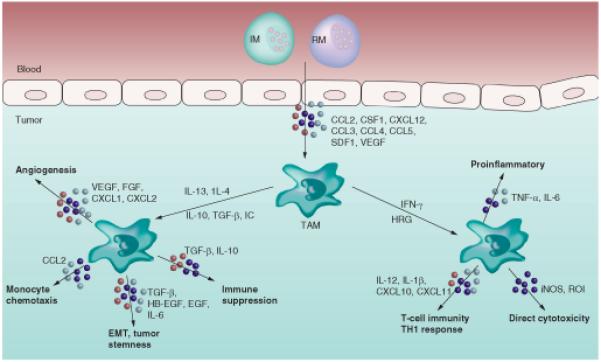
EMT: Epithelial-mesenchymal transition; IC: Immune complex; IM: Inflammatory monocyte; iNOS: Inducible nitric oxide synthase; ROI: Reactive oxygen intermediate; RM: Resident monocyte; TAM: Tumor-associated macrophage.
The interaction between macrophages and cancer cells can facilitate changes in tumor cell differentiation including the development of epithelial-mesenchymal transition (EMT) and cancer stem-like phenotypes. EMT is a process that allows epithelial cells to separate from their neighbors and migrate to distant regions resulting in invasion and metastasis [41,42]. In some tumors, macrophages have been shown to mediate EMT, which can be blocked by EGF receptor inhibitors and SRC family kinase inhibitors [43]. Work by Tahara et al. identified MFGE-8 as a macrophage-derived factor, which can potently increase the tumor initiating properties of murine colon and lung carcinoma cell lines [44]. This activity was attributed to both activation of STAT3 signaling and Hedgehog signaling, which are major contributors in triggering tumorigenicity and resistance to anticancer therapy. It has been shown that the crosstalk between TAMs and tumor cells can regulate the induction of pluripotency gene SOX-2 through EGF receptor-mediated activation of STAT3 signaling [45]. These data suggest that TAMs play a key role in cancer stem cell maintenance and/or expansion, and chemotherapeutic resistance. Blocking TAM recruitment also decreases cancer stem cell population in a pancreas cancer model by activating STAT3 [46]. These properties make macrophages independent targets within the tumor microenvironment. Therefore, targeting macrophages is likely to improve response to conventional chemotherapy in solid tumors that are chemoresistant.
Targeting macrophages
As discussed above there is strong evidence of tumor promotion by macrophages in different cancer models and increased macrophage prevalence correlates with poor overall survival in many human cancers. This provides a strong basis for targeting macrophages independently at different levels and comparing responses to different targeting strategies (Figure 2 & Table 1) . In this section, we will discuss how different properties of macrophages can be targeted in order to achieve a therapeutic benefit and will review the experimental data in animal models for each.
Figure 2. Strategies to target macrophages in blood and at different levels within the tumor.
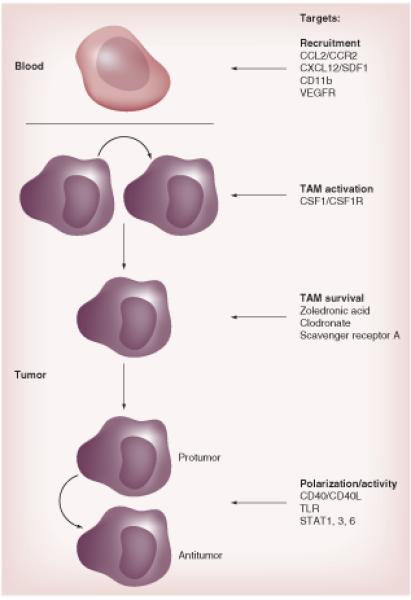
CD40L: CD40 ligand; CSF1R: CSF1 receptor; TAM: Tumor-associated macrophage; TLR: Toll-like receptor; VEGFR: VEGF receptor.
Table 1.
Targeting strategies for tumor-associated macrophages in mouse models.
| Target or drug | Mechanism of action |
|---|---|
| CCL2-CCR2 axis | Prevents monocyte recruitment |
| CSF1-CSF1R axis | Inhibits/reprograms TAMs |
| CXCL12-CXCR4 axis | Prevents recruitment of macrophages |
| DNA repair mechanisms (trabectedin) | Targets TAMs |
| Clodronate and zoledronic acid | Induces macrophage apoptosis |
| Anti-CpG and IL-10 Ab | Prevents antitumor to protumor macrophage polarization |
| CD40 agonist | Restores tumor immunity |
| Sibilin | Suppresses NF-κB and STAT3 phosphorylation, blocks angiogenesis |
Ab: Antibody; CSF1R: CSF1 receptor; TAM: Tumor-associated macrophage.
Inhibiting monocyte/macrophage recruitment
One strategy is to block the recruitment or infiltration of monocytes into tumors. On approach to this is blockade of CCL2 or its receptor CCR2. The CCL2–CCR2 axis plays an important role in monocyte recruitment in the tumor in many cancer types. CCL2 is produced by tumor cells and stroma and it is a major chemoattractant for monocytes, which can then develop into macrophages and promote invasiveness, metastasis and correlate with poor prognosis [47,48]. Targeting the CCL2–CCR2 axis is promising as it results in blocking mobilization of monocytes from the bone marrow to the blood, which results in preventing their recruitment to the tumor [49-52]. CCL2 also promotes a protumor phenotype and its blockade leads to decreased tumor growth and necrosis. Recently, Sanford et al. demonstrated that a CCR2 antagonist (PF-04136309) can block the mobilization of CCR2-positive monocytes from the bone marrow to the tumor in a mouse model of pancreatic cancer and lead to TAM depletion, causing slower tumor growth and preventing distant metastasis [12]. CCL2-blocking agents have also been shown to promote initiation and promotion of colon carcinogenesis [49]. Neutralization of CCL2 reduces tumor growth in prostate cancer [53,54], breast cancer [55] and lung cancer [56] in mice.
In a recent study, trabectedin was used to selectively deplete monocytes and TAMs by targeting downstream TNF-related apoptosis, inducing ligand receptors and activating a caspase-8-mediated extrinsic apoptotic pathway in a fibrosarcoma model [57]. Trabectedin also targets CCL2, which suppresses the recruitment of monocytes and inhibits TAM development, as has been shown in ovarian cancer and myxoid liposarcoma in humans [57]. Other pharmacological inhibitors have also been shown to exhibit negative effects on macrophage migration and many CCL2/CCR2 humanized monoclonal antibodies (mAbs) are under clinical investigation (Table 2).
Table 2.
Summary of NIH clinical trials.
| Target | Phase | Trial number | Tumor type | Agent name | Effect | Ref. |
|---|---|---|---|---|---|---|
| CSF1/CSF1R | I/II | NCT01346358 | Advanced solid tumors | IMC-CS4 | CSF1 R-blocking antibody | [201] |
| NCT01444404 | Advanced solid tumors | AMG 820 | CSF1 R-blocking antibody | [202] | ||
| NCT01804530 | Pancreatic cancer | PLX7486 | Kinase inhibitor of CSF1R and Trk |
[203] | ||
| NCT01004861 | Advanced solid tumors | PLX3397 | Kinase inhibitor of CSF1R and cKit |
[204] | ||
|
| ||||||
| CCL2/CCR2 | II | NCT01015560 | Bone metastasis | MLN1202 | Anti-CCR2 antibody | [205] |
| NCT01413022 | Locally advanced pancreatic cancer |
PF-04136309 | CCR2 antagonist | [206] | ||
|
| ||||||
| IL-6R | I/II | NCT01637532 | Ovarian cancer | Tocilizumab and Peg-Intron |
IL-6R monoclonal antibody | [207] |
|
| ||||||
| DNA repair mechanisms |
III | NCT01692678 | Liposarcoma and leimyosarcoma |
YONDELIS (Trabectedin) |
DNA backbone cleavage and cell apoptosis |
[208] |
| II | NCT01772979 | Ovarian cancer | [209] | |||
| I | NCT01426633 | Liposarcoma and leimyosarcoma |
[210] | |||
|
| ||||||
| CD40 mAb | I/II | NCT01433172 | Lung cancer | (GM.CD40L) vaccine in combination with CCL21 |
Boosts the immune system | [211] |
| NCT01103635 | Metastatic melanoma | Tremelimumab and CP-870, CP-893 |
CD40 agonist mAb | [212] | ||
|
| ||||||
| STAT3 | I | NCT01839604 | Metastatic hepatocellular carcinoma |
AND9150 (ISIS-STAT3 Rx) |
Antisense oligonucleotide inhibitor of STAT3 |
[213] |
CD40L: CD40 ligand; CSF1R: CSF1 receptor; mAb: Monoclonal antibody; R: Receptor; Rx: Prescription.
Use of systemic CD11b-neutralizing monoclonal antibodies also prevents the recruitment of myeloid cells to tumors. In squamous cell carcinoma xenografts in mice, it has been shown that the use of Mac-1 (CD11b/CD18) antibodies leads to an improved response to radiation therapy, which is accompanied by a reduced infiltration of myeloid cells expressing MMP-9 and S100A8 into the tumors [58]. Hypoxia within the tumor microenvironment leads to the production of hypoxia inducible factors (HIFs), which increase vascularization and directly increase macrophage recruitment. It has been shown in a glioblastoma model that HIF-1α deficiency can lead to a decrease in macrophage density in the tumor [59]. Therefore, HIF inhibitors decrease vascularity and macrophage density within the tumor. These factors have been shown to be transcriptional activators of VEGF and CXCR4 genes [60]. The CXCR4−SDF1 axis and VEGF receptor 1 pathway are also important in recruitment of macrophages and their targeting leads to reduced macrophage counts [61,62]. Inhibition of the VEGF receptor 2 pathway results in reduced macrophage infiltration and decreased angiogenesis in breast and pancreatic cancer models [63,64]. While targeting monocyte/macrophage recruitment before they arrive to the tumors is effective in various cancer models, macrophages can also be directly targeted by other approaches once they invade the tumors.
Targeting macrophage activation
TAMs can be targeted at the level of activation by various strategies. Targeting CSF1 or CSF1R may be one approach. CSF1 is highly expressed by several tumor types and in some cancer types, its expression correlates with poor survival [17]. CSF1/CSF1R signaling is critical for the generation of monocyte progenitors in the bone marrow. In the tumor microenvironment CSF1 can act as a chemoattractant; however, other factors, such as CCL2, may play a dominant role in monocyte migration. Nonetheless, blockade of CSF1/CSF1R signaling can rapidly result in reduced numbers of TAMs within 24−48 h [46,65]. This effect is likely due to a prosurvival role of CSF1R signaling in macrophages within the tumors. In addition to regulation of macrophage numbers, CSF1R signaling appears to also regulate the protumor properties of TAMs. Elegant work by several groups has shown that CSF1R signaling can be critical for the invasion-promoting behavior of macrophages, by upregulating their EGF production [66]. Genetic loss of CSF1 (op/op mice) results in significantly reduced metastasis in mammary tumors and delayed tumor progression in breast and neuroendocrine tumor models [14,67]. For these reasons, CSF1/ CSF1R has been an attractive target and has been tested in several mouse models. While less effective as a single agent as compared with op/op mice, neutralizing CSF1 in breast cancer xenografts decreased tumor growth [68]. Even more strikingly, CSF1R signaling blockade appears to enhance the efficacy of several other standard therapies. As such CSF1R blockade has been shown to increase the efficacy of chemotherapy in murine mammary and pancreatic tumors [46,65], radiation therapy in prostate tumor models [69] and improve responses to antiangiogenic therapies [70]. Based on these results, several Phase I clinical trials of CSF1/CSF1R inhibitors have been initiated (Table 2).
Decreasing survival of TAMs
Another attractive strategy for targeting of TAMs within the tumor is to trigger apoptosis. Clodronate and zoledronic acid are two bisphosphonates that have been investigated for their role in macrophage depletion [71]. Clodronate, which has been shown to destroy macrophages and other phagocytic cells, also depletes TAM population and this can result in the regression of tumor growth, angiogenesis and metastasis as shown in lung cancer models [72-74]. In breast cancer, zoledronic acid was shown to selectively deplete MMP-9-expressing TAM, as well as impair differentiation of myeloid cells into TAM, which improves tumoricidal activity of macrophages and some trials have shown prolonged survival in cancer patients [75-77]. This tumoricidal activity can also be seen in prostate cancer and cervical cancer models [78,79]. In chronic myelogenous leukemia, the use of Src kinase inhibitor (dasatinib) has been demonstrated to decrease the density of MMP9+ macrophages [80].
Macrophage surface markers are very important as these can act as useful targets. Targeting markers such as scavenger receptor A and CD52 by using immunotoxin-conjugated mAbs is an attractive approach and has been studied in ovarian cancer [81,82]. Folate receptor β is another surface specific marker for protumor macrophages and their density positively correlates with tumor vascularity and poor prognosis in patients with pancreatic cancer [83]. By inhibiting this receptor using a folate immunotoxin conjugate, it was observed that protumor macrophages were significantly depleted whereas the antitumor macrophage population was maintained [84,85]. Specific bacteria that target the macrophage population can be used to induce macrophage apoptosis. Important ones that have been tested in mouse models are Shigella flexneri. A single injection of an attenuated strain of Shigella was shown to induce TAM apoptosis and >70% reduction in the size of tumor [86]. In addition, certain bacteria that harbor in macrophages, such as Listeria monocytogenes, Chlamydia psittaci and Legionella pneumophila, are also being considered for TAM-targeted immunotherapy [87]. Macrophage destruction within the tumor is being studied further in the setting of preclinical models and clinical trials in different cancer models.
Increasing antitumor macrophages
As discussed earlier, one of the key features of macrophages is their plasticity, which enables them to change their phenotype in the tumor. Thus, reprogramming tumor-infiltrating myeloid cells towards an antitumor phenotype is an attractive therapeutic strategy in targeting macrophages. CD40 is a macrophage cell surface marker that inhibits cytotoxic functions, and anti-CD40 mAb results in upregulation of expression of MHC-II and costimulatory molecule CD86 on TAMs. The combination of a CD40 agonist with gemcitabine in unresectable pancreatic cancer patients showed tumor regression by promoting antitumor macrophages [88]. CD40 mAb also promotes TLR9 to respond to CpG-oligodeoxynucleotide (ODN) in macrophages and polarizes them towards an antitumor phenotype [89]. NF-κB pathway activation also plays an important role in modulating Th1 immune response and this can polarize the macrophages towards an antitumor phenotype, which have antitumor properties [74]. The NF-κB pathway can be activated by using TLR agonists, anti-CD40 mAbs and IL-10 mAbs [90]. There are many types of TLR agonists including PolyI:C, a dsRNA that reverses protumor macrophages to antitumor phenotype by binding TLR-3 [90,91]. CpG-ODN (for TLR-9) promotes production of IL-12, IFN-α and TNF-α by the macrophages and can upregulate TLR-9 activity of NF-κB in macrophages [92]. Imiquimod (for TLR-7) enhances antigen presentation by the tumor responses of lymphocytes and in cutaneous squamous cell carcinoma, it has been shown to polarize macrophages towards an antitumor phenotype [93]. Combination of one or more of these agents has been shown to cause rapid switch from a protumor to an antitumor phenotype; for example, CpG-ODN in combination with anti-IL-10 receptor mAb [94]. In a mouse ovarian cancer model, inhibition of NF-κB activity has been shown to significantly polarize macrophages towards a tumoricidal phenotype [95]. Further exploration of applicability of NF-κB mediators to re-educate macrophages is essential.
Modulation of STAT1 activity is an attractive target to induce an antitumor phenotype in macrophages [6]. IFN-γ is an activator of STAT1 and has been approved by the US FDA for its role in promoting antitumor activities [96]. STAT1 deficiency has been shown to enhance IL-12 induced tumor regression by a T-cell-dependent mechanism in a murine squamous cell carcinoma model. STAT1-positive TAMs are also associated with adverse survival in human follicular lymphomas [97]. Therefore, the effects of STAT1 on the modulation of TAM properties have to be carefully studied before they can be used for therapy. STAT3 and STAT6 pathways have an important role in protumor-like macrophage polarization. A small molecule inhibitor of STAT3 (WP1066) was found to reverse immune tolerance in patients with malignant glioma, correlating with selectively induced expression of costimulatory molecules, CD80 and CD86 on peripheral macrophages and tumor-infiltrating microglias and cytokines such as IL-12 [98]. Other cytokines, such as GM-CSF, have been shown to polarize macrophages towards an antitumor phenotype and are used as an immunotherapy for human cancers; for example, neuroblastoma [99]. Tyrosine kinase inhibitors, sorafenib and sunitinib, have also been demonstrated to inhibit STAT3 in macrophages in vitro [100,101]. Sorafenib can restore IL-12 production, but suppresses IL-10 expression in prostaglandin E2-conditioned macrophages, which shows that the immunosuppressive cytokine profile of TAMs is reversed [101]. STAT1 deficiency enhances IL-12 induced tumor regression by a T-cell-dependent mechanism in a murine squamous cell cancer model [102]. Pathways that promote a protumor phenotype include peroxisome proliferator-activated receptor (PPAR)-γ and HIF-d. PPAR promotes the protumor macrophages and antagonizes antitumor macrophage polarization [103]. Therefore, the role of synthetic inhibitors of PPAR-α and -γ in targeting TAMs should be evaluated.
Many drugs suppress TAMs by various off-target activities and may be effectively utilized as combinatorial therapies. The most common include histidine-rich glycoprotein and copper chelate. Histidine-rich glycoprotein polarizes macrophages towards an antitumor phenotype by the downregulation of placental growth factor [104]. Copper chelate-CuNG, has been shown to increase IFN-γ, IL-12 and also decreases the production of TGF-β thus, promoting an antitumor phenotype of macrophages [105]. 5,6-dimethy XAA xanthenone-4-acetic acid and Vadimezan (ASA404) increase immune stimulation in innate immune cells and CD8 infiltration in the tumor. Other chemotherapeutic agents, such as silibinin [106] and proton pump inhibitors [107], have been shown to target different functional properties of protumor macrophages. It is important to understand the direct and synergistic effects of these drugs on TAMs in preclinical cancer models so that they can be effectively used in clinics.
TAM targeting & radiation therapy
Radiation therapy is a useful treatment modality in many cancer types and studies have demonstrated that myeloid infiltrate increases after tumor irradiation. However, the interactions between the tumor cells and stroma after tumor radiation remain poorly defined. It has been demonstrated in multiple animal models that DNA damage, cell death and increased hypoxia in the tissue after radiation therapy leads to macrophage recruitment, which can promote tumor growth [108]. The SDF1/CXCR4, HIF-1 pathways are stimulated by radiation-induced tumor hypoxia. Using a HIF-1 inhibitor results in decreased infiltration of myeloid cells to the tumor [109]. Blocking the interaction of SDF-1 with its receptor in irradiated tumors has been shown to inhibit regrowth of tumor after irradiation [110]. Additionally, CSF1/CSF1R signaling has recently been implicated in the recruitment of myeloid cells to tumors during radiation. Xu et al. have recently demonstrated that a selective inhibitor of CSF1R combined with radiation therapy suppressed tumor growth in murine prostate cancer model compared with radiation alone. According to their model, radiation-induced DNA damage leads to activation and translocation of ABL kinase into the nucleus, which binds to the CSF1 gene promoter and increases CSF1 gene expression [111].
TAMs isolated from irradiated tumors have increased expression of arginase-1, COX-2 and inducible nitric oxide synthase, and promoted tumor growth as compared with tumors that are not irradiated [112]. The location of these macrophages is variable in different models depending on the level of hypoxia in the tumor. This suggests that they play an important role in tissue repair after tumor irradiation [113]. Evidence supports that TAMs can promote tumor growth and survival; therefore, targeting TAMs within the tumor will improve the overall effectiveness of radiation therapy.
Conclusion
TAMs orchestrate tumor progression in many malignancies and targeting these cells offers a novel therapeutic approach to improve anticancer therapy. Many targeting strategies mentioned earlier have been shown to improve outcome and efficacy of chemotherapeutic response in experimental models and some of these strategies are being tested in clinical trials. The challenge of shifting the balance between protumor and antitumor responses to achieve maximum responses to these therapies remains unexplored. In addition, there are practical issues associated with determining the most appropriate patient population, timing and therapeutic combination in various cancer patients because it affects the therapeutic efficacy of these strategies.
Future perspective
We anticipate that emerging technologies will develop novel therapeutics that will effectively target macrophages in human cancers and will be a part of future chemotherapeutic regimens in many human cancers. Targeting recruitment of monocyte/macrophages and/or reprogramming their activity after invading the tumor will be key areas to investigate clinically. Development of effective therapeutic agents in order to achieve an optimal balance between pro- and anti-tumor macrophage activities will help us achieve maximal therapeutic responses.
Executive summary.
Origin & subsets of macrophages
-
■
Bone marrow-derived and nonbone marrow-derived macrophages contribute to malignancy.
Macrophages can promote tumor progression by multiple mechanisms
-
■
These include enhancing angiogenesis, invasion and metastasis, immunosuppression, promotion of epithelial-mesenchymal transition and increasing cancer stem cells.
Diversity of tumor-associated macrophage responses
-
■
Protumor/M2 macrophages have protumor activity that can be driven by IL-4, IL-13, IL-10 and other factors. These tumor-associated macrophages (TAMs) suppress T-cell immunity, increase angiogenesis and promote invasion.
-
■
Antitumor/M1 macrophages are activated by Toll-like receptor agonists and IFN-γ and secrete superoxide anions, oxygen and nitrogen radicals to increase their tumor killing ability.
Strategies to target TAMs for therapeutic benefit
-
■Strategies to target TAMs for therapeutic benefit include:
-
-Blocking monocyte recruitment;
-
-Targeting macrophage activation;
-
-Decreasing macrophage survival;
-
-Reprogramming TAMs toward antitumor immune responses;
-
-Radiation therapy and macrophages.
-
-
Conclusion
-
■
Shifting the balance between pro- and anti-tumor responses is important to improve clinical outcome.
-
■
Appropriate timing of macrophage targeted therapy will be important.
-
■
Appropriate combination therapy needs to be investigated.
Footnotes
Financial & competing interests disclosure
Funding for these researchers was provided by the Lustgarten Foundation, V Foundation, A Edward Mallinckrodt Jr Award, the Cancer Research Foundation and the Cancer Frontier Fund. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
■ of interest
- 1.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357(9255):539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 2.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bui JD, Schreiber RD. Cancer immunosurveillance, immunoediting and inflammation: independent or interdependent processes? Curr. Opin. Immunol. 2007;19(2):203–208. doi: 10.1016/j.coi.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005;5(12):953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 5.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 6.Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J. Clin. Invest. 2007;117(5):1155–1166. doi: 10.1172/JCI31422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hashimoto D, Chow A, Noizat C, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38(4):792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Auffray C, Fogg D, Garfa M, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317(5838):666–670. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- 10.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19(1):71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 11.Nahrendorf M, Swirski FK, Aikawa E, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007;204(12):3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanford DE, Belt BA, Panni RZ, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin. Cancer Res. 2013;19(13):3404–3415. doi: 10.1158/1078-0432.CCR-13-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Porembka MR, Mitchem JB, Belt BA, et al. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol. Immunother. 2012;61(9):1373–1385. doi: 10.1007/s00262-011-1178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 2001;193(6):727–740. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Groblewska M, Mroczko B, Wereszczynska-Siemiatkowska U, Mysliwiec P, Kedra B, Szmitkowski M. Serum levels of granulocyte colony-stimulating factor (G-CSF) and macrophage colony-stimulating factor (M-CSF) in pancreatic cancer patients. Clin. Chem. Lab. Med. 2007;45(1):30–34. doi: 10.1515/CCLM.2007.025. [DOI] [PubMed] [Google Scholar]
- 16.Mroczko B, Groblewska M, Wereszczynska-Siemiatkowska U, et al. Serum macrophage-colony stimulating factor levels in colorectal cancer patients correlate with lymph node metastasis and poor prognosis. Clin. Chim. Acta. 2007;380(1-2):208–212. doi: 10.1016/j.cca.2007.02.037. [DOI] [PubMed] [Google Scholar]
- 17.Zhu XD, Zhang JB, Zhuang PY, et al. High expression of macrophage colony-stimulating factor in peritumoral liver tissue is associated with poor survival after curative resection of hepatocellular carcinoma. J. Clin. Oncol. 2008;26(16):2707–2716. doi: 10.1200/JCO.2007.15.6521. [DOI] [PubMed] [Google Scholar]
- 18.Passlick B, Flieger D, Ziegler-Heitbrock HW. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood. 1989;74(7):2527–2534. [PubMed] [Google Scholar]
- 19.Wijffels JF, de Rover Z, Beelen RH, Kraal G, van Rooijen N. Macrophage subpopulations in the mouse spleen renewed by local proliferation. Immunobiology. 1994;191(1):52–64. doi: 10.1016/s0171-2985(11)80267-6. [DOI] [PubMed] [Google Scholar]
- 20.Cortez-Retamozo V, Etzrodt M, Newton A, et al. Origins of tumor-associated macrophages and neutrophils. Proc. Natl Acad. Sci. USA. 2012;109(7):2491–2496. doi: 10.1073/pnas.1113744109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front. Biosci. 2008;13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 22.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25(12):677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 23.Gerber JS, Mosser DM. Reversing lipopolysaccharide toxicity by ligating the macrophage Fc γ receptors. J. Immunol. 2001;166(11):6861–6868. doi: 10.4049/jimmunol.166.11.6861. [DOI] [PubMed] [Google Scholar]
- 24.Mantovani A. From phagocyte diversity and activation to probiotics: back to Metchnikoff. Eur. J. Immunol. 2008;38(12):3269–3273. doi: 10.1002/eji.200838918. [DOI] [PubMed] [Google Scholar]
- 25.Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat. Rev. Immunol. 2008;8(7):533–544. doi: 10.1038/nri2356. [DOI] [PubMed] [Google Scholar]
- 26.Swann JB, Vesely MD, Silva A, et al. Demonstration of inflammation-induced cancer and cancer immunoediting during primary tumorigenesis. Proc. Natl Acad. Sci. USA. 2008;105(2):652–656. doi: 10.1073/pnas.0708594105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pollard JW. Macrophages define the invasive microenvironment in breast cancer. J. Leukoc. Biol. 2008;84(3):623–630. doi: 10.1189/jlb.1107762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin EY, Li JF, Gnatovskiy L, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006;66(23):11238–11246. doi: 10.1158/0008-5472.CAN-06-1278. [DOI] [PubMed] [Google Scholar]
- 29.Ohri CM, Shikotra A, Green RH, Waller DA, Bradding P. The tissue microlocalisation and cellular expression of CD163, VEGF, HLA-DR, iNOS, and MRP 8/14 is correlated to clinical outcome in NSCLC. PLoS ONE. 2011;6(7):e21874. doi: 10.1371/journal.pone.0021874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ohri CM, Shikotra A, Green RH, Waller DA, Bradding P. Macrophages within NSCLC tumour islets are predominantly of a cytotoxic M1 phenotype associated with extended survival. Eur. Respir. J. 2009;33(1):118–126. doi: 10.1183/09031936.00065708. [DOI] [PubMed] [Google Scholar]
- 31.Ma J, Liu L, Che G, Yu N, Dai F, You Z. The M1 form of tumor-associated macrophages in non-small cell lung cancer is positively associated with survival time. BMC Cancer. 2010;10:112. doi: 10.1186/1471-2407-10-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr. Opin. Immunol. 2010;22(2):231–237. doi: 10.1016/j.coi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 33.Mantovani A, Garlanda C, Allavena P. Molecular pathways and targets in cancer-related inflammation. Ann. Med. 2010;42(3):161–170. doi: 10.3109/07853890903405753. [DOI] [PubMed] [Google Scholar]
- 34.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012;12(4):253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reilkoff RA, Bucala R, Herzog EL. Fibrocytes: emerging effector cells in chronic inflammation. Nat. Rev. Immunol. 2011;11(6):427–435. doi: 10.1038/nri2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mason SD, Joyce JA. Proteolytic networks in cancer. Trends Cell. Biol. 2011;21(4):228–237. doi: 10.1016/j.tcb.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vasiljeva O, Papazoglou A, Kruger A, et al. Tumor cell-derived and macrophage-derived cathepsin B promotes progression and lung metastasis of mammary cancer. Cancer Res. 2006;66(10):5242–5250. doi: 10.1158/0008-5472.CAN-05-4463. [DOI] [PubMed] [Google Scholar]
- 39.Heuff G, Oldenburg HS, Boutkan H, et al. Enhanced tumour growth in the rat liver after selective elimination of Kupffer cells. Cancer Immunol. Immunother. 1993;37(2):125–130. doi: 10.1007/BF01517045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greten FR, Karin M. The IKK/NF-κB activation pathway-a target for prevention and treatment of cancer. Cancer Lett. 2004;206(2):193–199. doi: 10.1016/j.canlet.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 41.Savagner P. Leaving the neighborhood: molecular mechanisms involved during epithelial-mesenchymal transition. Bioessays. 2001;23(10):912–923. doi: 10.1002/bies.1132. [DOI] [PubMed] [Google Scholar]
- 42.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer. 2002;2(6):442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 43.Lin CY, Lin CJ, Chen KH, Wu JC, Huang SH, Wang SM. Macrophage activation increases the invasive properties of hepatoma cells by destabilization of the adherens junction. FEBS Lett. 2006;580(13):3042–3050. doi: 10.1016/j.febslet.2006.04.049. [DOI] [PubMed] [Google Scholar]
- 44.Jinushi M, Chiba S, Yoshiyama H, et al. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc. Natl Acad. Sci. USA. 2011;108(30):12425–12430. doi: 10.1073/pnas.1106645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang J, Liao D, Chen C, et al. Tumor associated macrophages regulate murine breast cancer stem cells through a novel paracrine EGFR/Stat3/Sox-2 signaling pathway. Stem Cells. 2013;31(2):248–258. doi: 10.1002/stem.1281. [DOI] [PubMed] [Google Scholar]
- 46.Mitchem JB, Brennan DJ, Knolhoff BL, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73(3):1128–1141. doi: 10.1158/0008-5472.CAN-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roca H, Varsos ZS, Sud S, Craig MJ, Ying C, Pienta KJ. CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. J. Biol. Chem. 2009;284(49):34342–34354. doi: 10.1074/jbc.M109.042671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang J, Lu Y, Pienta KJ. Multiple roles of chemokine (C-C motif) ligand 2 in promoting prostate cancer growth. J. Natl Cancer Inst. 2010;102(8):522–528. doi: 10.1093/jnci/djq044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Popivanova BK, Kostadinova FI, Furuichi K, et al. Blockade of a chemokine, CCL2, reduces chronic colitis-associated carcinogenesis in mice. Cancer Res. 2009;69(19):7884–7892. doi: 10.1158/0008-5472.CAN-09-1451. [DOI] [PubMed] [Google Scholar]
- 50.Gazzaniga S, Bravo AI, Guglielmotti A, et al. Targeting tumor-associated macrophages and inhibition of MCP-1 reduce angiogenesis and tumor growth in a human melanoma xenograft. J. Invest. Dermatol. 2007;127(8):2031–2041. doi: 10.1038/sj.jid.5700827. [DOI] [PubMed] [Google Scholar]
- 51.Qian BZ, Li J, Zhang H, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu X, Fujita M, Snyder LA, Okada H. Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy. J. Neurooncol. 2011;104(1):83–92. doi: 10.1007/s11060-010-0473-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Loberg RD, Ying C, Craig M, et al. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res. 2007;67(19):9417–9424. doi: 10.1158/0008-5472.CAN-07-1286. [DOI] [PubMed] [Google Scholar]
- 54.Lu Y, Cai Z, Xiao G, et al. Monocyte chemotactic protein-1 mediates prostate cancer-induced bone resorption. Cancer Res. 2007;67(8):3646–3653. doi: 10.1158/0008-5472.CAN-06-1210. [DOI] [PubMed] [Google Scholar]
- 55.Lu X, Kang Y. Chemokine (C-C motif) ligand 2 engages CCR2+ stromal cells of monocytic origin to promote breast cancer metastasis to lung and bone. J. Biol. Chem. 2009;284(42):29087–29096. doi: 10.1074/jbc.M109.035899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fridlender ZG, Kapoor V, Buchlis G, et al. Monocyte chemoattractant protein-1 blockade inhibits lung cancer tumor growth by altering macrophage phenotype and activating CD8+ cells. Am. J. Respir. Cell. Mol. Biol. 2011;44(2):230–237. doi: 10.1165/rcmb.2010-0080OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Germano G, Frapolli R, Belgiovine C, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23(2):249–262. doi: 10.1016/j.ccr.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 58.Ahn GO, Tseng D, Liao CH, Dorie MJ, Czechowicz A, Brown JM. Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc. Natl Acad. Sci. USA. 2010;107(18):8363–8368. doi: 10.1073/pnas.0911378107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Du R, Lu KV, Petritsch C, et al. HIF1α induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13(3):206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fang HY, Hughes R, Murdoch C, et al. Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood. 2009;114(4):844–859. doi: 10.1182/blood-2008-12-195941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Welford AF, Biziato D, Coffelt SB, et al. TIE2-expressing macrophages limit the therapeutic efficacy of the vascular-disrupting agent combretastatin A4 phosphate in mice. J. Clin. Invest. 2011;121(5):1969–1973. doi: 10.1172/JCI44562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fischer C, Jonckx B, Mazzone M, et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell. 2007;131(3):463–475. doi: 10.1016/j.cell.2007.08.038. [DOI] [PubMed] [Google Scholar]
- 63.Dineen SP, Lynn KD, Holloway SE, et al. Vascular endothelial growth factor receptor 2 mediates macrophage infiltration into orthotopic pancreatic tumors in mice. Cancer Res. 2008;68(11):4340–4346. doi: 10.1158/0008-5472.CAN-07-6705. [DOI] [PubMed] [Google Scholar]
- 64.Roland CL, Dineen SP, Lynn KD, et al. Inhibition of vascular endothelial growth factor reduces angiogenesis and modulates immune cell infiltration of orthotopic breast cancer xenografts. Mol. Cancer Ther. 2009;8(7):1761–1771. doi: 10.1158/1535-7163.MCT-09-0280. [DOI] [PubMed] [Google Scholar]
- 65.DeNardo DG, Brennan DJ, Rexhepaj E, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1(1):54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Patsialou A, Wyckoff J, Wang Y, Goswami S, Stanley ER, Condeelis JS. Invasion of human breast cancer cells in vivo requires both paracrine and autocrine loops involving the colony-stimulating factor-1 receptor. Cancer Res. 2009;69(24):9498–9506. doi: 10.1158/0008-5472.CAN-09-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pyonteck SM, Gadea BB, Wang HW, et al. Deficiency of the macrophage growth factor CSF-1 disrupts pancreatic neuroendocrine tumor development. Oncogene. 2011;31(11):1459–1467. doi: 10.1038/onc.2011.337. [DOI] [PubMed] [Google Scholar]
- 68.Paulus P, Stanley ER, Schafer R, Abraham D, Aharinejad S. Colony-stimulating factor-1 antibody reverses chemoresistance in human MCF-7 breast cancer xenografts. Cancer Res. 2006;66(8):4349–4356. doi: 10.1158/0008-5472.CAN-05-3523. [DOI] [PubMed] [Google Scholar]
- 69.Xu J, Escamilla J, Mok S, et al. Abrogating the protumorigenic influences of tumor-infiltrating myeloid cells by CSF1R signaling blockade improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013;73(9):2782–2794. doi: 10.1158/0008-5472.CAN-12-3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Priceman SJ, Sung JL, Shaposhnik Z, et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: combating tumor evasion of antiangiogenic therapy. Blood. 2010;115(7):1461–1471. doi: 10.1182/blood-2009-08-237412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rogers TL, Holen I. Tumour macrophages as potential targets of bisphosphonates. J. Transl. Med. 2011;9:177. doi: 10.1186/1479-5876-9-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zeisberger SM, Odermatt B, Marty C, Zehnder-Fjallman AH, Ballmer-Hofer K, Schwendener RA. Clodronate-liposome-mediated depletion of tumour-associated macrophages: a new and highly effective antiangiogenic therapy approach. Br. J. Cancer. 2006;95(3):272–281. doi: 10.1038/sj.bjc.6603240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hiraoka K, Zenmyo M, Watari K, et al. Inhibition of bone and muscle metastases of lung cancer cells by a decrease in the number of monocytes/macrophages. Cancer Sci. 2008;99(8):1595–1602. doi: 10.1111/j.1349-7006.2008.00880.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Miselis NR, Wu ZJ, van Rooijen N, Kane AB. Targeting tumor-associated macrophages in an orthotopic murine model of diffuse malignant mesothelioma. Mol. Cancer Ther. 2008;7(4):788–799. doi: 10.1158/1535-7163.MCT-07-0579. [DOI] [PubMed] [Google Scholar]
- 75.Gnant M, Mlineritsch B, Schippinger W, et al. Endocrine therapy plus zoledronic acid in premenopausal breast cancer. N. Engl. J. Med. 2009;360(7):679–691. doi: 10.1056/NEJMoa0806285. [DOI] [PubMed] [Google Scholar]
- 76.Eidtmann H, de Boer R, Bundred N, et al. Efficacy of zoledronic acid in postmenopausal women with early breast cancer receiving adjuvant letrozole: 36-month results of the ZO-FAST study. Ann. Oncol. 2010;21(11):2188–2194. doi: 10.1093/annonc/mdq217. [DOI] [PubMed] [Google Scholar]
- 77.Coleman R, Cook R, Hirsh V, Major P, Lipton A. Zoledronic acid use in cancer patients: more than just supportive care? Cancer. 2011;117(1):11–23. doi: 10.1002/cncr.25529. [DOI] [PubMed] [Google Scholar]
- 78.Tsagozis P, Eriksson F, Pisa P. Zoledronic acid modulates antitumoral responses of prostate cancer-tumor associated macrophages. Cancer Immunol. Immunother. 2008;57(10):1451–1459. doi: 10.1007/s00262-008-0482-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Giraudo E, Inoue M, Hanahan D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J. Clin. Invest. 2004;114(5):623–633. doi: 10.1172/JCI22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kantarjian H, Shah NP, Hochhaus A, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 2010;362(24):2260–2270. doi: 10.1056/NEJMoa1002315. [DOI] [PubMed] [Google Scholar]
- 81.Bak SP, Walters JJ, Takeya M, Conejo-Garcia JR, Berwin BL. Scavenger receptor-A-targeted leukocyte depletion inhibits peritoneal ovarian tumor progression. Cancer Res. 2007;67(10):4783–4789. doi: 10.1158/0008-5472.CAN-06-4410. [DOI] [PubMed] [Google Scholar]
- 82.Pulaski HL, Spahlinger G, Silva IA, et al. Identifying alemtuzumab as an anti-myeloid cell antiangiogenic therapy for the treatment of ovarian cancer. J. Transl. Med. 2009;7:49. doi: 10.1186/1479-5876-7-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Puig-Kroger A, Sierra-Filardi E, Dominguez-Soto A, et al. Folate receptor β is expressed by tumor-associated macrophages and constitutes a marker for M2 anti-inflammatory/regulatory macrophages. Cancer Res. 2009;69(24):9395–9403. doi: 10.1158/0008-5472.CAN-09-2050. [DOI] [PubMed] [Google Scholar]
- 84.Kurahara H, Takao S, Kuwahata T, et al. Clinical significance of folate receptor β-expressing tumor-associated macrophages in pancreatic cancer. Ann. Surg. Oncol. 2012;19(7):2264–2271. doi: 10.1245/s10434-012-2263-0. [DOI] [PubMed] [Google Scholar]
- 85.Nagai T, Tanaka M, Tsuneyoshi Y, et al. Targeting tumor-associated macrophages in an experimental glioma model with a recombinant immunotoxin to folate receptor β. Cancer Immunol. Immunother. 2009;58(10):1577–1586. doi: 10.1007/s00262-009-0667-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Galmbacher K, Heisig M, Hotz C, et al. Shigella mediated depletion of macrophages in a murine breast cancer model is associated with tumor regression. PLoS ONE. 2010;5(3):e9572. doi: 10.1371/journal.pone.0009572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Weigert A, Sekar D, Brune B. Tumor-associated macrophages as targets for tumor immunotherapy. Immunotherapy. 2009;1(1):83–95. doi: 10.2217/1750743X.1.1.83. [DOI] [PubMed] [Google Scholar]
- 88.Beatty GL, Chiorean EG, Fishman MP, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Buhtoiarov IN, Lum HD, Berke G, Sondel PM, Rakhmilevich AL. Synergistic activation of macrophages via CD40 and TLR9 results in T cell independent antitumor effects. J. Immunol. 2006;176(1):309–318. doi: 10.4049/jimmunol.176.1.309. [DOI] [PubMed] [Google Scholar]
- 90.Seya T, Shime H, Matsumoto M. TAMable tumor-associated macrophages in response to innate RNA sensing. Oncoimmunology. 2012;1(6):1000–1001. doi: 10.4161/onci.19894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shime H, Matsumoto M, Oshiumi H, et al. Toll-like receptor 3 signaling converts tumor-supporting myeloid cells to tumoricidal effectors. Proc. Natl Acad. Sci. USA. 2012;109(6):2066–2071. doi: 10.1073/pnas.1113099109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hemmi H, Takeuchi O, Kawai T, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408(6813):740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 93.Smith KJ, Hamza S, Skelton H. Topical imidazoquinoline therapy of cutaneous squamous cell carcinoma polarizes lymphoid and monocyte/macrophage populations to a Th1 and M1 cytokine pattern. Clin. Exp. Dermatol. 2004;29(5):505–512. doi: 10.1111/j.1365-2230.2004.01593.x. [DOI] [PubMed] [Google Scholar]
- 94.Guiducci C, Vicari AP, Sangaletti S, Trinchieri G, Colombo MP. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res. 2005;65(8):3437–3446. doi: 10.1158/0008-5472.CAN-04-4262. [DOI] [PubMed] [Google Scholar]
- 95.Hagemann T, Lawrence T, Mcneish I, et al. “Re-educating” tumor-associated macrophages by targeting NF-κB. J. Exp. Med. 2008;205(6):1261–1268. doi: 10.1084/jem.20080108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 2006;6(11):836–848. doi: 10.1038/nri1961. [DOI] [PubMed] [Google Scholar]
- 97.Alvaro T, Lejeune M, Camacho FI, et al. The presence of STAT1-positive tumor-associated macrophages and their relation to outcome in patients with follicular lymphoma. Haematologica. 2006;91(12):1605–1612. [PubMed] [Google Scholar]
- 98.Hussain SF, Kong LY, Jordan J, et al. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 2007;67(20):9630–9636. doi: 10.1158/0008-5472.CAN-07-1243. [DOI] [PubMed] [Google Scholar]
- 99.Yu AL, Gilman AL, Ozkaynak MF, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N. Engl. J. Med. 2010;363(14):1324–1334. doi: 10.1056/NEJMoa0911123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009;69(6):2506–2513. doi: 10.1158/0008-5472.CAN-08-4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Edwards JP, Emens LA. The multikinase inhibitor sorafenib reverses the suppression of IL-12 and enhancement of IL-10 by PGE(2) in murine macrophages. Int. Immunopharmacol. 2010;10(10):1220–1228. doi: 10.1016/j.intimp.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Torrero MN, Xia X, Henk W, Yu S, Li S. Stat1 deficiency in the host enhances interleukin-12-mediated tumor regression. Cancer Res. 2006;66(8):4461–4467. doi: 10.1158/0008-5472.CAN-05-3554. [DOI] [PubMed] [Google Scholar]
- 103.Van Ginderachter JA, Movahedi K, Van den Bossche J, De Baetselier P. Macrophages, PPARs, and cancer. PPAR Res. 2008;2008:169414. doi: 10.1155/2008/169414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rolny C, Mazzone M, Tugues S, et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell. 2011;19(1):31–44. doi: 10.1016/j.ccr.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 105.Chakraborty P, Chatterjee S, Ganguly A, et al. Reprogramming of TAM toward proimmunogenic type through regulation of MAP kinases using a redox-active copper chelate. J. Leukoc. Biol. 2012;91(4):609–619. doi: 10.1189/jlb.0611287. [DOI] [PubMed] [Google Scholar]
- 106.Tyagi A, Singh RP, Ramasamy K, et al. Growth inhibition and regression of lung tumors by silibinin: modulation of angiogenesis by macrophage-associated cytokines and nuclear factor-κB and signal transducers and activators of transcription 3. Cancer Prev. Res. (Phila.) 2009;2(1):74–83. doi: 10.1158/1940-6207.CAPR-08-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vishvakarma NK, Singh SM. Immunopotentiating effect of proton pump inhibitor pantoprazole in a lymphoma-bearing murine host: implication in antitumor activation of tumor-associated macrophages. Immunol. Lett. 2010;134(1):83–92. doi: 10.1016/j.imlet.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 108.De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. 2013;23(3):277–286. doi: 10.1016/j.ccr.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 109.Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J. Clin. Invest. 2010;120(3):694–705. doi: 10.1172/JCI40283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kozin SV, Kamoun WS, Huang Y, Dawson MR, Jain RK, Duda DG. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res. 2010;70(14):5679–5685. doi: 10.1158/0008-5472.CAN-09-4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xu J, Escamilla J, Mok S, et al. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013;73(9):2782–2794. doi: 10.1158/0008-5472.CAN-12-3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tsai CS, Chen FH, Wang CC, et al. Macrophages from irradiated tumors express higher levels of iNOS, arginase-I and COX-2, and promote tumor growth. Int. J. Radiat. Oncol. Biol. Phys. 2007;68(2):499–507. doi: 10.1016/j.ijrobp.2007.01.041. [DOI] [PubMed] [Google Scholar]
- 113.Chiang CS, Fu SY, Wang SC, et al. Irradiation promotes an m2 macrophage phenotype in tumor hypoxia. Front. Oncol. 2012;2:89. doi: 10.3389/fonc.2012.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Websits
- 201.A Study of IMC-CS4 in Subjects With Advanced Solid Tumors. www.clinicaltrials.gov/show/NCT01346358.
- 202.A Study of AMG 820 in Subjects With Advanced Solid Tumors. www.clinicaltrials.gov/show/NCT01444404.
- 203.Phase 1 Study of PLX7486 as Single Agent and With Gemcitabine Plus Nab-Paclitaxel in Patients With Advanced Solid Tumors. www.clinicaltrials.gov/show/NCT01804530.
- 204.Safety Study of PLX108-01 in Patients With Solid Tumors. www.clinicaltrials.gov/show/NCT01004861.
- 205.S0916, MLN1202 in Treating Patients With Bone Metastases. www.clinicaltrials.gov/show/NCT01015560.
- 206.FOLFIRINOX Plus PF-04136309 in Patients With Borderline Resectable and Locally Advanced Pancreatic Adenocarcinoma. www.clinicaltrials.gov/show/NCT01413022.
- 207.Feasibility of the Combination of Chemotherapy (Carbo/Caelyx or Carbo/ Doxorubicin) With Tocilizumab (mAb IL-6R) and Peg-Intron in Patients With Recurrent Ovarian Cancer (PITCH) www.clinicaltrials.gov/show/NCT01637532.
- 208.A Study of Trabectedin (YONDELIS) in Patients With Locally Advanced or Metastatic Liposarcoma or Leiomyosarcoma. www.clinicaltrials.gov/show/NCT01692678.
- 209.Study With Trabectedin in BRCA1 and BRCA2 Mutation Carrier and BRCAness Phenotype Ovarian Cancer (MITO15) www.clinicaltrials.gov/show/NCT01772979.
- 210.Combination Therapy of Gemcitabine and Trabectedin in L-sarcomas. www.clinicaltrials.gov/show/NCT01426633.
- 211.Combination Immunotherapy of GM.CD40L Vaccine With CCL21 in Lung Cancer. www.clinicaltrials.gov/show/NCT01433172.
- 212.Tremelimumab and CP-870,893 in Patients With Metastatic Melanoma. www.clinicaltrials.gov/show/NCT01103635.
- 213.A Phase I/Ib Study of AZD9150 (ISIS-STAT3Rx) in Patients With Advanced/Metastatic Hepatocellular Carcinoma. www.clinicaltrials.gov/show/NCT01839604.